
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo active organometallic-containing antimycotic agents†
Riccardo
Rubbiani
*a,
Tobias
Weil
 *b,
Noemi
Tocci
b,
Luciano
Mastrobuoni
a,
Severin
Jeger
a,
Marco
Moretto
c,
James
Ng
d,
Yan
Lin
d,
Jeannine
Hess
a,
Stefano
Ferrari
e,
Andres
Kaech
f,
Luke
Young
g,
John
Spencer
g,
Anthony L.
Moore
h,
Kevin
Cariou
*d,
Giorgia
Renga
i,
Marilena
Pariano
i,
Luigina
Romani
i and
Gilles
Gasser
*d
*b,
Noemi
Tocci
b,
Luciano
Mastrobuoni
a,
Severin
Jeger
a,
Marco
Moretto
c,
James
Ng
d,
Yan
Lin
d,
Jeannine
Hess
a,
Stefano
Ferrari
e,
Andres
Kaech
f,
Luke
Young
g,
John
Spencer
g,
Anthony L.
Moore
h,
Kevin
Cariou
*d,
Giorgia
Renga
i,
Marilena
Pariano
i,
Luigina
Romani
i and
Gilles
Gasser
*d
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: rubbianiric@gmail.com
bDepartment of Food Quality and Nutrition, Research and Innovation Centre, Fondazione Edmund Mach, Via E. Mach 1, 38010 San Michele all'Adige, Italy. E-mail: tobias.weil@fmach.it
cUnit of Computational Biology, Research and Innovation Centre, Fondazione Edmund Mach, Via E. Mach 1, 38010 San Michele all'Adige, Italy
dChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: kevin.cariou@chimieparistech.psl.eu; gilles.gasser@chimieparistech.psl.eu
eInstitute of Molecular Cancer Research, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
fCenter for Microscopy and Image Analysis, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
gDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK
hBiochemistry & Biomedicine, School of Life Sciences, University of Sussex, Brighton BN1 9QG, UK
iUniversity of Perugia, Department of Medicine and Surgery, Piazzale Lucio Severi – Polo Unico Sant’Andrea delle Fratte, 06132 Perugia, Italy
First published on 8th July 2021
Abstract
Fungal infections represent a global problem, notably for immunocompromised patients in hospital, COVID-19 patient wards and care home settings, and the ever-increasing emergence of multidrug resistant fungal strains is a sword of Damocles hanging over many healthcare systems. Azoles represent the mainstay of antifungal drugs, and their mode of action involves the binding mode of these molecules to the fungal lanosterol 14α-demethylase target enzyme. In this study, we have prepared and characterized four novel organometallic derivatives of the frontline antifungal drug fluconazole (1a–4a). Very importantly, enzyme inhibition and chemogenomic profiling demonstrated that lanosterol 14α-demethylase, as for fluconazole, was the main target of the most active compound of the series, (N-(ferrocenylmethyl)-2-(2,4-difluorophenyl)-2-hydroxy-N-methyl-3-(1H-1,2,4-triazol-1-yl)propan-1-aminium chloride, 2a). Transmission electron microscopy (TEM) studies suggested that 2a induced a loss in cell wall integrity as well as intracellular features ascribable to late apoptosis or necrosis. The impressive activity of 2a was further confirmed on clinical isolates, where antimycotic potency up to 400 times higher than fluconazole was observed. Also, 2a showed activity towards azole-resistant strains. This finding is very interesting since the primary target of 2a is the same as that of fluconazole, emphasizing the role played by the organometallic moiety. In vivo experiments in a mice model of Candida infections revealed that 2a reduced the fungal growth and dissemination but also ameliorated immunopathology, a finding suggesting that 2a is active in vivo with added activity on the host innate immune response.
Introduction
Mycoses are one of the most common opportunistic infections worldwide, affecting poor as well as industrialized countries. Skin infections (e.g. athlete's foot), for example, affect 20–25% of the world's population and systemic opportunistic infections are the 4th common cause of bloodstream infections with a lifetime incidence of about 75% (e.g. in the case of Candidiasis).1–6 Even if such extensive morbidity does not lead to a high mortality rate, local opportunistic infections together with invasive fungal infections (IFI) have a deep impact on the entire health system because they suppress the immune system and thus, increase the risk to develop other/additional pathologies, which can be fatal to immunosuppressed people.7–16 Although potent drugs (e.g. clotrimazole) are applied in antifungal therapy, the infection rate is worryingly increasing every year. Moreover, incidences of resistance to conventional antimycotics are steadily increasing, especially for long term therapies that last from several weeks to months.17 Main mechanisms necessary for antifungal resistance are drug target mutations (e.g. mutations of CYP51A1), upregulation of efflux channels (e.g. major facilitator super-family (MFS) and ATP-binding cassette (ABC channels)), aneuploidy and loss of heterozygosity (LOH) and modulation of the stress response (e.g. translational signal pathways, RNA dependent response).18–21 Surprisingly, despite a pressing need for novel antifungal agent research and drug development, there is an apparent downturn in such activities.22 However, the recent COVID-19 pandemic has witnessed numerous examples of COVID-19 and C. auris co-infections, adding further pressure to healthcare systems.23Cell wall targeting is at the basis of modern antifungal therapy since it is absent in mammalian cells allowing for selectivity. A selective destabilization or inhibition of cell wall biosynthesis leads to metabolic onset, growth arrest and culminates in fungal death. For decades, Amphotericin B deoxycholate (AmB) has been the only available treatment for invasive fungal infections (IFIs).24 However, during the last ten years, the growing number of IFI incidences has stimulated the introduction of four new classes of antifungal agents, namely polyene-based compounds, azoles, allylamines and echinocandins.25–28 Inhibition of cell wall biosynthesis is associated with an accumulation of sterol toxic precursors, cell wall destabilization and cellular stress. During the second half of the 20th century, antifungal research mainly focused on an intensive derivatization of accepted compounds (e.g. from ketoconazole to fluconazole of from AmB to Nystatin and AmB liposomes).19–21 Despite these efforts, to date, only seven compounds are included in the World Health Organization Essential Medicine list (WHO-EM, data of October 2013).1–6 Moreover, the benchmark drugs were all discovered almost 20 or more years ago and accounts of ineffective therapies have been reported for several of these agents especially towards Candida albicans (C. albicans) and Aspergillus terreus.1–6 For this reason, and encouraged by the spectacular results obtained with metal complexes (e.g. salvarsan, cisplatin, auranofin, ferroquine, ferrocifen, etc.) in other medicinal fields,29–38 some groups have decided to assess the potential of such compounds to fight fungal infections.39,40 Despite such a wealth of promising examples of metal-based drugs, there is a paucity of examples of such compounds applied in the field of antifungal therapy. Here, just sporadic reports confirmed the effectiveness of different transition metal complexes (e.g. Co(II), Ni(II), Cu(II), Pd(II) complexes) bearing a variety of ligands (e.g. azole moieties, thiosemicarbazones, carboxamides, indoles) against several fungal strains or some organometallic moieties bound to active antifungal agents.41–53 However, all these studies generally provided only MIC50 values without any further biological investigation such as target engagement and toxicity. Willing to grasp this opportunity, we embarked on a research program to unveil potent complexes against fungal infections with an emphasis on addressing cellular targets and mode of action. Herein, we present the identification of a metallocene-based lead compound against such infections.
