
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysine succinylation on non-histone chromosomal protein HMG-17 (HMGN2) regulates nucleosomal DNA accessibility by disrupting the HMGN2–nucleosome association†
Yihang
Jing‡
,
Gaofei
Tian‡
,
Xiaoyu
Qin
,
Zheng
Liu
* and
Xiang David
Li
 *
*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: lz0418@hku.hk; xiangli@hku.hk
First published on 27th May 2021
Abstract
Lysine succinylation (Ksucc) is a novel posttranslational modification that frequently occurs on chromatin proteins including histones and non-histone proteins. Histone Ksucc affects nucleosome dynamics by increasing the DNA unwrapping rate and accessibility. However, very little is known about the regulation and functions of Ksucc located on non-histone chromosomal proteins. Here, we site-specifically installed a succinyl lysine analogue (Kcsucc) onto the non-histone chromosomal protein HMG-17 (HMGN2) to mimic the natural succinylated protein. We found that the incorporation of Kcsucc into HMGN2 at the K30 site (HMGN2Kc30succ), which is located within the nucleosome-binding domain (NBD), leads to significantly decreased HMGN2 binding to the mononucleosome. HMGN2Kc30succ also increased the nucleosomal DNA accessibility by promoting nucleosomal DNA unwrapping in the entry/exit region. This study reveals a novel mechanism of non-histone protein succinylation on altering chromatin recruitment, which can further affect nucleosome and chromatin dynamics.
Introduction
In eukaryotic cells, genomic DNA is packaged with histones and non-histone proteins into nucleosomes that form chromatin, a highly dynamic structure of repeating nucleosome units. Non-histone chromosomal proteins play vital roles in the maintenance of the chromatin structure and the regulation of chromatin-templated biological processes such as gene expression, DNA replication, and DNA damage repair.High mobility group nucleosome-binding (HMGN) protein is a family of architectural non-histone chromosomal proteins ubiquitously expressed in vertebrate cell nuclei. HMGNs bind specifically to nucleosome core particles with high affinity. The binding of HMGNs to nucleosomes is mediated by their evolutionarily conserved and highly positively charged nucleosome binding domain (NBD).1 By weakening the binding of linker histone H1 to nucleosomes, HMGNs destabilise higher-order chromatin structures to promote the binding of nuclear regulatory factors and enhance chromatin-templated transcription activity.2,3 Moreover, HMGNs can modulate the level of histone PTMs and facilitate the repair of DNA lesions.4,5
Similar to histone, a variety of PTMs have been identified in HMGNs including acetylation,6,7 phosphorylation,8 and SUMOylation,9 which regulate their biological activities. These PTMs can often facilitate the dissociation of HMGNs from the nucleosome, as they alter the physical and chemical properties of the modified residues. For example, phosphorylation at S24 and S28 of HMGN2, a member of the HMGN family, will induce the dissociation of HMGN2 from the nucleosome due to an unfavourable electrostatic repulsion between NBD and the nucleosomal acidic patch.8 Both acetylation and SUMOylation on HMGN2 result in a lower binding affinity to nucleosome core particles, suggesting that PTMs can disrupt the interaction between HMGN2 and the nucleosome.
Besides the aforementioned PTMs, lysine succinylation (Ksucc) has been recently discovered on the K17 and K30 residues of HMGN2, but the biological significance of these non-histone protein PTMs remains unclear.10 Ksucc frequently occurs on multiple mitochondrial and nuclear proteins and has been shown to have widespread roles in regulating diverse metabolic pathways.11,12 Importantly, Ksucc on core histones (e.g., H2BK34 and H4K77) change the lysine charge status from +1 to −1 due to the presence of a carboxylate group, disrupting DNA–histone ionic interactions, leading to reduced nucleosome stability and promoting DNA unwrapping.13,14 However, whether and how Ksucc on non-histone chromosomal proteins affects nucleosome dynamics has not yet been studied.
To fill this knowledge gap, we focused on the succinylation of the K17 and K30 sites of HMGN2 to investigate their effects on HMGN2 binding to nucleosomes and their potential roles in regulating nucleosome dynamics. Specifically, we employed a well-developed “thiol–ene” reaction to generate succinylated HMGN2 mimics by incorporating a Ksucc analogue (Kcsucc) at K17 and K30 sites to yield HMGN2Kc17succ and HMGN2Kc30succ, respectively. We performed biochemical and biophysical characterizations of the interactions between succinylated HMGN2 mimics and nucleosomes, which showed that the K30 succinylation disrupted the binding of HMGN2 to the nucleosome, resulting in increased nucleosomal DNA accessibility and exposing the DNA to chromatin binding proteins such as transcription factors.
