
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of a G protein-coupled receptor through indirect antibody-mediated tethering of ligands†
Ross W.
Cheloha‡
 *a,
Fabian A.
Fischer§
a,
Thomas J.
Gardella
b and
Hidde L.
Ploegh
*a
*a,
Fabian A.
Fischer§
a,
Thomas J.
Gardella
b and
Hidde L.
Ploegh
*a
aProgram in Cellular and Molecular Medicine, Boston Children's Hospital, 1 Blackfan Circle, Boston, MA 02115, USA. E-mail: ross.cheloha@nih.gov; hidde.ploegh@childrens.harvard.edu
bMassachusetts General Hospital and Harvard Medical School, 50 Blossom Street, Boston, MA 02114, USA
First published on 11th October 2021
Abstract
Antibodies raised against many cell surface proteins, including G protein-coupled receptors, remain important tools for their functional characterization. By linking antibodies to ligands for cell surface proteins, such adducts can be targeted to the surface of a cell type of choice. Site-specific functionalization of full-size antibodies with synthetic moieties remains challenging. Here we present new approaches in which single domain antibodies (known as VHHs or nanobodies) that target either cell surface proteins or conventional antibodies are used to indirectly deliver ligands for GPCRs to their sites of action. The combination of high yield production of nanobodies, facile site-specific functionalization, and compatibility with commercially available mouse and rabbit antibodies should enable wide application of this approach.
Introduction
Antibodies remain the gold standard for the selective detection and targeting of proteins in biological applications. Many proteins can be detected with commercially available antibodies, which have become essential tools in biology. Antibodies are often used to block the action of ligands on cell surface proteins. Antibodies can also deliver bioactive compounds to cells that express the relevant antigen. Antibody–drug conjugates (ADCs) are typically composed of an antibody functionalized with cytotoxic compounds. Upon internalization into endosomal compartments and proteolysis, ADCs release cytotoxic payloads that act inside the cell.1 Ligands for cell surface receptors can also be linked to antibodies; however, such efforts often suffer from a lack of specificity or difficulties in producing active, stable conjugates in sufficient yield.2–6The use of single domain antibodies (VHHs or nanobodies) can overcome some of these complications.7 Nanobodies are the recombinantly expressed variable regions of camelid heavy chain only antibodies. They are the smallest antibody fragments that retain the capacity to bind antigen. Because nanobodies do not require pairing with the light chain through disulfide linkage, they can be expressed recombinantly in bacteria in high yield. Sortase-mediated labeling offers a straightforward method for site-specifically labeling recombinant proteins, including nanobodies.8 Nanobodies have been applied in a variety of settings, including as imaging agents in immuno-positron emission tomography,9 as crystallization chaperones,10 and as inhibitors or agonists of cell surface receptor signaling.11–14 The first nanobody-based therapeutic has now been approved for clinical application.15
Nanobody conjugates have also been used to deliver bioactive compounds to specific cell types. A nanobody was applied as a conventional ADC to deliver a cytotoxic compound to kill murine lymphoma cells in vitro and in mice.16 In an example of targeting cell surface proteins,17 a conjugate between a green fluorescent protein (GFP)-specific nanobody and a photo-switchable agonist for the metabolic glutamate receptor enabled light-mediated activation of receptor signaling, but required the use of an engineered receptor-GFP fusion.14 A variant of glucagon-like peptide-1 (GLP1) was fused to a GLP1-receptor-specific nanobody to provide a conjugate with activity in mice.18 We showed that the conjugation of weakly active fragments of parathyroid hormone (PTH) to nanobodies that bound the receptor or that recognized epitope tags incorporated into type-1 PTH-receptor (PTHR1) augmented the biological activity and receptor specificity of these otherwise suboptimal fragments.2 This approach was dubbed “conjugation of ligands and antibody for membrane protein”, or CLAMP. One of these CLAMPs induced a physiological response in mice, whereas the corresponding free peptide did not.2
These applications relied in large part on the use of target-specific nanobodies. While the pool of nanobodies continues to expand,19 there are many targets for which appropriately specific nanobodies are not available. Here we describe new approaches that rely on nanobodies for delivery of ligands for cell surface receptors independent of the availability of target-specific nanobodies.
Experimental
General
HEK293 cell lines were cultured in DMEM medium containing 10% (v/v) fetal bovine serum and penicillin/streptomycin. Cell lines were routinely tested for mycoplasma infection. LC/MS was performed in the positive ionization mode. Masses for proteins and conjugates were calculated by analysis of multiply charged ions using the MaxEnt feature on MassLynx software. Protein and conjugate concentrations were calculated using absorbance at 280 nm for VHHs. Peptide concentrations were determined gravimetrically, assuming that the weighed mass consisted of ∼50% peptide (w/w). Transfections of HEK293 were performed using Lipofectamine2000 (Invitrogen, 11668019) using manufacturer's instructions. Commercial antibodies used in these studies are listed in Table S1 (ESI†).Plasmids and DNA
HEK293-derived cell lines stably expressing human PTHR1 (GP2.3) and PTHR16E (G6E19) along with a cAMP-responsive luciferase variant have been previously reported.2 HEK293 cells expressing only the cAMP-responsive luciferase (GS22a) were transiently transfected with a plasmid encoding PTHR1 lacking its extracellular domain with or without an N-terminal HA-tag extension. Annotated sequence data for all PTHR1 constructs are found in Fig. S1 (ESI†). Sequences for nanobodies targeting human β2-microglobulin (MHC-I), MHC-II, UBC6E (6E tag), α5β1, mouse Igκ, and rabbit IgG have been published previously.20–23Peptide synthesis
Peptides were prepared using conventional solid-phase synthesis methods with Fmoc-protection of backbone amines as described previously.2 Synthesis was performed on Rink-amide linker resin to yield C-terminal amides. Identity and purity of peptides were confirmed by LC/MS. All peptides were of a purity of >85%. Purified products were dissolved in water (10 mM stock concentration) and stored at −20 °C. Purified peptides with C-terminal cysteines were functionalized with DBCO-maleimide (Click Chemistry Tools) as described previously.2Protein expression, purification, and functionalization
The production and purification of VHHs and sortase A pentamutant was performed as described previously.24 VHHs were labeled using sortase A pentamutant as described previously.25Flow cytometry
Adherent cells were dissociated from the substrate with trypsin, washed and resuspended in PBS, followed by staining for 1 h on ice in the presence of the indicated concentrations of VHH probes functionalized with biotin. Cells were pelleted by centrifugation and washed with PBS prior to staining with streptavidin–PE (BioLegend, 405204). Cells were again pelleted by centrifugation, washed, and analyzed by flow cytometry (BD Accuri C6).Conventional antibodies were also used for staining cells for flow cytometry as described above, with the following exceptions. In one staining method, primary antibodies (IgGs) were incubated with cells at the indicated concentrations in the presence of a 3 fold molar excess of secondary (anti-IgG) nanobodies site-specifically conjugated with biotin. Biotin was detected with streptavidin–PE as described above. In an alternative method, cells were stained with unlabeled primary antibodies and detected following secondary staining, using commercial secondary anti-IgG antibodies conjugated with Alexafluor647 (ThermoFisher, A-21235). Data were analyzed using FlowJo version 7.6. The median fluorescence intensity (MFI) of stained cells was used to generate VHH binding dose response curves.