Results and discussion
Synthesis and characterization
The two most common antifungal drugs inscribed in the List of Essential Medicine of the WHO and commercially available, namely clotrimazole and fluconazole (Fig. 1), belong to the azole class. The molecular target for both those azoles was found to be the enzyme lanosterol-14α-demethylase.47 Of utmost interest, mammalian cells use cholesterol instead of ergosterol. This important difference secures high selectivity for the reported antimycotics.54 Recently, the broad therapeutic applications of clotrimazole as well as fluconazole against skin, genital and invasive fungal infections have generated comparison of primary or cross resistance, especially for Candidiasis.55,56 Motivated by the promising results obtained from the ferrocenyl derivatization of organic drugs such as tamoxifen and chloroquine to give ferrocifen and ferroquine, respectively, we envisaged applying the same concept to fight fungal infections.32,49,57–59 Fluconazole has been chosen as the parent drug because of its broad spectrum applications (e.g. systemic and topic administration, active against a large number of mycoses). The drug design took into consideration the mode of action of fluconazole and its interaction with the active site of the target enzyme. Computing binding geometry data showed how fluconazole activity is exerted by the interaction of the triazole moiety (i.e. the nitrogen atom highlighted in blue in Fig. 1) with the haem group present in the active site of the target enzyme (i.e. the iron atom).60 This interaction is also favoured by the presence of the difluorophenyl group that is located in the enzyme active site, in (close) proximity to the hydrophobic binding cleft and interacts with it via π–π stacking.60 On the contrary, the second triazole moiety is involved in non-bonding interactions with several prosthetic groups present in the enzyme cavity. Interesting work by Sheng and co-workers, involving protein docking studies, demonstrated that organic modification with different functional groups at the C3 atom of fluconazole resulted in an increased activity in relation to the parent drug. This was supported by MIC80 values. Therefore, in the present case, we aimed at derivatizing the fluconazole core on the triazole not involved in the binding pocket interaction, replacing it with a ferrocenyl moiety. Ferrocenyl derivatization is proposed to play an important role for the overall biodistribution and uptake (e.g. increasing the lipophilicity of the parent drug) as well as in potentially allowing for an additional redox-induced mode of action as observed for ferrocifen and ferroquine.32,57,58,61 Finally, different alkyl substituents were inserted at the bridging nitrogen in order to garner Structure–Activity Relationship (SAR) information. The target ferrocenyl compounds of this study, namely 1a–4a can be seen in Fig. 1. Herein, we name them fluconazenes.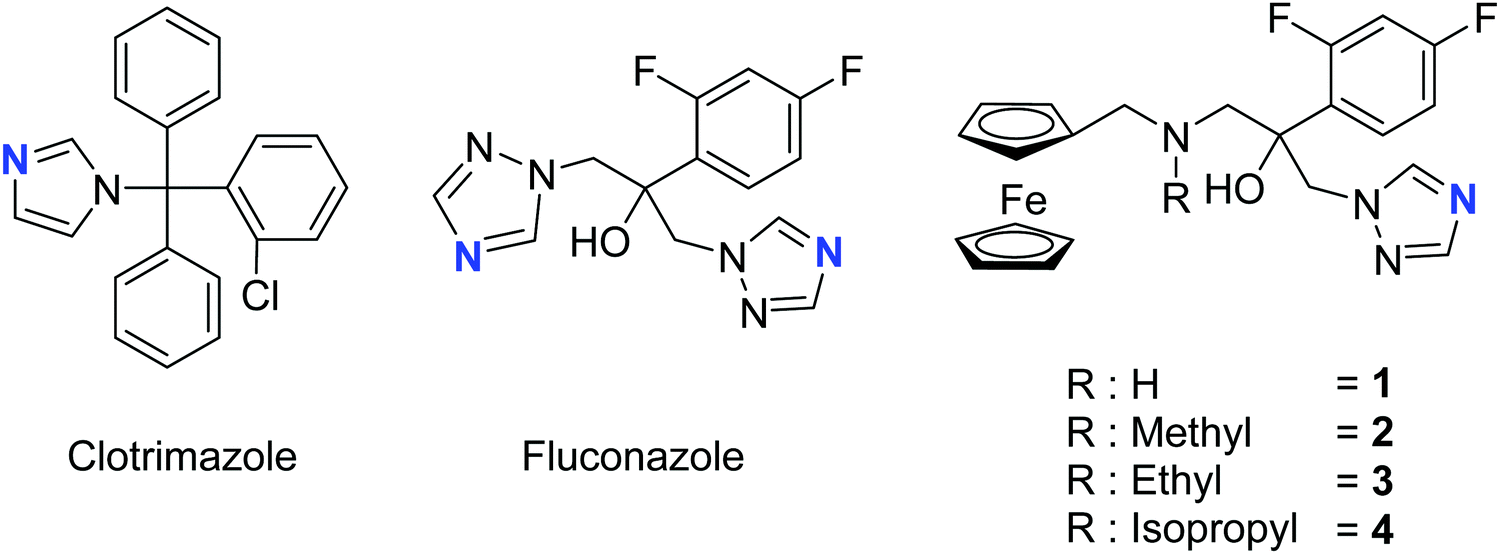 | ||
| Fig. 1 Chemical structures of the main antifungal therapeutics present on the market (clotrimazole and fluconazole) and our fluconazenes 1–4, highlighted in blue are the nitrogen responsible for the binding with the haem group. | ||
The synthesis of the new fluconazenes 1–4 can be visualized in Scheme 1. In the envisioned retrosynthesis (Scheme 1A), the target compounds would be obtained from the addition of a ferrocenylamine (5) on the epoxide 6, easily accessible from 1,2,4-triazolo-ketone 7, which is commercially available, by a Corey–Chaykovski epoxidation. The ferrocenylmethanamines (5a–d) were prepared from ferrocene carboxaldehyde (8) in two steps, following adapted procedures by Tice et al. and Baramee et al.62,63 Reaction of the aldehyde (8) with hydroxylamine hydrochloride under basic conditions yielded the oxime (9a). The imines (9b–d) were obtained by stirring 8 in the presence of a solution of the corresponding alkyl amines. The intermediates (9a–d) were reduced to amines (5a–d) with LiAlH4 and NaBH4, respectively. Spectroscopic data of 5a–d matched those reported in the literature (Scheme 1B).62–64 Epoxide 6 was obtained in 76% yield by treating ketone 5 with a solution of trimethylsulfoxonium iodide ylide (Scheme 1C).60,65 The subsequent epoxide ring opening of 6 with the primary and secondary ferrocenemethanamines (5a–d) yielded compounds 1–4.64,65 The derivative carrying the methylamine linker (2) was obtained in noticeably higher yield (46%) than the other derivatives (1, 30%; 3, 31%; 4, 20%). This can be attributed to the donating inductive effect of the alkyl groups on the N atom, which increases the nucleophilic strength of 9b–d. In contrast, the growing steric hindrance of the ethyl and isopropyl group can explain the decreasing yield within the series of tertiary amines (2 > 3 > 4).
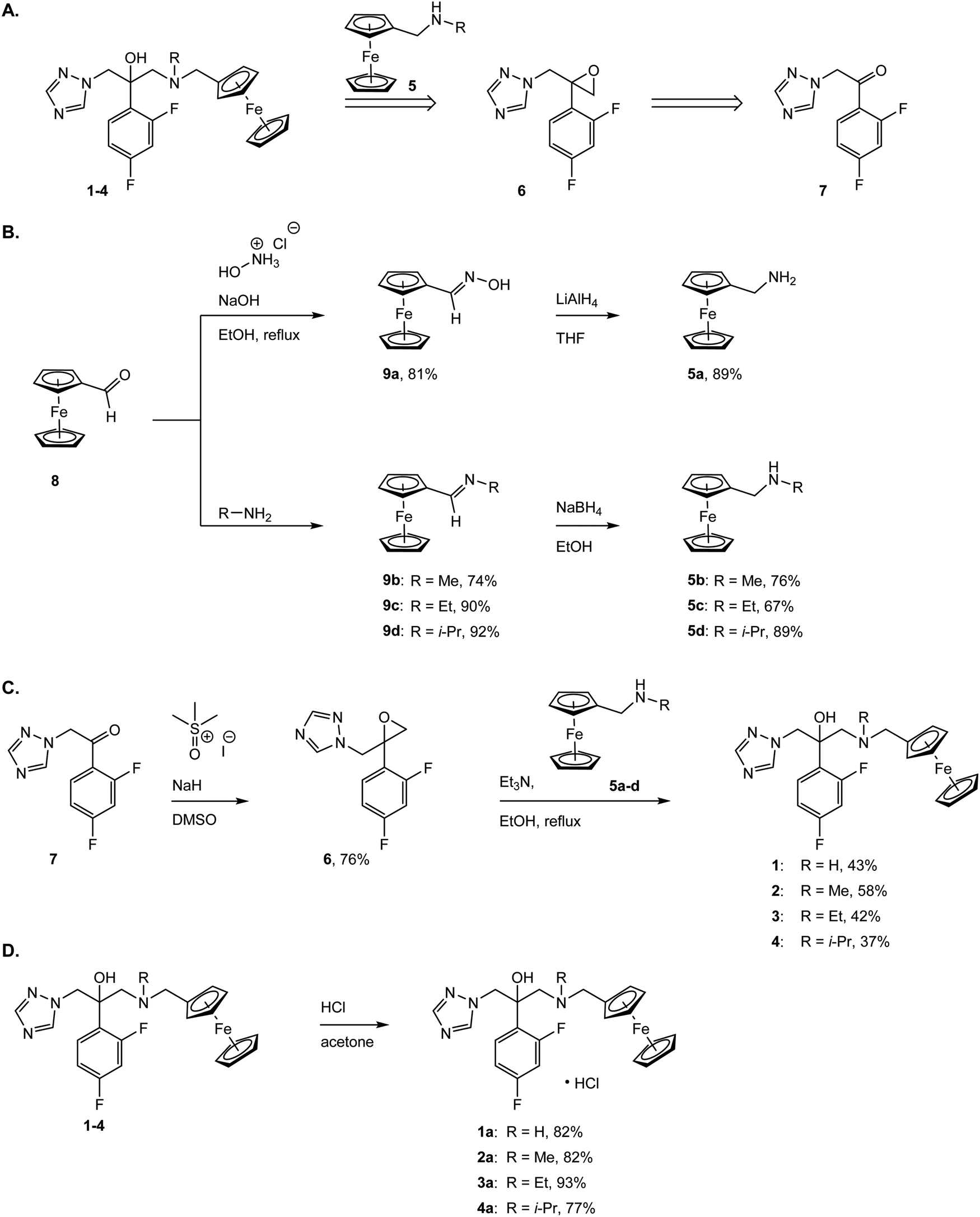 | ||
| Scheme 1 (A) Retrosynthetic analysis; (B) preparation of the primary (7a) and secondary (7b–d) ferrocenylmethanamines via the oxime (9a) and imine (9b–d) intermediates. Overall yields, comprising both steps: 7a, 71%; 7b, 56%; 7c, 60%; 7d, 81%; (C) synthesis of the organometallic fluconazole derivatives (1–4), which feature an amine-linked ferrocene moiety; (D) synthesis of the hydrochloride salts of 1–4 (1a–4a). | ||
While fluconazole is achiral, with two identical 1,2,4-triazolylmethyl side chains attached to the C2 carbon, our derivatives 1–4 are chiral. However, according to molecular docking experiments of chiral fluconazole derivatives by Sheng et al., both R and S isomers, can interact with the active site of the C. albicans CYP51 through a similar binding mode,66 thus avoiding the need for a chiral separation, or an asymmetric synthesis of a specific enantiomer.67 The derivatives 1–4 were converted to the corresponding hydrochloride salts by treatment with HCl in acetone, following an adapted procedure of Bader et al. (Scheme 1D).68 Overall, the new complexes 1a–4a were all characterized via1H, 13C, 19F NMR, MS, IR and their purity was confirmed via elemental analysis and UPLC-MS (see Fig. S2–S29 in ESI†).
