
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution NMR and racemic crystallography provide insights into a novel structural class of cyclic plant peptides†
Colton D.
Payne
 a,
Grishma
Vadlamani
bcd,
Fatemeh
Hajiaghaalipour
a,
Taj
Muhammad
e,
Mark F.
Fisher
bcd,
Håkan S.
Andersson
ef,
Ulf
Göransson
e,
Richard J.
Clark
a,
Charles S.
Bond
c,
Joshua S.
Mylne
bcd and
K. Johan
Rosengren
*a
a,
Grishma
Vadlamani
bcd,
Fatemeh
Hajiaghaalipour
a,
Taj
Muhammad
e,
Mark F.
Fisher
bcd,
Håkan S.
Andersson
ef,
Ulf
Göransson
e,
Richard J.
Clark
a,
Charles S.
Bond
c,
Joshua S.
Mylne
bcd and
K. Johan
Rosengren
*a
aThe University of Queensland, School of Biomedical Sciences, Brisbane, QLD 4072, Australia. E-mail: j.rosengren@uq.edu.au
bCurtin University, Centre for Crop and Disease Management, School of Molecular and Life Sciences, Bentley, WA 6102, Australia
cThe University of Western Australia, School of Molecular Sciences, Crawley, WA 6009, Australia
dThe University of Western Australia, ARC Centre of Excellence in Plant Energy Biology, School of Molecular Sciences, Crawley, WA 6009, Australia
eUppsala University, Division of Pharmacognosy, Department of Pharmaceutical Biosciences, 75124 Uppsala, Sweden
fKarolinska Institute, Department of Medical Biochemistry and Biophysics, 17177 Stockholm, Sweden
First published on 13th September 2021
Abstract
Head-to-tail cyclic and disulfide-rich peptides are natural products with applications in drug design. Among these are the PawS-Derived Peptides (PDPs) produced in seeds of the daisy plant family. PDP-23 is a unique member of this class in that it is twice the typical size and adopts two β-hairpins separated by a hinge region. The β-hairpins, both stabilised by a single disulfide bond, fold together into a V-shaped tertiary structure creating a hydrophobic core. In water two PDP-23 molecules merge their hydrophobic cores to form a square prism quaternary structure. Here, we synthesised PDP-23 and its enantiomer comprising D-amino acids and achiral glycine, which allowed us to confirm these solution NMR structural data by racemic crystallography. Furthermore, we discovered the related PDP-24. NMR analysis showed that PDP-24 does not form a dimeric structure and it has poor water solubility, but in less polar solvents adopts near identical secondary and tertiary structure to PDP-23. The natural role of these peptides in plants remains enigmatic, as we did not observe any antimicrobial or insecticidal activity. However, the plasticity of these larger PDPs and their ability to change structure under different conditions make them appealing peptide drug scaffolds.
Introduction
Head-to-tail macrocyclic peptides have been discovered in all kingdoms of life.1–3 One family of macrocycles known as the PawS-Derived Peptides (PDPs) is produced within seeds from the Asteraceae family of daisies.4 The PDPs have evolved within genes encoding precursors to seed storage albumins. This was originally evidenced by the discovery of the sequence for the prototypic PDP, sunflower trypsin inhibitor-1 (SFTI-1), within a gene consequently named Preproalbumin with SFTI-1 (PawS1).5 PDPs are excised from the larger albumin precursor sequence and head-to-tail cyclised in a transpeptidation reaction during post-translational processing by asparaginyl endopeptidases.6Currently, a large number of unique peptide sequence inserts have been identified in daisy albumins via transcriptomics, while 23 have been described in more detail and annotated as members of the PDP family.4,7 Most PDPs are 14–17 residues long with a cyclic backbone, which results from the formation of a peptide bond between a C-terminal Asp and an N-terminal Gly residue. Some PDPs do have an Asn residue rather than an Asp residue at the C-terminal position, and are simply cleaved from the precursor yielding an acyclic hairpin peptide.8 Until recently, it was thought that all PDPs contained a single disulfide bond and were structurally limited to a small β-sheet structure and turns. However, a new member of the PDP family was discovered that contains 28 amino acids and two disulfide bonds in a I–II/III–IV ladder-like configuration.9 NMR spectroscopy showed that this peptide, PDP-23, adopts a unique tertiary structure wherein two loops, each containing a β-sheet bridged by a single disulfide bond, fold together into a V-shaped tertiary structure enclosing a hydrophobic core, more akin to the structures of larger proteins.9 Additionally, in water PDP-23 self-associates and forms a square prism-shaped symmetrical homodimer quaternary structure by increasing the distance between the loop regions and merging the hydrophobic cores of the two monomers.9 The PDP-23 symmetrical homodimer disassociates into two well-defined monomers in less polar environments like 20% acetonitrile or membrane-mimicking micelles. In each case the β-sheet structure is retained but the hinges allow opening and closing of the ‘V’.
SFTI-1 and other cyclic plant peptides have been explored as scaffolds for grafting of molecular functionalities due to their inherent stability.8,10 PDP-23 may be a superior scaffold to other members of the family due to its unique size, structure and ability to adopt different conformations under different conditions. These features could allow for more accessible regions to graft bioactive epitopes or conjugate small molecule payloads without sacrificing stability. An analogue of rhodamine, a family of fluorescent dyes known to be rapidly effluxed from cells by the drug efflux pump P-glycoprotein,11 was successfully attached to an analogue of PDP-23. The conjugate demonstrated effective cell uptake and inhibition of this pump, thereby restoring drug sensitivity in a cancer cell line resistant to chemotherapeutics due to overexpression of P-glycoprotein.9
In this study we employ racemic crystallisation to independently determine the structure of PDP-23 using X-ray crystallography. NMR spectroscopy is notoriously difficult for homomeric proteins given the inability to distinguish between inter- and intra-molecular distance restraints. However, the crystal structure confirms the unique dimeric assembly. Furthermore, we describe the identification, chemical synthesis and NMR structure of a second example of a PDP containing two disulfide bonds, PDP-24. PDP-24 also forms a tertiary V-shaped structure in less polar environments, but despite high sequence identity to PDP-23 does not form an ordered, symmetrical homodimer in water. We also investigate some potential native functions of PDP-23 and PDP-24.