Experimental
General methods
All the buffers were prepared with ddH2O using the standard methods. The synthesized proteins were purified on a preparative high performance liquid chromatography (HPLC) system with a Waters 2535 Quaternary Gradient Module, a Waters 515 HPLC pump, a Waters SFO system Fluidics Organizer and a Waters 2767 Sample Manager. The synthesis of compound 1 was performed following a previously reported method.14Preparation of Xenopus laevis histones and HMGN2 and LexA proteins
Mutations on K119C in H2A and on K17 or K30 in HMGN2 were introduced by site-directed mutagenesis. All recombinant histones (H2A, H2B, H3, H4, and H2A K119C) and HMGN2 proteins were expressed and purified as previously described.15,16 The LexA protein was expressed and purified from the pJWL288 plasmid (kindly gifted by Prof. Michael G. Poirier, Department of Physics, Department of Chemistry & Biochemistry, Ohio State University) as previously described.17Preparation of succinylated HMGN2 mimics
Mutated HMGN2 (HMGN2K17C, HMGN2K30C, and HMGN2K17/30C) were dissolved in 0.1 M phosphate buffer (pH 7.0) containing 6 M guanidinium hydrochloride (GdnHCl) to a final concentration of 1 mM. Dithiothreitol (DTT) was added to the reaction mixture to a final concentration of 5 mM, followed by 100 equivalents of compound 1. Finally, 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) and dimethyl sulphide were added to the reaction mixture at the final concentrations of 20 mM and 100 mM, respectively. The reaction was allowed to take place in a 42 °C water bath for 2 h. The reaction products were analyzed by LC-MS using a Vydac 218 TP C4 column (4.6 mm × 250 mm, Grace) and ESI-MS. The tBu-protected succinylated HMGN2 proteins were purified by preparative reverse phase HPLC (22 mm × 250 mm, Vydac C4, Grace). The pure fractions were pooled and freeze-dried. The lyophilized tBu-protected succinylated HMGN2 proteins were dissolved in 0.1 M phosphate buffer (pH 7.0) containing 6 M GdnHCl to a final concentration of 5 mM. In the deprotection step, 70% trifluoroacetic acid (TFA) was added dropwise to the mixture in an ice-cold bath and left for 1 h at room temperature with stirring. The deprotected succinylated HMGN2 proteins were then purified by preparative reverse phase HPLC (Vydac C4 column, Grace). The purity and identity of final succinylated HMGN2 mimics were confirmed by ESI-MS (Fig. 2).Preparation of histone H2A–H2B dimers and (H3–H4)2 tetramers
The histone dimers and tetramers were prepared following a previously reported method.15 Briefly, for H2A–H2B dimers, equal amounts of H2A and H2B were mixed and dissolved in unfolding buffer (6 M GdnHCl, 10 mM Tris–HCl, pH 7.5, 1.0 mM EDTA, and 10 mM DTT) to a final histone concentration of 1 mg mL−1. The mixture was incubated on ice for 30 min, followed by dialysis in a histone refolding buffer (2 M NaCl, 10 mM Tris–HCl, pH 7.5, 1.0 mM EDTA, and 5 mM 2-mercaptoethanol) at 4 °C. The refolded dimers were purified using a Superdex 200 pg column (HiLoad 16/60, GE). The same method was used to prepare (H3–H4)2 tetramers and Cy5-labelled H2A–H2B dimers (H2AK119Cy5–H2B).Isothermal titration calorimetry (ITC) experiments
All ITC experiments were performed at room temperature (25 °C) in a buffer (10 mM KCl, 10 mM Tris, pH 7.5, and 0.1 mM EDTA) using a MicroCal PEAQ-ITC. The reaction cell containing 200 μL of 15 μM purified nucleosomes was titrated with the corresponding HMGN2 proteins (450 μM). The titration was started with an initial injection volume of 0.4 μL over 0.8 s, followed by 18 injections (2.0 μL) over 4.0 s spaced at intervals of 150 s for each injection. The reference power was set at 10.0 μcal s−1 and the stirring speed was 750 rpm. The binding isotherm fit was performed using MicroCal PEAQ-ITC analysis software with a single set of sites to determine the thermodynamic binding constants and stoichiometry.Electrophoretic mobility shift assay (EMSA) to examine the HMGN2–nucleosome association
The DNA and histone dimers and tetramers were assembled into nucleosomes by step-wise dialysis as previously reported.18 Briefly, purified histone H2A–H2B dimers and tetramers were incubated with 153 bp of SELEX-generated “Widom 601” DNA (molar ratio of dimer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tetramer:DNA = 2:1:1) at a concentration of 2 μM in 50 μL of reconstitution buffer (2 M KCl, 10 mM Tris, pH 7.8, 0.1 mM EDTA). The samples were transferred into a Slide-A-Lyzer MINI dialysis unit (Thermo) and dialyzed at 4 °C against the reconstitution buffer (concentrations: 1.6 M KCl, 1.4 M KCl, 1.2 M KCl, 1.0 M KCl, 0.8 M KCl, 0.5 M KCl, and 0.2 M KCl) with 50 min for each buffer, followed by 10 mM KCl overnight. The reconstituted nucleosomes were incubated with different amounts of the various HMGN2 proteins in 0.5× TE buffer containing 75 mM NaCl for 15 min at room temperature. The HMGN2–nucleosome mixture was resolved by native-PAGE (5% TBE gel, acrylamide:bis-acrylamide = 29:1). The gel was stained with ethidium bromide (EB) for 15 min and the nucleosome or HMGN2–nucleosome complexes were visualized under UV light using a MyECL Imaging system (Thermo Fisher Scientific).