Stability
As potential drug candidates, the hydrochloride salts 1a–4a need to be stable in DMSO, which is the administering medium for in vitro and in vivo studies, as well as in the biological medium, namely water. It was previously demonstrated that this can be problematic for metal-based drugs.69–71 The stability of 1a–4a was assayed by dissolving the salts in deuterated solvents, namely neat DMSO-d6 and neat D2O, and by monitoring the samples using 1H NMR spectroscopy over a period of 48 hours (see ESI† for details). It is worth mentioning that 1a and 4a are only sparingly soluble in D2O. Generally, all four hydrochloride salts were stable for up to 24 hours and only little decomposition was observed for 2a and 4a in water after 48 h.In vitro biological screening
To assess the antifungal activity of the new drug candidates, we employed a well-plate based assay recently developed in our group.39 The first antimycotic investigation was performed on the solid and largely used model system S. cerevisiae. Therefore, S. cerevisiae was cultured and diluted to reach the beginning of the growing phase just before treatment. YPD agar terrain at increasing concentrations of the target complexes were poured on a 6 or 12-wells plate and the S. cerevisiae culture were then spotted on them and incubated for 24 h at 30 °C. Interestingly, all compounds displayed an improved antifungal activity compared to the parent drug (e.g. see Fig. 2). Small alkyl group-substituted tertiary amines (complexes 2a and 3a) as linkers, showed an improved profile, between 9–17-fold, compared with secondary amines (see Table 1).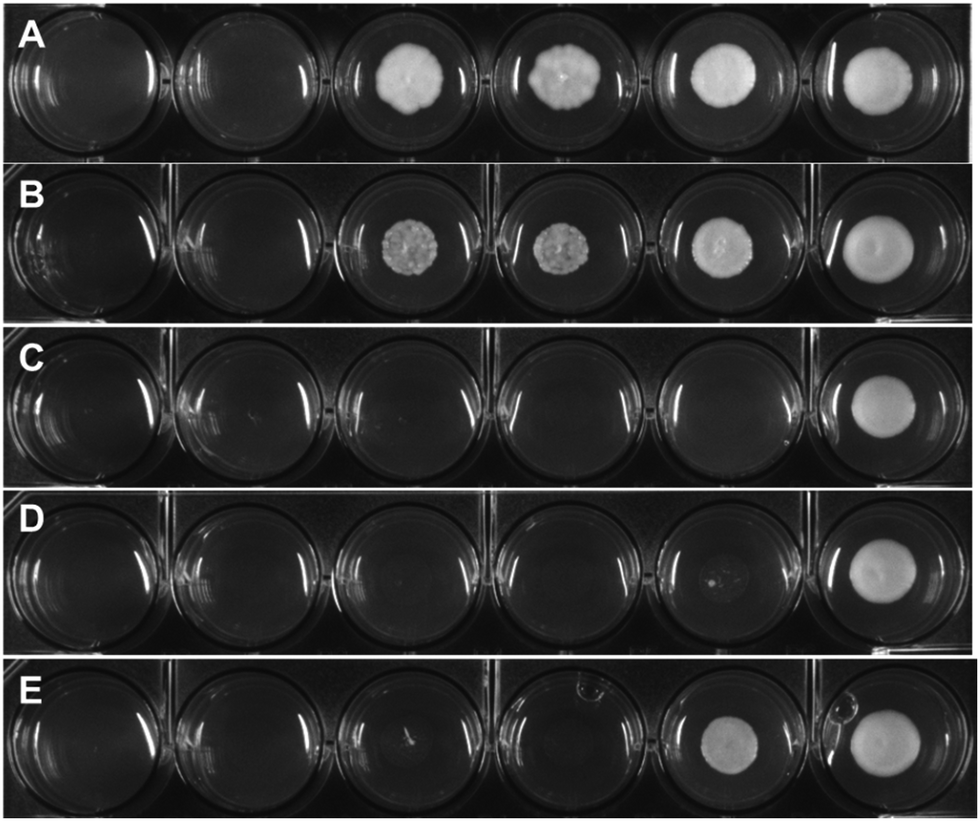 | ||
| Fig. 2 Depicted example of a colony formation inhibition experiment; (A) S. cerevisiae treated with fluconazole; (B) S. cerevisiae treated with 1a; (C) S. cerevisiae treated with 2a; (D) S. cerevisiae treated with 3a; (E) S. cerevisiae treated with 4a. S. cerevisiae has been treated with increasing concentrations of the target drug candidates ranging from 2.5 up 200 μM (higher concentration not depicted) and incubated at 30 °C for 24 h. The experiments with the drug candidates and with negative control of untreated colony have been performed in triplicate. | ||
| Compound | MRC5 | RPE | S. cerevisiae |
|---|---|---|---|
| Fluconazole | >100 | >100 | 51.0 ± 3.7 |
| 1a | 94.6 ± 2.4 | >100 | 13.2 ± 3.0 |
| 2a | 32.2 ± 0.1 | 74.7 ± 5.2 | 3.02 ± 2.18 |
| 3a | 70.4 ± 17.9 | 51.2 ± 0.1 | 5.86 ± 0.41 |
| 4a | 59.4 ± 2.8 | 48.4 ± 2.9 | 9.62 ± 0.56 |
To further evaluate the potential of the compounds, we investigated their host toxicity, in vitro mycotoxicity and cellular uptake. An effective inhibitor would need a good therapeutic window, with low host toxicity versus effective nM fungal toxicity The first investigation on the new antimycotic drug candidates involved their possible influence on host cells and their efficiency towards a common fungal model. We investigated the antiproliferative effect of 1a–4ain vitro on human retinal pigment epithelial cell (RPE-1-hTert) and human fibroblast (MRC-5). As expected, fluconazole displayed only moderate effects on cell viability. All complexes displayed IC50 values in the mid-high micromolar range, following the order of potency: isopropyl (4a) > ethyl (3a) > methyl (2a) > H (1a) (see Table 1). This suggests that a decrease in the size of the N-substituent (i.e., a decrease in lipophilicity) correlates with a decrease in host toxicity. Overall, while the organometallic compounds are slightly more toxic than fluconazole, the toxicity remains relatively moderate, especially taking into account that the toxicity towards yeast is much higher (see below).
In order to investigate the pharmaco-dynamic profile of the target antimycotic drug candidates, a series of uptake studies in S. cerevisiae were performed. The uptake measurements were performed via ICP-MS by detecting the free iron content in the culture medium and normalizing it against the colony density at different incubation times (1 h, 6 h and 18 h). The results were then compared with the survival rate measured via OD (see Fig. 3). The uptake of the target complexes followed the order 4a ≪ 1a < 3a < 2a and fits very well with their time-dependent mycotoxicity. This suggests that complexes with sterically-hindered groups like an isopropyl or the less lipophilic complex 1a showed a lower efficacy than ones with short chain alkyl groups (2a, 3a).