Methods
Chemical synthesis
The peptides PDP-23, PDP-24 and the enantiomeric form of PDP-23, D-PDP-23, were assembled in full on 2-chlorotrityl chloride resin at a 0.25 mM scale by Fmoc-based solid phase peptide synthesis using a CS336X peptide synthesiser (CSBio, CA, USA). Resin was pre-swollen in dichloromethane (DCM) for 1 h, prior to the loading of the C-terminal residue. Loading was achieved by applying a solution of 1 M eq. of Fmoc-protected glycine and 4 M eq. of N,N′-diisopropylethylamine in minimal DCM to the resin. Standard deprotection using 2 × 5 min reactions of 20% v/v piperidine in dimethylformamide (DMF) was conducted between couplings of amino acids. Amino acid couplings were achieved by activating 4 M eq. of the Fmoc protected amino acid in a solution of DMF containing 4 M eq. of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate and 8 M eq. of DIPEA, this solution was then applied to the resin. These couplings were performed twice for all amino acids containing a branched β-carbon. All amino acids, including D variants, were purchased from Mimotopes (VIC, AUS). Regio-selective disulfide bond formation was employed to ensure the correct I–II/III–IV disulfide configuration, this was achieved by protecting the sidechains of Cys III & IV with acetamidomethyl protecting groups with Cys I & II having trityl protecting groups. Upon completion of synthesis PDP-23, -24 & D-PDP-23 were cleaved from resin using 10 × 3 min treatments of 2% trifluoroacetic acid (TFA) in DCM to maintain sidechain protection of amino acids. CH3CN was added and the solution was rotary evaporated to remove the DCM and TFA before lyophilisation. Cyclisation of the peptide backbone was conducted by dissolving peptide in DMF at a concentration of 10 mM before the addition of 1 M eq. of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, followed by the slow addition of 10 M eq. of DIPEA. The reaction was monitored using electrospray mass spectrometry (ESI-MS) over 2 h by dilution of a sample of the cyclization mixture in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 CH3CN/H2O followed by direct injection. Upon completion, phase separation using DCM was used to isolate the peptide from the DMF. The DCM phase containing the peptide was diluted with CH3CN before rotary evaporation to remove the DCM and subsequent lyophilisation. The remaining sidechain protecting groups were removed from the peptide by treatment with a solution of 96:2:1:1 TFA/triisopropylsilane/3,6-dioxa-1,8-octanedithiol/H2O for 2 h. The peptide was removed from the deprotection solution by precipitation with cold diethyl ether, followed by filtration and re-solvation in 50:50 CH3CN/H2O before lyophilisation.
:50 CH3CN/H2O followed by direct injection. Upon completion, phase separation using DCM was used to isolate the peptide from the DMF. The DCM phase containing the peptide was diluted with CH3CN before rotary evaporation to remove the DCM and subsequent lyophilisation. The remaining sidechain protecting groups were removed from the peptide by treatment with a solution of 96:2:1:1 TFA/triisopropylsilane/3,6-dioxa-1,8-octanedithiol/H2O for 2 h. The peptide was removed from the deprotection solution by precipitation with cold diethyl ether, followed by filtration and re-solvation in 50:50 CH3CN/H2O before lyophilisation.
Peptide purification and folding
Formation of the first disulfide bond was conducted by dissolving the peptide in ammonium bicarbonate (pH 8.3) at a concentration of 0.25 mg mL−1. To this 0.1 mL mg−1 of 2 mM 2,2′-dipyridyldisulfide in methanol was added and the solution left stirring for 24 h. Formation of the second disulfide bond was conducted by iodolysis. This was done by dissolving lyophilised, partially oxidised, peptide in a solution of 50:50 CH3CN/H2O at a concentration of 0.25 mg mL−1 before the slow addition of a solution of 0.1 M iodine in 50:50 CH3CN/H2O until the solution was coloured orange. This solution was stirred under nitrogen in the dark for 2 h, prior to quenching with ascorbic acid. Purification was conducted via reverse-phase high performance liquid chromatography on a Prominence (Shimadzu, Rydalmere, AUS) using a solution of 90% CH3CN and 0.05% TFA at a gradient of 1% min−1. A preparative C18 column (300 Å, 10 μm, 21.20 mm i.d. × 250 mm, Phenomenex) was used for purification before and after cyclisation as well as after each disulfide bond formation step. A semi-preparative C18 column (300 Å, 5 μm, 10 mm i.d. × 250 mm, Vydac) was used to achieve final purity greater than 95% for all peptides. Purity was assessed using a C18 analytical column (300 Å, 5 μm, 2.1 mm i.d. × 150 mm, Vydac). Final product identity was confirmed by ESI-MS.