:tetramer:DNA = 2:1:1) at a concentration of 2 μM in 50 μL of reconstitution buffer (2 M KCl, 10 mM Tris, pH 7.8, 0.1 mM EDTA). The samples were transferred into a Slide-A-Lyzer MINI dialysis unit (Thermo) and dialyzed at 4 °C against the reconstitution buffer (concentrations: 1.6 M KCl, 1.4 M KCl, 1.2 M KCl, 1.0 M KCl, 0.8 M KCl, 0.5 M KCl, and 0.2 M KCl) with 50 min for each buffer, followed by 10 mM KCl overnight. The reconstituted nucleosomes were incubated with different amounts of the various HMGN2 proteins in 0.5× TE buffer containing 75 mM NaCl for 15 min at room temperature. The HMGN2–nucleosome mixture was resolved by native-PAGE (5% TBE gel, acrylamide:bis-acrylamide = 29:1). The gel was stained with ethidium bromide (EB) for 15 min and the nucleosome or HMGN2–nucleosome complexes were visualized under UV light using a MyECL Imaging system (Thermo Fisher Scientific).
Preparation of fluorophore-labelled nucleosomal DNA
The fluorophore-labelled DNA fragments were prepared by PCR using primers labelled with Cy3 as previously reported.14,19–21 The template for the Cy3-labelled DNA used “Widom 601” DNA with base pairs 8–27 replaced with a LexA recognition sequence (TACTGTATGAGCATACAGTA). The PCR products were precipitated with isopropyl alcohol, washed with ethanol dissolved in water, and then purified using a size-exclusion column (GE Superose 6 increase 10/300). The DNA concentration was determined using a UV-vis spectrophotometer (260 nm, NanoDrop 2000, Thermo).Preparation and purification of fluorophore-labelled nucleosomes
The H2AK119Cy5–H2B dimers and (H3–H4)2 tetramers were incubated with Cy3-labelled nucleosomal DNA (molar ratio of dimer:tetramer:DNA = 4:1: 1, with excess amounts of dimers to ensure a fully compacted nucleosome is generated) at a concentration of 2 μM in 50 μL of reconstitution buffer. The dialysis procedure was the same as that for the nucleosomes described above. The reconstituted nucleosomes were purified by native-PAGE (5% TBE gel, acrylamide:bis-acrylamide = 60:1) using a Mini Prep Cell (Bio-Rad, Model 491). The purity of nucleosomes used for the FRET assay was checked by native-PAGE (Fig. S1a, ESI†).
FRET measurement of nucleosomal DNA unwrapping by LexA titration
The accessibility of DNA in the entry/exit region was determined by the reduction in FRET efficiency as LexA binds to its target sites on the nucleosome.21–23 In these experiments, 10 nM Cy3–Cy5-labelled nucleosomes were incubated with HMGN2 in 0.5× TE buffer containing 75 mM NaCl. The LexA protein was titrated into the system and then incubated at room temperature for 10 min. The Cy3–Cy5-labelled nucleosomes were excited using a 531/25 nm bandpass filter for Cy3 and a 635/25 nm bandpass filter for Cy5. The acceptor (Cy5) emission was collected using a 670/25 nm bandpass filter. For each sample, the fluorescence intensity was measured using a plate reader (Victor X5, PerkinElmer) in triplicate. The FRET efficiency was calculated using the (ratio)A method as previously reported24 using the equation:| E = 2[εA(v′′)FA(v′)/FA(v′′) − εA(v′)]/[εD(v′)d+] |