VHH–peptide conjugation reactions
VHH–biotin–azide conjugates were mixed with a three-fold molar excess of PTH-DBCO in TBS with 10% (v/v) glycerol. The reaction mixture was shaken at 22 °C until any unreacted VHH–biotin–azide had been completely consumed. The desired conjugate was purified from free PTH-DBCO using a PD-10 size exclusion column (Cytiva, 17085101). A summary of the characterization of these conjugates by LC/MS is shown in Table S2 (ESI†).Measurement of cAMP response
Assays were performed using a luminescence-based readout with cells expressing cAMP Glosensor as previously described26 with exceptions. A modified protocol was followed for assays that include commercial IgGs. IgG concentrations were calculated based on protein concentrations listed on manufacturer data sheets and assuming an average IgG molecular weight of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. VHH–PTH1–11 conjugates were premixed with indicated IgGs at a 3:1 molar ratio and incubated for at least 10 minutes at room temperature prior to addition to cells expressing PTHR1 or variants.
000 g mol−1. VHH–PTH1–11 conjugates were premixed with indicated IgGs at a 3:1 molar ratio and incubated for at least 10 minutes at room temperature prior to addition to cells expressing PTHR1 or variants.
Intercellular receptor activation assays
Murine B cell lymphoma A20 cells (ATCC TIB-208) were incubated with PTH1–34 or VHH–PTH1–34 conjugates in PBS containing 1% (w/v) bovine serum albumin (BSA) at the indicated concentrations for 1 h on ice. These A20 cells were then washed with additional PBS with BSA and pelleted by centrifugation through four wash cycles. The loaded and washed A20 cells were then distributed (50000 cells per well) to a 96 well plate containing HEK293 cells expressing PTHR1 and cAMP Glosensor as described above. Luminescence readouts at time points prior to 10 minutes after addition of A20 cells were unreliable, so luminescence was measured between 12–16 minutes after addition of the A20 cells.
Data calculations
Data were processed using Microsoft Excel and GraphPad Prism 6. Data from cAMP dose–response assays were analyzed using a sigmoidal dose–response model with variable slope. Data sets were statistically compared by using one-way ANOVA with multiple comparisons. Statistically significant differences between data sets were inferred at p < 0.05.Results and discussion
Nanobody–ligand conjugates bind surface markers and are capable of indirectly activating PTHR1
Several approaches for tethering ligands for cell surface receptors at or near their site of action are shown in Fig. 1. Previous work from our lab focused on activation of the type-1 parathyroid hormone receptor (PTHR1), which is activated under physiological conditions by the peptide parathyroid hormone (PTH).27 Parathyroid hormone/parathyroid hormone-related protein receptor (PTH/PTHrP type 1 receptor; commonly known as PTHR1) is a family B G-protein-coupled receptor (GPCR) that regulates skeletal development, bone turnover and mineral ion homeostasis.28 Conjugation of weakly active peptide agonists for PTHR1 to VHHs that bind to the same receptor (Fig. 1a) augments the biological activity of the ligand and avoids off-target activation in vitro and in vivo.2 This observation prompted us to ask whether other mechanisms for tethering ligands for PTHR1 near their site of action through conjugation with nanobodies would also enable activation of PTHR1 (Fig. 1). This scheme depends on the spatial disposition of PTHR1 relative to the target recognized by the relevant nanobody or antibody, as it requires an interaction of the ligand-modified nanobody with a surface molecule other than PTHR1 (Fig. 1c and d). | ||
| Fig. 1 Schematic of methods for receptor activation using ligand-nanobody fusions with PTHR1 as an example. (a) Direct tethering of ligand to its target with a VHH as previously described.2 (b) Indirect targeting using a VHH that binds another protein (MHC-I) on the cell surface. (c) Antibody-mediated direct tethering using a receptor-specific (or fusion protein-specific) full-size antibody (IgG) and a VHH that binds IgG. (d) Antibody-mediated indirect tethering using a full-size antibody that binds another protein (MHC-I) on the cell surface and a VHH that binds IgG. | ||
We used a cell-based system in which human PTHR1 or its variants are stably expressed in HEK293 cells as a test bed for alternative tethering strategies. In the first iteration, VHHs that target cell surface proteins naturally expressed on HEK293 cells were prepared and evaluated (Fig. 1b). We used VHHs that target human β2-microglobulin, the light chain of cell surface Class I major histocompatibility complex products (VHHMHC-I, previously named Nb30),20 the integrin α5β1 (VHHα5β1), and a short peptide epitope taken from the intracellular protein UBC6E (VHH6E).22 These VHHs were labeled at their C-terminus by means of sortagging with a peptide functionalized with biotin for detection and with an azide for conjugation to appropriately functionalized peptides of choice (Fig. 2a).2 Conjugates comprised of VHHs, biotin, and PTH1–11 were used to label HEK293 cells, followed by detection with a streptavidin–phycoerythrin conjugate (Fig. 2b–d). The HEK293 cell line (PTHR1-6E-HEK) expresses PTHR1 with the 6E tag inserted into a disordered portion of the receptor extracellular domain2 (Fig. 2b), whereas HEK293 serves as a negative control (neg-HEK) (Fig. 2c and d). VHHMHC-I and VHHα5β1 conjugates labeled PTHR1-6E-HEK and neg-HEK cells at low concentrations, indicating tight binding. VHHMHC-I labeling provided a higher maximal intensity of staining (approximately 10 fold) than VHHα5β1 (Table 1).29 Staining of PTHR1-6E-HEK cells with the VHH6E conjugate provided a stronger signal than staining with VHHMHC-I (Table 1), consistent with a high level of expression of PTHR1-6E on PTHR1-6E-HEK cells. We then evaluated whether the VHH–PTH conjugates shown to bind to PTHR1-6E-HEK cells stimulated receptor activation and the production of cyclic adenosine monophosphate (cAMP), a second messenger produced upon PTHR1 activation. Consistent with previous results, VHH6E–PTH1–11 potently stimulated cAMP responses on PTHR1-6E-HEK cells (Fig. 2e and Table 1).2 VHHMHC-I–PTH1–11 was essentially inactive in stimulating cAMP responses, whereas VHHα5β1–PTH1–11 induced a weak but detectable response. This observation was unexpected given that staining of HEK cell lines was stronger with VHHMHC-I than VHHα5β1. The origin of this difference is unknown, but it may relate to the larger size and expanded flexibility of the extracellular portions of α5β1 relative to β2-microglobulin as a more compact subunit of MHC-I. Another possibility is that PTHR1 may localize to membrane domains closer to α5β1 relative to the β2-microglobulin-MHC-I complex. In this context, the use of a longer flexible linker between the receptor agonist and nanobody may improve activation efficacy. A conjugate consisting of a nanobody and ligand that both target the same receptor was relatively insensitive to inclusion of a short polyethylene glycol linker.2 Collectively, VHH–PTH conjugates that directly target other proteins on the cell surface (MHC-I, α5β1) are only weak agonists of PTHR1.