PDP-23 crystallisation and X-ray crystal structure determination
Crystals of PDP-23 were grown at 20 °C using the hanging drop vapour-diffusion method by mixing equal volumes (1 μL each) of reservoir buffer (0.2 M trimethylamine N-oxide dihydrate, 0.1 M Tris pH 8.5, 20% PEG monomethyl ether 2000; Index HR2-144 # 62, Hampton Research) with 14 mg mL−1 of racemic peptide solution (50:50 PDP-23/D-PDP-23) dissolved in water. PDP-23 crystals were cryoprotected in reservoir solution supplemented with 25% glycerol and flash-cooled in liquid nitrogen. X-ray data were collected using beamline MX2 (ANSTO)12 at the Australian Synchrotron. The X-ray data were indexed using XDS,13 followed by space group search using Pointless14,15 (CCP4 package) with parameters extended to include centrosymmetric space groups. The data were scaled and averaged using Aimless14,15 (CCP4 package). The PDP-23 structure was determined by molecular replacement using Molrep15,16 (CCP4 package) using the NMR structure of the PDP-23 dimer (PDB ID: 7L51) as a search model. The resulting solution was refined using Refmac15,17 (CCP4 package) and Coot,18 and the final model and structure factors were deposited in the Protein Data Bank under the ID 7MMY. A composite omit map for L-PDP-23 was generated using PHENIX.19
NMR spectroscopy
The NMR sample of PDP-24 was prepared by dissolving 2 mg of the peptide in 500 μL of H2O/CD3CN (80:20) at pH ∼ 3.5. 1H 1D data as well as 1H–1H 2D Total Correlation Spectroscopy (TOCSY;20 mixing time of 80 ms), and Nuclear Overhauser Spectroscopy (NOESY;21 mixing time of 200 ms) were recorded at 298 K on a 900 MHz Bruker Avance III spectrometer equipped with a cryoprobe. TOCSY experiments were recorded with 8 scans and 512 increments and NOESY experiments were recorded with 40 scans and 512 increments, with a sweep width of 12 ppm. 1H–13C and 1H–15N Heteronuclear Single Quantum Coherence (HSQC) experiments were recorded at natural abundance. The 13C HSQC data were recorded with 128 scans and 256 increments with a sweep width of 10 ppm in the F2 dimension and 80 ppm in the F1 dimension in D2O/CD3CN (80:20) to minimise the overlap of residual water with the Hα–Cα resonances. The 15N experiments were recorded with 256 scans and 128 increments, with a sweep width of 10 ppm in the F2 dimension and 32 ppm in the F1 dimension. The data were processed using Topspin 4.0.4 (Bruker), with the CH3CN solvent signal at 2.031 ppm (298 K) used as reference. The data were assigned using sequential assignment strategies in the program CARA (Computer Assisted Resonance Assignment).22 Secondary structural features were determined by comparison of the secondary 1Hα shifts generated by PDP-24 to that of equivalent values generated in a random coil peptide.23 Additional TOCSY experiments were recorded at 288 K, 293 K, 298 K, 303 K and 308 K to monitor the temperature dependence of the amide protons.
Solution structure calculation of PDP-24
Interproton distance restraints for PDP-24 were generated from the cross-peak volumes in the NOESY spectra. TALOS-N was used to predict dihedral ϕ(C−1–N–Cα–C) and ψ(N–Cα–C–N+1) backbone angles.24 These TALOS-N dihedral angles as well as chemical shifts were used to predict the χ1(N–Cα–Cβ–SX) and χ2(Cα–Cβ–SX–SY) dihedral angles for the disulfide bonded Cys residues using the program DISH (di-sulfide and di-hedral prediction).25 Hydrogen bonds donors were identified from backbone amide temperature coefficients. The chemical shift of the backbone 1HN proton of each residue was plotted against temperature with values >−4.6 ppb K−1 for the coefficient of the linear relationship being taken as indicative of a hydrogen bond being donated by that particular 1HN.26 Hydrogen bond acceptors were determined by preliminary calculations using simulated annealing in the program CYANA 3.98.5.27 Distance restraints and starting coordinates generated by CYANA through automatic assignment, along with the dihedral angle restraints from TALOS-N and DISH, and the hydrogen bond restraints from temperature coefficients and preliminary calculations were all used as input for the program CNS 1.21.28 Final structures were generated by CNS via simulated annealing using torsion angle dynamics. Fifty final structures were then minimised in explicit water using Cartesian dynamics. MolProbity29 was used to analyse the stereochemical quality of the structures by comparison to published structures. MOLMOL30 was used to display and generate images of the secondary and tertiary structure of the ensemble of the best 20 structures, which contained no violations of distances or dihedral angles above 0.2 Å or 2°, and had low energy. The structures, NMR restraints, and chemical shift information have been submitted to the Protein Data Bank (PDB) and Biological Magnetic Resonance Bank (BMRB). The accession codes for PDP-24 are 7M3U and 30889 respectively.Assembly of Zinnia haageana transcriptome
Raw Illumina RNA-seq reads from seeds of Z. haageana were downloaded from the Sequence Read Archive (BioSample No. SAMN02933922)31 and a transcriptome assembled using CLC Genomics Workbench (version 21.0.3; QIAGEN Aarhus A/S). The reads were first trimmed using parameters Q = 30 and minimum length 50, then assembled with word size 64 and minimum contig length 200. Other parameters were left at their default values. A previously assembled and translated sequence for the transcript Zh_PawS1h, which encodes PDP-24,31 was used to locate a contig coding for PDP-24 in the new assembly by means of a search with tBLASTn.32 Raw reads were mapped to the contig with CLC Genomics Workbench, masked to the area coding for PDP-24.Insecticidal assay
Juvenile specimens of Blaptica dubia cockroaches (90) were acquired at Herper's Choise pet shop (Uppsala, Sweden), and toxicity was assessed following the strategy of Jacobsson et al.33 Single doses consisting of 10 μL PDP-23 solution (0.4–3897 μg kg−1) were injected between the 4th and 5th sternite, using a pointed GC Hamilton syringe (25 μL model nr. 702). The cockroaches were subdivided into groups of five individuals per concentration and housed in ventilated plastic containers containing dandelion leaves for 24 h post injection. Cockroaches that were able to flip over from an upside-down placement, 24 h post injection, were considered unaffected. Before the experiment all cockroaches were weighed, and average doses were calculated. A nemertide α-1 mutant,33 synthesised with two hydroxyprolines replaced by prolines (MW: 3277.77 kDa), was used as a positive control (α-1 NOHYP). All solutions were prepared in water, including the control.Antimicrobial assays
Two bacterial strains: Escherichia coli ATCC 25922, and Staphylococcus aureus ATCC 29213, as well as one fungal strain Candida albicans ATCC 90028 were obtained from the Department of Clinical Bacteriology, Lund University Hospital, Sweden. The antimicrobial activity of PDP-23 and PDP-24 was evaluated using a two-step microdilution assay,34 which is designed for testing anti-microbial peptides without activity-inhibiting components. Briefly, bacteria were grown to mid-logarithmic phase at 37 °C in 3% (w/v) tryptic soy broth (Merck KGaA, Darmstadt, Germany). The microbial culture was washed twice by centrifugation and re-suspended in fresh 10 mM Tris buffer (pH 7.4 at 37 °C, adjusted by HCl). The microbes were quantified at OD600, diluted and transferred to 96-well microtiter plates (U-shaped untreated polystyrene) prepared with peptides in 2-fold serial dilutions, ranging from 80 μM to 78 nM. The resulting wells had 50000 cfu suspended in 100 μL of Tris buffer. After a 5 h incubation with the peptides, 5 μL of 20% (w/v) tryptic soy broth was added to each well and the plates were re-incubated for an additional 6–12 h (depending on the growth rate of each organism). At least two replicates per experiment and three independent experiments (biological replicates) were performed.