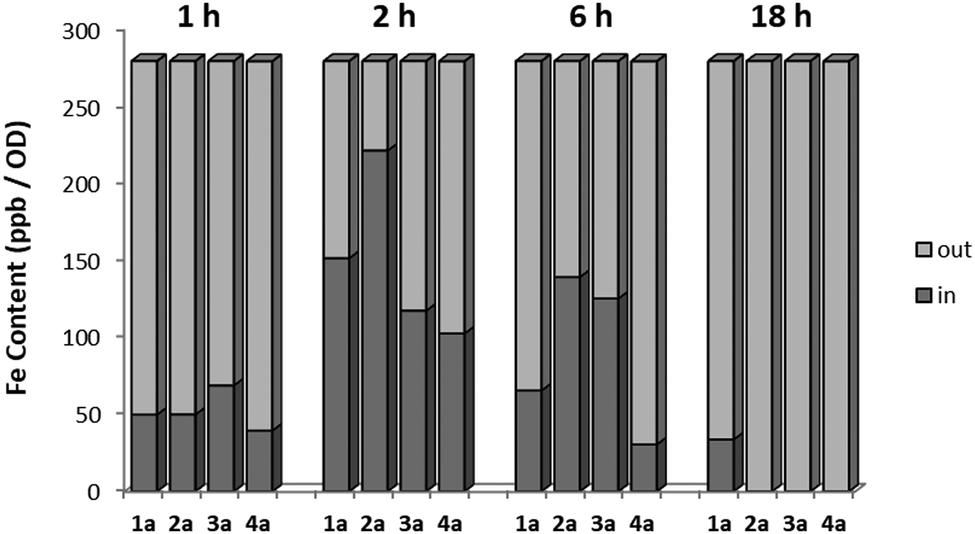 | ||
| Fig. 3 Uptake studies compared to the colony survival rate measured in OD. The experiment has been performed on the medium of incubation and corrected with OD in order to take into account of the possible biological material loss due to toxicity of the compounds and which would lead to decrease in uptake. Negative controls of the untreated and of the medium alone have been also performed. | ||
In order to assess the possible increase in reactive oxygen species (ROS) level induced by 1a–4a, possibly due to the presence of the ferrocenyl moiety, we performed a series of in vitro experiment to measure the ROS level upon treatment on S. cerevisiae. Briefly, S. cerevisiae was cultured in YPD medium overnight before treatment. After that, the fungal concentration was measured and normalized and the cultures were treated with YPD containing 5 μM of the different drug candidates at different time frames (1 h, 6 h and 18 h). After incubation, the colonies were washed, fresh medium containing H2DCF was added and the fluorescence of the hydrolyzed DCF was measured (see Fig. S1, ESI†). While the parent drug fluconazole displayed weak action at early stages, which increased with incubation time, the new drug candidates showed a higher ROS levels at an early stage. However, although the complexes proved to induce an increase of ROS levels, the relative ROS induction still remains moderate and it is probable that ROS production is not responsible for fungi death.
Morphology studies
The activity of 2a at the biochemical level is also mirrored by morphologic features at the ultra-structural level. The morphologic effect on S. cerevisiae upon treatment with the target complex could be conveniently monitored by transmission electron microscopy (TEM). As can be seen in Fig. 4, the resulting images (selection of a large pool) suggested that 2a induces a loss in wall integrity as well as intracellular features ascribable to late apoptosis or necrosis.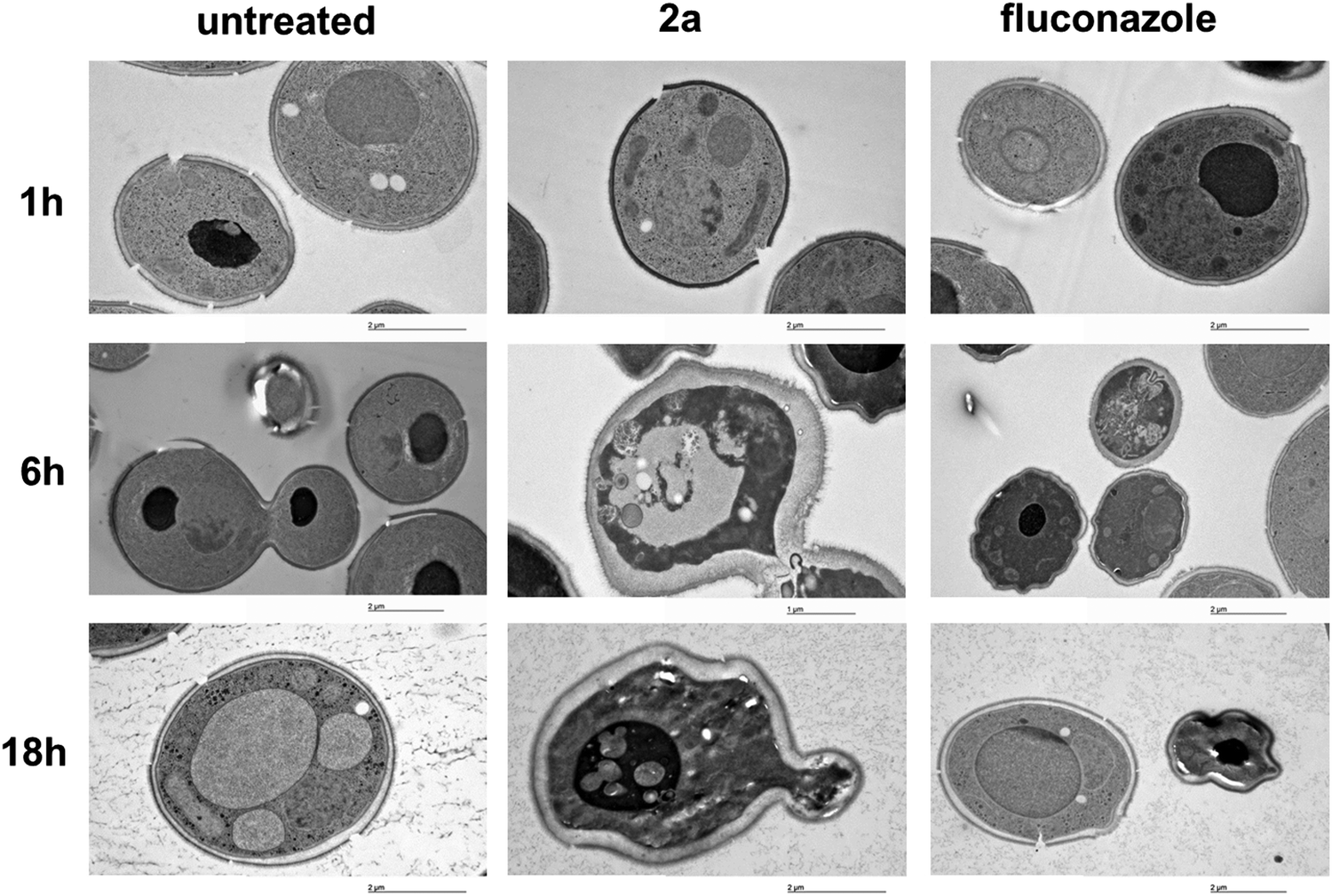 | ||
| Fig. 4 Ultra-structural studies of untreated S. cerevisiae, treated with 2a or with fluconazole tested at 5 μM and 30 °C incubation temperature. The pictures showed starting with a 6 h incubation formation of vesicles, ascribable as apoptotic bodies and loss of membrane integrity. After longer incubation times a shrinking effect as well as nuclear blebbing could be also noted. | ||
In vitro antifungal screening on clinical isolates
Motivated by the promising results obtained for the in vitro antifungal activities against the non-pathogenic model system S. cerevisiae, we performed further screening on clinical isolates. We again chose complex 2a since it displayed the best mycotoxicity and, on the other side, showed low cytotoxicity against human cells. We tested this antifungal drug candidate towards C. albicans and non albicans strains, including fluconazole-resistant (MIC50 > 100 μM) and other mycotoxin-producing and pathogenic fungi, such as Penicillum paneum, Aspergillus glaucus and Trichosporon asahi (see Table 2). Of utmost interest, the new antifungal drug candidate displayed a very strong activity towards almost all investigated strains (only in the case of A. glaucus, C. parapsilosis, and C. albicans MFB005 and SC5314 was the MIC50 not in the nanomolar range). Moreover, the antimycotic potency of the complex did overcome the values of the parent drug up to a factor of 400-fold (e.g. P. paneum) and also showed activity towards azole resistant strains.| Strains | MIC502a (μM) | MIC50 fluconazole (μM) |
|---|---|---|
| C. albicans SC5314 | >1 | 0.82 |
| C. albicans MFB005 FS3 | >1 | 0.41 |
| C. albicans YMS 102-2 | 1.00 | >100 |
| C. albicans YMS 102-6 | 0.06 | 0.82 |
| C. glabrata MFB005 FS4 | 0.06 | 0.41 |
| C. glabrata RTT 199_3 | 1.00 | >100 |
| C. parapsilosis MFB005 FS5 | 0.50 | 0.41 |
| C. parapsilosis MFB070 N1 | >1 | >100 |
| C. tropicalis RTT35-1 | 0.69 | >100 |
| C. tropicalis RTT35-3 | 0.50 | >100 |
| P. paneum MFB042 N1 | 0.25 | >100 |
| A. glaucus MFB027 N1 | >1 | 0.82 |
| T. asahii MFB034 N1 | 0.13 | 1.63 |
Fluconazole is a fungistatic drug and growth curve analysis performed on the C. albicans reference strain SC5314, a frequently used wild-type control, indicate a fungistatic effect of compound 2a at MIC50 concentration for this specific strain (1.98 μM, Fig. S2, ESI†).