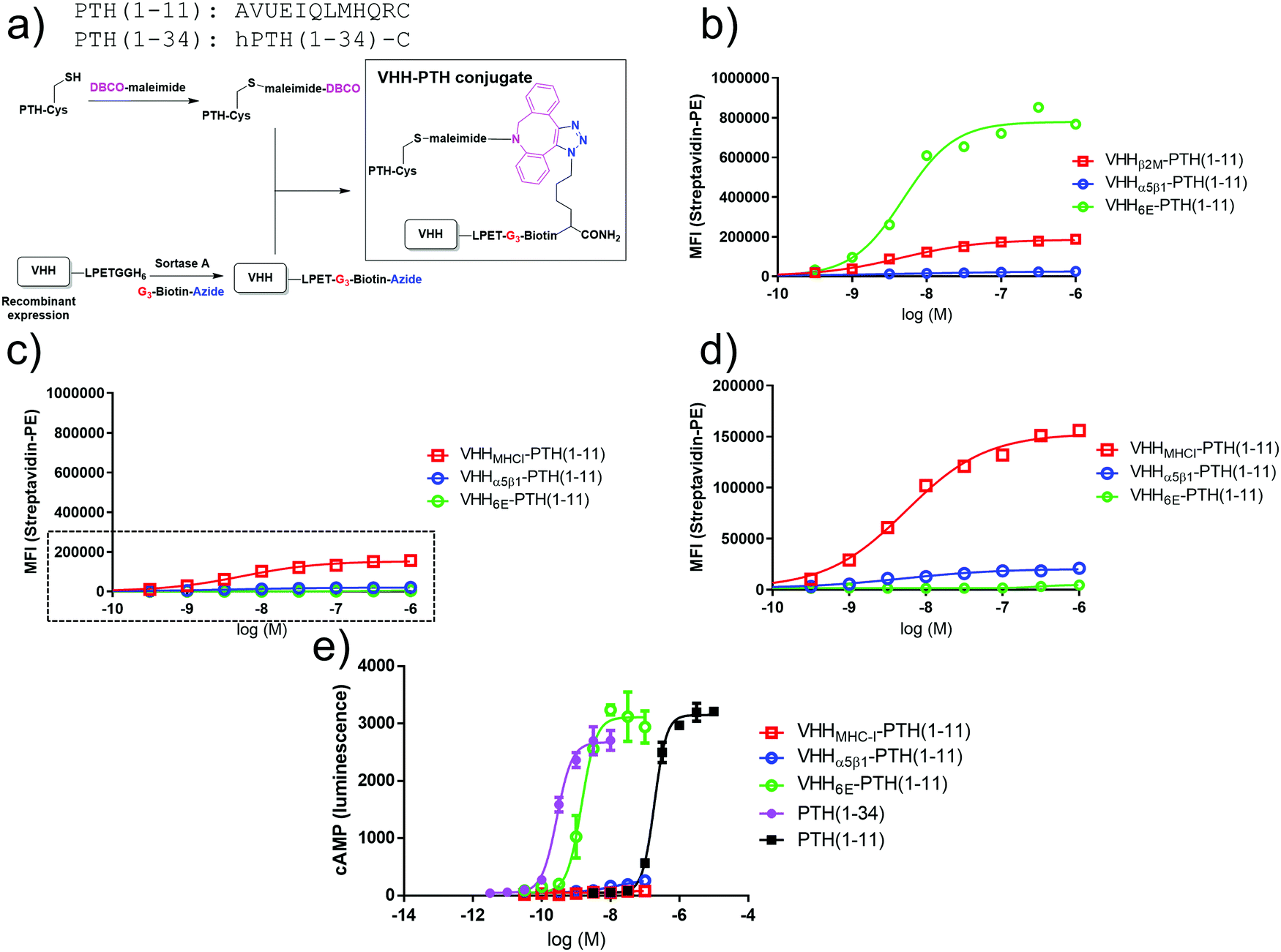 | ||
| Fig. 2 Synthesis and evaluation of PTH–nanobody conjugates. (a) Synthetic scheme for production of PTH–nanobody conjugates. PTH-peptide synthesis and functionalization, VHH production and functionalization, and conjugation was performed as described in methods. (b) Representative (one of three experiments) dose–response curve for binding of VHH–biotin–PTH conjugates to PTHR1-6E-HEK cells. The plots were produced by fitting a sigmoidal dose–response curve. Staining, detection, and quantitation were performed as described in methods. Representative data from one of three independent experiments is shown, with compiled data shown in Table 1. (c) Binding of VHH–biotin–PTH conjugates to neg-HEK cells with a zoomed inset (boxed region) shown in panel d. Representative data from one of three independent experiments is shown with compiled data shown in Table 1. (e) Representative dose–response assay for induction of cAMP responses in PTHR1-6E-HEK cells. Error bars correspond to standard deviation from replicate conditions in a single experiment. Plots were generated by fitting a sigmoidal dose–response curve. Representative data from one of three independent experiments is shown with compiled data shown in Table 1. | ||
| PTHR1-6E-HEK | Binding EC50 | Max. stain | ||
|---|---|---|---|---|
| nM | SD | Intensity (relative to MHC-I) | SD | |
| a The maximal staining for VHH6E–PTH1–11 on HEK-neg cells was very weak, so binding EC50 values were not determined (ND). | ||||
| VHH6E–PTH1–11 | 5.1 | 0.3 | 5.6 | 2.5 |
| VHHMHC-I–PTH1–11 | 3.2 | 1.2 | 1 | 0 |
| VHHa5b1–PTH1–11 | 4.6 | 3.3 | 0.11 | 0.04 |
| PTHR1-6E-HEK | Signaling cAMP EC50 | Max. cAMP signal | ||
|---|---|---|---|---|
| nM | SD | Normalized (fold over no ligand) | SD | |
| PTH1–34 | 0.18 | 0.090 | 120 | 23 |
| PTH1–11 | 132 | 51 | 150 | 17 |
| VHH6E–PTH1–11 | 0.71 | 0.64 | 104 | 17 |
| VHHMHC-I–PTH1–11 | >100 nM | 3.8 | 3.1 | |
| VHHa5b1–PTH1–11 | >100 nM | 33 | 29 | |
| Neg-HEK | Binding EC50 | Max. stain | ||
|---|---|---|---|---|
| nM | SD | Intensity (relative to MHC-I) | SD | |
| VHH6E–PTH1–11 | NDa | ND | 0.020 | 0.016 |
| VHHMHC-I–PTH1–11 | 4.3 | 1.7 | 1.0 | 0 |
| VHHa5b1–PTH1–11 | 3.0 | 2.8 | 0.10 | 0.05 |
Combination of primary antibodies with VHHmIg–PTH1–11 strongly increases PTHR1 activation
We also tested whether targeting a receptor of interest using a conventional monoclonal antibody (immunoglobulin, Ig) and a nanobody–PTH conjugate in which the nanobody binds to Ig could serve to activate the receptor (Fig. 1c). We used a nanobody (previously23 named TP1170, named here VHHmIg) that binds to the kappa light chain found in 95% of mouse Igs.23 We prepared VHHmIg–PTH1–11 and evaluated its agonist activity on HEK293 cells that stably express PTHR1 fused to GFP30 in the presence or absence of a mouse monoclonal IgG antibody specific for GFP (α-GFP). As a control we used non-specific polyclonal mouse IgG (isotype) in place of α-GFP. We mixed VHHmIg–PTH1–11 at a 3:1 molar ratio with mouse IgGs prior to addition to HEK293 cells and then recorded cAMP responses at VHHmIg–PTH1–11 doses of 100 nM and 10 nM (Fig. 3a and b). The combination of VHHmIg–PTH1–11 and α-GFP led to a robust cAMP response at 100 nM and a weak response at 10 nM, whereas the combination of VHHmIg–PTH1–11 and isotype was inactive. The combination of VHHmIg–PTH1–11 and α-GFP was more potent than PTH1–11 alone. In a separate experiment, we used versions of PTHR1 in which the extracellular portion of the receptor was absent or replaced with the HA-epitope tag (Fig. S2, ESI†).31 The combination of VHHmIg–PTH1–11 and α-HA mouse monoclonal IgG lead to a cAMP response that far exceeded the response seen with PTH1–11 alone, whereas the combination of VHHmIg–PTH1–11 and isotype mouse IgG was inactive. These data show that indirect targeting of PTHR1 using an Ig-specific nanobody–PTH conjugate and a bridging IgG (Fig. 1c) can augment the biological activity of a weakly active ligand.