Results and discussion
Racemic crystallography of PDP-23 confirms its unique quaternary structure
Small disulfide-rich peptides can be difficult to study by crystallography, but racemic crystallography in which a mixture of chemically synthesised peptides assembled with all L or all D amino acids improves crystallisation by allowing access to more favoured space groups.35,36 Thus, to independently determine the tertiary and quaternary structure of PDP-23 for comparison with the water-based solution NMR spectroscopy,9 we attempted racemic X-ray crystallography. Both PDP-23 and its mirror image, D-PDP-23, were synthesised as described previously using (Fmoc)-based solid phase peptide synthesis.9 After assembly on resin, sidechain protected peptide was liberated and cyclised in solution before sidechain deprotection. Disulfide bonds were regio-selectively formed in the I–II/III–IV configuration in two steps before extensive purification to >99% purity using RP-HPLC. The peptides were quantified using Direct Detect (Merck Millipore) to ensure accurate racemic mixing.A racemic mixture of PDP-23 and D-PDP-23 was subjected to crystallisation screening and diffraction quality crystals were obtained. Synchrotron radiation yielded crystal diffraction to a 1.46 Å resolution. The space group for PDP-23 racemic crystals was P21/c (indexed as P121/n1) with unit cell dimensions a, b, c = 27.53, 49.01, 29.40 Å and β = 92.28°. The data are 98.8% complete to 1.46 Å with the overall merging Rsym being 7.1%. The crystallographic statistics for the structure are shown in Table 1. The crystal structure of PDP-23 was determined by molecular replacement using the PDP-23 homodimer in water (PDB code: 7L51) as the search model. Molecular replacement using a monomeric structure was unsuccessful.
| Data collection | |
| Resolution (Å) | 49.01–1.46 |
| Space group | P121/n1 |
| Total reflections | 85984 (3128) |
| Unique reflections | 13346 (515) |
| Multiplicity | 6.4 (6.1) |
| Completeness (%) | 98.8 (80.8) |
| I/σI | 10.6 (2.5) |
| R merge | 0.071 (0.712) |
| CC1/2 | 0.998 (0.833) |
| Unit cell dimensions | |
| a, b, c (Å) | 27.53, 49.01, 29.40 |
| α, β, γ (°) | 90.00, 92.28, 90.00 |
| Refinement | |
| R work/Rfree | 0.188/0.276 |
| No. of atoms: protein | 476 |
| water | 45 |
| B-Factors (Å2): protein | 22.3 |
| water | 35.8 |
| Ramachandran plot (%) | |
| Favoured | 100 |
| Allowed | 0 |
| Outliers | 0 |
| RMSD | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.82 |
The asymmetric unit of racemic PDP-23 crystals contains a single non-crystallographic dimer of L-/L- (or D-/D-) PDP-23 monomers. Crystal symmetry in the centrosymmetric space group P121/n1 results in a unit cell containing two L-dimers and two D-dimers, such that crystal contacts exist between L- and D-dimers (Fig. 1A). The structure confirms both the V-shaped tertiary structure and the dimeric quaternary structure originally determined using solution NMR spectroscopy. The structural similarity is high (RMSD of 0.64 Å for 53 of 56 Cα atoms) which confirms the NMR model was a suitable molecular replacement search model. The observed electron density is of high quality, as expected for a structure at <1.5 Å resolution (Fig. 1B), with all amino acids well resolved except for His5 and His6 in each subunit which have disordered sidechains. For a high-resolution structure, our reported Rwork/Rfree values (19%/28%) appear high. We note however that higher R-values are observed in centrosymmetric diffraction data than in non-centrosymmetric data for equivalent degrees of coordinate error, for statistical reasons.37 To validate the quality of our data, we generated a composite omit map and found that it is clearly resolved around individual residues in the L-PDP-23 dimer, except His5 and His6 as noted above (Fig. S1, ESI†).
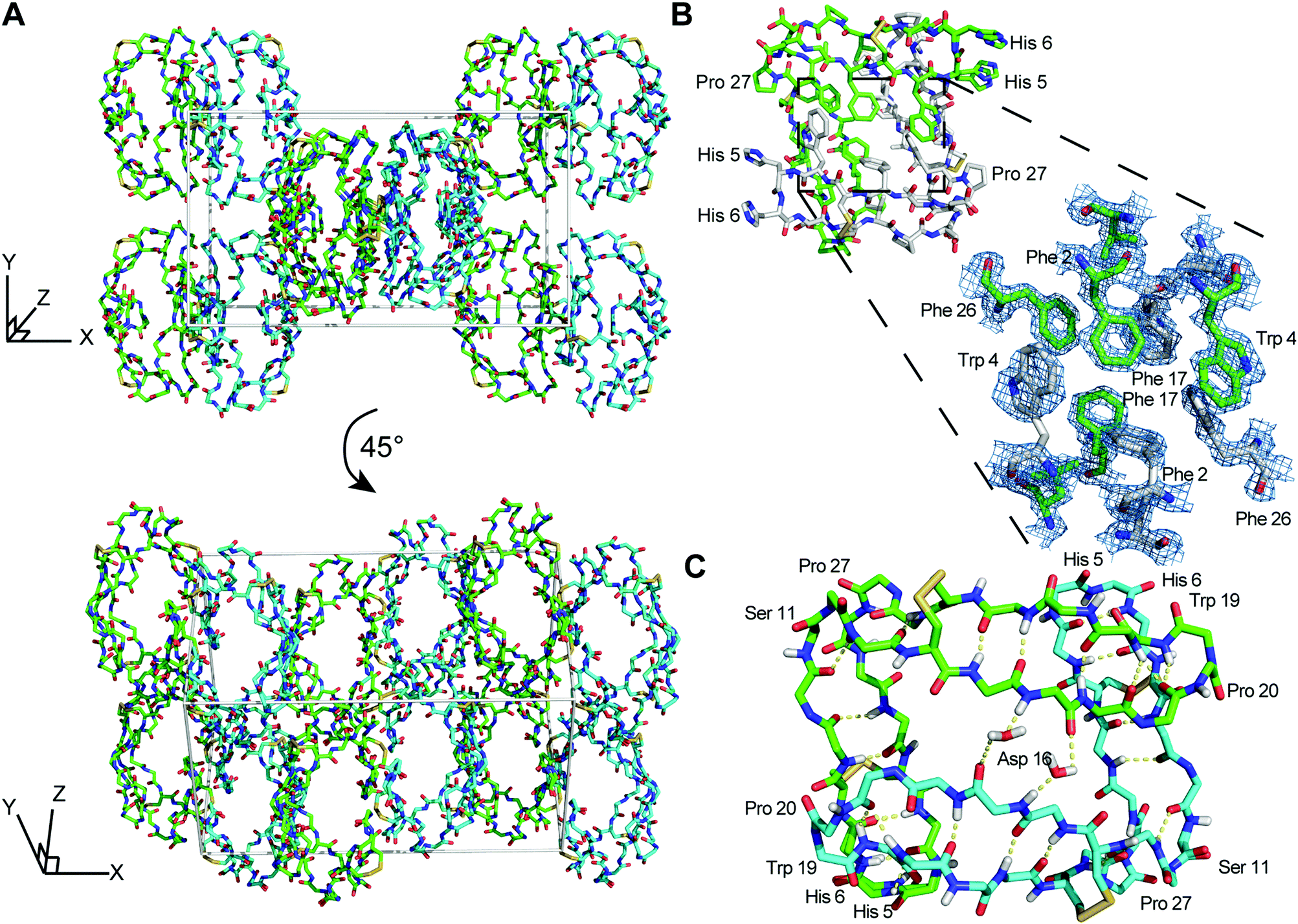 | ||