AOX enzyme inhibition
In order to further define the inhibitory capabilities of the compounds, they were also tested in the mitochondrial complex II and III pathway (succinate quinol reductase pathway (SQR)), two common targets for fungicidal treatment.72,73 As can be seen in Fig. 5, compound 1a demonstrated no inhibitory activity towards either complex, confirming that the antifungal properties of these compounds are not due to off-target effects within mitochondrial respiration. However, while the other analogues demonstrate slightly improved inhibitory properties against the pathway when compared to 1a, it is unlikely to be the source of fungicidal activity.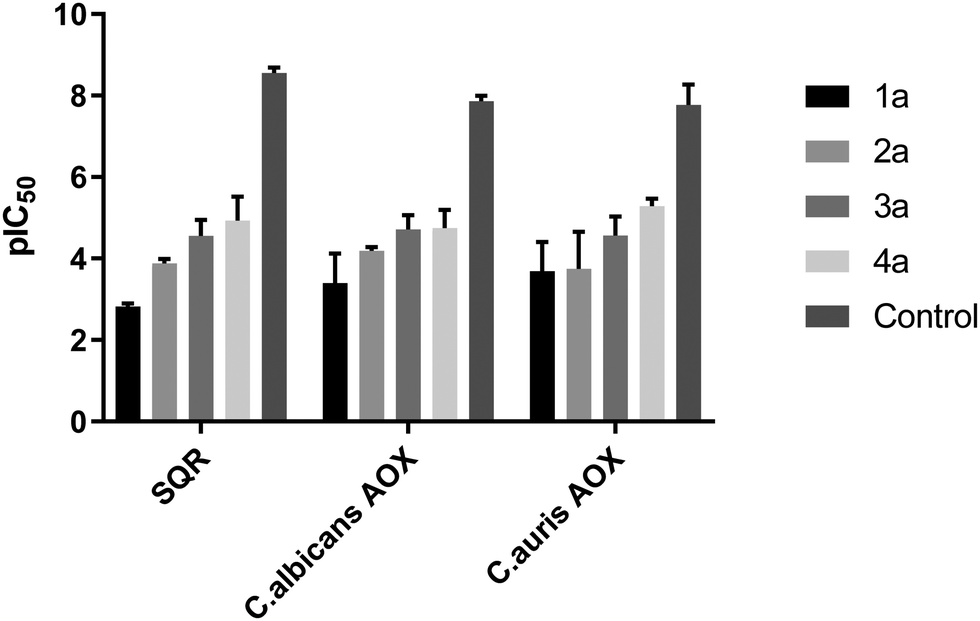 | ||
Fig. 5 pIC50 values (−log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50) for all compounds against the mitochondrial complex II and III pathway (SQR) from rat liver mitochondria, and rAOX's from C. albicans and C. auris. Control denotes control compounds for each specific pathway, with antimycin A for SQR and ascofuranone for the AOX pathways. Data was collected in a 96-well plate format, following cytochrome c reduction (ΔA550) for the SQR pathway, and NADH oxidation (ΔA340) for the AOX pathway. All data are a mean average of 3 biological replicates ±SEM. IC50) for all compounds against the mitochondrial complex II and III pathway (SQR) from rat liver mitochondria, and rAOX's from C. albicans and C. auris. Control denotes control compounds for each specific pathway, with antimycin A for SQR and ascofuranone for the AOX pathways. Data was collected in a 96-well plate format, following cytochrome c reduction (ΔA550) for the SQR pathway, and NADH oxidation (ΔA340) for the AOX pathway. All data are a mean average of 3 biological replicates ±SEM. | ||
The compounds were also tested against alternative oxidases (AOX) from both C. albicans and C. auris. The proteins were expressed in a recombinant E. coli system to allow for more accurate data acquisition due to their low expression levels proving problematic from a detection standpoint. Again, compound 1a demonstrated minimal inhibitory activity towards the AOX's, however compounds 3a and 4a showed moderate activity against both AOX's, comparable to values typically seen with non-competitive AOX inhibitors such as salicylhydroxamic acid (SHAM).74 The parent compound, fluconazole, displayed no inhibition towards either pathway up to a concentration of 100 μM.
Cytochrome P450 enzyme inhibition
Based on the very promising data obtained with complex 2a, further biological experiments were performed on this complex to understand further its mechanism of action. Enzyme inhibition studies were performed on different cytochrome P450 enzymes by the service provider Cyprotex GmbH. The new fluconacene 2a displayed an extremely strong inhibition profile throughout all the series of enzymes, with a larger efficacy in comparison with the parent drug fluconazole, between 4 and 150 fold (see Table 3) suggesting that, if this series of molecule are optimised towards clinical use, like fluconazole,75 potential drug–drug interactions will need to be taken into account.| Compound | CYP2C9 | CYP2C8 | CYP2C19 | CYP3A4 | CYP1A | CYP2D6 |
|---|---|---|---|---|---|---|
| Fluconazole | 13.9 ± 1.63 | 97.2 ± 27.9 | 3.02 ± 0.34 | 25.8 ± 2.45 | >100 | >100 |
| 2a | 3.33 ± 0.71 | 0.64 ± 0.09 | <0.4 | 0.4 ± 0.06 | 10.4 ± 0.41 | 2.71 ± 0.12 |
Chemogenomic studies
In order to investigate the mechanism of action of the new organometallic complex 2a, chemogenomic assays were performed. The strength of the genome-wide chemogenomic screen is reflected in the clear identification of ERG11, encoding lanosterol 14-alpha-demethylase, as the main target of the drug (see Fig. 6 and Table S1, ESI†). ERG11 is a key enzyme of the ergosterol biosynthesis pathway and the primary target of azole antifungals to which the tested compound 2a belongs.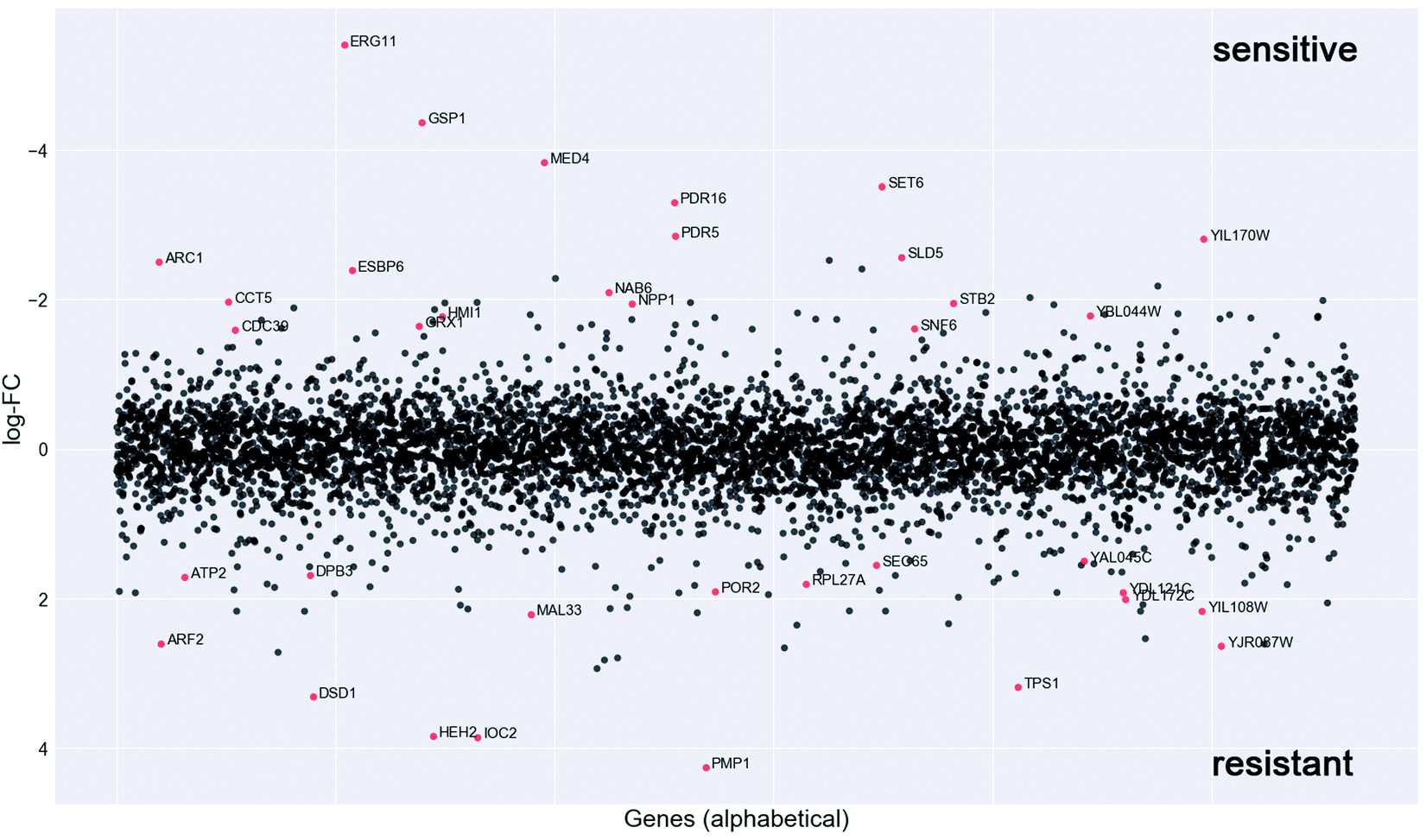 | ||
| Fig. 6 Chemogenomic screen. To identify potential drug targets the pool of tagged 5936 S. cerevisiae heterozygous deletion mutants was grown competitively for 20 generations in the presence and absence of compound 2a. Strain fitness was determined via high-throughput barcode sequencing and normalization to the untreated control. log2 ratio (control intensity/treatment intensity) was calculated and plotted as a function of gene. The genome-wide readout of heterozygous mutants highly sensitive to compound 2a included the known target of azoles, ERG11 and novel targets such as the RAN GTPase GSP1 (red dots: logFC > 1.5 and P value >0.01). | ||
Azoles are known to induce ERG11 gene expression,76 which also holds true for compound 2a yet to a lower extent than fluconazole as revealed by a screen in both susceptible C. albicans strains (Fig. S3A, ESI†) and in the highly fluconazole resistant strain YMS102-2 (Fig. S3B, ESI†). The latter strain showed a high expression of ERG11 when grown in the absence of drug and ERG11 gene expression was significantly (FC = 1.69; P = 0.000086) increased upon fluconazole exposure, while decreased (FC = −1.81; P = 0.000193) when grown in the presence of compound 2a (Fig. S3B, ESI†). Hence, the improved antifungal activity of compound 2a for the strain YMS102-2 might be partially explained by the significant reduction of ERG11 gene expression, because overexpression of ERG11 is known as an azole resistance mechanism in Candida.16,77 Hence, future work should be directed to study if compound 2a is able to reduce ERG11 gene expression also in other ERG11 overexpressing Candida isolates in a similar way.