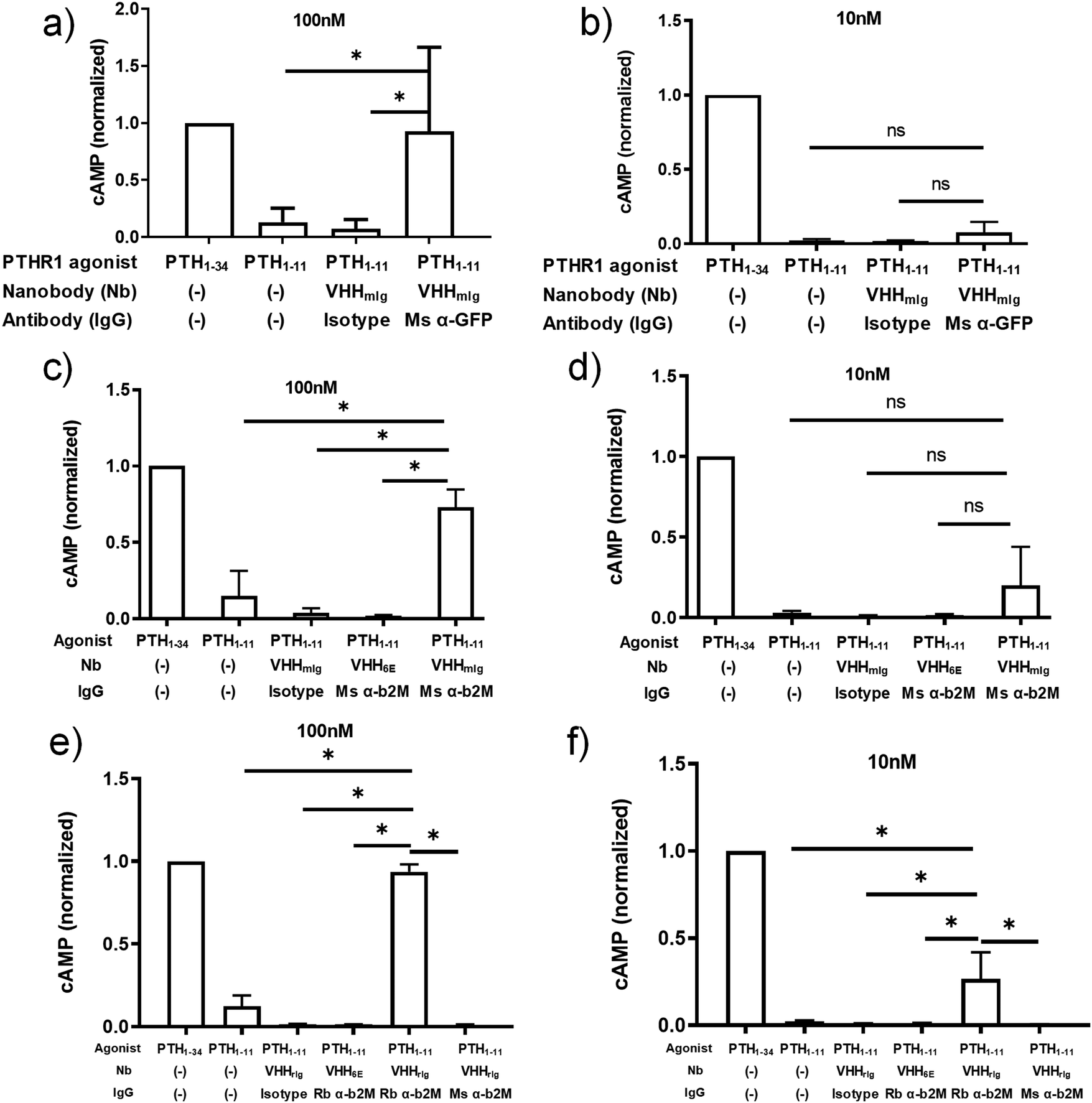 | ||
| Fig. 3 Assessment of tethered VHH–PTH conjugates in activating PTHR1 signaling. PTH peptides or conjugates prepared as described in Fig. 2a were either mixed with commercially available full-size antibodies or added directly to HEK293 cells that express PTHR1 or its variant as described in methods. Data sets were compared for statistically significant differences using one-way ANOVA with multiple comparisons; *p < 0.05. (a and b) HEK293 cells expressing a GFP-PTHR1 fusion protein (see Fig. S1, ESI†) were treated with PTH agonist peptides (columns 1 + 2) or VHH–PTH conjugates mixed with commercial antibodies (columns 3 + 4). PTH agonist peptides and VHH–PTH conjugates were applied at a dose of 100 nM (panel a) or 10 nM (panel b). VHH–PTH conjugates were used a 3 fold molar excess over commercial antibodies. ‘Isotype’ refers to a polyclonal mouse IgG preparation. The magnitude of the cAMP responses is normalized to that of PTH1–34. (c and d) Experiments were performed as in panels a and b, except that HEK293 cells expressed WT hPTHR1. Bars and error bars represent mean ± SD from 6 independent experiments. (e and f) HEK293 cells expressing WT hPTHR1 were treated with PTH agonist peptides (columns 1 + 2) or VHH–PTH conjugates mixed with commercial antibodies (columns 3–6). “Rb” indicates a monoclonal antibody derived from rabbit. “Ms” indicates monoclonal antibody derived from mouse. Isotype refers to a polyclonal IgG antibody preparation derived from WT mouse. Bars and error bars represent mean ± SD from 3 independent experiments. | ||
The ability to use antibodies to target proteins on the cell surface other than the receptor of interest (Fig. 1d) provides opportunities for the delivery of ligands to cell types of choice, based on the presence of an IgG-recognized surface marker. As a proof of concept, we deployed commercial antibodies that target β2-microglobulin (α-β2M) as a component of cell surface MHC-I, a ubiquitously expressed type I membrane protein. This also allowed a direct comparison with the use of a nanobody that targets β2-microglobulin (Fig. 1a and 2). Because we were targeting PTHR1 indirectly, we used a HEK293 cell line that stably expresses wild-type PTHR1. The combination of VHHmIg–PTH1–11 and mouse α-β2M induces a robust cAMP response at a concentrations of 100 nM whereas the combination of VHHmIg–PTH1–11 and a control antibody (“isotype”) was inactive (Fig. 3c and d). The use of a control nanobody–PTH conjugate that targets the 6E epitope tag (VHH6E–PTH1–11) in combination with α-β2M was also inactive. Neither the binding of IgG to PTHR1 nor the presence of an irrelevant VHH–PTH1–11 conjugate leads to activation. The combination of VHHmIg–PTH1–11 and α-β2M was more potent than PTH1–11 alone. Indirect targeting (as in Fig. 1d) can therefore substantially increase the biological activity of a weakly active ligand. This stands in sharp contrast to parallel efforts with conjugates that comprise nanobodies that target β2M, which failed to activate PTHR1 (Fig. 1b and 2). The cause of this divergence is unclear but may be related to the additional flexibility and/or distance provided by the bridging full-size antibody.