| Fig. 1 X-ray structure and racemic crystal packing of PDP-23. (A) Crystal packing for the racemic PDP-23 crystal structure with L-dimers (cyan) and D-dimers (green). (B) Stick diagram of an L-PDP-23 homodimer, with the individual monomers shown with green and white carbon atoms, respectively. The zoomed region depicts a maximum-likelihood weighted 2Fo − Fc electron density map (blue mesh) contoured at 1.7σ for the hydrophobic core formed by inter-facing monomers. (C) The L-enantiomer of PDP-23 shown in stick format, the backbone is coloured by atom with carbons in green or cyan to distinguish monomers. H-bonds are shown with yellow dashed lines. Two water molecules create bridged hydrogen bonds between the monomers. The dense hydrogen bond network proposed for PDP-23 by solution NMR9 is fully supported by the crystal data. | ||
Comparison of the crystal dimer and the solution NMR structure revealed noteworthy features and differences: the crystallographic dimer displayed asymmetry in the packing of its hydrophobic core. While the backbones of both chains were highly similar (RMSD 0.32 Å for 24 pairs of Cα atoms), Phe2, Trp4, Leu21 and Phe26 differed in conformation between subunits (Fig. 2A). This was in contrast to the solution NMR data and structure, which was fully symmetric, and these sidechains appeared in conformations intermediate between crystal structure extremes (Fig. 2A). In particular, the χ1 angles of both Phe2 and Leu21 differed between monomers within the crystal dimer (Fig. 2A). The sidechains of these two residues form critical contacts and establish the NOE network of the solution NMR structure of PDP-23. Leu21 has, because of its network of unambiguous intermolecular NOEs, been used as a signature to determine whether PDP-23 is in a dimeric or monomeric state when in solution. The methyl groups of the Leu21 generate NOEs to the sidechains of Trp4 and Val9, which cannot be explained by a monomeric structure. The absence of duplicate resonances in the homonuclear and heteronuclear NMR data is consistent with a fully symmetrical structure, however, structural rearrangements on a fast time scale could mean the NMR data are averaged over local fluctuations rather than reflecting a singular conformation. In the NMR structural ensemble Leu21 did adopt multiple different χ1 angles, consistent with this notion. Phe2 however only appears in a single conformation in the NMR structure, nonetheless there may be breathing motions in the homodimeric core in solution that is reduced in the crystalised form explaining the asymmetry in the X-ray structure. As we have previously reported that the homodimeric structure of PDP-23 could be separated by temperature or by the addition of a small amount of aprotic polar solvent, it is possible the homodimeric complex is somewhat dynamic. Nevertheless, the dimer interfaces of crystal and NMR structures appear similarly favourable, with buried areas of 779 Å2 and 740 Å2, respectively.38
 | ||
| Fig. 2 Comparison of the X-ray and NMR structures of PDP-23. (A) Overlap of the backbone and hydrophobic core of the PDP-23 homodimer in solution (magenta), the crystal structure (blue) and a rotated crystal structure (green) in line format; this overlap highlights the consistency of the backbone structure and highlights the minute difference in sidechain projection for the hydrophobic core constituents. (B) Overlap of the backbone of the solution structure of PDP-23 with a member in the crystal lattice using the same colouring scheme as (A), plus neighbouring members of the PDP-23 crystal lattice (grey, partial structures). This overlap highlights the difference in sidechain projection of Trp19 between the crystal and solution NMR structures. In the PDP-23 crystal lattice Trp19 forms an interaction between two unique L- and two unique D-PDP-23 homodimers, whereas in the solution NMR structure Trp19 folds on top of Pro20 to form a cis-Pro turn. | ||
It is possible that lattice contacts influence the conformations observed in crystal structures. A conspicuous interaction between dimers in the lattice involves Trp19. Each individual L- and D-homodimer extends the Trp19 sidechain from both its monomers. The Trp19 residue of one form of homodimer, L- or D-, then associates with a Trp19 residue from the adjacent homodimer of the same form in the lattice (Fig. 2B). These two Trp sidechains are sandwiched between two Cys14–Cys25 disulfide bonds from separate homodimers of the opposing form; L-Trp19 residues between D-Cys14–Cys25 disulfide bonds and vice versa.