| E = E0 + (EF − E0)/(1 + S1/2/[LexA]), |
Results and discussion
The synthesis of site-specific succinylated HMGN2 mimics
The study of HMGN2 succinylation in vitro requires the generation of homogeneous HMGN2 proteins that carry site-specific succinylations at the stoichiometric level. In our previous study, we developed a cysteine-selective ‘thiol–ene reaction’ to prepare histone H2B carrying a site-specific lysine succinylation analogue (Kcsucc) at K34 (H2BKc34succ) (Fig. 1A). Inspired by our successful incorporation of Kcsucc onto this histone, we aimed to install Kcsucc onto HMGN2, a non-histone protein without cysteine residues. To this end, we focused on two reported succinylation sites on HMGN2, lysine 17 (HMGN2K17) and lysine 30 (HMGN2K30), both located on the nucleosome binding domain (NBD) (Fig. 1B). Therefore, succinylation at these two sites will likely interfere with the interactions between NBD and the nucleosome, resulting in altered nucleosome dynamics.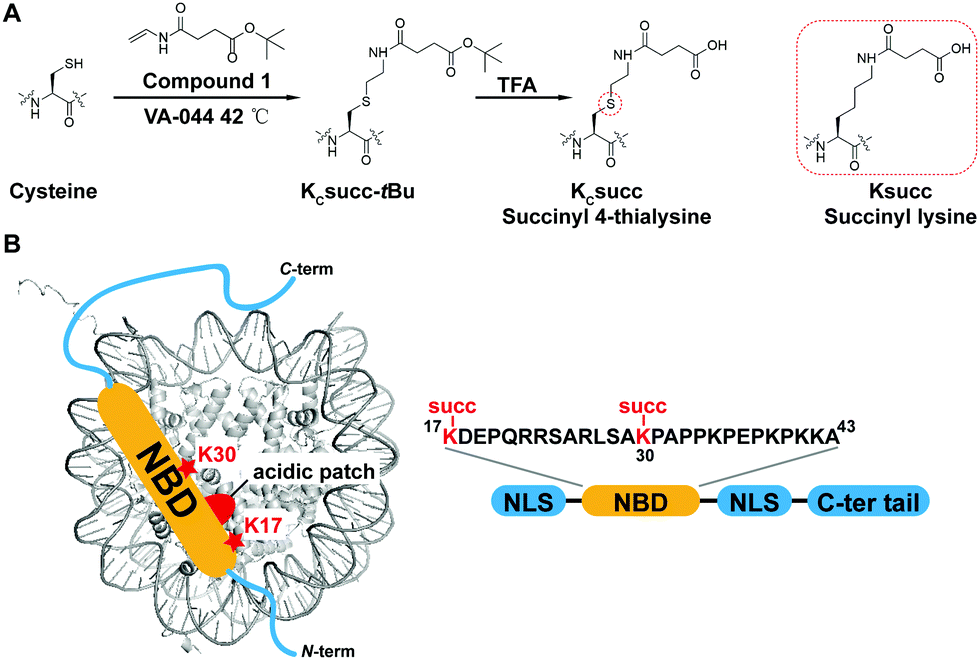 | ||
| Fig. 1 Illustration of the ‘thiol–ene reaction’ for the installation of lysine succinylation marks. (A) Scheme for the installation of the succinyl lysine analogue (Kcsucc) at a cysteine residue. (B) The association of HMGN2 is mediated by its nucleosome-binding domain (NBD). The two reported succinylation sites of HMGN2, K17succ and K30succ, are located within the NBD. | ||
To introduce succinylation into HMGN2, we first obtained recombinant HMGN2 mutants with the site-specific lysines being replaced by cysteines (i.e., HMGN2K17C, HMGN2K30C, and HMGN2K17/30C double K-to-C mutation). The subsequent thiol–ene reaction on the HMGN2 mutants using compound 1 yielded the tBu-protected succinylated HMGN2 mimic. Deprotection of the tBu-protected product with 70% TFA afforded the desired HMGN2 mimic. The purity and identity of the modified HMGN2Kc17succ, HMGN2Kc30succ, and HMGN2Kc17/30succ proteins were examined and confirmed by LC-MS (Fig. 2).
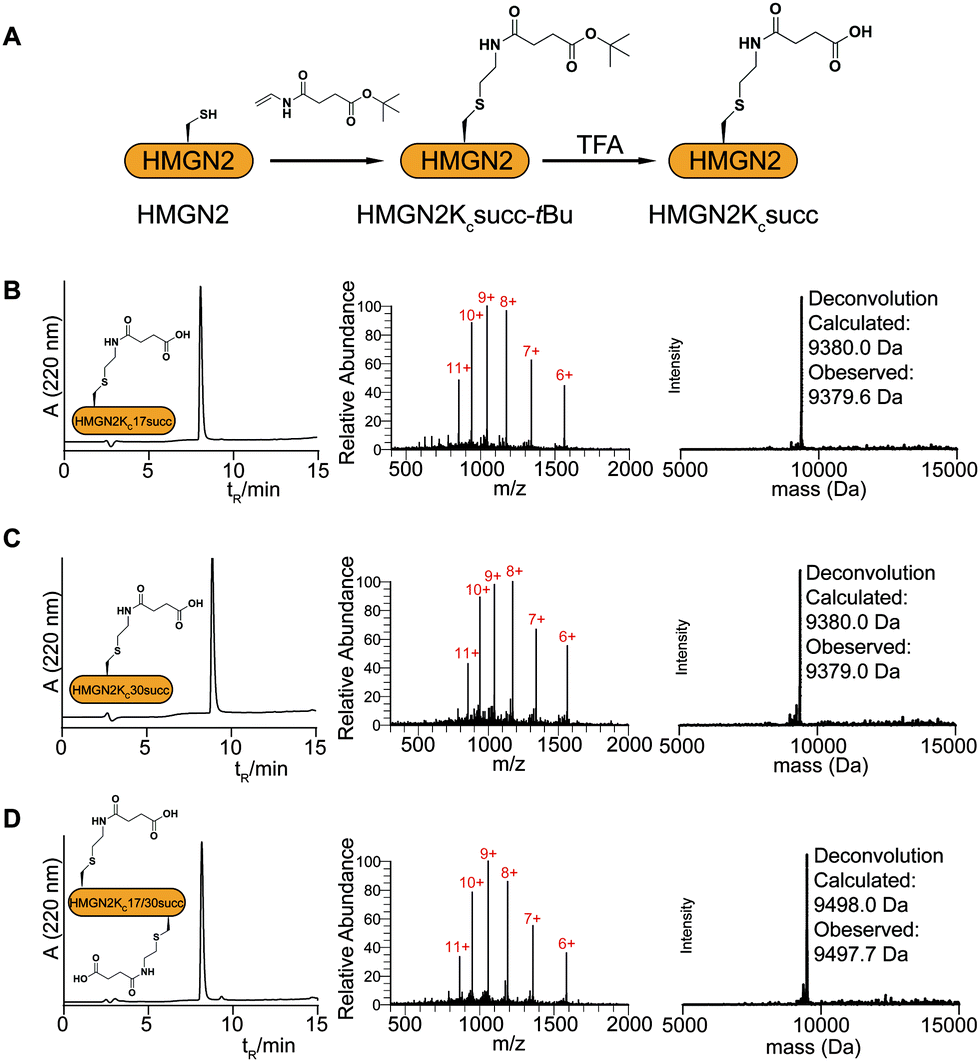 | ||
| Fig. 2 Installation of the succinyl lysine analogue into recombinant HMGN2. (A) Scheme for the installation of the succinyl lysine analogue (Kcsucc) onto HMGN2. LC-MS and deconvolution results for the ‘thiol–ene reaction’ products HMGN2Kc17succ (B), HMGN2Kc30succ (C), and HMGN2Kc17/30succ (D). | ||
Succinylation reduces the HMGN2 binding affinity to nucleosomes
We next investigated the effects of HMGN2 succinylation on its interaction with nucleosomes. The non-histone protein HMGN2 normally binds to mononucleosomes to form a complex containing one nucleosome and two molecules of HMGN2.25 The extent of the assembly of the HMGN2–nucleosome complex can be examined by the gel electrophoresis mobility shift assay (EMSA), in which the inverse proportional mobility can reveal differences in the mass of the particles. For this study, unmodified