Any attempt to apply VHHmIg conjugates in mice would likely be stymied by the high concentration of mIgs found in the mouse blood stream. To circumvent this problem, we turned to a nanobody (previously23 named TP897, named here VHHrIg) that binds to rabbit Igs, but not mouse. We prepared a VHHrIg–PTH1–11 conjugate as described above and evaluated its performance in PTHR1 activation assays. VHHrIg–PTH1–11 activated PTHR1 signaling when combined with rabbit α-β2M but not mouse α-β2M (Fig. 3e and f). This finding offers a path towards using this approach in vivo in mice.
These findings raised the question whether targeting other cell surface proteins for antibody tethering would likewise facilitate receptor activation. We therefore expanded the approach described above by inclusion of monoclonal antibodies that target the cell surface markers CD63 and CD81, both widely expressed. CD63 and CD81 are members of the tetraspanin family of proteins, naturally expressed on HEK293 cells and on most other cell types, at modest levels.32,33 We confirmed by flow cytometry that these monoclonal antibodies bound to HEK293 cells (Fig. S3, ESI†). The α-CD63 and α-CD81 antibodies labeled HEK293 cells following secondary staining with either α-mouse Ig-alexafluor 647 or with VHHmIg–biotin and streptavidin–phycoerythrin (streptavidin–PE, Fig. S3, ESI†). However, neither of the α-CD63 and α-CD81 antibodies stained HEK293 cells as strongly as the anti β2M antibody, consistent with the high level of β2M found on most cells (Fig. S3, ESI†).29 We then tested whether the α-CD63 and α-CD81 antibodies could provide tethering to facilitate activation of PTHR1 signaling (Fig. 4). Whereas α-CD81 facilitated activation comparable to α-β2M at both concentrations tested, α-CD63 failed to promote activation above background levels. The source of this difference between the α-CD81 and α-CD63 antibodies in these assays is not clear, but it may be related to differences in target expression levels or differences in membrane protein localization.33 These findings open the door to identifying other cell surface markers, potentially expressed specifically on cell types of interest, that could allow cell-type specific activation of widely expressed receptors.
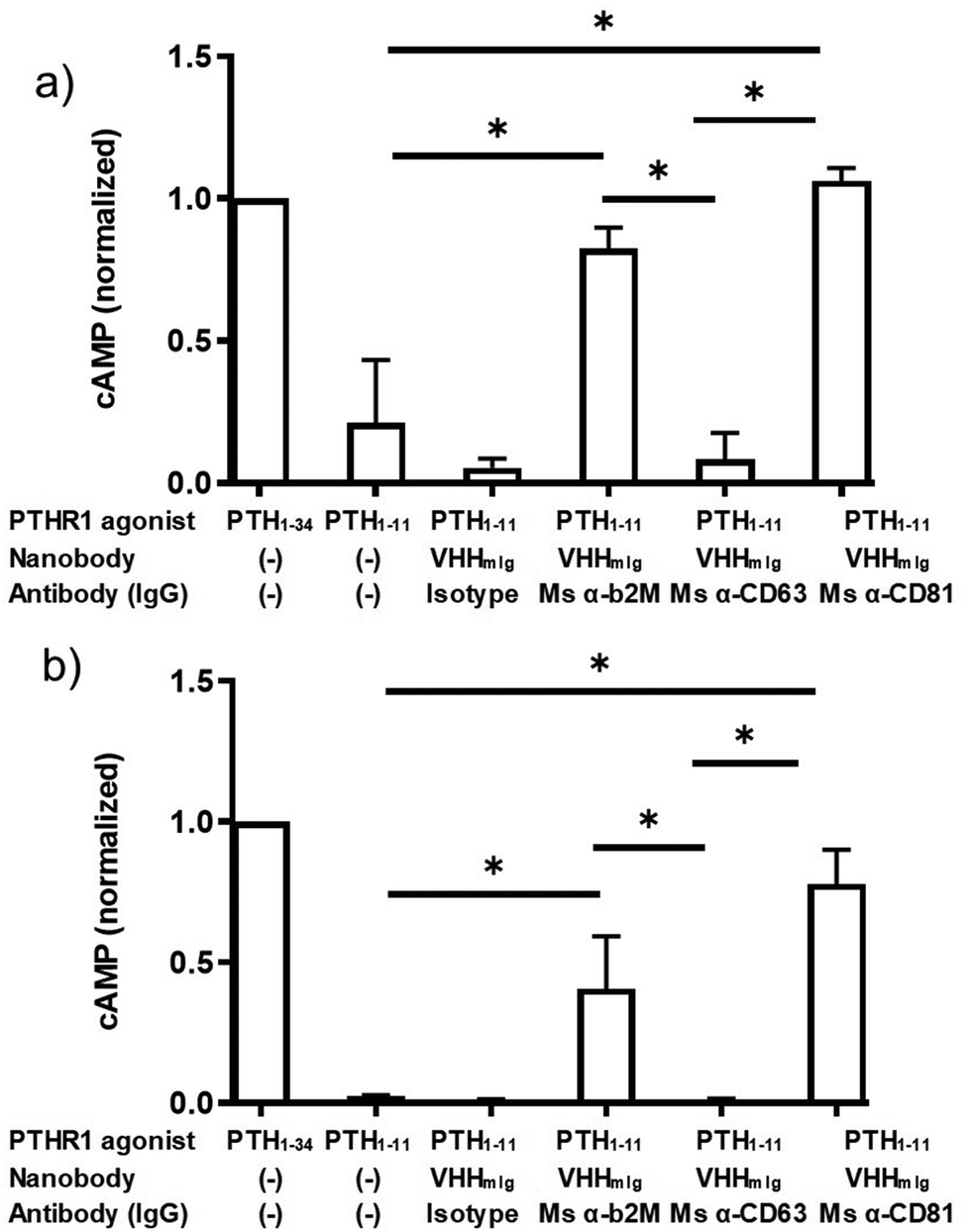 | ||
| Fig. 4 Assessment of indirect activation efficiency through targeting varied surface proteins. PTH peptides or conjugates prepared as described in Fig. 2a were either mixed with commercially available antibodies or added directly to HEK293 cells expressing PTHR1 or its variant as described in methods. (a and b) HEK293 cells that express PTHR1 were treated with PTH agonist peptides (columns 1 + 2) or VHH–PTH conjugates mixed with commercial antibodies (columns 3–6). PTH agonist peptides and VHH–PTH conjugates were applied at a dose of 100 nM (panel a) or 10 nM (panel b). VHH–PTH conjugates were used at a 3 fold molar excess over commercial antibodies. ‘Isotype’ refers to a polyclonal mouse IgG preparation. The magnitude of the cAMP responses is normalized to that of PTH1–34. Bars and error bars represent mean ± SD from 3 independent experiments that are distinct from those shown in Fig. 3. Data sets were compared for statistically significant differences using one-way ANOVA with multiple comparisons; *p < 0.05. | ||