Our chemogenomic assay identified five other heterozygous deletion strains that in previous chemogenomic screens have been shown to be extremely sensitive to azole antifungals; these are SET6, PDR5, PDR16, MED4 and CDC39. The latter two are essential nuclear genes involved in the regulation of transcription. CDC39 acts as a component of the CCR4-NOT core complex78 and MED4 encodes a subunit of the RNA polymerase II mediator complex. In Saccharomyces cerevisiae and C. glabrata the mediator complex is a coactivator of the multiple drug resistance regulator PDR1 that controls the activation of PDR5 and PDR16.79–81 Similarly, also in C. albicans the mediator activator complex is recruited to the CDR1 promotor,80 which is the C. albicans ortholog of PDR5. PDR5 is a multidrug ABC efflux pump that confers resistance to several chemicals including azoles and it mediates transmembrane transport of steorids.82PDR16 (ortholog of the C. albicans PDR17) is a phosphatidylinositol transfer protein involved in sterol biosynthetic process and in resistance to azole drugs.83 Notably, the phosphatidylinositol signal transduction pathway that controls amongst others cell membrane and cell wall remodelling as well as downstream targets such as DNA repair, was recently suggested to play an important role in phenotypic antifungal drug resistance related to protein synthesis.18 Indeed many of the identified deletion strains that are highly sensitive to compound 2a were heterozygous for genes involved in protein synthesis (e.g. ARC1, RPS5, MED4, CDC39, SNF6, CCT5, GSP1) (Fig. 4 and Table S1, ESI†).
Furthermore, compound 2a targets nuclear genes that are not only essential for RNA transcription, processing and transport (GSP1, MED4, CDC39) but also essential for DNA replication (SLD5), and these together with gene targets functioning in the correct folding of actin and tubulin (CCT5) (Fig. 6) point to a possible interaction of compound 2a with cell division.
Taken together, the genome-wide profiling of the in vivo cellular response to compound 2a identified candidate protein targets (Fig. 6 and Table S1, ESI†), which hint to a mechanism of drug action that goes beyond the main targets of azole antifungals, which might explain its elevated activity towards azole resistant strains (Table 1).
In vivo studies
Motivated by these promising results, we decided to perform in vivo experiments in a mice model of Candida infection. As positive control, fluconazole was used. It significantly improved pathology and cured mice from the infection, as judged by the decreased fungal growth in the kidney and dissemination to the liver (see Fig. 7A), reduction of the inflammatory pathology in the kidney and colon (see Fig. 7B–D) and downregulation of pro-inflammatory cytokine (IL-1β and TNF-α) levels (Fig. 7D). Similar results were obtained with compound 2a that proved to be effective in reducing fungal growth and dissemination (see Fig. 7A) as well as tissue inflammatory pathology (see Fig. 6B for histological scores) at a dose as low as 1 mg kg−1 (Fig. 7C and D). Of great interest, at variance with fluconazole, compound 2a also promoted a vigorous pro-inflammatory cytokine response promptly controlled by the associated high levels IL-10 (Fig. 7E). These findings indicate that compound 2a is active in vivo and that its activity may encompasse an action on innate immune response, consistent with the well known synergistic activity of azoles, including fluconazole, with antifungal effector activities, such as phagocytosis, radicals production and Toll-like receptors activation.84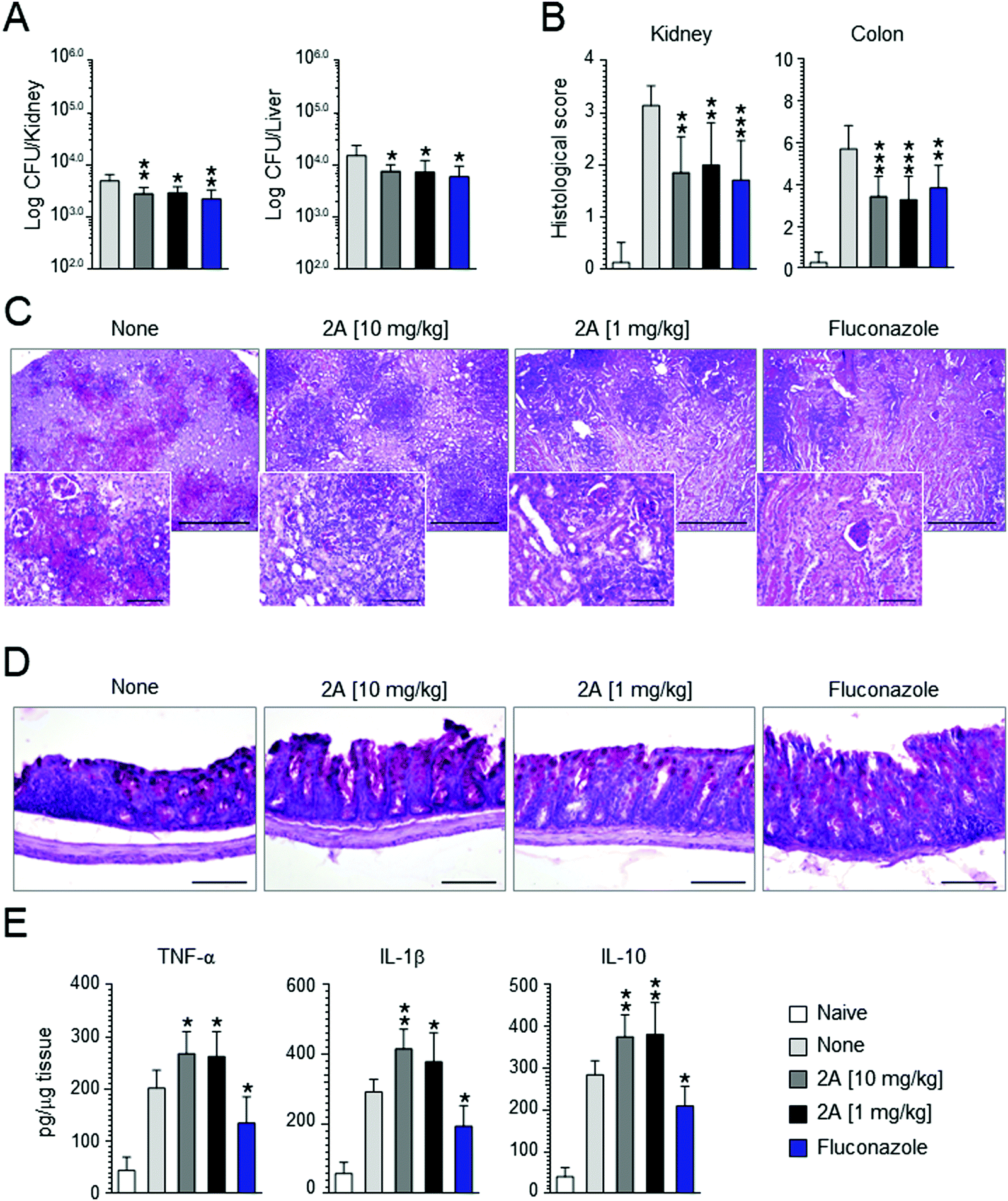 | ||
| Fig. 7 Effect of the compound 2a on C. albicans (SC-5314) systemic infection. C57BL/6 mice were infected via systemic route with 1 × 106C. albicans yeasts. Fluconazole and compound 2a were administered intragastrically daily, beginning the day of the infection until mice sacrifice (4 days after the infection). Control mice received the diluent alone. (A) Fungal growth. (B) Histological scores and (C and D) analysis (10× magnification and 40× in the inset) of the kidneys and colon of infected and treated mice. (E) Levels of cytokines in kidney homogenates. For histological scores of kidneys, score 0 = no inflammation, score 1 ≤ 3 foci of inflammation, score 2 = 4 to 6 foci of inflammation, score 3 ≥ 6 foci of inflammation, but less than 25% of kidney affected, score 4 ≥ 25% of kidney affected. For histological scores of colon, the total histological score was obtained by summing the four histological components scores: ‘inflammation extent’, ‘damage in crypt architecture’, ‘hyperemia/edema’, ‘grade of accumulation with inflammatory cells’. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.005, treated vs. untreated C57BL/6 mice (n = 7 mice per group, representative of three experiments). Naïve, uninfected mice. None, untreated mice. | ||
Conclusion
Four novel ferrocene-based derivatives (1a–4a) of the frontline antifungal drug fluconazole have been synthesized and characterized. All four organometallic derivatives were tested for antifungal activity against the model system S. cerevisiae and showed an impressively improved activity compared to fluconazole. Importantly, this activity was confirmed on a panel of clinical isolates with the best candidate of this study (complex 2a). An improvement of the minimum inhibitory concentration of up to 400 times compared to fluconazole was observed and activity against azole-resistant strains was clearly demonstrated for 2a. During in vivo experiments in mice model of Candida infections, compound 2a was found to diminish the growth of the fungus and to greatly improve the inflammatory pathology in the kidney and colon. Overall, this study further demonstrates the potential of organometallic compounds in medicine. By merely inserting a ferrocenyl moiety in a well-known drug, namely fluconazole, resistance can be overcome, although the main target of the novel antimycotic agent is the same as that of the organic drug (lanosterol 14α-demethylase), as demonstrated by chemogenomic profiling. The ferrocenyl insertion allows for an additional mode of action.Ethical statement
Mouse experiments were performed according to Italian Approved Animal Welfare Authorization 360/2015-PR and Legislative Decree 26/2014 regarding the animal license obtained by the Italian Ministry of Health lasting for 5 years (2015–2020). Animals were treated as the guidelines of EAMC.Author contributions
All authors were involved with the design and interpretation of experiments and with the writing of the manuscript. The synthesis and characterization of the compounds were performed by R. R., L. M., S. J., J. N., Y. L., and J. H. TEM studies were carried out by A. K. Biological experiments were performed by R. R., T. W., N. T., A. L. M., and L. Y. Chemogenomic studies were carried out by T. W. and M. M. Animal testing was carried out by R. G., M. P. and L. R. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was funded by the Swiss National Science Foundation (Grant Sinergia CRSII5_173718, Professorships No. PP00P2_133568 and PP00P2_157545 to G. G.), the University of Zurich (G. G.), the Stiftung für Wissenschaftliche Forschung of the University of Zurich (G. G.), the Novartis Jubilee Foundation (G. G. and R. R.), the Forschungskredit of the University of Zurich (R. R.) and the UBS Promedica Stiftung (G. G. and R. R.). This work has received support under the program “Investissements d’Avenir” launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). This research was supported by the Autonomous Province of Trento (Accordo di Programma P1611051I (M. M.)). Work in A. L. M.'s laboratory is supported by BBSRC (BB/L022915/1 and BB/NO10051/1). The authors thank Monica Borghi for her technical assistance for the mice experiments. We would also like to thank the University of Sussex (HEIF COVID-19 emergency funds) and the Ewart Bequest Fund for this work (J. S.).References
- T. Ohyama, S. Miyakoshi and F. Isono, Antimicrob. Agents Chemother., 2004, 48, 319–322 CrossRef CAS PubMed.
- G. Maschmeyer, Int. J. Antimicrob. Agents, 2006, 27S, S3–S6 CrossRef.
- A. Mikolajewska, S. Schwartz and M. Ruhnke, Mycoses, 2012, 55, 2–16 CrossRef CAS PubMed.
- R. Pelletier, J. Peter, C. Antin, C. Gonzalez, L. Wood and T. J. Walsh, J. Clin. Microbiol., 2000, 38, 1563–1568 CrossRef CAS.
- K. Donhuijsen, P. Petersen and W. K. Schmid, Dtsch. Arztebl. Int., 2008, 105, 501–506 Search PubMed.
- P. N. Lipke and R. Ovalle, J. Bacteriol., 1998, 180, 3735–3740 CrossRef CAS.
- S. M. Bowman and S. J. Free, BioEssays, 2006, 28, 799–808 CrossRef.
- S. Perea, M. J. Ramos, M. Garau, A. Gonzalez, A. R. Noriega and A. del Palacio, J. Clin. Microbiol., 2000, 38, 3226–3230 CrossRef CAS.
- M. P. English and J. Turvey, Br. Med. J., 1968, 4, 228–230 CrossRef CAS PubMed.
- B. Havlickova, V. A. Czaika and M. Friedrich, Mycoses, 2008, 51(Suppl. 4), 2–15 CrossRef PubMed.
- R. P. Hobson, J. Hosp. Infect., 2003, 55, 159–168 CrossRef CAS.
- D. A. Enoch, H. A. Ludlam and N. M. Brown, J. Med. Microbiol., 2006, 55, 809–818 CrossRef CAS PubMed.
- J. Menzin, J. L. Meyers, M. M. Friedman, J. R. Korn, J. R. Perfect, A. A. Langston, R. P. Danna and G. Papadopoulos, Am. J. Infect. Control, 2011, 39, 15–20 CrossRef.
- R. J. Wang, R. F. Miller and L. Huang, Clin. Chest Med., 2017, 38, 465–477 CrossRef PubMed.
- G. D. Brown, D. W. Denning, N. A. Gow, S. M. Levitz, M. G. Netea and T. C. White, Sci. Transl. Med., 2012, 4, 165rv113 Search PubMed.
- J. Berman and D. J. Krysan, Nat. Rev. Microbiol., 2020, 18, 319–331 CrossRef CAS PubMed.
- H. Hof, Drug Resist. Updates, 2008, 11, 25–31 CrossRef CAS PubMed.
- T. Weil, R. Santamaría, W. Lee, J. Rung, N. Tocci, D. Abbey, A. R. Bezerra, L. Carreto, G. R. Moura, M. Bayés, I. G. Gut, A. Csikasz-Nagy, D. Cavalieri, J. Berman and M. A. S. Santos, mSphere, 2017, 2, e00167 CrossRef CAS PubMed.
- L. E. Cowen and W. J. Steinbach, Eukaryotic Cell, 2008, 7, 747–764 CrossRef CAS.
- M. A. Pfaller, Am. J. Med., 2012, 125, S3–S13 CrossRef CAS PubMed.
- C. M. Martel, J. E. Parker, A. G. S. Warrilow, N. J. Rolley, S. L. Kelly and D. E. Kelly, Antimicrob. Agents Chemother., 2010, 54, 4920–4923 CrossRef CAS.
- A. Batta, B. S. Kalra and R. Khirasaria, J. Family Med Prim Care, 2020, 9, 105–114 CrossRef.
- A. Chowdhary and A. Sharma, J. Global Antimicrob. Resist., 2020, 22, 175–176 CrossRef PubMed.
- R. S. Shapiro, N. Robbins and L. E. Cowen, Microbiol. Mol. Biol. Rev., 2011, 75, 213–267 CrossRef CAS.
- D. J. Woods and T. M. Williams, Invertebr. Neurosci., 2007, 7, 245–250 CrossRef CAS PubMed.
- R. Laniado-Laborin and M. N. Cabrales-Vargas, Rev. Iberoam. Micol., 2009, 26, 223–227 CrossRef PubMed.
- S. J. Kashyap, V. K. Garg, P. K. Sharma, N. Kumar, R. Dudhe and J. K. Gupta, Med. Chem. Res., 2012, 21, 2123–2132 CrossRef CAS.
- M. A. Ghannoum and L. B. Rice, Clin. Microbiol. Rev., 1999, 12, 501–517 CrossRef CAS.
- C. Biot, G. Glorian, L. A. Maciejewski and J. Brocard, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC , and references therein.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS , and references therein.
- Y. C. Ong, S. Roy, P. C. Andrews and G. Gasser, Chem. Rev., 2019, 119, 730–796 CrossRef CAS.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- M. Patra, G. Gasser and N. Metzler-Nolte, Dalton Trans., 2012, 41, 6350–6358 RSC.
- E. Boros, P. J. Dyson and G. Gasser, Chem, 2020, 6, 41–60 CAS.
- R. Rubbiani, O. Blacque and G. Gasser, Dalton Trans., 2016, 45, 6619–6626 RSC.
- E. W. Hunsaker, K. J. McAuliffe and K. J. Franz, J. Biol. Inorg. Chem., 2020, 25, 729–745 CrossRef CAS.
- A. Garoufis, S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 1384–1397 CrossRef CAS.
- N. V. Loginova, T. V. Koval’chuk, N. P. Osipovich, G. I. Polozov, V. L. Sorokin, A. A. Chernyavskaya and O. I. Shadyro, Polyhedron, 2008, 27, 985–991 CrossRef CAS.
- T. Mangamamba, M. C. Ganorkar and G. Swarnabala, Int. J. Inorg. Chem., 2014, 2014, 22 Search PubMed.
- M. R. Karekal, V. Biradar and M. Bennikallu Hire Mathada, Bioinorg. Chem. Appl., 2013, 2013, 16 Search PubMed.
- M. Kaya, E. Demir and H. Bekci, J. Enzyme Inhib. Med. Chem., 2013, 28, 885–893 CrossRef CAS PubMed.
- E. Pahontu, V. Fala, A. Gulea, D. Poirier, V. Tapcov and T. Rosu, Molecules, 2013, 18, 8812–8836 CrossRef CAS PubMed.
- E. Rodriguez-Fernandez, J. L. Manzano, J. J. Benito, R. Hermosa, E. Monte and J. J. Criado, J. Inorg. Biochem., 2005, 99, 1558–1572 CrossRef CAS PubMed.
- Z. H. Chohan, H. Pervez, K. M. Khan and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2005, 20, 81–88 CrossRef CAS PubMed.
- C. Biot, N. François, L. Maciejewski, J. Brocard and D. Poulain, Bioorg. Med. Chem. Lett., 2000, 10, 839–841 CrossRef CAS.
- Z. H. Chohan, Appl. Organomet. Chem., 2006, 20, 112–116 CrossRef CAS.
- G. Golbaghi, M.-C. Groleau, Y. López de los Santos, N. Doucet, E. Déziel and A. Castonguay, ChemBioChem, 2020, 21, 3112–3119 CrossRef CAS PubMed , and references therein.
- J. A. d. Azevedo-França, R. Granado, S. T. de Macedo Silva, G. d. Santos-Silva, S. Scapin, L. P. Borba-Santos, S. Rozental, W. de Souza, É. S. Martins-Duarte, E. Barrias, J. C. F. Rodrigues and M. Navarro, Antimicrob. Agents Chemother., 2020, 64, e01980 CrossRef PubMed.
- N. L. Stevanović, I. Aleksic, J. Kljun, S. Skaro Bogojevic, A. Veselinovic, J. Nikodinovic-Runic, I. Turel, M. I. Djuran and B. Đ. Glišić, Pharmaceuticals, 2021, 14, 24 CrossRef.
- T. Vago, G. Baldi, D. Colombo, M. Barbareschi, G. Norbiato, F. Dallegri and M. Bevilacqua, Antimicrob. Agents Chemother., 1994, 38, 2605 CrossRef CAS PubMed.
- K. Shalini, N. Kumar, S. Drabu and P. K. Sharma, Beilstein J. Org. Chem., 2011, 7, 668–677 CrossRef CAS PubMed.
- A. Stütz, Angew. Chem., Int. Ed. Engl., 1987, 26, 320–328 CrossRef.
- G. Jaouen, A. Vessieres and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- D. Dive and C. Biot, Curr. Top. Med. Chem., 2014, 14, 1684–1692 CrossRef CAS , and references therein.
- S. Sansook, S. Hassell-Hart, C. Ocasio and J. Spencer, J. Organomet. Chem., 2019, 905, 121017 CrossRef.
- X. Chai, J. Zhang, S. Yu, H. Hu, Y. Zou, Q. Zhao, Z. Dan, D. Zhang and Q. Wu, Bioorg. Med. Chem. Lett., 2009, 19, 1811–1814 CrossRef CAS PubMed.
- C. A. Ocasio, S. Sansook, R. Jones, J. M. Roberts, T. G. Scott, N. Tsoureas, P. Coxhead, M. Guille, G. J. Tizzard, S. J. Coles, H. Hochegger, J. E. Bradner and J. Spencer, Organometallics, 2017, 36, 3276–3283 CrossRef CAS.
- A. Baramee, A. Coppin, M. Mortuaire, L. Pelinski, S. Tomavo and J. Brocard, Bioorg. Med. Chem., 2006, 14, 1294–1302 CrossRef CAS PubMed.
- N. C. Tice, S. Parkin and J. P. Selegue, J. Organomet. Chem., 2007, 692, 791–800 CrossRef CAS.
- H. Zhang, F. Xin, W. An, A. Hao, X. Wang, X. Zhao, Z. Liu and L. Sun, Colloids Surf., A, 2010, 363, 78–85 CrossRef CAS.
- R. S. Upadhayaya, S. Jain, N. Sinha, N. Kishore, R. Chandra and S. K. Arora, Eur. J. Med. Chem., 2004, 39, 579–592 CrossRef CAS PubMed.
- C. Sheng, W. Zhang, H. Ji, M. Zhang, Y. Song, H. Xu, J. Zhu, Z. Miao, Q. Jiang, J. Yao, Y. Zhou, J. Zhu and J. Lü, J. Med. Chem., 2006, 49, 2512–2525 CrossRef CAS PubMed.
- M. K. Lindvall and A. M. Koskinen, J. Org. Chem., 1999, 64, 4596–4606 CrossRef CAS PubMed.
- T. Bader, H.-U. Bichsel, B. GilomenImeld, I. Meyer-Wilmes and M. Sundermeier, Polymorphic forms of olopatadine hydrochloride and methods for producing olopatadine and salts thereof, US Pat., WO2007110761A8, 2007 Search PubMed.
- S. Keller, Y. C. Ong, Y. Lin, K. Cariou and G. Gasser, J. Organomet. Chem., 2020, 906, 121059 CrossRef CAS.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, Chem. – Eur. J., 2013, 19, 14768–14772 CrossRef CAS PubMed.
- M. D. Hall, K. A. Telma, K. E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed.
- H. Sierotzki and G. Scalliet, Phytopathology, 2013, 103, 880–887 CrossRef CAS PubMed.
- H. Balba, J. Environ. Sci. Health, Part B, 2007, 42, 441–451 CrossRef CAS PubMed.
- L. Young, B. May, A. Pendlebury-Watt, J. Shearman, C. Elliott, M. S. Albury, T. Shiba, D. K. Inaoka, S. Harada, K. Kita and A. L. Moore, Biochim. Biophys. Acta, 2014, 1837, 1219–1225 CrossRef CAS PubMed.
- T. Niwa, T. Shiraga and A. Takagi, Biol. Pharm. Bull., 2005, 28, 1805–1808 CrossRef CAS PubMed.
- K. W. Henry, J. T. Nickels and T. D. Edlind, Antimicrob. Agents Chemother., 2000, 44, 2693–2700 CrossRef CAS PubMed.
- Y. Lee, E. Puumala, N. Robbins and L. E. Cowen, Chem. Rev., 2020, 121, 3390–3411 CrossRef PubMed.
- H.-Y. Liu, V. Badarinarayana, D. C. Audino, J. Rappsilber, M. Mann and C. L. Denis, EMBO J., 1998, 17, 1096–1106 CrossRef CAS PubMed.
- Z. Liu and L. C. Myers, Antimicrob. Agents Chemother., 2017, 61, e01344–01317 CAS.
- G. P. Moran, M. Z. Anderson, L. C. Myers and D. J. Sullivan, Curr. Genet., 2019, 65, 621–630 CrossRef CAS PubMed.
- J. L. Nishikawa, A. Boeszoermenyi, L. A. Vale-Silva, R. Torelli, B. Posteraro, Y.-J. Sohn, F. Ji, V. Gelev, D. Sanglard, M. Sanguinetti, R. I. Sadreyev, G. Mukherjee, J. Bhyravabhotla, S. J. Buhrlage, N. S. Gray, G. Wagner, A. M. Näär and H. Arthanari, Nature, 2016, 530, 485–489 CrossRef CAS PubMed.
- Y. Mahé, Y. Lemoine and K. Kuchler, J. Biol. Chem., 1996, 271, 25167–25172 CrossRef PubMed.
- H. B. van den Hazel, H. Pichler, M. A. do Valle Matta, E. Leitner, A. Goffeau and G. Daum, J. Biol. Chem., 1999, 274, 1934–1941 CrossRef CAS PubMed.
- R. B. Ami, R. E. Lewis and D. P. Kontoyiannis, Clin. Infect. Dis., 2008, 47, 226–235 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00123j |
| This journal is © The Royal Society of Chemistry 2021 |