or succinylated recombinant HMGN2 was incubated with in vitro reconstituted mononucleosomes in different molar ratios, respectively. As shown in Fig. 3, the mononucleosomes were predominantly assembled into the HMGN2–nucleosome complex when titrated at two equivalents of unmodified HMGN2. In contrast, under the same conditions, the HMGN2Kc30succ showed a lower binding affinity to mononucleosomes compared to unmodified HMGN2 and HMGN2Kc17succ, which had similar binding affinities. The HMGN2 with dual succinylation (HMGN2Kc17/30succ) did not show further lowering of the binding affinity compared to HMGN2Kc30succ (Fig. 3). These results suggest that the succinylation of K30 reduces the binding affinity of HMGN2 to the nucleosome.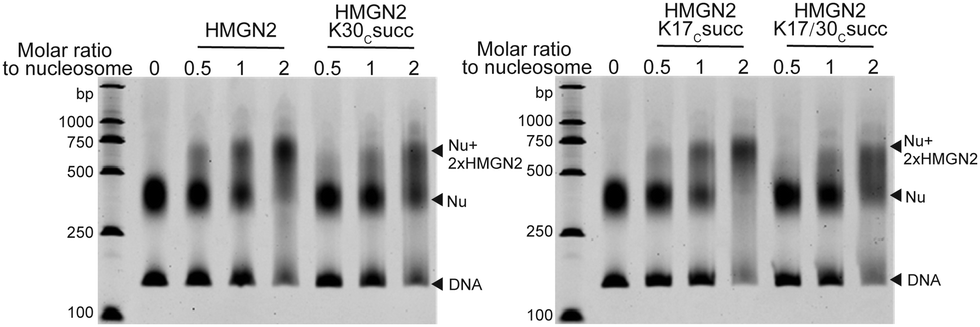 | ||
| Fig. 3 The EMSA assay showing that the association of HMGN2 with the nucleosome was disrupted by HMGN2Kc30succ. | ||
To quantitatively characterize how succinylation marks on HMGN2 affect its interaction with nucleosomes, we performed isothermal titration calorimetry (ITC) to determine the disassociation constants (KD). Compared with unmodified HMGN2, HMGN2Kc30succ showed a four-fold higher KD toward the nucleosome, whereas HMGN2Kc17succ did not alter the binding affinity (Fig. 4). The ITC results reflected our observation from the EMSA that succinylation on HMGN2 K30, which is located within the NBD, plays an essential role in regulating the association of HMGN2 to the nucleosome.
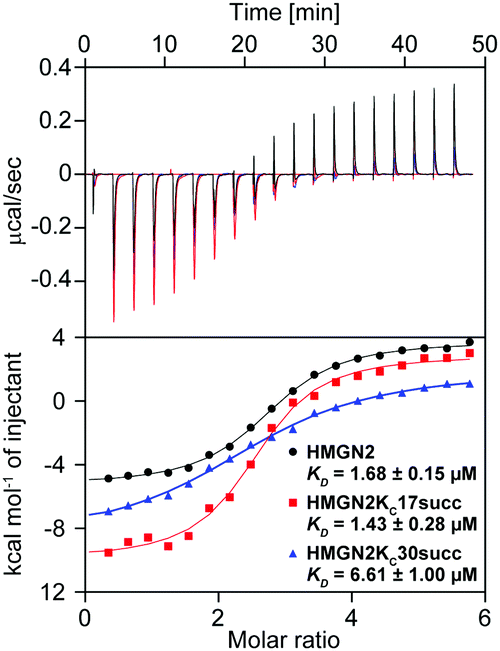 | ||
| Fig. 4 The succinylation at K30 on HMGN2 interferes with HMGN2 binding to the nucleosome as measured by ITC. The purified reconstituted nucleosome was titrated by unmodified HMGN2 (black curve), HMGN2Kc17succ (red curve), and HMGN2Kc30succ (blue curve) at 25 °C. | ||
Succinylation on K30 of HMGN2 regulates nucleosomal DNA accessibility
The NBD of HMGN2, which is enriched with positively charged Lys/Arg residues, forms extensive coulombic interactions with both the negatively charged residues in the nucleosomal acidic patch and DNA in the entry/exit region.25 This is an important feature of HMGN2 NBD, which is expected to decrease the accessibility of DNA at the entry/exit site. To test this hypothesis, we used a fluorescence resonance energy transfer (FRET)-based assay to examine nucleosomal DNA accessibility. This assay can determine the DNA accessibility by measuring the binding of the DNA-binding protein LexA to its target sequence buried within the nucleosome entry/exit region.23,26 Specifically, a LexA target sequence was inserted between base pairs 8 and 27 of the ‘Widom 601’ nucleosomal DNA. The DNA was labelled with a FRET donor Cy3 fluorophore at the 5′ end near the LexA binding site, whereas a FRET acceptor Cy5 fluorophore was linked to K119C on histone H2A (Fig. 5A). In a fully wrapped nucleosome, the distance between these two fluorophores is shorter than their Förster radius, which results in an efficient FRET. However, when