Cell-bound nanobody–PTH conjugates can activate adjacent cells in co-culture experiments.
Finally, we tested whether antibody-based ligand tethering could bridge the space between distinct cells (Fig. 5a). In this approach the antibody targeting portion of the CLAMP binds to one cell type (targeting cell) and the ligand targets a receptor expressed on another cell type (receptor cell). We combined HEK293 cells that express PTHR1 (receptor cell) and a murine B cell lymphoma line, A20, which expresses high levels of class II major histocompatibility complex (MHC-II, targeting cell33). Of note, A20 are cells in suspension in contrast to HEK293 cells, which grow adherently. We used a well-charactered VHH that binds murine MHC-II (VHHMHC-II, previously17 called VHH7). We predicted that intercellular tethering would prove more challenging than intracellular tethering, so we used a larger, more potent PTHR1 peptide agonist (PTH1–34), instead of PTH1–11, in the CLAMPs that we tested.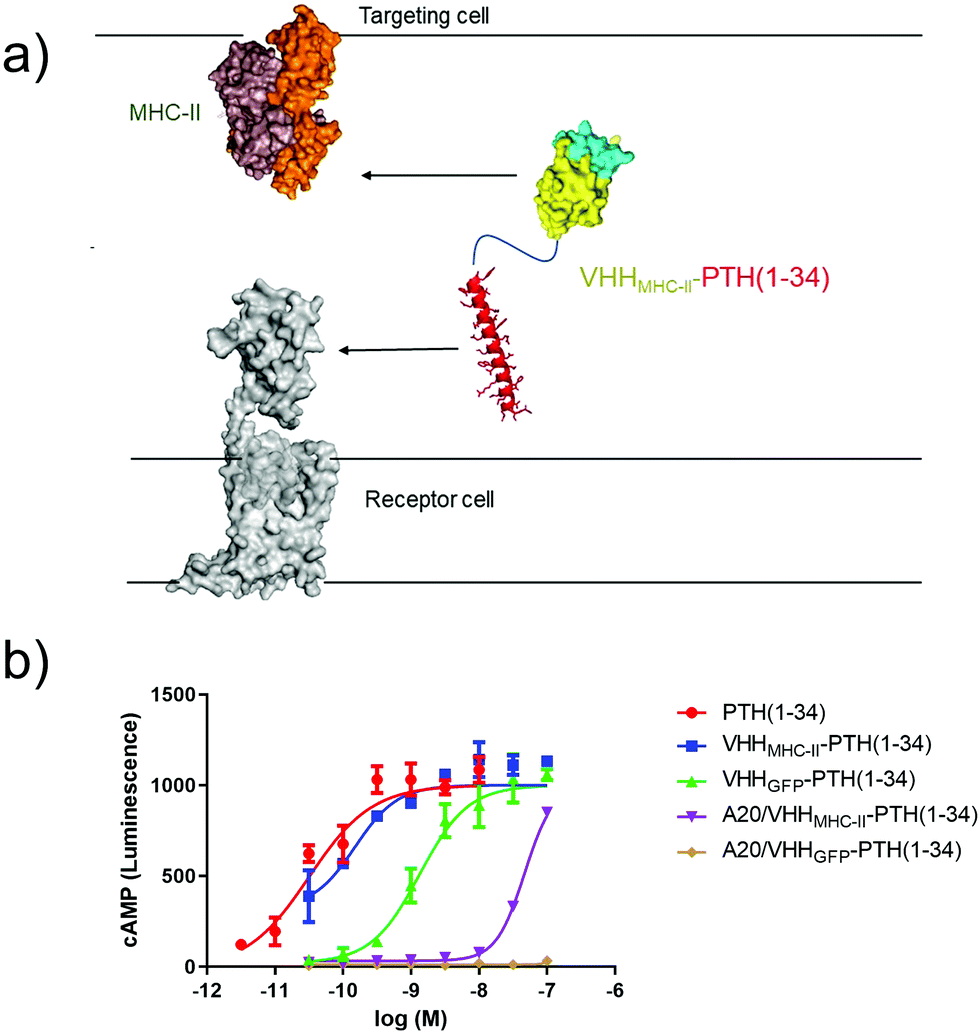 | ||
| Fig. 5 Intercellular activation of GPCR signaling. (a) Schematic of intercellular antibody tethering. (b) Representative dose–response assay for induction of cAMP responses in PTHR1-6E-HEK cells. Error bars correspond to standard deviation from replicate conditions in a single experiment. Data were fitted to a sigmoidal dose–response model with variable slope. Representative data from one of three independent experiments is shown. | ||
PTH1–34 can be conjugated to a nanobody with only a minor impact on PTHR1 agonist activity.2 We confirmed this: CLAMPs consisting of PTH1–34 were conjugated to VHHs that target either GFP34 (VHHGFP–PTH1–34) or MHC-II (VHHMHC-II–PTH1–34) using the chemistry described in Fig. 2a. These CLAMPs performed similarly to PTH1–34 itself in cAMP stimulation assays on HEK293 cells (receptor cells) that express human PTHR1 (Fig. 5). This assay was also run using A20 cells (targeting cells) treated with CLAMPs and then washed to remove unbound ligand. Application of targeting cells pre-treated with VHHMHC-II–PTH1–34 but not VHHGFP–PTH1–34 stimulated robust cAMP responses. These responses were observed when loading A20 cells with CLAMP concentrations of >10 nM. VHHMHC-II binds to MHC-II with half-maximal staining at concentrations of approximately 50 nM.35 This similarity in concentrations suggests a correlation between the amount of CLAMP loaded on the target cell and the extent of receptor activation on the receptor cell. Whether CLAMPs can simultaneously bind to both a targeting cell and receptor cell, of if receptor activation first requires dissociation from the targeting cell, is not clear from this study, although past work with immunocytokines suggests that such dissociation is not required.3 This type of correlation may provide guidelines for future efforts to use intercellular antibody tethering.
Conclusions
We have demonstrated several approaches for activation of GPCR signaling with CLAMPs that do not rely on direct targeting of the receptor. The ability to use conventional monoclonal antibodies as tethering agents should allow expansion of the CLAMP methodology to a variety of targets. This approach is of practical value, as it avoids the need for target-specific nanobodies. Efforts to identify nanobodies that bind to the exposed extracellular face of GPCRs have been successful in some cases11,36–40 but for many cell surface targets nanobodies of appropriate specificity do not yet exist. In contrast, there is a wide array of commercially available monoclonal antibodies that recognize cell surface proteins in mouse and man. Seen from this perspective, the ability to use tethering by commercially available antibodies to activate a receptor of interest only on those cell types that express the antibody target is a major asset. This could offer a means to engineer “AND” logic gates41 that target receptor signaling only in a subset of cells. The availability of a nanobody that selectively targets rabbit IgG, but not mouse IgGs, provides a path to extend this approach in vivo in mice. Intercellular tethering could further expand applications of this approach to interrogate cell-to-cell signaling within a given organ or biological compartment.Author contributions
Project conceptualization (R. W. C., F. A. F., H. L. P.), research resources (R. W. C., T. J. G., H. L. P.), funding acquisition (R. W. C., T. J. G., H. L. P.), laboratory experimentation (R. W. C., F. A. F., T. J. G.), data visualization (R. W. C., F. A. F.), methodology development (R. W. C., F. A. F.), and manuscript writing (R. W. C., F. A. F., T. J. G., H. L. P.).Conflicts of interest
R. W. C., T. J. G., and H. L. P. have filed a patent on the use of nanobodies to target GPCRs for signaling (US application number 16810783).Acknowledgements
Funding was provided by the National Institutes of Health grant numbers P01-DK011794 (T. J. G.), 1DP1AI150593 (H. L. P.) and R01-AI087879 (H. L. P.). R. W. C. was supported in part by funding from the Cancer Research Institute Irvington Postdoctoral Fellowship. We acknowledge Thomas Dean at Massachusetts General Hospital for technical assistance with cell bioassays.References
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef PubMed.
- R. W. Cheloha, F. A. Fischer, A. W. Woodham, E. Daley, N. Suminski, T. J. Gardella and H. L. Ploegh, Nat. Commun., 2020, 11, 2087 CrossRef PubMed.
- A. Tzeng, B. H. Kwan, C. F. Opel, T. Navaratna and K. D. Wittrup, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3320–3325 CrossRef PubMed.
- L. Sweeny, Y. E. Hartman, K. R. Zinn, J. R. Prudent, D. J. Marshall, M. S. Shekhani and E. L. Rosenthal, Oral Oncol., 2013, 49, 991–997 CrossRef CAS PubMed.
- D. J. Marshall, S. S. Harried, J. L. Murphy, C. A. Hall, M. S. Shekhani, C. Pain, C. A. Lyons, A. Chillemi, F. Malavasi, H. L. Pearce, J. S. Thorson and J. R. Prudent, Mol. Ther., 2016, 24, 1760–1770 CrossRef CAS PubMed.
- A. C. Cheng, E. M. Doherty, S. Johnstone, E. F. DiMauro, J. Dao, A. Luthra, J. Ye, J. Tang, T. Nixey, X. Min, P. Tagari, L. P. Miranda and Z. Wang, Sci. Rep., 2018, 8, 7570 CrossRef PubMed.
- R. W. Cheloha, T. J. Harmand, C. Wijne, T. U. Schwartz and H. L. Ploegh, J. Biol. Chem., 2020, 295, 15307–15327 CrossRef PubMed.
- N. Pishesha, J. R. Ingram and H. L. Ploegh, Annu. Rev. Cell Dev. Biol., 2018, 34, 163–188 CrossRef CAS PubMed.
- T. J. Harmand, A. Islam, N. Pishesha and H. L. Ploegh, RSC Chem. Biol., 2021, 2, 685 RSC.
- E. Pardon, T. Laeremans, S. Triest, S. G. F. Rasmussen, A. Wohlkönig, A. Ruf, S. Muyldermans, W. G. J. Hol, B. K. Kobilka and J. Steyaert, Nat. Protoc., 2014, 9, 674–693 CrossRef CAS PubMed.
- R. Heukers, T. W. M. De Groof and M. J. Smit, Curr. Opin. Cell Biol., 2019, 57, 115–122 CrossRef CAS PubMed.
- Y. Ma, Y. Ding, X. Song, X. Ma, X. Li, N. Zhang, Y. Song, Y. Sun, Y. Shen, W. Zhong, L. A. Hu, Y. Ma and M.-Y. Zhang, Sci. Adv., 2020, 6, eaax7379 CrossRef CAS PubMed.
- C. McMahon, D. P. Staus, L. M. Wingler, J. Wang, M. A. Skiba, M. Elgeti, W. L. Hubbell, H. A. Rockman, A. C. Kruse and R. J. Lefkowitz, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20284–20291 CrossRef CAS PubMed.
- H. Ren, J. Li, N. Zhang, L. A. Hu, Y. Ma, P. Tagari, J. Xu and M.-Y. Zhang, Commun. Biol., 2020, 3, 146 CrossRef CAS PubMed.
- C. Morrison, Nat. Rev. Drug Discovery, 2019, 18, 485–487 CrossRef CAS PubMed.
- T. Fang, J. N. Duarte, J. Ling, Z. Li, J. S. Guzman and H. L. Ploegh, Angew. Chem., Int. Ed., 2016, 55, 2416–2420 CrossRef CAS PubMed.
- T. Fang, C. H. M. J. Van Elssen, J. N. Duarte, J. S. Guzman, J. S. Chahal, J. Ling and H. L. Ploegh, Chem. Sci., 2017, 8, 5591–5597 RSC.