The Trp19 interactions are seemingly critical to the arrangement of the crystal lattice, and also highlight a key difference between the structures determined by crystallography and solution NMR spectroscopy. In solution, instead of being extended outwards away from the homodimer, the Trp19 sidechain is packed against the adjacent Pro20 sidechain creating a cis-Pro turn. This cis-Pro conformation of the turn is clearly evident from electron density, chemical shifts and NOE patterns, and the packing of the aromatic ring against the Pro is evident from the ring current effect creating a large upfield shift of one of the 1Hβ protons of the Pro20 sidechain, a hallmark classically used to evidence this type of turn in the cyclotides.39
Overall, the consistency between the solution NMR structure and the crystal form is remarkable, given the complexity of resolving an NMR structure of a homodimer where every NOE contact can reflect either an intra or intermolecular interaction.
Transcriptomic discovery, chemical synthesis and NMR studies of PDP-24
The interesting structural features and potential applications of PDP-239 provoked a desire to find more examples of this unique PDP among daisies, and to determine how it might have evolved. To look for closely related peptides we inspected previously assembled and translated PawS transcripts for other members of the daisy family.31 Looking for the hallmark features of a PawS gene containing not two but four cysteine residues in the inserted domain N-terminal to the albumin small subunit, we identified a 27 amino acid sequence sharing 81% sequence identity with PDP-23 in another Zinnia species, Z. haageana.To confirm this sequence, we reassembled the raw reads produced by Jayasena et al.31 and identified a 744 nucleotide contig coding for PDP-24 and its adjacent seed storage albumin. We mapped the raw RNA-seq reads to this contig, concentrating on the 81-nucleotide sequence encoding PDP-24, and found coverage of 216 reads to the peptide-encoding sequence, including about 30 reads with single nucleotide mismatches at random positions, likely caused by sequencing errors (Fig. 3).
 | ||
| Fig. 3 (A) Nucleotide and amino acid sequences of the incomplete PawS1h transcript with the PDP-24 peptide sequence highlighted in aqua. (B) Mapping of raw RNA-seq reads to the PDP-24 transcript in Zinnia haageana. The deeper colours represent the part of the transcript coding for PDP-24. Reads are shown in green (forward) and red (reverse). | ||
Given the intriguing features of PDP-23, we sought to determine whether the symmetrical homodimer quaternary structure is a common fold for bi-disulfide bonded PDPs. PDP-24 was thus synthesised and purified using the same protocol as PDP-23. Solution NMR spectroscopy was used to study PDP-24 and determine if it adopts the aforementioned features present in PDP-23. In contrast to PDP-23, PDP-24 dissolved poorly in water with signs of non-specific aggregation with an opaque appearance of the sample, and the 1D 1H NMR spectrum showed broad lines. However, similar to PDP-23, PDP-24 dissolved readily in 80:20 H2O/CD3CN and 1D 1H NMR gave a spectrum with excellent dispersion and sharp lines indicating a well-folded, structured peptide. Because of this, extensive NMR data were recorded for PDP-24 under these conditions, and assigned manually using sequential assignment strategies.40 The quality of the datasets allowed complete assignment of the backbone and sidechain resonances using TOCSY and NOESY spectra. HSQC data were recorded at natural abundance and the 13C and 15N backbone and sidechain resonances were assigned with assistance from the 1H assignments. PDP-24 contains four proline residues with Pro19 adopting a cis conformation and Pro10, Pro17, and Pro26 all adopting trans conformations based on NOE patterns and 13C chemical shifts. These conformational states are identical to what is observed in PDP-23.9 Additionally, the secondary Hα shifts, which are sensitive indicators of secondary structure, of PDP-24 are remarkably similar to those observed in PDP-23 when in either H2O/D2O or H2O/CD3CN,9 indicating PDP-24 adopts the same secondary structural features as PDP-23. In water PDP-23 contains key NOEs that indicate a symmetrical homodimer structure. These NOEs disappear in less polar solutions, or when PDP-23 is in the presence of micelles, indicating dissociation of the symmetrical homodimer.9 Notably, PDP-24 contains no NOEs suggestive of a symmetrical homodimer when in 80:20 H2O/CD3CN, consistent with what was observed with PDP-23. No additional spin systems suggesting conformational inhomogeneity were identified in the data, indicating the fold adopted is preferred in this environment.
To determine the 3D structure of PDP-24, structural restraints were derived from the NMR data. These included inter-proton distances based on NOE volumes, dihedral angles derived from chemical shifts using TALOS-N, as well as hydrogen bonds based on temperature coefficients and preliminary structure calculations. Initial structures were calculated using automated NOE assignment with CYANA and final structures were calculated and refined in explicit water using CNS. All structures were analysed using MolProbity to determine the quality of the structural geometry and atom packing. The best 20 structures from the 50 calculated based on MolProbity scores, low energy as well as containing no significant violations were chosen to represent the solution structures of PDP-24. The structures calculated for PDP-24 are of good stereochemical quality, with minimal clashes >0.4 Å and all residues being in favoured Ramachandran regions. The data also highlights the well-defined backbone of PDP-24, which has an RMSD of 0.85 Å. The structures generated for PDP-24 are above the 92nd percentile of all structures according to MolProbity (Table 2).