the nucleosome partially unwraps, LexA can access and bind to its target site and prevent the rewrapping of the nucleosome, leading to a reduction in the FRET efficiency (Fig. 5B). We titrated LexA into the fluorophore-labelled nucleosome with or without the unmodified HMGN2 and monitored the change in FRET efficiency. The titration curves were then fitted to a non-cooperative binding isotherm to determine S1/2, which is the LexA concentration at which half of the nucleosomes are bound by LexA. As shown in Fig. 5C, compared with the untreated nucleosome, the S1/2 increased 2.6-fold in the nucleosome treated with 10 nM HMGN2. Besides, when the concentration of HMGN2 was increased (30 and 300 nM), the S1/2 values of HMGN2-treated nucleosomes also increased (Fig. S1B, ESI†). These results verified our hypothesis that the nucleosomal DNA at the entry/exit site in the HMGN2–nucleosome complex is constrained by HMGN2, resulting in decreased DNA accessibility.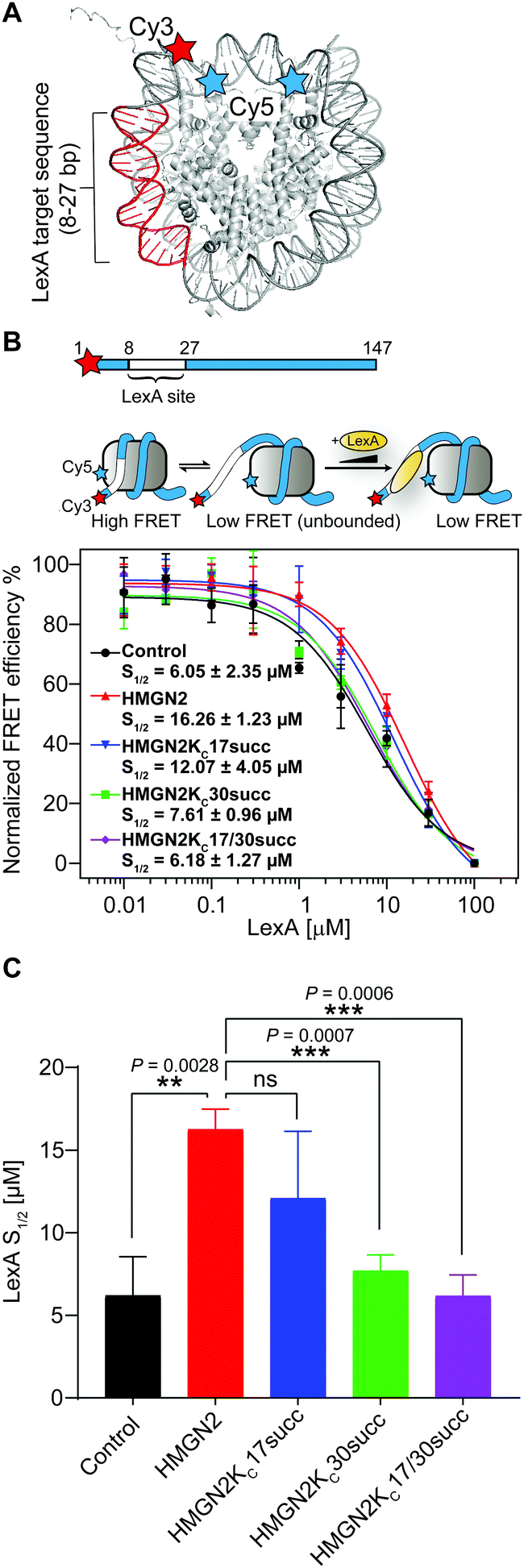 | ||
| Fig. 5 The succinylation at K30 on HMGN2 increases nucleosomal DNA accessibility. (A) Crystal structure of the nucleosome (PDB: 3LZ1). The locations of the Cy3/Cy5 FRET pair and LexA binding sites. (B) The nucleosomal DNA used for the FRET assay is a 147 bp Widom 601 DNA with a LexA binding site from 8–27 bp and labelled with Cy3 at the 5′ end. The three-state model showed decreasing FRET efficiency as the LexA is titrated in and binds to its target site. The plot shows the normalised FRET intensity as a function of LexA concentration for the nucleosome without HMGN2 treatment (Black) or treated with unmodified HMGN2 (red), HMGN2Kc17succ (blue), HMGN2Kc30succ (green), and HMGN2Kc17/30succ (purple). The LexA concentration at which the FRET decreases by 50% is denoted as S1/2. For visualization, all curves were normalized between 100% at the maximum FRET value and 0% at the highest LexA concentration. Titrations were repeated in triplicate for each experiment (n = 3, S1/2 = mean ± SD). (C) The histogram shows the S1/2 values of nucleosomes (n = 3, S1/2 = mean ± SD). The P values were based on a two-tailed Student's t test. **P < 0.01. ***P < 0.001. ns, no significance. | ||
We then investigated the effects of HMGN2 succinylation on regulating nucleosomal DNA accessibility using the same experimental design with the three succinylated HMGN2 (i.e., HMGN2Kc17succ, HMGN2Kc30succ, and HMGN2Kc17/30succ). The results showed that the presence of the Kcsucc mark at K30 of HMGN2 substantially decreased the nucleosome S1/2 value to 7.61 μM compared with the unmodified HMGN2, and also approached the level in HMGN2-free nucleosomes (6.05 μM). This finding suggests that the HMGN2Kc30succ shifts the nucleosomal DNA breathing equilibrium toward the unwrapped state, thereby increasing the accessibility of DNA in the entry/exit region. On the other hand, the introduction of succinylation on K17 of HMGN2 (HMGN2Kc17succ) caused a moderate decrease in the S1/2 value of the treated nucleosome compared with the unmodified HMGN2-treated nucleosome. Meanwhile, HMGN2Kc17/30succ with dual Kcsucc marks resulted in only a slight decrease in the S1/2 value compared to the HMGN2Kc30succ-treated nucleosome (6.18 μM vs. 7.61 μM). These results suggest that lysine succinylation on K30 of HMGN2 plays a predominant role in increasing nucleosomal DNA accessibility.