- H. Pan, Y. Su, Y. Xie, W. Wang, W. Qiu, W. Chen, W. Lu, Z. Lu, W. Wang and A. Shang, Artif. Cells, Nanomed., Biotechnol., 2020, 48, 854–866 CrossRef CAS PubMed.
- E. E. Wilton, M. P. Opyr, S. Kailasam, R. F. Kothe and H.-J. Wieden, ACS Synth. Biol., 2018, 7, 2480–2484 CrossRef CAS PubMed.
- K. Domanska, S. Vanderhaegen, V. Srinivasan, E. Pardon, F. Dupeux, J. A. Marquez, S. Giorgetti, M. Stoppini, L. Wyns, V. Bellotti and J. Steyaert, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1314–1319 CrossRef CAS PubMed.
- M. Rashidian, E. J. Keliher, A. M. Bilate, J. N. Duarte, G. R. Wojtkiewicz, J. T. Jacobsen, J. Cragnolini, L. K. Swee, G. D. Victora, R. Weissleder and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6146–6151 CrossRef CAS PubMed.
- J. Ling, R. W. Cheloha, N. McCaul, Z.-Y. J. Sun, G. Wagner and H. L. Ploegh, Mol. Immunol., 2019, 114, 513–523 CrossRef CAS PubMed.
- T. Pleiner, M. Bates and D. Görlich, J. Cell Biol., 2018, 217, 1143–1154 CrossRef CAS PubMed.
- R. W. Cheloha, Z. Li, D. Bousbaine, A. W. Woodham, P. Perrin, J. Volarić and H. L. Ploegh, ACS Chem. Biol., 2019, 14, 1836–1844 CrossRef CAS PubMed.
- J. N. Duarte, J. J. Cragnolini, L. K. Swee, A. M. Bilate, J. Bader, J. R. Ingram, A. Rashidfarrokhi, T. Fang, A. Schiepers, L. Hanke and H. L. Ploegh, J. Immunol., 2016, 197, 4838–4847 CrossRef CAS PubMed.
- R. W. Cheloha, A. Maeda, T. Dean, T. J. Gardella and S. H. Gellman, Nat. Biotechnol., 2014, 32, 653–655 CrossRef CAS PubMed.
- R. W. Cheloha, S. H. Gellman, J.-P. Vilardaga and T. J. Gardella, Nat. Rev. Endocrinol., 2015, 11, 712–724 CrossRef CAS PubMed.
- T. J. Gardella and J.-P. Vilardaga, Pharmacol. Rev., 2015, 67, 310–337 CrossRef CAS PubMed.
- T. Geiger, A. Wehner, C. Schaab, J. Cox and M. Mann, Mol. Cell. Proteomics, 2012, 11, M111.014050 CrossRef PubMed.
- M. J. Mahon, Adv. Biosci. Biotechnol., 2011, 2, 132–137 CrossRef CAS PubMed.
- M. D. Luck, P. H. Carter and T. J. Gardella, Mol. Endocrinol., 1999, 13, 670–680 CAS.
- M. D. Kalemera, J. Cappella-Pujol, A. Chumbe, A. Underwood, R. A. Bull, J. Schinkel, K. Sliepen and J. Grove, Optimised cell systems for the investigation of hepatitis C virus E1E2 glycoproteins, Microbiology, 2020, 102(1) Search PubMed.
- F. Fordjour, S. Owiredu, H. Muendlein, J. Plange, J. Morrell, J. Han and S. Gould, Matters, 2016 DOI:10.19185/matters.201604000004.
- A. Kirchhofer, J. Helma, K. Schmidthals, C. Frauer, S. Cui, A. Karcher, M. Pellis, S. Muyldermans, C. S. Casas-Delucchi, M. C. Cardoso, H. Leonhardt, K.-P. Hopfner and U. Rothbauer, Nat. Struct. Mol. Biol., 2010, 17, 133–138 CrossRef CAS PubMed.
- M. Rashidian, E. Keliher, M. Dougan, P. K. Juras, M. Cavallari, G. R. Wojtkiewicz, J. Jacobsen, J. G. Edens, J. M. G. Tas, G. Victora, R. Weissleder and H. Ploegh, ACS Cent. Sci., 2015, 1, 142–147 CrossRef CAS PubMed.
- D. Maussang, A. Mujić-Delić, F. J. Descamps, C. Stortelers, P. Vanlandschoot, M. Stigter-van Walsum, H. F. Vischer, M. van Roy, M. Vosjan, M. Gonzalez-Pajuelo, G. A. M. S. van Dongen, P. Merchiers, P. van Rompaey and M. J. Smit, J. Biol. Chem., 2013, 288, 29562–29572 CrossRef CAS PubMed.
- S. Jähnichen, C. Blanchetot, D. Maussang, M. Gonzalez-Pajuelo, K. Y. Chow, L. Bosch, S. De Vrieze, B. Serruys, H. Ulrichts, W. Vandevelde, M. Saunders, H. J. De Haard, D. Schols, R. Leurs, P. Vanlandschoot, T. Verrips and M. J. Smit, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20565–20570 CrossRef PubMed.
- X. Peyrassol, T. Laeremans, V. Lahura, M. Debulpaep, H. El Hassan, J. Steyaert, M. Parmentier and I. Langer, Front. Endocrinol., 2018, 9, 153 CrossRef PubMed.
- P. Scholler, D. Nevoltris, D. de Bundel, S. Bossi, D. Moreno-Delgado, X. Rovira, T. C. Møller, D. El Moustaine, M. Mathieu, E. Blanc, H. McLean, E. Dupuis, G. Mathis, E. Trinquet, H. Daniel, E. Valjent, D. Baty, P. Chames, P. Rondard and J.-P. Pin, Nat. Commun., 2017, 8, 1967 CrossRef PubMed.
- C. Hong, N. J. Byrne, B. Zamlynny, S. Tummala, L. Xiao, J. M. Shipman, A. T. Partridge, C. Minnick, M. J. Breslin, M. T. Rudd, S. J. Stachel, V. L. Rada, J. C. Kern, K. A. Armacost, S. A. Hollingsworth, J. A. O’Brien, D. L. Hall, T. P. McDonald, C. Strickland, A. Brooun, S. M. Soisson and K. Hollenstein, Nat. Commun., 2021, 12, 815 CrossRef CAS PubMed.
- J. Z. Williams, G. M. Allen, D. Shah, I. S. Sterin, K. H. Kim, V. P. Garcia, G. E. Shavey, W. Yu, C. Puig-Saus, J. Tsoi, A. Ribas, K. T. Roybal and W. A. Lim, Science, 2020, 370, 1099–1104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00118c |
| ‡ Current affiliation: National Institute of Health, National Institutes of Diabetes, Digestive and Kidney Disease, Laboratory of Bioorganic Chemistry, Bethesda, MD, 20894, USA. |
| § Current affiliation: Kennedy Institute of Rheumatology, University of Oxford, OX3 7FY, Oxford, UK. |
| This journal is © The Royal Society of Chemistry 2021 |