| Energies (kcal mol−1) | |
| Overall | −813.06 ± 13.18 |
| Bonds | 13.05 ± 1.48 |
| Angles | 29.03 ± 2.11 |
| Improper | 10.76 ± 1.45 |
| Dihedral | 114.00 ± 0.89 |
| van der Waals | −93.43 ± 3.43 |
| Electrostatic | −887.04 ± 10.61 |
| NOE | 0.065 ± 0.007 |
| cDih | 0.508 ± 0.206 |
| MolProbity statistics | |
| Clashes (>0.4 Å/1000 atoms) | 12.66 ± 1.65 |
| Poor rotamers | 0 |
| Ramachandran outliers (%) | 0 |
| Ramachandran favoured (%) | 100 |
| MolProbity score | 1.61 ± 0.05 |
| MolProbity score percentile | 92.05 ± 1.57 |
| Atomic RMSD (Å) | |
| Mean global backbone | 0.85 ± 0.33 |
| Mean global heavy | 1.51 ± 0.38 |
| Experimental restraints | |
| Distance restraints | |
| Short range (i–j < 2) | 282 |
| Medium range (i–j < 5) | 75 |
| Long range (i–j ≥ 5) | 130 |
| Hydrogen bond restraints | 24 (12 bonds) |
| Total | 511 |
| Dihedral angle restraints | |
| ϕ | 15 |
| ψ | 17 |
| χ1 | 5 |
| χ2 | 8 |
| Total | 45 |
| Violations from experimental restraints | |
| NOE violations exceeding 0.2 Å | 0 |
| Dihedral violations exceeding 2.0° | 0 |
PDP-24 adopts identical secondary structure to PDP-23 (Fig. 4), with an almost identical hydrogen bond network, evident from amide temperature coefficients. However, there is a key difference, this being the absence of the hydrogen bond between residues 13–10, which stabilises a type II β-turn in PDP-23. Residue 13 in PDP-23, a Thr, is not present in PDP-24, making the hinge between its two β-sheets shorter and preventing formation of a well-defined turn. The change in the hinge region appears to have a flow-on effect to the hydrophobic core, altering its composition and packing of sidechains. The ‘V’ shape is more open overall in PDP-24 and the hydrophobic core is packed to give a more twisted interface between the two β-hairpins compared to the more planar interaction in PDP-23 (Fig. 4). In PDP-23 the Val and Leu residues at positions 9 and 21, respectively, are a key part of the hydrophobic core. In PDP-24 these two residues are switched with a Leu at position 9 and Val at 20 (the equivalent positions in PDP-24); neither residue interacts strongly with the hydrophobic core in contrast to what is observed in PDP-23. This is evidenced by an absence of NOEs generated by the Leu and Val residues to hydrophobic core residues on the opposing β-hairpin. Conversely, in PDP-23 a multitude of NOEs were observed from these residues to almost all members of the hydrophobic core, both locally and across to the opposing β-hairpin. Overall the sequence changes present in PDP-24 cause the peptide to adopt a tertiary structure more akin to what is observed in the dimeric form of PDP-23 rather than the monomeric form (Fig. S2, ESI†). Based on the high sequence identity and structural similarity to the dimeric form of PDP-23 it is perhaps surprising that PDP-24 is unable to form the same quaternary square prism homodimeric structure of PDP-23. Despite the clear similarities, there are also obvious differences, particularly in the hairpin containing the substitutions H5Q, S7T and V9L. While His5 is surface exposed, both the S7T and V9L changes would cause clashes that would require structural rearrangement relative to the PDP-23 dimer. Alternatively, the shorter turn resulting from the deletion of T13 is forcing a different alignment of the hairpins, incompatible with a dimer. While the experimental data clearly shows that PDP-24 does not adopt a homodimeric structure, it is difficult to pinpoint which sequence change is primarily preventing the formation of a dimer. Whether the ability to form a dimer is functionally significant is not known. Nonetheless, identification of another scaffold adopting a tertiary V-shaped structure may be useful for protein engineering. Hydrophobicity and solvation are easily overcome by simple modifications,41–43 allowing this particular PDP to remain available for drug conjugation.
 | ||
| Fig. 4 Comparison of PDP-23 and PDP-24. (A) 3D ensembles of PDP-24 (above) and PDP-23 (below), PDP-24 is displayed with a magenta backbone and PDP-23 with a cyan backbone, sidechains are coloured based on properties: disulfide bonds are in orange, hydrophobic residues are in green, basic residues in blue, acidic residues in red, and polar residues in grey. (B) Singular structures of PDP-24 (magenta) and PDP-23 (cyan) in ribbon form. (C) Schematic of the network of hydrogen bonds (dashed lines) and disulfide bonds (solid lines) with antiparallel β-sheets as arrows. | ||
Are PDP-23 and PDP-24 involved in plant defence?
As PDP-23 is a naturally occurring peptide from plant seeds, it is potentially involved in host defense. To test PDP-23 for insecticidal bioactivity we used cockroaches as a model with single injections of PDP-23 administered between the 4th and 5th sternite. No effect was observable following injection of up to 3.9 mg kg−1 (1.25 μmol kg−1) doses and insects remained unaffected 24 h later (Table S1, ESI†). The positive control, α-1 NOHYP, killed 80% of cockroaches at 23.1 μg kg−1 (7.04 nmol kg−1), and all insects at higher concentrations. The EC50 of the α-1 NOHYP was estimated as 21 nmol kg−1, a result equivalent to that reported for nemertide α-1.33As another potential function in defense may be antimicrobial activity, PDP-23 and PDP-24 were tested against two bacterial species and one fungus in a two-step microdilution assay protocol. PDP-23 and PDP-24 showed no inhibition of growth at all tested concentrations up to 80 μM (Table S2, ESI†). The human cathelicidin LL-3734 was used as a positive control and inhibited growth at sub-micro molar concentrations.
Conclusion
In conclusion, we have confirmed the homodimeric structure of PDP-23 in water by racemic X-ray crystallography, reported the synthesis and structural characterization of a second bi-disulfide member of the PDP family, and probed potential bioactivities of PDP-23 and PDP-24. X-ray crystallography revealed that the PDP-23 hydrophobic core is not symmetrical between monomers in the crystal form, which might indicate structural plasticity. As predicted from sequence homology, PDP-24 folds into the same secondary and tertiary structure observed in PDP-23 with an almost identical hydrogen bond network. Interestingly, PDP-24 is unable to adopt the same symmetrical homodimer quaternary structure adopted by PDP-23 in water, instead dissolving poorly and aggregating. PDP-23 was tested for potential insecticidal activity, but demonstrated none. Both PDP-23 and PDP-24 were tested for antimicrobial activity but again were not found to be active. These two peptides highlight the unique tertiary fold available to bi-disulfide bonded members of the PDP family and present as excellent candidates for drug scaffolding.Author contributions
C. D. P.: peptide chemistry, NMR data analysis, solution NMR structure calculation. G. V. and C. S. B.: X-ray crystallography. F. H. and R. J. C.: peptide chemistry. T. M., H. S. A. and U. G.: biological assays. M. F. F. transcriptomics data analysis. J. S. M. and K. J. R.: project design and funding. C. D. P. wrote the paper together with J. S. M., C. S. B. and K. J. R. with input from all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work, and G. V. and M. F. F. were supported by Australian Research Council (ARC) grants DP120103369 and DP190102058 to J.S.M. and K.J.R. K. J. R. and J.S.M were supported in part by ARC Future Fellowships FT130100890 and FT120100013 respectively. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.Notes and references
- U. Göransson, R. Burman, S. Gunasekera, A. A. Strömstedt and K. J. Rosengren, J. Biol. Chem., 2012, 287, 27001–27006 CrossRef PubMed.
- R. I. Lehrer, A. M. Cole and M. E. Selsted, J. Biol. Chem., 2012, 287, 27014–27019 CrossRef CAS.
- M. Montalbán-López, M. Sánchez-Hidalgo, R. Cebrián and M. Maqueda, J. Biol. Chem., 2012, 287, 27007–27013 CrossRef.
- A. G. Elliott, C. Delay, H. Liu, Z. Phua, K. J. Rosengren, A. H. Benfield, J. L. Panero, M. L. Colgrave, A. S. Jayasena, K. M. Dunse, M. A. Anderson, E. E. Schilling, D. Ortiz-Barrientos, D. J. Craik and J. S. Mylne, Plant Cell, 2014, 26, 981–995 CrossRef CAS PubMed.
- J. S. Mylne, M. L. Colgrave, N. L. Daly, A. H. Chanson, A. G. Elliott, E. J. McCallum, A. Jones and D. J. Craik, Nat. Chem. Biol., 2011, 7, 257–259 CrossRef CAS PubMed.
- J. S. Mylne, L. Y. Chan, A. H. Chanson, N. L. Daly, H. Schaefer, T. L. Bailey, P. Nguyencong, L. Cascales and D. J. Craik, Plant Cell, 2012, 24, 2765 CrossRef CAS.
- A. S. Jayasena, D. Secco, K. Bernath-Levin, O. Berkowitz, J. Whelan and J. S. Mylne, Plant Methods, 2014, 10, 34 CrossRef PubMed.
- B. Franke, J. S. Mylne and K. J. Rosengren, Nat. Prod. Rep., 2018, 35, 137–146 RSC.
- C. D. Payne, B. Franke, M. F. Fisher, F. Hajiaghaalipour, C. E. McAleese, A. Song, C. Eliasson, J. Zhang, A. S. Jayasena, G. Vadlamani, R. J. Clark, R. F. Minchin, J. S. Mylne and K. J. Rosengren, Chem. Sci., 2021, 12, 6670–6683 RSC.
- C. K. Wang and D. J. Craik, Nat. Chem. Biol., 2018, 14, 417–427 CrossRef CAS.
- C. Loetchutinat, C. Saengkhae, C. Marbeuf-Gueye and A. Garnier-Suillerot, Eur. J. Biochem., 2003, 270, 476–485 CrossRef CAS PubMed.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS.
- P. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 282–292 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. Vagin and A. Teplyakov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 22–25 CrossRef CAS.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkóczi, V. B. Chen, T. I. Croll, B. Hintze, L. W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2019, 75, 861–877 CrossRef CAS PubMed.
- L. Braunschweiler and R. R. Ernst, J. Magn. Reson., 1983, 53, 521–528 CAS.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS.
- R. L. J. Keller, The computer aided resonance assignment tutorial, CANTINA Verlag, 1st edn, 2004 Search PubMed.
- D. Wishart, C. Bigam, A. Holm, R. Hodges and B. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS.
- Y. Shen and A. Bax, J. Biomol. NMR, 2013, 56, 227–241 CrossRef CAS PubMed.
- D. A. Armstrong, Q. Kaas and K. J. Rosengren, Chem. Sci., 2018, 9, 6548–6556 RSC.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249–261 CrossRef CAS PubMed.
- P. Güntert, Methods Mol. Biol., 2004, 278, 353–378 Search PubMed.
- A. T. Brünger, Nat. Protoc., 2007, 2, 2728–2733 CrossRef PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS.
- R. Koradi, M. Billeter and K. Wüthrich, J. Mol. Graphics, 1996, 14, 51–55 CrossRef CAS.
- A. S. Jayasena, M. F. Fisher, J. L. Panero, D. Secco, K. Bernath-Levin, O. Berkowitz, N. L. Taylor, E. E. Schilling, J. Whelan and J. S. Mylne, Mol. Biol. Evol., 2017, 34, 1505–1516 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS.
- E. Jacobsson, H. S. Andersson, M. Strand, S. Peigneur, C. Eriksson, H. Lodén, M. Shariatgorji, P. E. Andrén, E. K. M. Lebbe, K. J. Rosengren, J. Tytgat and U. Göransson, Sci. Rep., 2018, 8, 4596 CrossRef.
- A. A. Strömstedt, S. Park, R. Burman and U. Göransson, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1986–2000 CrossRef PubMed.
- T. O. Yeates and S. B. Kent, Annu. Rev. Biophys., 2012, 41, 41–61 CrossRef CAS PubMed.
- S. W. Wukovitz and T. O. Yeates, Nat. Struct. Biol., 1995, 2, 1062–1067 CrossRef CAS PubMed.
- B. W. Matthews, Protein Sci., 2009, 18, 1135–1138 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774–797 CrossRef CAS.
- K. J. Rosengren, N. L. Daly, M. R. Plan, C. Waine and D. J. Craik, J. Biol. Chem., 2003, 278, 8606–8616 CrossRef CAS.
- C. I. Schroeder and K. J. Rosengren, in Snake and Spider Toxins: Methods and Protocols, ed. A. Priel, Springer US, New York, NY, 2020, pp. 129–162 Search PubMed.
- X. Han, W. Ning, X. Ma, X. Wang and K. Zhou, Metab. Eng. Commun., 2020, 11, e00138 CrossRef PubMed.
- J. Xiao, A. Burn and T. J. Tolbert, Bioconjugate Chem., 2008, 19, 1113–1118 CrossRef CAS PubMed.
- C. T. Mant, J. M. Kovacs, H.-M. Kim, D. D. Pollock and R. S. Hodges, Pept. Sci., 2009, 92, 573–595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00155h |
| This journal is © The Royal Society of Chemistry 2021 |