Conclusions
In this study, we expanded the application of the ‘thiol–ene reaction’ to prepare site-specific succinylated non-histone proteins (HMGN2Kc17succ, HMGN2Kc30succ, and HMGN2Kc17/30succ). The results of EMSA and ITC assays revealed the introduction of negatively charged lysine succinylation on K30 of HMGN2 changing the net charge of the modified lysine residues from +1 to −1 and significantly disrupting the association of HMGN2 with the mononucleosome. Besides, the FRET-based assay demonstrated that the NBD domain of HMGN2 was required in binding to the acidic patch on the histones and the DNA in the entry/exit region, which decreased the nucleosomal DNA accessibility by constraining the nucleosomal DNA breathing. On the contrary, succinylation on K30 of HMGN2 introduces a negatively charged Ksucc mark, leading to increased nucleosomal DNA accessibility (Fig. 6). Besides K17 and K30 sites on HMGN2, many other lysine succinylation sites have been identified in several non-histone proteins.12,27 A comprehensive investigation of non-histone lysine succinylation will help us to better understand the novel functions of this PTM in regulating chromatin-templated biological processes.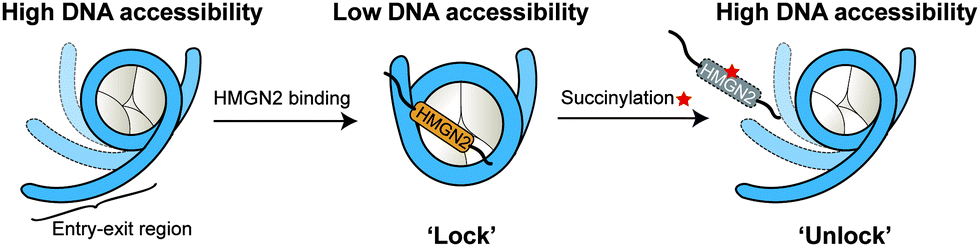 | ||
| Fig. 6 The function of lysine succinylation in regulating the nucleosomal DNA accessibility by promoting the release of HMGN2 from nucleosomes. In the HMGN2–nucleosome complex (middle), the DNA in the entry/exit region is constrained by HMGN2, which results in a decrease of DNA accessibility. The succinylation on K30 induces the dissociation of HMGN2 from the nucleosome and ‘unlocks’ the DNA to be accessible. | ||
Author contributions
Z. L. and X. D. L. conceived and designed the project. Y. J. synthesized the small-molecule compounds and performed the protein chemistry, nucleosome reconstitution, and FRET experiments. G. T. purified the LexA protein. X. Q. performed the ITC experiments. Z. L. and X. D. L. wrote the manuscript with input from G. T.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the support from the Excellent Young Scientists Fund of China (Hong Kong and Macau) (21922708), the Hong Kong Research Grants Council Collaborative Research Fund (CRF C7028-19G, C7058-18G, and C7009-20G), Areas of Excellence Scheme (AoE/P-705/16) and General Research Fund (GRF 17125917, 17126618, and 17121120).Notes and references
- J. E. Kugler, T. Deng and M. Bustin, Biochim. Biophys. Acta, 2012, 1819, 652–656 Search PubMed.
- D. J. Tremethick and L. Hyman, J. Biol. Chem., 1996, 271, 12009–12016 Search PubMed.
- M. Rochman, Y. Postnikov, S. Correll, C. Malicet, S. Wincovitch, T. S. Karpova, J. G. McNally, X. Wu, N. A. Bubunenko, S. Grigoryev and M. Bustin, Mol. Cell, 2009, 35, 642–656 Search PubMed.
- Y. V. Postnikov, G. I. Belova, J. H. Lim and M. Bustin, Biochemistry, 2006, 45, 15092–15099 Search PubMed.
- Y. Birger, K. L. West, Y. V. Postnikov, J. H. Lim, T. Furusawa, J. P. Wagner, C. S. Laufer, K. H. Kraemer and M. Bustin, EMBO J., 2003, 22, 1665–1675 Search PubMed.
- M. Bergel, J. E. Herrera, B. J. Thatcher, M. Prymakowska-Bosak, A. Vassilev, Y. Nakatani, B. Martin and M. Bustin, J. Biol. Chem., 2000, 275, 11514–11520 Search PubMed.
- J. E. Herrera, K. Sakaguchi, M. Bergel, L. Trieschmann, Y. Nakatani and M. Bustin, Mol. Cell. Biol., 1999, 19, 3466–3473 Search PubMed.
- M. Prymakowska-Bosak, T. Misteli, J. E. Herrera, H. Shirakawa, Y. Birger, S. Garfield and M. Bustin, Mol. Cell. Biol., 2001, 21, 5169–5178 Search PubMed.
- J. Wu, S. Kim, M. S. Kwak, J. B. Jeong, H. J. Min, H. G. Yoon, J. H. Ahn and J. S. Shin, J. Biol. Chem., 2014, 289, 20000–20011 Search PubMed.
- B. T. Weinert, C. Scholz, S. A. Wagner, V. Iesmantavicius, D. Su, J. A. Daniel and C. Choudhary, Cell Rep., 2013, 4, 842–851 Search PubMed.
- J. Park, Y. Chen, D. X. Tishkoff, C. Peng, M. Tan, L. Dai, Z. Xie, Y. Zhang, B. M. Zwaans, M. E. Skinner, D. B. Lombard and Y. Zhao, Mol. Cell, 2013, 50, 919–930 Search PubMed.
- Z. Zhang, M. Tan, Z. Xie, L. Dai, Y. Chen and Y. Zhao, Nat. Chem. Biol., 2011, 7, 58–63 Search PubMed.
- Y. H. Jing, D. B. Ding, G. F. Tian, K. C. J. Kwan, Z. Liu, T. Ishibashi and X. D. Li, Nucleic Acids Res., 2020, 48, 9538–9549 Search PubMed.
- Y. Jing, Z. Liu, G. Tian, X. Bao, T. Ishibashi and X. D. Li, Cell Chem. Biol., 2018, 25, 166–174 Search PubMed.
- K. Luger, T. J. Rechsteiner and T. J. Richmond, Methods Mol. Biol., 1999, 119, 1–16 Search PubMed.
- J. H. Lim, F. Catez, Y. Birger, Y. V. Postnikov and M. Bustin, Methods Enzymol., 2004, 375, 323–342 Search PubMed.
- J. W. Little, B. Kim, K. L. Roland, M. H. Smith, L. L. Lin and S. N. Slilaty, Methods Enzymol., 1994, 244, 266–284 Search PubMed.
- K. Luger, T. J. Rechsteiner, A. J. Flaus, M. M. Y. Waye and T. J. Richmond, J. Mol. Biol., 1997, 272, 301–311 Search PubMed.
- A. Gansen, K. Toth, N. Schwarz and J. Langowski, Nucleic Acids Res., 2015, 43, 1433–1443 Search PubMed.
- V. Bohm, A. R. Hieb, A. J. Andrews, A. Gansen, A. Rocker, K. Toth, K. Luger and J. Langowski, Nucleic Acids Res., 2011, 39, 3093–3102 Search PubMed.
- M. Simon, J. A. North, J. C. Shimko, R. A. Forties, M. B. Ferdinand, M. Manohar, M. Zhang, R. Fishel, J. J. Ottesen and M. G. Poirier, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12711–12716 Search PubMed.
- J. C. Shimko, J. A. North, A. N. Bruns, M. G. Poirier and J. J. Ottesen, J. Mol. Biol., 2011, 408, 187–204 Search PubMed.
- G. Li and J. Widom, Nat. Struct. Mol. Biol., 2004, 11, 763–769 Search PubMed.
- R. M. Clegg, Methods Enzymol., 1992, 211, 353–388 Search PubMed.
- H. Kato, H. van Ingen, B. R. Zhou, H. Feng, M. Bustin, L. E. Kay and Y. Bai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12283–12288 Search PubMed.
- G. Li, M. Levitus, C. Bustamante and J. Widom, Nat. Struct. Mol. Biol., 2005, 12, 46–53 Search PubMed.
- C. Peng, Z. Lu, Z. Xie, Z. Cheng, Y. Chen, M. Tan, H. Luo, Y. Zhang, W. He, K. Yang, B. M. Zwaans, D. Tishkoff, L. Ho, D. Lombard, T. C. He, J. Dai, E. Verdin, Y. Ye and Y. Zhao, Mol. Cell. Proteomics, 2011, 10, M111 012658 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Purity of nucleosomes, the result of FRET assay. See DOI: 10.1039/d1cb00070e |
| ‡ Y. J. and G. T. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |