
Hydrogel facilitated bioelectronic integration
Richard
Vo†
 ,
Huan-Hsuan
Hsu†
* and
Xiaocheng
Jiang
*
,
Huan-Hsuan
Hsu†
* and
Xiaocheng
Jiang
*
Department of Biomedical Engineering, Tufts University, Medford, MA 02155, USA. E-mail: Xiaocheng.Jiang@tufts.edu; Huan-Hsuan.Hsu@tufts.edu
First published on 29th September 2020
Abstract
The recent advances in bio-integratable electronics are creating new opportunities for investigating and directing biologically significant processes, yet their performance to date is still limited by the inherent physiochemical and signaling mismatches at the heterogeneous interfaces. Hydrogels represent a unique category of materials to bridge the gap between biological and electronic systems because of their structural/functional similarity to biological tissues and design versatility to accommodate cross-system communication. In this review, we discuss the latest progress in the engineering of hydrogel interfaces for bioelectronics development that promotes (1) structural compatibility, where the mechanical and chemical properties of hydrogels can be modulated to achieve coherent, chronically stable biotic-abiotic junctions; and (2) interfacial signal transduction, where the charge and mass transport within the hydrogel mediators can be rationally programmed to condition/amplify the bioderived signals and enhance the electrical/electrochemical coupling. We will further discuss the application of functional hydrogels in complex physiological environments for bioelectronic integration across different scales/biological levels. These ongoing research efforts have the potential to blur the distinction between living systems and artificial electronics, and ultimately decode and regulate biological functioning for both fundamental inquiries and biomedical applications.
 Xiaocheng Jiang | Xiaocheng Jiang is the John A. and Dorothy M. Adams Faculty Development Assistant Professor at Tufts University. He received his Ph.D. in physical chemistry from Harvard University in 2011. Prior to joining Tufts, he was an American Cancer Society postdoctoral fellow at Harvard Medical School and Massachusetts General Hospital. He is the recipient of NSF CAREER award (2017) and AFOSR young investigator award (2018). His lab is interested in exploring the unique biomaterial interface between living and electronic systems, with top priorities on (1) developing bio-integratable platforms for probing, interrogating, and directing biologically significant processes; and (2) pursuing bio-derived materials and bio-inspired approaches for various engineering applications. |
1. Introduction
The relentless evolution of modern electronics is enabling unprecedented capability for information processing and storage. When integrated with biosystems, it allows quantitative interpretation of complex bio-derived signals and dynamic modulation of critical biological functions, empowering influential innovations in glucose monitoring, electrocardiogram and electroencephalogram, cardiac pacemakers, neurostimulators and more.1–5 Central to bioelectronic development is the effective and reliable signal transduction across the biotic/abiotic interface – a fundamental requisite that continues to challenge current bioelectronic design and operation, as a result of the intrinsic structural and signaling mismatch between the two distinct systems.Structurally, traditional electronics are composed of solid-state materials (e.g. metals and semiconductors) that are chemically inert and orders of magnitude stiffer as compared with soft, bioactive components.6 This mismatch can adversely affect cell behavior and development, and also lead to insufficient electrode interaction, large contact impedance and poor signal coupling.7,8 Particularly, for in vivo applications, these stiff materials can cause vascular and tissue damage during implantation, and induce foreign body responses and fibrous encapsulation, thus further impeding the quality of cross-system communication.9,10 Recent progress in nano- and flexible electronics has shown promising improvement for bio-integration through the reduction of device dimension11 and/or substrate stiffness,12 enabling less-invasive probe design with intimate and chronically stable bio-contact for implantable/wearable applications.13 These research efforts will continue to benefit from localized biomaterial engineering at the active recording/stimulation interfaces to achieve ultimate structural coherence across the boundary.
Functionally, biological and electrical circuits are processing signals in a completely different modality. Biosystems are capable of transmitting highly complex and dynamic physiochemical signals via water-compliant carriers (such as ions and biomolecules), while conventional electronics represent deterministic systems that rely on the controlled transport of delocalized electrons/holes. The cross-system signaling, which can be achieved either passively (e.g. with conductive electrodes) or actively (e.g. with field-effect transistors, or FETs), remains a limiting factor in device functioning, especially under physiologically relevant conditions. For example, electrophysiological recording by microelectrode arrays (MEAs) can only detect attenuated, spatially averaged and temporarily filtered field-potential as a result of poor electrical coupling at the device interface.14 Similarly, FET biosensors, which convert biologically induced potential variation into conductance changes, typically suffer from compromised signal transduction in physiological fluids, as a result of charge screening (Debye length <1 nm in high-ionic strength solutions),15 signal decay (due to diffusion/neutralization), and nonspecific binding (by overwhelming background molecules).
Overall, the intrinsic mismatch at the bio-/electronic interface, both structurally and functionally, continuously challenges the efficiency and stability of existing devices. To accommodate the mismatch, hydrogels, three-dimensional polymeric networks with great structural similarity to biological tissues, have been extensively studied as a bridging material (Fig. 1). In this review we will discuss the unique properties of hydrogel materials that can be rationally designed and programmed to enhance the structural integration and interfacial signaling between biological and electronic systems, and highlight the latest progress in hydrogel-mediated bioelectronic development at molecular, cellular, tissue, and body levels.
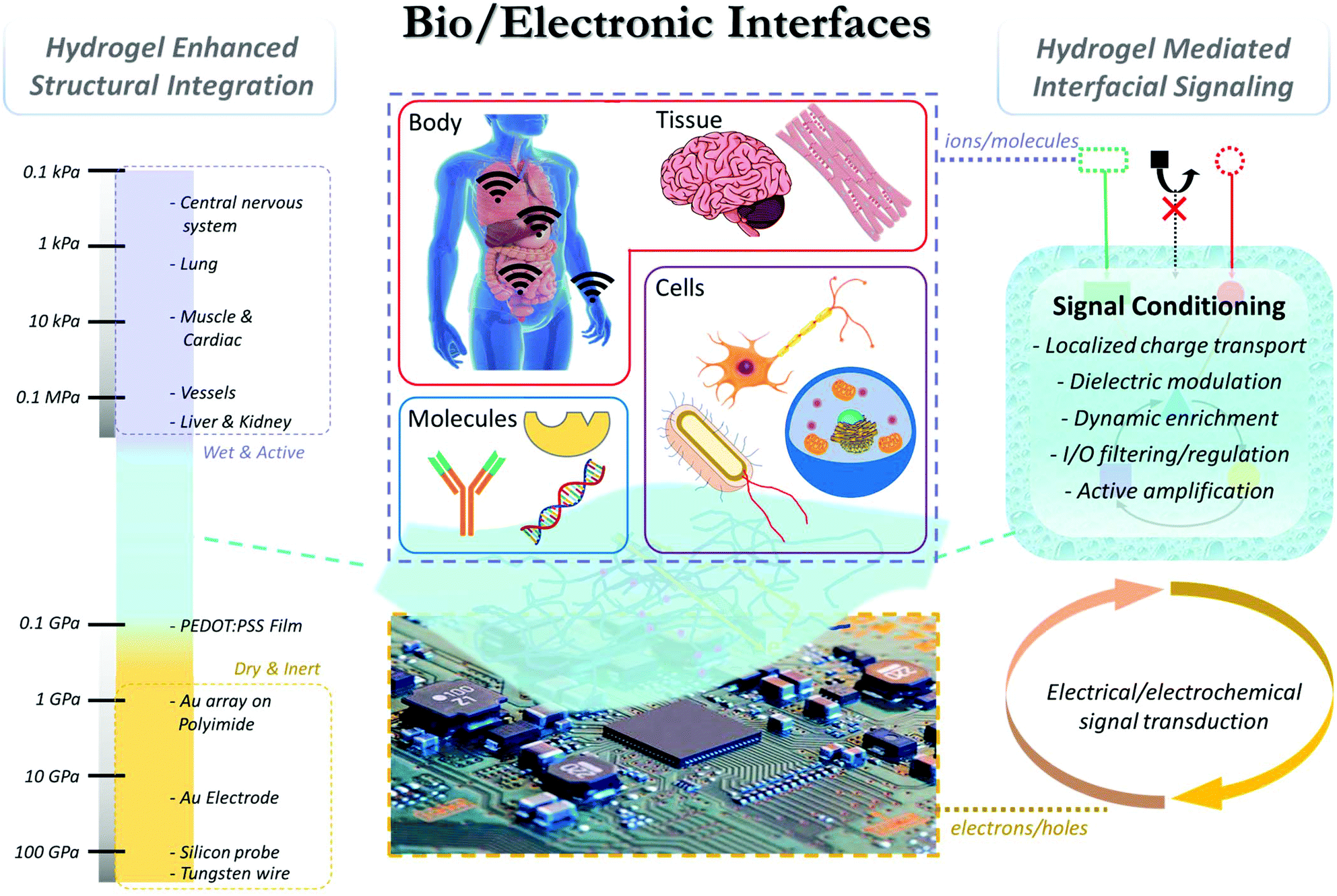 | ||
| Fig. 1 Hydrogel facilitated bioelectronic integration. (Left) Structural integration: hydrogel has unique mechanical and chemical properties to bridge soft, wet, and chemically active biological components with rigid, dry, and inert electronics. Young's moduli of: different biological components (e.g. central nervous system; 0.1–10 kPa; lungs: 1–5 kPa; muscle and cardiac: 10–20 kPa; vessels: 125 kPa; liver and kidney: 190 kPa), common hydrogels (hydrogel: 0.1–100 kPa; composite hydrogels: 1–100's of kPa, tough hydrogel: ∼MPa) and electronic materials/devices. (Right) Functional integration: rationally designed hydrogel interfaces enhance the cross-system signal coupling through: (i) facilitating the electron and/or ionic transport; (ii) modulation of local dielectric environment and Debye screening; (iii) dynamic enrichment of molecular biosignals via mass transport control; (iv) regulation/filtering of biological inputs/outputs via programmable hydrogel properties (e.g. pore size/surface charge/chemical affinity); and (v) active signal transduction/amplification via stimuli-responsive hydrogel design. | ||
2. Hydrogel enhanced structural integration
Hydrogels are hydrophilic polymer networks that contain up to thousands of times their dry weight in water.16 They have been widely recognized for their unique physiochemical properties in favor of bio-integration. Mechanically, the stiffness/Young's modulus of hydrogels is usually in the range of 0.1–100 kPa.17 Tough hydrogels with stiffness up to MPa have also be generated by regulating the composition and crosslinking mechanism.18,19 This range accommodates various types of cells and tissues20 to bridge the gap with stiff electronics (Fig. 1). Chemically, intrinsic or modified surface functional groups on hydrogels can provide strong adhesion to biological components through non-covalent (e.g. hydrogen bonds, π–π stacking, and cation–π interaction) or covalent interactions.21 Leveraging strategies from emerging biomedical research, additional hydrogel features such as porosity/pore size,16 stretchability,22 water content, topology,23 and conductivity24 can also be tailored to further control the interfacial properties. In general, two types of materials have been exploited to form hydrogels: (1) naturally derived polymers and (2) synthetic macromolecules. Due to their improved uniformity, stability and simplified synthesis/purification, synthetic hydrogels provide rational control over physical and chemical properties, enabling extensive flexibility in designing bioelectronic interfaces based on specific demands.25,26 For the structural integration of bioelectronics, hydrogels have been exploited as the interfacing material between biological and electronic components27,28 to improve the structural compatibility. For example, hydrogel coatings have been extensively applied in epidermal bioelectronics to ensure conformal and stable device-epidermis contacts. This hydrogel-mediated intimate interface also leads to enhancement in both stimulation and recording performances due to the reduced gap junction, which will be extensively discussed in the next section. Similarly, hydrogels have found extensive applications in many other bioelectronic designs, such as electroencephalogram, electrocardiogram, transcutaneous electrical nerve stimulation, electronic skin, and highly stretchable wearable devices.29–31Different from skin, the integration of bioelectronic devices with internal biological systems typically requires invasive procedures, where immune responses and scar formation around electronics are common barriers to electrical recording and stimulation. Soft cells/tissues have a Young's modulus in the range of 0.5 to 100's of kPa,32,33 whereas that of typical electronic materials (e.g. gold, silicon, etc.) are closer to 100's of GPa.34 These differences cause considerable damage to surrounding tissue after electronic implantation due to local mechanical strain.35 Furthermore, immediately after contact, proteins adsorb to the electronic surface due to their hydrophobicity and lack of bioactive functional groups. The protein adsorption then activates immune signaling cascades and pro-inflammatory responses, inducing complex cellular responses to the devices. This foreign body response can increase the impedance at the tissue/electrode interface that challenges the electrical signal transduction.36–38 Therefore, harmonizing the mechanical mismatch between tissue and electronics is important for improving device performance. Recently, hydrogel coatings have been utilized to improve the long-term biocompatibility of stiff electronic devices by reducing the large mechanical mismatch to minimize the immune response.39,40 Furthermore, the physical properties of the hydrogel may be tuned to match the local biological environment in order to elicit normal behavior after integration with electronics. As the mechanical forces acting on cells and tissues can greatly affect their function and behavior,41,42 by modifying composition and crosslinking density, hydrogels have been engineered to have tissue-like mechanical properties for improving bioelectronic integration. For example, polyethylene glycol dimethacrylate hydrogel with a stiffness similar to brain tissue (1.6 kPa to 171.5 kPa) has been coated on implanted electrodes of brain tissues.43 These hydrogel coatings significantly reduced the local strain caused by the large mechanical mismatch between brain tissue and metal electrodes, and micromotion of brain tissue relative to the stationary implanted device. The decrease in strain resulted in a reduction of the glial scar formation surrounding the implantation site compared to uncoated devices.
Overall, hydrogels provide a wide selection in compositions, structures, and functions, which offers unique advantages in the customization of bioelectronic interfaces for modifying electronics to accommodate various biological components, hence, advancing the quality and satiability of existing tools for the physiological signal recording/simulation of human tissues. Recent developments in hydrogel-coated bioelectronics for in vivo applications were systematically reviewed by Yuk et al.17
3. Hydrogel mediated bio-signal transduction
The functions of living systems rely on highly sensitive, dynamic, and error-tolerant transduction of complex bio-signals through: (1) bioelectrical signaling (e.g. in brain, heart, and muscles), which is mediated by ion fluxes and cell membrane potential changes; and (2) biochemical signaling, where (bio)molecules transmit and trigger internal reaction cascades (e.g. metabolism, immune response, tissue regeneration). Coupling these two distinct signaling pathways at the bioelectronic interface will allow comprehensive modulation/interrogation of biofunctions through electrical inputs/outputs. However, challenges remain in establishing an effective yet reliable cross-system signal coupling at bio-electronic interfaces, which can be summarized into the following three aspects: (1) the physiochemical mismatch between both systems can prohibit intimate contacts and lead to signal attenuation (ion/molecule diffusions); (2) the physiological fluid presents a high-ionic strength environment with a large amount of background molecules that jeopardizes the efficiency and accuracy in signal transduction; (3) bio-recognition components (such as enzymes, antibodies, bio-receptors) that have been used to facilitate biochemical signal transduction usually hold limited lifespans owing to bio-incompatible immobilization techniques. Toward overcoming these challenges, hydrogels represent unique interfacing materials as they provide a biologically relevant microenvironment with tunable mass and/or charge transport properties. The state-of-the-art achievements of the implementation of hydrogels in improving bioelectronic signal coupling are reviewed in the following sections.3.1 Bioelectrical signaling
In electrically active cells and tissues (e.g. neurons, muscle cells, cardiomyocytes, etc.), the selective ion transport across cell membranes and the corresponding membrane potential changes are central to the generation and transmission of bioelectrical signals. The continuous recoding and comprehensive interpretation of these signals can greatly elevate our understanding in important biological processes,44,45 while stimulation of these tissues finds critical importance for both physiological studies and disease treatments.46 Hence, many state-of-the-art developments in bioelectronics are targeted at improving the bi-directional communication between these tissues and external electronics. Generally, the electrical recording/simulation of excitable tissues is completed by the conversion between ion- and electron-mediated electrical signals. At the tissue-electronic interface, equilibrant electrolyte-electrode interactions (ion diffusion, redox reaction, electrical double layer, etc.) can establish a semi-stable electrical potential. During recording, the ion flux varies the electrical potential and consequently induces the electron flow in electronics to be detected. In contrast, during stimulation, applying an external electric field can trigger ion re-distribution at the tissue-electronic interface, altering the membrane potential of excitable cells, and activating ion channels.As both bioelectrical recording and stimulation are associated with the highly localized, transient ion flux, an intimate and chronically stable tissue-electronic contact becomes critically important for effective interfacial signaling. However, as discussed earlier, the intrinsic mismatch in mechanical and chemical properties limits the quality of tissue-electronic contacts. Hydrogels have been used as coatings or encapsulating materials on the electrode surface to bridge the structural mismatch between electronics and electrically active tissues17 While demonstrating improved biocompatibility, the insulating nature of hydrogels impedes the signal transduction between bio- and electronic-systems. Although hydrogels hold certain degrees of ionic conductivity47 that can be further enhanced by introducing high concentration ionic solutions such as ionic liquids and buffers into hydrogel matrixes,48,49 the stability of such ionic conductivity can be disturbed by continuous ion diffusion. Consequently, the performance of hydrogel-coated devices is limited, especially for chronic applications. To overcome this limitation, conductive hydrogels that display both tissue-like mechanical properties and electrical conductivity have been developed by incorporating different conductive fillers such as graphene, carbon nanotubes, gold/silver nanoparticles, or conductive polymers into the hydrogel network.50–54 In particular, PEDOT:PSS has been widely used in fabricating conductive hydrogels for bioelectronic applications due to its high electrical conductivity and solution-based processing capabilities.22,55,56 Liu et al. reported soft micropillar electrodes composed of an electrically conductive hydrogel with tissue-like stiffness for electrophysiological recording of HL-1 cardiomyocytes.55 The soft conductive hydrogel electrodes were composed of PEDOT:PSS modified with ionic liquid and exhibited a Young's modulus of 13.4 kPa. The soft nature of the electrodes allowed for accommodation of the movements of cardiomyocytes during beating (Fig. 2a). Furthermore, this conductive hydrogel reduced the impedance at the tissue-electronic interface to improve transduction of electrophysiological signals (Fig. 2b). Altogether, this hydrogel electrode demonstrated a greater quality in recorded signals in terms of both amplitude and larger signal-to-noise ratio compared to metal electrodes with a stiffness of 100 GPa (Fig. 2c). Moreover, Yuk et al. developed a method for 3D printing PEDOT:PSS polymers that can be used to form conductive hydrogels.57 After printing and annealing, the dry 3D-printed polymer exhibits a conductivity over 155 S cm−1. The conductive polymer can be converted into hydrogel by swelling in aqueous solution. In the hydrogel state, the Young's modulus was reported to be 1.1 MPa with an electrical conductivity of 28 S cm−1. This approach was utilized to fabricate soft probes for in vivo recording of neurons over a 2-week period (Fig. 3a–c). Dalrymple et al. demonstrated the advantages of conductive hydrogel coated platinum electrodes versus bare platinum electrodes implanted in rat cochlea.54 PEDOT was incorporated into a PVA hydrogel as a conductive hydrogel coating and electrodes were implanted over a 5-week period. The coated electrodes showed significant improvement of electrical properties, displaying significantly higher charge storage capacity, charge injection limit and lower impedance. The effective long-term integration of bioelectronic devices in vivo is vital for communication with the body. These studies present the use of hydrogel to facilitate structural integration and improve signal coupling at the bioelectronic interface. Thus, engineering of both hydrogel and device properties to match the biological environment offers the potential to overcome the challenges of immune response caused device failure.
 | ||
| Fig. 2 Comparison between soft hydrogel probes and rigid metal electrodes for interfacing with beating HL-1 cardiomyocytes. (a) Schematic of soft conductive micropillars for electrophysiological recording of HL-1 cardiomyocytes during spontaneous beating. (b) Impedance measurements of metal micropillars (blue) compared to conductive hydrogel micropillars (red). (c) Extracellular recording of cardiomyocyte activity from the conventional metal electrode (top) and soft conductive hydrogel micropillar (bottom). Reproduced from ref. 55 with permission from National Academy of Sciences, Copyright © 2018. | ||
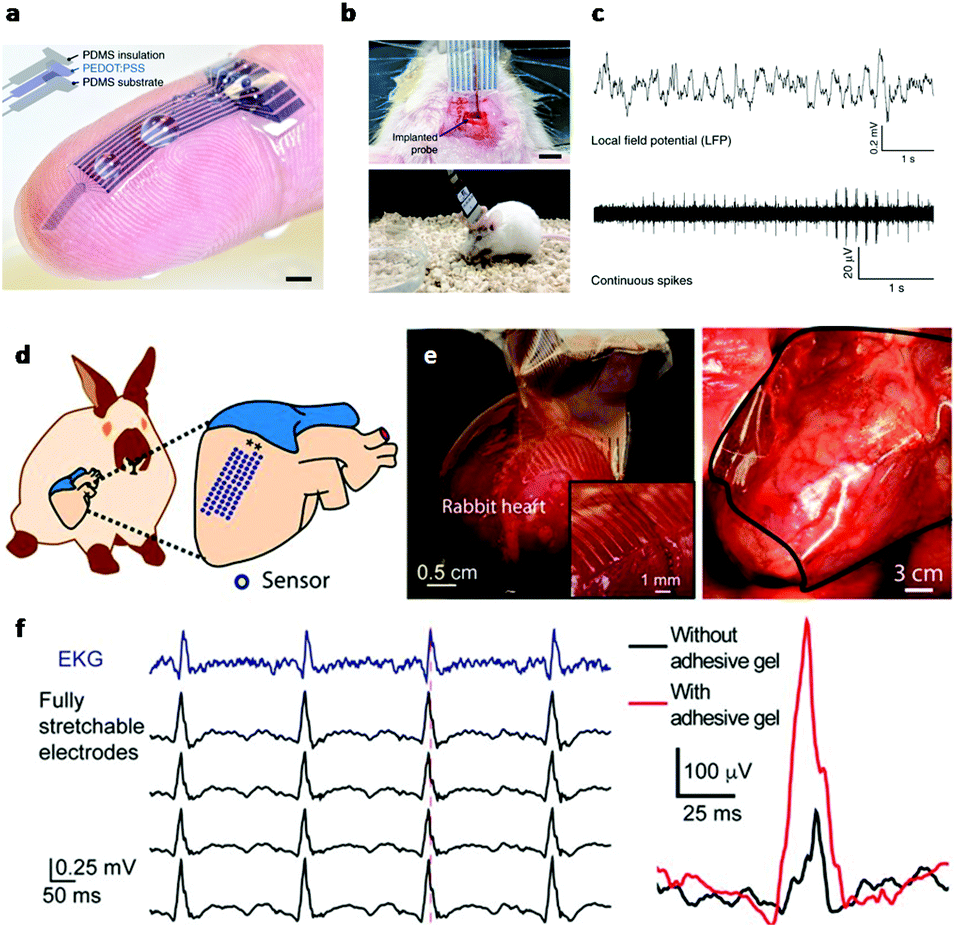 | ||
| Fig. 3 Hydrogels for in vivo tissue-electronics interfacing. (a) Image of 3D printed soft neural probe. Scale bar, 2 mm. (b) Images of probes implanted in mouse. (c) Continuous measurement of local field potential (top) and extracellular action potentials (bottom). Reproduced from ref. 57 with permission from Springer Nature, Copyright © 2020. (d) Schematic of the stretchable hydrogel electrode array placed on heart. (e) Images of the hydrogel electrode array conforming to a rabbit heart. (f) Left: Voltage traces from electrocardiogram and hydrogel electrodes. Right: Voltage trace from hydrogel electrodes with (red) and without (black) bioadhesive gel. Reproduced from ref. 58 with permission from National Academy of Sciences, Copyright © 2020. | ||
In addition to common conductive hydrogels, composite hydrogels have been developed to provide additional versatility in bio-integration due to their tunable soft, conductive, and elastic properties. For example, an interpenetrating hydrogel network composed of both poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) and polyacrylic acid hydrogels was electrically conductive and highly elastic, capable of stretching over 100% strain while maintaining conductivity.56 The stiffness could be tuned between 8 and 374 kPa by changing the polymer concentrations, making it applicable to match a wide range of biological tissue. Similarly, Liu et al. demonstrated a 64 channel array of hydrogel electrodes for interfacing with beating hearts for electrophysiological recording in vivo (Fig. 3d).58 The electrodes of this array are designed to be <100 μm for potential single cardiomyocyte recording and possess tissue-like Young's modulus and elasticity, which enable a stable interface with beating cardiac tissue in vivo (Fig. 3e). Additionally, the device was glued to the heart using a bioadhesive for strengthening hydrogel-heart integrations. This strategy can provide stable signal recording during heart beating and leads to the improvement in signal quality (Fig. 3f). Moving forward, composite hydrogels may be further engineered for additional functions, such as eluting bioactive substances (i.e. growth factors or drugs). For example, a multifunctional hydrogel coating incorporated with both conducting polymers and anti-inflammatory drugs was used for improving the interface of neural cuff electrodes.59 The device displayed significantly increased axon density and decreased scar tissue formation in the surround area compared to control groups, and was capable of recording and stimulating over 5 weeks. These studies demonstrate the potential of hydrogel bioelectronics for long-term in vivo use by matching the mechanical properties of the device to the in vivo environment and attenuating the immune response. Overall, the extensive tunability of electrically conductive hydrogels endows them with great potential for use in implantable bioelectronics. By utilizing the tissue-like properties of hydrogels with the electrical properties of conducting polymers, conductive hydrogels enable improved structural integration and signal coupling.
3.2 Biochemical signaling
Many biological functions including sensation, metabolism, immune response, etc., are mediated by a series of biomolecular interactions such as enzymes, membrane/nuclear receptors, and antibodies/immunoglobulin receptors. The precise interpretation of these complex biochemical signals in the quantitative electrical language will provide unique insights into the underlying biological function. Electrochemical methods have been widely used for bio-to-electronic signal transduction. In particular, with the incorporation of bio-recognition molecules that either (1) selectively convert the target analyte into electroactive species or (2) selectively bind to the target analyte, the electrochemical sensors can specifically translate corresponding biological events in the form of current, potential or impedance changes. A comprehensive review in electrochemical bioelectronics was presented by Ronkainen et al.60 Alternatively, FETs possess unique capability to actively sense and amplify the variation of electrical potential at the device surface. When integrated with bio-recognition molecules such as enzymes, antibodies, and single-strand DNA, the selective binding of the target molecule, or the generation of biologically derived species induces a change in local charge and the biological event is transduced into an electrical signal in real time. This capacity makes FETs an excellent candidate for coupling electronic- and living-signals. While both types of detection mechanisms have been widely investigated, further improving the signal transduction at bio-electronic interfaces, especially under physiologically relevant conditions, remains challenging.First, interfacial signal attenuation becomes significant as the bioderived molecules are quickly diluted and/or neutralized before meaningful information can be transmitted to the electronics, demanding extremely intimate bio-electronic interfaces.61–63 In particular, for FET sensing, signal attenuation is aggravated by the presence of a high-concentration of electrolytes, which induce electrostatic screening.64 The strength of the electrical field generated by charged analytes is diminished at a distance of 0.75 nm in physiological environments. Although diluted buffer solutions, desalting, or purification can increase the Debye screening length, post-processing compromises the real time sensing capabilities of bioelectronics.65 Shorter bioreceptors such as truncated antibodies66 and aptamers67,68 have also been exploited to overcome the charge screening effect, but their application is typically limited by their complex design/synthesis.
Second, nonspecific binding of background species such as serum albumin can induce significant false signals or biofouling interfering with the functioning of bioelectronics. Effective filtering of competing biochemical signals has the potential to improve the device performance in both sensitivity and selectivity. Existing strategies (e.g. pre-absorption of blocker proteins69 or hydrophilic/hydrophobic modifications) could reduce the non-specific binding of certain biomolecules, but lack the capability to regulate the accessibility of dynamic biochemical signals in general.70
Lastly, the chronic performance of bioelectronics is compromised by the limited lifetime of bio-recognition components, which lose their activity quickly as a result of fast and progressive chemical/structural degradation in non-native environments. This issue is further amplified by the bio-incompatible functionalization strategies such as physical adsorption or chemical conjugation.61 Physical adsorption usually relies on van der Waals or electrostatic interactions.62 However, these weak interactions can lead to desorption of biomolecules and loss of sensitivity over time.63 Chemical conjugation generates a strong and stable biomolecule attachment through covalent bonding,71 but typically compromises the bioactivity due to the disturbance to the native structure.72
Toward overcoming these mismatches, hydrogels have been utilized to immobilize molecular biomachinery such as enzymes or antibodies for functionalizing electronics.73 The “hydrogel biotransducer” demonstrates abilities in (1) modifying the local dielectric environment thus increasing the Debye screening length;15,74 (2) regulating the “input” and “output” biosignals through mass transport control,75 which reduces nonspecific absorption/interactions of interference species75,76 while enriching/amplifying the bio-transformed signal; and (3) providing a biologically relevant microenvironment for maintaining the functions of immobilized biomachinery, through mild, biocompatible fabrication processes. Recent developments in hydrogel enabled structural integration and signal coupling between biomolecules and electronics are summarized in the following sections.
Enzymatic transformation has been widely explored in electrochemical based sensor design, where hydrogels can preserve the activity of encapsulated enzymes77 while providing sufficient porosity to facilitate the contact between electrodes and enzymatic products. Furthermore, the 3-D matrix of hydrogels can also increase the encapsulation efficiency of enzymes compared to planar electrodes, increasing the amplitude of generated biosignals. These features make hydrogels an excellent candidate for enzyme-electronic integration. For example, by immobilizing lactate oxidase inside a dimethylferrocene-modified poly(ethylenimine) hydrogel while incorporating a bilirubin oxidase-based cathode, Hickey et al. fabricated a self-powered lactate biosensor with a detection range between 0–5 mM with a sensitivity of 45 ± 6 μA mM−1 cm−2.78 Additionally, Wang et al. immobilized alcohol oxidase and glucose oxidase onto the electrodes using a chitosan hydrogel. These hydrogel-based biosensors present the ability to detect alcohol and glucose in bodily fluids by measuring electric currents produced by the enzymatic reactions.79,80
To enable multiplexed sensing capability, Yan et al. fabricated a biosensor array through a multistep photopolymerization to immobilize glucose oxidase and lactate oxidase on separated microelectrodes. This device demonstrates simultaneous detection of glucose and lactate with a sensitivity of 0.9 μA cm−2 mM−1 and 1.1 μA cm−2 mM−1, respectively.27 Li et al. also demonstrated the multiplex detection of different biomarkers by functionalizing electrodes with hydrogels through multi-step inkjet printing.81 By loading the printer cartridges with different bio-inks, electrodes were independently functionalized with different enzymes sensitive to glucose, lactate, and triglycerides. The sensors perform similarly in both phosphate buffer and serum solutions, which indicates that hydrogels can minimize the interference from background metabolites and molecules. Besides, the fabrication using ink-jet printing represents the possibility for mass production of biosensors with customized biomarker functionalization.
Similarly, Bay et al. created a multi-functional FET array using projection microlithography with diffraction-limited spatial resolution. In this design, enzyme functionalized polyethylene diacrylate (PEGDA) hydrogels were individually crosslinked on the top of graphene FET by controlling the area of light exposure with an inverted microscope and a computer-controlled photomask (Fig. 4a). Multiplex detection was demonstrated by sequential photopolymerization of hydrogels containing enzymes for the specific detection of penicillin or acetylcholine (Fig. 4a). The hydrogel encapsulation was also shown to extend the activity of penicillinase up to 7 days compared to only several hours in solution. Additionally, the PEGDA hydrogel was found to significantly reduce the nonspecific absorption of bovine serum albumin (BSA, MW 6.65 × 104 g mol−1) to the FET surface.76 To further improve the design flexibility, Dai et al. demonstrated the modular version of hydrogel-gate FETs made of independently fabricated enzyme functionalized hydrogels and an electronic transducer that can be reversibly assembled/disassembled.75 In this work, hydrogels containing urease and penicillinase were fabricated in a mold and then integrated onto the FET. The enzymatic reaction is highly confined within the hydrogel environment, accumulating within and slowing the diffusion to the external buffer environment. This local signal amplification allows for sensing without the permanent surface modification of the FET device and enables the ability to reprogram or replenish the bioreceptors by switching hydrogels without affecting the device sensitivity (Fig. 4b).
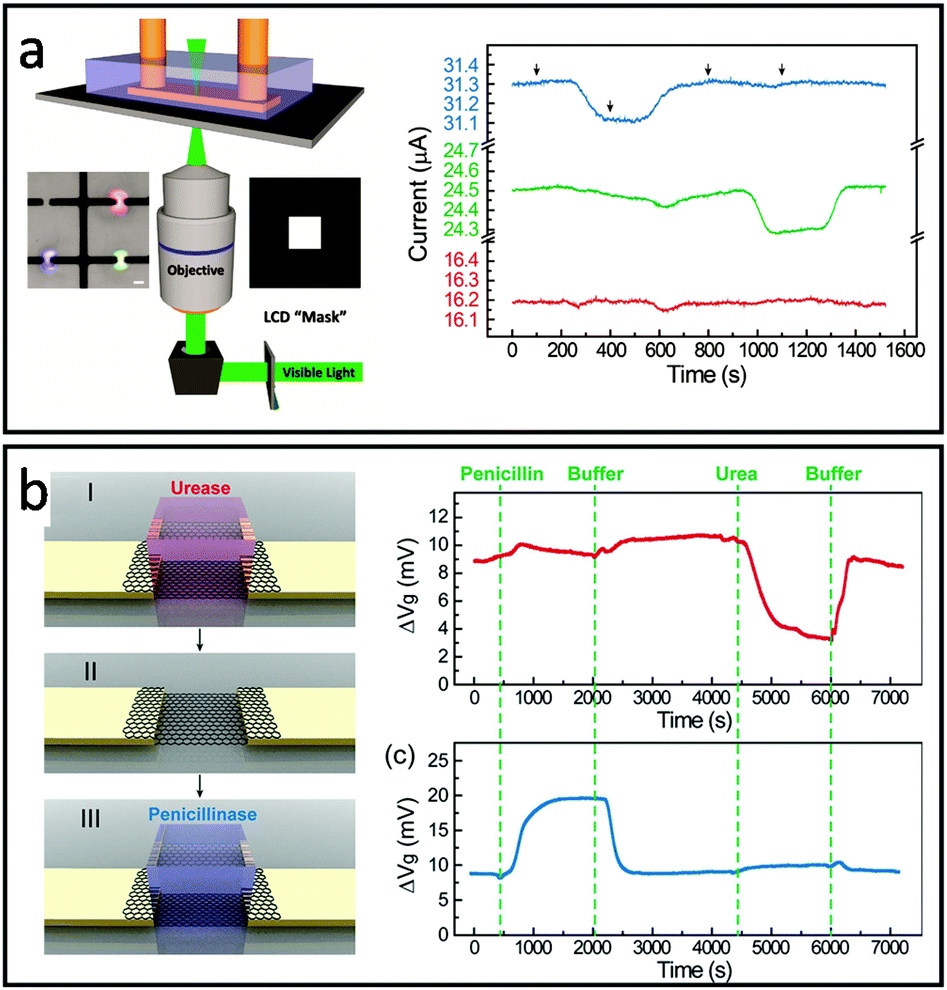 | ||
| Fig. 4 Designs of multifunctional-hydrogel-based-bioelectronics: (a) Left: schematic of projection lithography setup for hydrogel patterning. (inset of (a)) Image of hydrogels containing red, blue, and green fluorescence dyes. Scale bar, 20 μm. Right: Multiplex sensing of penicillin (blue), acetylcholine (green) and no-enzyme control (red). Reproduced from ref. 76 with permission from American Chemical Society, Copyright © 2019. (b) Left: Schematic of hydrogel-enabled modularized FET. Right: Performance of a modularized FET biosensor functionalized by the urease-encoded hydrogel (red) and penicillinase-encoded hydrogel (blue). Reproduced from ref. 75 with permission from American Chemical Society, Copyright © 2019. | ||
For active transducers like FET, another critical challenge is associated with electrostatic screening as the signal transduction is achieved through biologically induced changes in the local electrical field. This becomes particularly challenging in the physiological environment, where the effective detection range (or Debye length) is within the nanometer length-scale.82 By modifying the local dielectric environment and modulating the charge distribution, hydrogels provide a promising solution to reduce electrostatic screening for high-sensitivity FET detection in physiological fluids without pre-processing. For example, Lieber and colleagues presented that the Debye screening length of both silicon nanowire- (SiNW) and graphene-based FET can be significantly increased by PEG hydrogel functionalization.15,74 First, SiNW-FET modified by the PEG hydrogel successfully detected prostate specific antigen (PSA) in phosphate buffer solutions (PBS) with concentrations as high as 150 mM, whereas FETs without PEG could only detect PSA in PBS concentrations lower than 10 mM (Fig. 5a). Concentration-dependent measurements also demonstrate that in 100 mM PBS, PEG modified SiNW-FET is able to hold linear response to PSA in the range of 10 to 1000 nM when implemented.15 Similarly, PEG-modified graphene FETs also exhibited real-time reversible detection of PSA from 1 to 1000 nM in 100 mM PBS. In addition, co-modification of graphene FET with PEG and PSA aptamers enabled the sensitive yet reversible detection of PSA since (1) the conformational changes of these highly charged aptamers upon PSA binding led to a significant change in the electric field of graphene gate and (2) aptamers have reversible binding ability with PSA without loss of activity (Fig. 5b).74 Additionally, recent advancements in bio-stimuli responsive smart hydrogels represent an alternative strategy to overcome the charge screening effect by actively transducing and amplifying the biomolecular binding within the hydrogel matrix. For example, hydrogels made of mannose and N,N-dimethylacrylamide that undergo volume change in response to the formation of lectin-mannose molecular complex are applied as gate materials for fabricating FET-based lectin sensors. The change in hydrogel volume can introduce a shift in the local electrical field at the gate electrode, which can be detected by the FET.83 Many smart hydrogels have been developed recently, including antigen-,84 nucleic acid-,85 and enzymatic reaction-responsive hydrogels.86 We believe that functions of molecular-level bioelectronics can be broadened to a new level through further exploring possibilities in smart hydrogel-electronics integration.
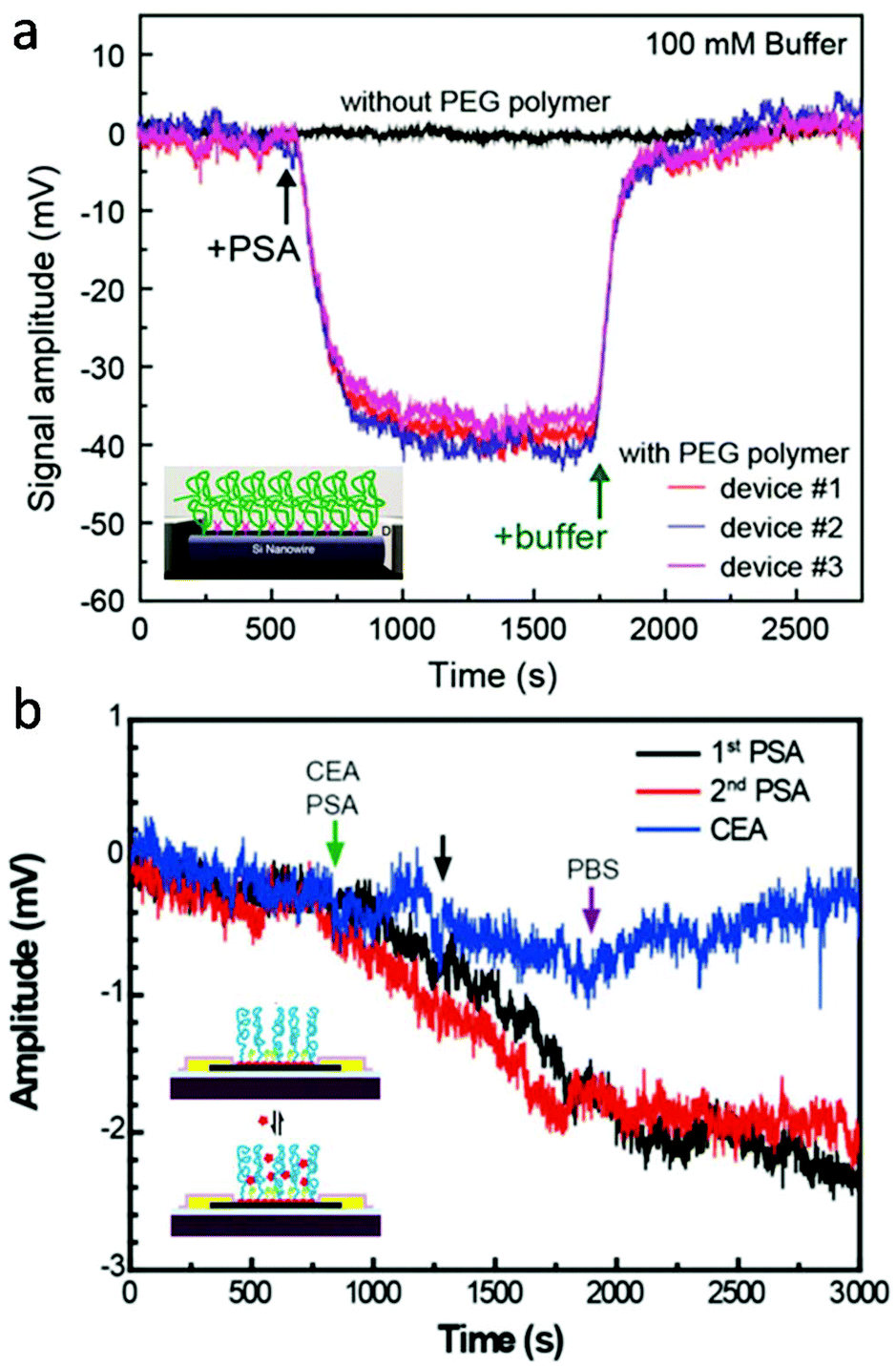 | ||
| Fig. 5 Hydrogel coating for reduced charge screening. (a) PEG modified SiNW-FET, which demonstrated reversible detection of PSA antigen in 150 mM PBS solution, while FET without PEG showed no signal. Reproduced from ref. 15 with permission from American Chemical Society, Copyright © 2015. (b) Graphene FET co-modified with PEG and PSA aptamers, which exhibited real-time reversible detection of PSA from 1 to 1000 nM in 100 mM PBS. Reproduced from ref. 74 with permission from National Academy of Sciences, Copyright © 2016. | ||
In hydrogel transducers, mass transport inside the hydrogel matrix determines the accessibility of ions and molecules to the FET gate, providing additional control over device sensitivity and selectivity based on specific demands. In general, the mass transport properties of the hydrogel material can be regulated by tuning the molecular weight of the monomer,87 cross-linking density,88 or through the introduction of specific-sized porogens.89 In the modular FET design presented earlier,94 for example, the diffusion of methylene blue (MB, MW 320 g mol−1) exhibits a substantially varied rate in hydrogels crosslinked from PEGDA, gelatin methacrylate (GelMA), and alginate, as a result of the difference in pore size (Fig. 6a insert).75 Correspondingly, the FET functionalized with GelMA shows a 4 mV signal after the introduction of poly-L-lysine (PLL) solution, while the same PLL solution cannot induce a detectable signal in the PEG functionalized FET (Fig. 6a).75 This difference in mass transport demonstrates a significant effect in preventing the nonspecific binding from large biomolecules with a hydrogel-gate design. Similar results have also been demonstrated in the research of Burrs et al., in which, alcohol oxidase was immobilized onto a nanoplatinum–graphene–modified electrode using hydrogel made of chitosan, poly–N–isopropylacrylamide (PNIPAAM), silk fibroin, and cellulose nanocrystals. The results demonstrated that high porosity of chitosan and PNIPAAM hydrogels can lead to better sensitivity and faster response time during alcohol sensing.90 Also, Kim et al. demonstrated the PEG hydrogel functionalization of interdigitated microelectrodes for the detection of amyloid beta 42 (Aβ42, 2.2 nm diameter) and prostate-specific antigen (PSA, 4.1 nm diameter) via antibody–antigen binding.91 The hydrogel porosity was adjusted between two sizes, “loose” and “dense”, by tuning the molecular weight of PEG monomers. The dense hydrogel enabled the diffusion of Aβ42 selectively, where the diffusion of PSA was inhibited. Detection of PSA was achieved on devices functionalized by a loose hydrogel where the diffusion of PSA results in signals greater than twice that of both dense hydrogel- and non-modified devices (Fig. 6b). Besides, the results indicated that the hydrogel functionalization also increased the device sensitivity, owing to its three orders of magnitude increase in immobilized antibodies as compared to electrodes without hydrogels.
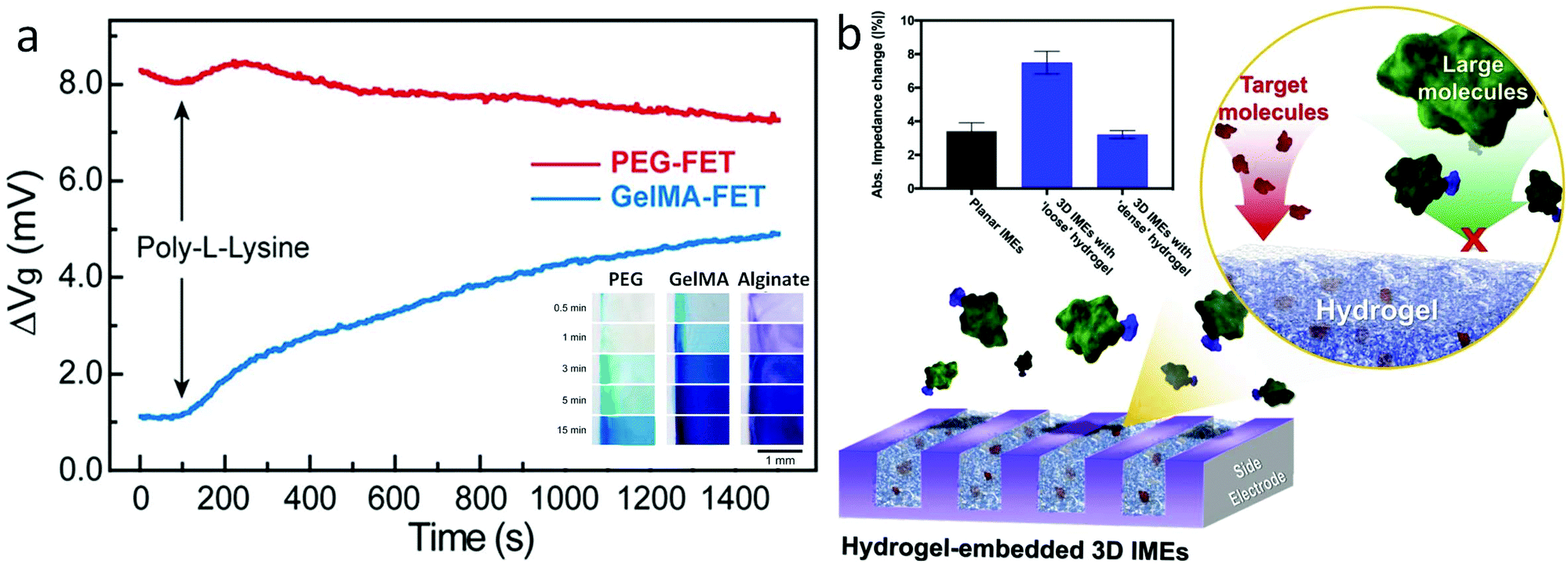 | ||
| Fig. 6 Bio-signal filtering by modulating the mass transport of the hydrogel matrix. (a) Compositional controls: poly-lysine nonspecific binding tests on FET passivated with PEG (red) and GelMA hydrogel. Results indicated that PEG can effectively prevent external noise from poly-lysine of due to its small pore size/low mass transport. Insert: Diffusion of methylene blue inside PEG, GelMA, and alginate hydrogel over time. Results present the influence of different hydrogel components in mass transport. Reproduced from ref. 75 with permission from American Chemical Society, Copyright ©2019. (b) Schematic of electrodes functionalized with the hydrogel. Inset: Impedance changes in planar electrodes and with dense and loose hydrogels by binding of Aβ42. Insert: Impedance changes in planar electrodes and with dense and loose hydrogel by binding of PSA. Reproduced from ref. 91 with permission from Elsevier, Copyright © 2020. | ||
In addition to mass transport, the chemical properties of hydrogels can be tuned to achieve selective diffusion of molecules with a certain charge or chemical affinity.92 This general strategy could serve to promote the real-time and label free detection of analytes in physiological solutions. The additional selectivity can increase the functionality of the bioelectronics for real-time sensing applications, potentially decreasing the need for pretreating samples to remove background species or significantly reducing biofouling and nonspecific adsorption for in vivo implantation. Besides, computational modeling could provide useful insights into the interfacial transport processes93,94 which will further assist the hydrogel design for both signal enrichment and reduced nonspecific binding. Due to these unique advantages in hydrogel functionalization, various bioreceptors have been incorporated with the hydrogel-based bioelectronics to transduce biological signals such as femtomolar levels of disease antibodies, nucleic acids, and single viruses.95–97 These approaches have opened many new opportunities in bioelectronics such as biosensing, implantable stimulators, drug screening, disease models, brain-machine interfaces and more.
4. State-of-the-art applications of hydrogel-based bioelectronics
4.1 Tissue-electronic interfaces
Hydrogels have been widely utilized as soft, bioactive coatings, or 3-D constructs to improve the integration of cells with synthetic substrates/scaffolds, which can promote cell adhesion, proliferation, and lifetime.98–100 In the context of bioelectronics, hydrogel mediators have been found to benefit cell functioning and bi-directional signaling for both electroactive- (e.g. neurons,101,102 cardiomyocytes103,104etc.) and non-electroactive-cells (macrophages,105 HeLa cancer cells,106etc.). In terms of electroactive cells, hydrogels offer superior biocompatibility to maintain their morphology and functions such as metabolism, proliferation and differentiation, while providing sufficient porosity to ensure the transduction of physiological signals. For example, a fibrin-based hydrogel was used as a soft substrate for integrating human induced pluripotent stem cell (iPSC) derived cardiomyocytes with nanomesh probes.103 The soft mechanical and elastic properties of both the hydrogel and probes allowed cardiomyocytes to perform contraction and relaxation motions comparable to the one without nanomesh attachment. This device enabled the recording of electrophysiological signals of the cardiomyocytes over 96 hours without significant cell damage. Moreover, Kujala et al. applied a micro-molded gelatin hydrogel to integrate cardiomyocytes with microelectrode arrays. On this device, the immobilized cells were able to develop normally to form laminar cardiac tissues, which were then exploited to investigate the pharmacological effects of the β-adrenergic agonist and terfenadine in human cardiac cells with electrophysiological recording.104 The latest developments in this direction have been discussed in the review articles published by Kitsara et al. and Fattahi et al.107,108In addition to cell/tissue recording on a planar substrate, there have been substantial on-going effort towards the construction of 3D electronic-innervated cells/tissues. Many studies suggested that the organization, development, and communication of cells are significantly different when cultured/immobilized on 2-D substrates as compared with their normal conditions in a native 3-D matrix.109,110 This difference can lead to bias/error in the in vitro studies in cellular behaviors and functions using planar bioelectronics. In tissue engineering, 3-D cell cultures are a popular approach, which provide a biological relevant microenvironment to ensure the normal behavior of cells.111 In order to enable the electrical access to these 3-D cultured cellular networks, many hydrogel-based 3-D electronics have been developed. In 2019, Kalmykov et al. demonstrated the use of self-rolling electrode arrays for interfacing with 3-D hydrogel cardiac models (Fig. 7a and b).112 The 3-D hydrogel creates a natural microenvironment by providing a scaffold that allows biologically relevant cell–cell and cell–matrix interaction, recapitulating the in vivo environment that cannot be achieved in 2-D cell culture.113,114 This allows for the detection of biologically relevant behavior from in vitro models. Self-rolling the electrode array around the hydrogel spheroid enables electrophysiological recording of 3-D signal propagation (Fig. 7c). Similarly, Soscia et al. reported the use of flexible 3-D microelectrode arrays for interfacing with and recording from 3-D neuron cultures in a collagen-based hydrogel (Fig. 7d and e). The hydrogel cell culture creates an environment that aims to recapitulate real brain function by facilitating cell–cell communication and interactions. The flexible electrodes could bend vertically 90 degrees in order to record in 3-D hydrogels. After vertical alignment of electrodes, the microelectrode arrays were seeded with human iPSC-derived neurons and astrocytes in a collagen hydrogel containing extracellular matrix proteins. Electrophysiological recordings were conducted (Fig. 7f) and neurons were found to be viable for over 30 days, demonstrating the potential for long-term studies in vitro.101,102
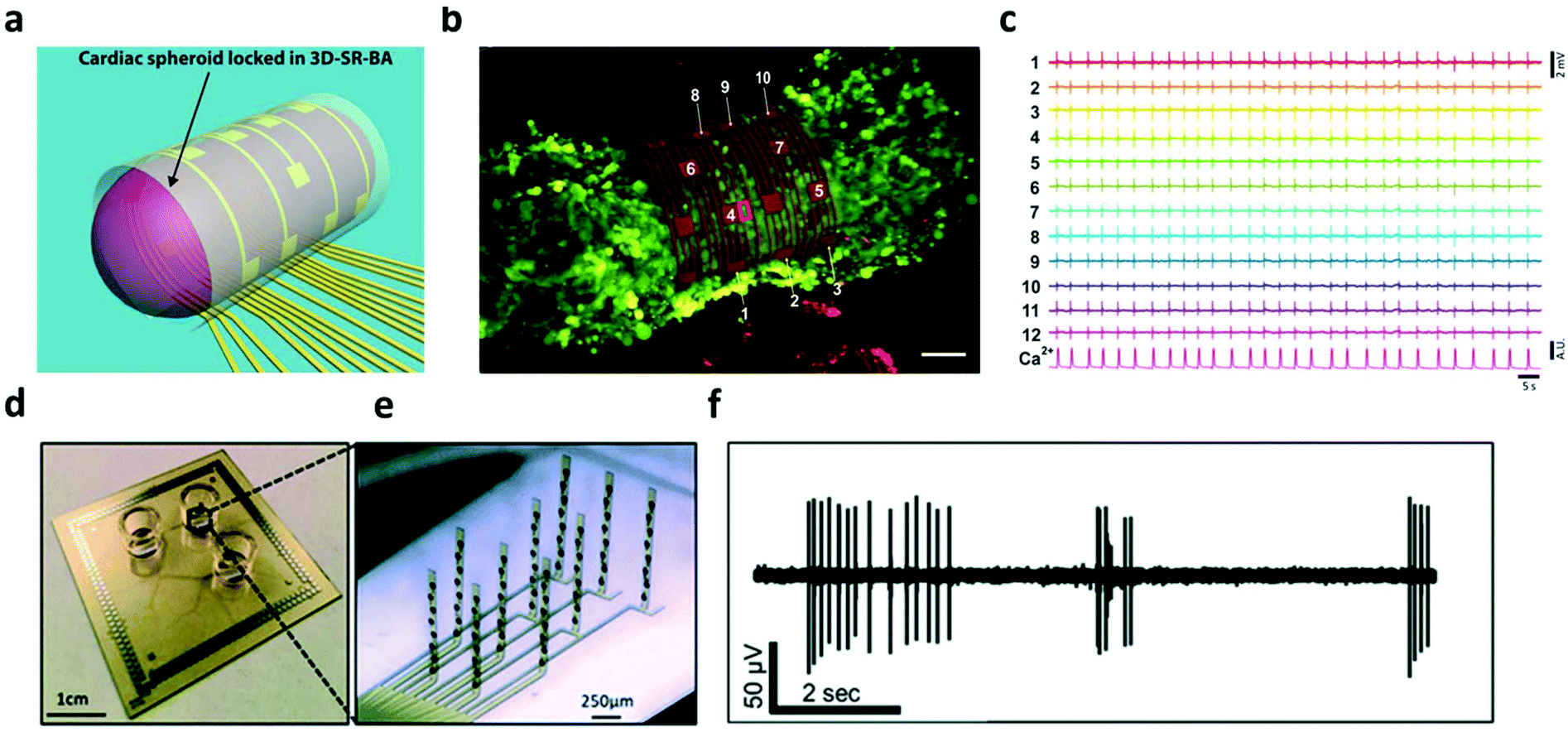 | ||
| Fig. 7 3D electrode interface with 3D in vitro models. (a) 3D schematic of organoid interfacing with self-rolled biosensor array. (b) Confocal image of cardiac spheroid labeled with the fluorescent calcium indicator. Scalebar, 50 μm. (c) Field potential measurements from recording elements around the spheroid. Reproduced from ref. 112 with permission from American Association for the Advancement of Science, Copyright © 2019. (d and e) Image of the device and close-up of bent electrodes. (f) Recording of neuronal activity in the 3D culture from a single electrode. Reproduced from ref. 101 with permission from Royal Society of Chemistry, Copyright © 2020. | ||
Hydrogel electronics have also been exploited to improve the electrical-to-biological signal transduction. Zhao et al. developed an electronic circuit made of salt/PEG two–phase hydrogels that is capable of effective modulation of cultured neuron cells (SH–SY5Y) and skeletal muscle tissue.48 In this design, high ionic conductivity salt–solutions were stably encapsulated within PEG hydrogel matrices. Patterning of the hydrogel circuit enables control over ionic current for high resolution stimulation both in vitro and in vivo. For in vitro neuron cell stimulation, a hydrogel based electronic circuit composed of four pairs of electrodes was applied, which delivered 3.6 V cm−1 electrical field to cells for stimulation (Fig. 8a and b). The results showed that the cells at the stimulated spots exhibited higher intracellular calcium increase compared to cells located at the resting spots, indicating successful cross-system signal transduction (Fig. 8c). For in vivo stimulation, a hydrogel ionic stimulator made of one pair of electrodes was interfaced with the tibialis anterior muscle at the knees of Sprague–Dawley rats. The stimulation results showed the force generated from stimulation increased slightly from 300 mN at a voltage of 0.9 V to a plateau of 380 mN with voltages of either 1.6 or 2.5 V. Additionally, compared with the gold electrode, a lower voltage (2.5 V vs. 4 V) was required to generate a similar force (1.38 N vs. 1.33 N) when a hydrogel stimulator was used, indicating more efficient electrical signal transmission/delivery.
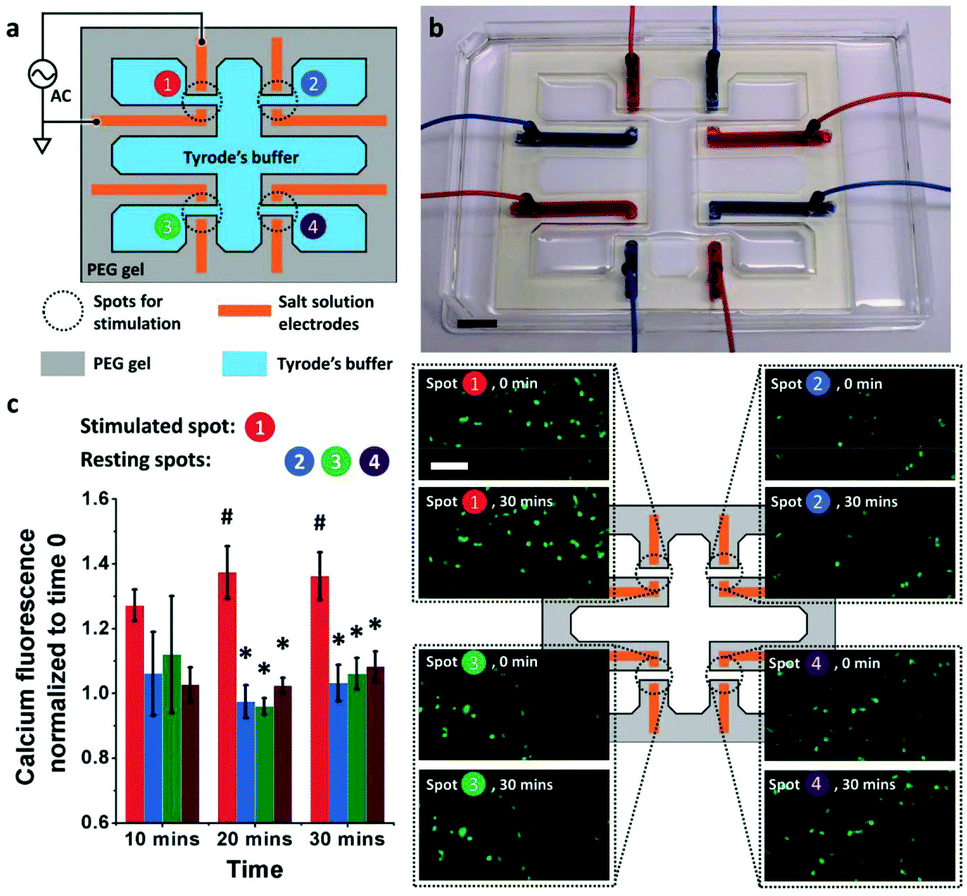 | ||
| Fig. 8 Hydrogel enabled bioelectronic interface for the manipulation of cellular functions (a) the schematic of the hydrogel ionic electrode array for in vitro neuron cell stimulation. (b) Image of the actual electronic circuit made of the PEG hydrogel with 20% w/w PEGDMA 8000, 20% w/w PEGDA 700, and 1% w/w irgacure 2959. Scale bar, 1 cm. (c) Left: The intracellular calcium fluorescence change during stimulation (error bars indicate standard deviation, N = 3). Spot 1 was stimulated, while the other spots were at rest. At 20 and 30 min, the fluorescence at stimulated spot (#) was significantly different from that at resting spots (*) (p < 0.05). Right: The corresponding fluorescence images at time 0 and 30 min at each spot. A higher fluorescence increase was seen at the stimulated spots. Scale bar is 100 μm. Reproduced from ref. 48 with permission from John Wiley & Sons, Inc., Copyright © 2018. | ||
Similarly, Liu et al. utilized micropatterned electrically conductive hydrogels (MECH) to fabricate microelectrodes for interfacing the nervous system of mice.22 Owing to their electrical and ionic conductivity as well as soft mechanical properties, the MECH-based microelectrodes feature a contact impedance >90% lower as compared with the conductive hydrogel coated Au electrode and >95% lower than the silane-crosslinked PEDOT:PSS coating. This low contact impedance enables the delivery of an excitation current density as high as 10 mA cm−2 at a low voltage of 50 mV, whereas the Pt electrode requires at least 500 mV to achieve observable leg movements. The experimental results demonstrated that MECH can locally stimulate the subgroups of peripheral nerve bundles to synchronize individual toe movements with the stimulation frequency.
In terms of non-electroactive cells, most of their functions are regulated by biochemical signals. The specific electrical/electrochemical transduction of these signals relies on the appropriate functioning and effective integration of bio-recognition molecules, where the hydrogel could enable unique possibilities to promote interfacial signaling as discussed earlier. For instance, Misun et al. demonstrated the amperometric detection of glucose consumption and lactate production from human colon carcinoma spheroids.115 The device consisted of two modular components: a microfluidic platform for media perfusion and glass plug-in with electrode components (Fig. 9a). The electrodes were functionalized with the enzymes: glucose oxidase or lactate oxidase immobilized in a hydrogel, enabling the real time detection of cell metabolism. The device measured the real time secretion/consumption of analytes from the perfused cell media (Fig. 9b). Lian et al. reported the amperometric detection of hydrogen peroxide secreted from HeLa cells utilizing horseradish peroxidase (HRP) functionalized hydrogel coating on a glassy carbon electrode (Fig. 9c).106 HeLa cells were cultured on top of bioactive hydrogels, showing activity for up to two weeks. Cells were stimulated with Phorbol 12-myristate 13-acetate (PMA) to trigger hydrogen peroxide production. Horseradish peroxidase (HRP) was immobilized in the hydrogel, enabling the real time detection of hydrogen peroxide (Fig. 9d). The hydrogel also served to inhibit the diffusion of hydrogen peroxide secreted from cells, effectively increasing the concentration that directly interacts with the HRP enzyme. A similar design has been applied by Yan et al. to study the metabolism of macrophages.105 These studies demonstrate the possibility for real time interpretation of cellular metabolic signals, which could be further expanded through incorporating different biomarkers and/or bioreceptors for real time drug screening, disease monitoring and personalized medicine.
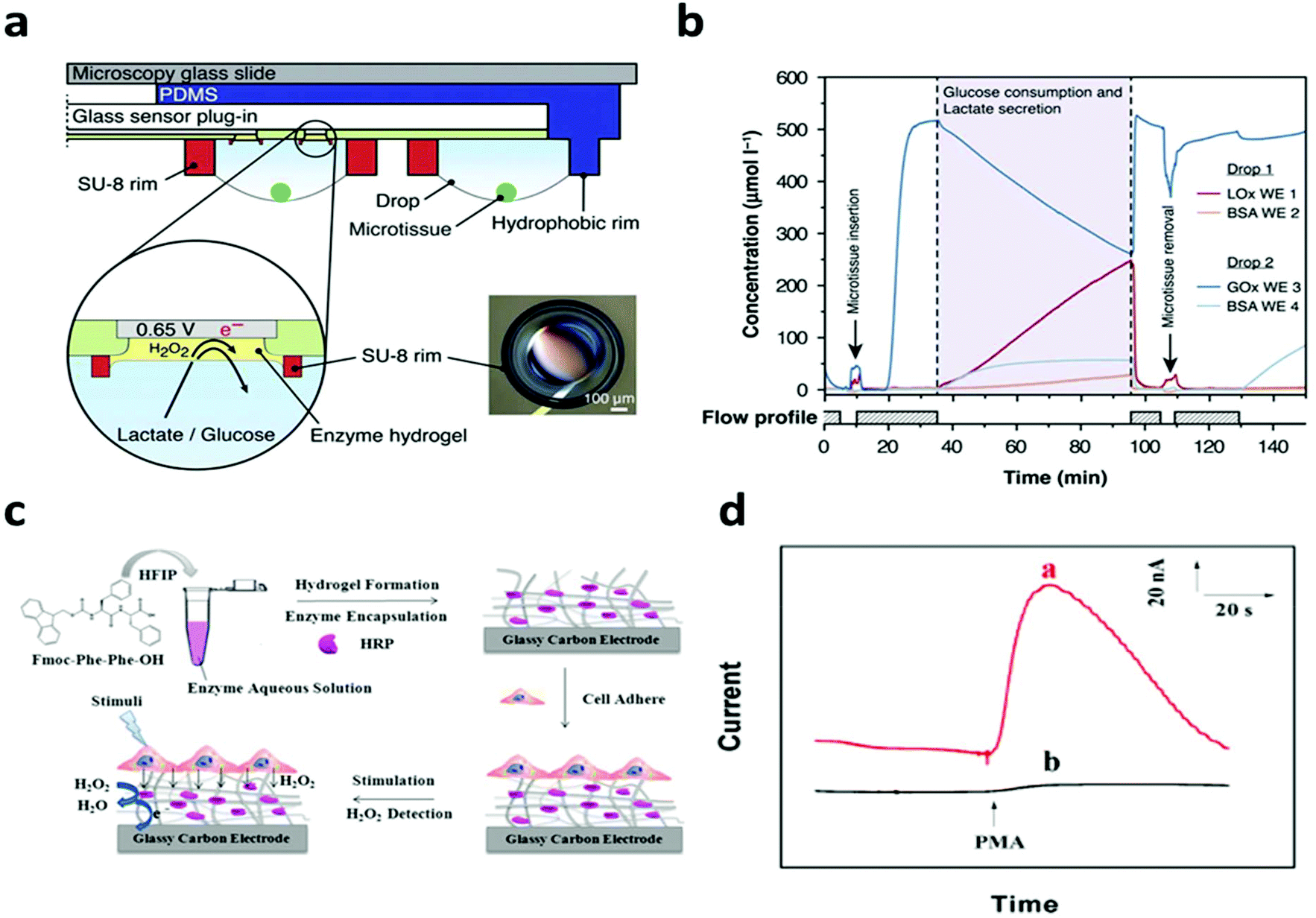 | ||
| Fig. 9 Hydrogel functionalization enables real time monitoring of cell metabolism. (a) Schematic of the biosensor device with hanging drop networks for cell culture and hydrogels functionalized with lactate oxidase and glucose oxidase. (b) Real time monitoring of glucose consumption and lactate production. Reproduced from ref. 115 with permission from Springer Nature, Copyright © 2016. (c) Schematic of hydrogel formation and cell integration for electrochemical biosensing of H2O2 after chemical stimuli. (d) Current response of the sensor with (red) and without (black) HeLa cells after chemical stimulation. Reproduced from ref. 106 with permission from American Chemical Society, Copyright © 2016. | ||
4.2 Wearable bioelectronics
Wearable bioelectronics are capable of real-time, noninvasive monitoring of physiological signals, and have become increasingly common in our everyday lives, e.g. in the form of smart watches/bands that can continuously measure heart rate or blood oxygen saturation.116 However, these commercially available wearable devices share some similar challenges with metal/semiconductor based bioelectronics with unstable body contact that is associated with low sensitivity and fluctuation in sensing results.117 To address this issue, flexible and stretchable electronics have been developed that comply with the curvatures of the human body; maintaining stable contacts to ensure consistent sensing results. Toward this goal, hydrogels are suggested as ideal body-electronics interfacing materials due to their superior mechanical properties and tunable bio-adhesiveness. For example, Pan et al. reported hydrogel-elastomer composites with low stiffness and high adhesiveness for interfacing with skin.118 Gold nanofilms were incorporated into the hydrogel structure for electrical conductivity and were demonstrated for on-skin electromyography and electrocardiography. The reported Young's modulus of the hydrogel composite was reported to be near 5.3 kPa and could stretch 25 times its length, enabling conformal contact with the skin. This work provides a general strategy for on-skin bioelectronics by engineering the hydrogel properties.In wearable electronics, body motions are one of the most common challenges that can lead to device detachment, abrasion, fracture, and eventually failure of device functions. Recent studies in stretchable-, tough- and healable-hydrogels provide potential solutions to this challenge.119,120 With further enhanced ionic conductivity, these novel hydrogels show potential to replace state-of-the-art substrates (e.g. metal, semiconductor, dry polymer, etc.) in the development of next generation wearable electronics. For example, Zhao et al. fabricated a conductive hydrogel from a supramolecular assembly of polydopamine decorated silver nanoparticles, polyaniline, and polyvinyl alcohol. The conductive hydrogel displayed tunable stiffness (132 Pa to 40 kPa), stretchiness (0.01–500%), self–adhesiveness and self-healing capacity, and has been successfully implemented as epidermal motion sensors and diabetic wound dressing.121 Also, Liu et al. created a microfluidic-based, ultra-stretchable hydrogel network with metallic conductivity using liquid metal as conductive fillers.122 This device showed good stretchability and flexibility, which remain functional under many types of deformations (e.g. up to 550% stretch, cyclic stretches, bends, and twists). Due to the metallic conductivity, this hydrogel can be applied to the fabrication of wireless bioelectronics for monitoring physiological conditions of the human body using near-field communication technology. Furthermore, a variety of functional hydrogel designs for wearable electronics have been comprehensively reviewed by Yang and Suo.123
Additionally, multifunctional wearable hydrogel bioelectronics has been developed for simultaneous monitoring of the physiological environment and delivery of drugs for treatment. For example, contact lenses are hydrogel-based medical devices that have long been used to correct vision. By embedding sensors within the lens, smart contact lenses have been used for monitoring diseases such as glaucoma and diabetes.124,125 Keum et al. demonstrated contact lenses capable of monitoring glucose levels from tears in rabbits and delivery of the drugs metformin and genistein for the treatment of hyperglycemia and diabetic retinopathy.126 Similarly, a smart bandage was developed for monitoring of the wound environment and delivery of antibiotics.127 Overall, hydrogels can create many new possibilities in wearable electronics owing to their programmable mechanical, electrical, and chemical properties.128,129
5. Conclusions
Engineered hydrogel interfaces have shown great promise towards the seamless structural and functional integration between biological and electronic systems, which is transforming the design and development of next-generation bioelectronics across molecular, cellular, tissue and body levels. The mismatch at the heterogeneous interface, both structurally and functionally, can be blurred by rationally programming the physiochemical parameters through controlled hydrogel synthesis/fabrication. In terms of structures, hydrogels provide a mechanically compliant, chemically active, and biologically favourable microenvironment for seamless bio-integration that is difficult to achieve on a traditional electronic interface. In terms of functions, hydrogels can facilitate the signal transduction between bio- (ions & molecules) and electrical-(electrons & holes) circuit by precisely regulating interfacial mass and transport, enabling localized amplification and/or filtering of bio-derived signals. At the molecular to the cellular level, the spatial organization and hierarchical assembling of functionalized hydrogels will create new signal transduction and energy conversion cascades with electrically controllable inputs and outputs for novel biosensor and biocatalyst developments.130 At the tissue to the body level, recent developments in stretchable-,131 biodegradable-,132 self-healing-,133 and bio-adhesive-hydrogels134 offer opportunities in designing new bioelectronic interfaces with intimate contact, minimal invasiveness, and maximized motion-compliance. Through these new bioelectronic interfaces, long term, continuous probing and regulation of human functions will be achieved, which are expected to contribute significantly to disease diagnosis and personalized medicine. Overall, we believe that hydrogel-mediated bio-integratable electronics can initiate an evolution in the way we communicate with biological systems by unambiguously decoding critical biological languages and precisely defining/regulating complex bio-functions.The future of hydrogel-based bioelectronics is anticipated to implement more advanced functions beyond the current scope of bioelectronics. However, before hydrogels can fully address the interfacing challenges, more validation and optimizations are required. Mainly, their long-term performance and biocompatibility demand further evaluation and optimization in order to obtain intimately integrated, yet chronically stable bio-interfaces, which are critically important to in vivo and implanted applications. Other concerns include degradation and potential cytotoxicity of different synthetic hydrogels, as well as additional complexity and variability in transducing and interpreting bioderived signals. In the long term, given the ability to tune the physical and chemical properties, biological interactions, and more, we are optimistic for hydrogels with the potential to address many challenges in bioelectronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the National Science Foundation (CBET-1803907 and DMR-1652095).References
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS.
- H. Berger, Arch. Psychiatr. Nervenkrankh., 1929, 87, 527–570 CrossRef.
- M. AlGhatrif and J. Lindsay, J. Community Hosp. Intern. Med. Perspect., 2012, 2, 14383 CrossRef.
- O. Aquilina, Images Paediatr. Cardiol., 2006, 8, 17–81 CAS.
- J. Gardner, Soc. Stud. Sci., 2013, 43, 707–728 CrossRef.
- J. P. Seymour, F. Wu, K. D. Wise and E. Yoon, Microsyst. Nanoeng., 2017, 3, 1–16 Search PubMed.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver and P. A. Janmey, Cell Motil., 2005, 60, 24–34 CrossRef.
- M. HajjHassan, V. Chodavarapu and S. Musallam, Sensors, 2008, 8, 6704–6726 CrossRef CAS.
- V. S. Polikov, P. A. Tresco and W. M. Reichert, J. Neurosci. Methods, 2005, 148, 1–18 CrossRef.
- N. Wisniewski, F. Moussy and W. M. Reichert, Fresenius. J. Anal. Chem., 2000, 366, 611–621 CrossRef CAS.
- R. C. Kelly, M. A. Smith, J. M. Samonds, A. Kohn, A. B. Bonds, J. A. Movshon and T. S. Lee, J. Neurosci., 2007, 27, 261–264 CrossRef CAS.
- A. Zhang and C. M. Lieber, Chem. Rev., 2016, 116, 215–257 CrossRef CAS.
- T. Zhou, G. Hong, T.-M. Fu, X. Yang, T. G. Schuhmann, R. D. Viveros and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5894–5899 CrossRef CAS.
- M. E. Spira and A. Hai, Nat. Nanotechnol., 2013, 8, 83–94 CrossRef CAS.
- N. Gao, W. Zhou, X. Jiang, G. Hong, T.-M. Fu and C. M. Lieber, Nano Lett., 2015, 15, 2143–2148 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- H. Yuk, B. Lu and X. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC.
- S. Lin, C. Cao, Q. Wang, M. Gonzalez, J. E. Dolbow and X. Zhao, Soft Matter, 2014, 10, 7519–7527 RSC.
- B. Xu, P. Zheng, F. Gao, W. Wang, H. Zhang, X. Zhang, X. Feng and W. Liu, Adv. Funct. Mater., 2017, 27, 1604327 CrossRef.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- Y. Liu, W. He, Z. Zhang and B. P. Lee, Gels, 2018, 4, 46 CrossRef.
- Y. Liu, J. Liu, S. Chen, T. Lei, Y. Kim, S. Niu, H. Wang, X. Wang, A. M. Foudeh, J. B.-H. Tok and Z. Bao, Nat. Biomed. Eng., 2019, 3, 58–68 CrossRef CAS.
- J. Yang, R. Bai, B. Chen and Z. Suo, Adv. Funct. Mater., 2020, 30, 1901693 CrossRef CAS.
- D. Gao, K. Parida and P. S. Lee, Adv. Funct. Mater., 2020, 30, 1907184 CrossRef CAS.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef.
- X. Li, Q. Sun, Q. Li, N. Kawazoe and G. Chen, Front. Chem., 2018, 6, 499 CrossRef CAS.
- J. Yan, V. A. Pedrosa, A. L. Simonian and A. Revzin, ACS Appl. Mater. Interfaces, 2010, 2, 748–755 CrossRef CAS.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- N. A. Alba, R. J. Sclabassi, M. Sun and X. T. Cui, IEEE. Trans. Neural. Syst. Rehabil. Eng., 2010, 18, 415–423 Search PubMed.
- S. Ji, C. Wan, T. Wang, Q. Li, G. Chen, J. Wang, Z. Liu, H. Yang, X. Liu and X. Chen, Adv. Mater., 2020, 32, 2001496 CrossRef CAS.
- M. Baumgartner, F. Hartmann, M. Drack, D. Preninger, D. Wirthl, R. Gerstmayr, L. Lehner, G. Mao, R. Pruckner, S. Demchyshyn, L. Reiter, M. Strobel, T. Stockinger, D. Schiller, S. Kimeswenger, F. Greibich, G. Buchberger, E. Bradt, S. Hild, S. Bauer and M. Kaltenbrunner, Nat. Mater., 2020, 1–8 Search PubMed.
- Z. Taylor and K. Miller, J. Biomech., 2004, 37, 1263–1269 CrossRef.
- W. Hiesinger, M. J. Brukman, R. C. McCormick, J. R. Fitzpatrick, J. R. Frederick, E. C. Yang, J. R. Muenzer, N. A. Marotta, M. F. Berry, P. Atluri and Y. J. Woo, J. Thorac. Cardiovasc. Surg., 2012, 143, 962–966 CrossRef.
- B. Wang, W. Huang, L. Chi, M. Al-Hashimi, T. J. Marks and A. Facchetti, Chem. Rev., 2018, 118, 5690–5754 CrossRef CAS.
- S. R. Goldstein and M. Salcman, IEEE Trans. Biomed. Eng., 1973, BME-20, 260–269 Search PubMed.
- J. W. Salatino, K. A. Ludwig, T. D. Y. Kozai and E. K. Purcell, Nat. Biomed. Eng., 2017, 1, 862–877 CrossRef CAS.
- A. Prasad and J. C. Sanchez, J. Neural Eng., 2012, 9, 026028 CrossRef.
- K. Woeppel, Q. Yang and X. T. Cui, Curr. Opin. Biomed. Eng., 2017, 4, 21–31 CrossRef.
- J. Goding, A. Gilmour, P. Martens, L. Poole-Warren and R. Green, Adv. Healthcare Mater., 2017, 6, 1601177 CrossRef.
- L. Ferlauto, A. N. D'Angelo, P. Vagni, M. J. I. Airaghi Leccardi, F. M. Mor, E. A. Cuttaz, M. O. Heuschkel, L. Stoppini and D. Ghezzi, Front. Neurosci., 2018, 12, 648 CrossRef.
- R. G. Wells, Hepatology, 2008, 47, 1394–1400 CrossRef CAS.
- A. J. Engler, C. Carag-Krieger, C. P. Johnson, M. Raab, H.-Y. Tang, D. W. Speicher, J. W. Sanger, J. M. Sanger and D. E. Discher, J. Cell Sci., 2008, 121, 3794–3802 CrossRef CAS.
- K. C. Spencer, J. C. Sy, K. B. Ramadi, A. M. Graybiel, R. Langer and M. J. Cima, Sci. Rep., 2017, 7, 1–16 CrossRef.
- J. D. Enderle, in Introduction to Biomedical Engineering, ed. J. D. Enderle and J. D. Bronzino, Academic Press, Boston, 3rd edn, 2012, ch. 8, pp. 447–508 Search PubMed.
- M. Escabí, in Introduction to Biomedical Engineering, ed. J. D. Enderle and J. D. Bronzino, Academic Press, Boston, 3rd edn, 2012, ch. 11, pp. 667–746 Search PubMed.
- J. D. Enderle, in Introduction to Biomedical Engineering, ed. J. D. Enderle and J. D. Bronzino, Academic Press, Boston, 3rd edn, 2012, ch. 12, pp. 747–815 Search PubMed.
- C.-J. Lee, H. Wu, Y. Hu, M. Young, H. Wang, D. Lynch, F. Xu, H. Cong and G. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 5845–5852 CrossRef CAS.
- S. Zhao, P. Tseng, J. Grasman, Y. Wang, W. Li, B. Napier, B. Yavuz, Y. Chen, L. Howell, J. Rincon, F. G. Omenetto and D. L. Kaplan, Adv. Mater., 2018, 30, 1800598 CrossRef.
- I. Noshadi, B. W. Walker, R. Portillo-Lara, E. S. Sani, N. Gomes, M. R. Aziziyan and N. Annabi, Sci. Rep., 2017, 7, 4345 CrossRef.
- H. Jo, M. Sim, S. Kim, S. Yang, Y. Yoo, J.-H. Park, T. H. Yoon, M.-G. Kim and J. Y. Lee, Acta Biomater., 2017, 48, 100–109 CrossRef CAS.
- X. Liu, A. L. Miller, S. Park, B. E. Waletzki, Z. Zhou, A. Terzic and L. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 14677–14690 CrossRef CAS.
- P. Baei, S. Jalili-Firoozinezhad, S. Rajabi-Zeleti, M. Tafazzoli-Shadpour, H. Baharvand and N. Aghdami, Mater. Sci. Eng., C, 2016, 63, 131–141 CrossRef CAS.
- L. Xu, X. Li, T. Takemura, N. Hanagata, G. Wu and L. L. Chou, J. Nanobiotechnol., 2012, 10, 16 CrossRef CAS.
- A. N. Dalrymple, U. A. Robles, M. Huynh, B. A. Nayagam, R. A. Green, L. A. Poole-Warren, J. B. Fallon and R. K. Shepherd, J. Neural Eng., 2020, 17, 026018 Search PubMed.
- Y. Liu, A. F. McGuire, H.-Y. Lou, T. L. Li, J. B.-H. Tok, B. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11718–11723 CAS.
- V. R. Feig, H. Tran, M. Lee and Z. Bao, Nat. Commun., 2018, 9, 2740 Search PubMed.
- H. Yuk, B. Lu, S. Lin, K. Qu, J. Xu, J. Luo and X. Zhao, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- J. Liu, X. Zhang, Y. Liu, M. Rodrigo, P. D. Loftus, J. Aparicio-Valenzuela, J. Zheng, T. Pong, K. J. Cyr, M. Babakhanian, J. Hasi, J. Li, Y. Jiang, C. J. Kenney, P. J. Wang, A. M. Lee and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14769 CrossRef CAS.
- D. N. Heo, S.-J. Song, H.-J. Kim, Y. J. Lee, W.-K. Ko, S. J. Lee, D. Lee, S. J. Park, L. G. Zhang, J. Y. Kang, S. H. Do, S. H. Lee and I. K. Kwon, Acta Biomater., 2016, 39, 25–33 CrossRef CAS.
- N. J. Ronkainen, H. B. Halsall and W. R. Heineman, Chem. Soc. Rev., 2010, 39, 1747–1763 RSC.
- W. Schramm, S.-H. Paek and G. Voss, ImmunoMethods, 1993, 3, 93–103 CrossRef CAS.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef CAS.
- U. Guzik, K. Hupert-Kocurek and D. Wojcieszyńska, Molecules, 2014, 19, 8995–9018 CrossRef.
- E. Stern, R. Wagner, F. J. Sigworth, R. Breaker, T. M. Fahmy and M. A. Reed, Nano Lett., 2007, 7, 3405–3409 CrossRef CAS.
- G. Zheng, F. Patolsky, Y. Cui, W. U. Wang and C. M. Lieber, Nat. Biotechnol., 2005, 23, 1294–1301 CAS.
- R. Elnathan, M. Kwiat, A. Pevzner, Y. Engel, L. Burstein, A. Khatchtourints, A. Lichtenstein, R. Kantaev and F. Patolsky, Nano Lett., 2012, 12, 5245–5254 CrossRef CAS.
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanović and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS.
- N. Kumar, M. Gray, J. C. Ortiz-Marquez, A. Weber, C. R. Desmond, A. Argun, T. van Opijnen and K. S. Burch, Med. Devices Sens., 2020, e10121 Search PubMed.
- J. E. Contreras-Naranjo and O. Aguilar, Biosensors, 2019, 9, 15 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS.
- Y. Li, T. L. Ogorzalek, S. Wei, X. Zhang, P. Yang, J. Jasensky, C. L. Brooks, E. N. G. Marsh and Z. Chen, Phys. Chem. Chem. Phys., 2018, 20, 1021–1029 RSC.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, 3 Biotech, 2013, 3, 1–9 CrossRef.
- J. Tavakoli and Y. Tang, Polymers, 2017, 9, 364 CrossRef.
- N. Gao, T. Gao, X. Yang, X. Dai, W. Zhou, A. Zhang and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14633–14638 CrossRef CAS.
- X. Dai, R. Vo, H.-H. Hsu, P. Deng, Y. Zhang and X. Jiang, Nano Lett., 2019, 19, 6658–6664 CrossRef CAS.
- H. H. Bay, R. Vo, X. Dai, H.-H. Hsu, Z. Mo, S. Cao, W. Li, F. G. Omenetto and X. Jiang, Nano Lett., 2019, 19, 2620–2626 CrossRef CAS.
- J. Kunkel and P. Asuri, PLoS One, 2014, 9, e86785 CrossRef.
- D. P. Hickey, R. C. Reid, R. D. Milton and S. D. Minteer, Biosens. Bioelectron., 2016, 77, 26–31 CrossRef CAS.
- J. Kim, I. Jeerapan, S. Imani, T. N. Cho, A. Bandodkar, S. Cinti, P. P. Mercier and J. Wang, ACS Sens., 2016, 1, 1011–1019 CrossRef CAS.
- A. J. Bandodkar, W. Jia, C. Yardımcı, X. Wang, J. Ramirez and J. Wang, Anal. Chem., 2015, 87, 394–398 CrossRef CAS.
- L. Li, L. Pan, Z. Ma, K. Yan, W. Cheng, Y. Shi and G. Yu, Nano Lett., 2018, 18, 3322–3327 CrossRef CAS.
- M. J. Schöning and A. Poghossian, Analyst, 2002, 127, 1137–1151 RSC.
- Y. Maeda, A. Matsumoto, Y. Miura and Y. Miyahara, Nanoscale Res. Lett., 2012, 7, 108 CrossRef.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS.
- T. Aoki, K. Nakamura, K. Sanui, A. Kikuchi, T. Okano, Y. Sakurai and N. Ogata, Polym. J., 1999, 31, 1185–1188 CrossRef CAS.
- K. Podual, F. J. Doyle and N. A. Peppas, J. Controlled Release, 2000, 67, 9–17 CrossRef CAS.
- P. B. Welzel, S. Prokoph, A. Zieris, M. Grimmer, S. Zschoche, U. Freudenberg and C. Werner, Polymers, 2011, 3, 602–620 CrossRef CAS.
- H. Chavda and C. Patel, Int. J. Pharm. Invest., 2011, 1, 17–21 CrossRef CAS.
- N. W. Choi, J. Kim, S. C. Chapin, T. Duong, E. Donohue, P. Pandey, W. Broom, W. A. Hill and P. S. Doyle, Anal. Chem., 2012, 84, 9370–9378 CrossRef CAS.
- S. L. Burrs, D. C. Vanegas, M. Bhargava, N. Mechulan, P. Hendershot, H. Yamaguchi, C. Gomes and E. S. McLamore, Analyst, 2015, 140, 1466–1476 RSC.
- H. J. Kim, W. Choi, J. Kim, J. Choi, N. Choi and K. S. Hwang, Sens. Actuators, B, 2020, 302, 127190 CrossRef CAS.
- R. J. Russell, A. C. Axel, K. L. Shields and M. V. Pishko, Polymer, 2001, 42, 4893–4901 CrossRef CAS.
- B. Amsden, Macromolecules, 1998, 31, 8382–8395 CrossRef CAS.
- E. Axpe, D. Chan, G. S. Offeddu, Y. Chang, D. Merida, H. L. Hernandez and E. A. Appel, Macromolecules, 2019, 52, 6889–6897 CrossRef CAS.
- A. Star, E. Tu, J. Niemann, J.-C. P. Gabriel, C. S. Joiner and C. Valcke, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 921–926 CrossRef CAS.
- E. Stern, J. F. Klemic, D. A. Routenberg, P. N. Wyrembak, D. B. Turner-Evans, A. D. Hamilton, D. A. LaVan, T. M. Fahmy and M. A. Reed, Nature, 2007, 445, 519–522 CrossRef CAS.
- F. Patolsky, G. Zheng, O. Hayden, M. Lakadamyali, X. Zhuang and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14017–14022 CrossRef CAS.
- G. L. Mario Cheong, K. S. Lim, A. Jakubowicz, P. J. Martens, L. A. Poole-Warren and R. A. Green, Acta Biomater., 2014, 10, 1216–1226 CrossRef CAS.
- J. O. Winter, S. F. Cogan and J. F. Rizzo, J. Biomed. Mater. Res., Part B, 2007, 81, 551–563 CrossRef.
- M. L. McCain, A. Agarwal, H. W. Nesmith, A. P. Nesmith and K. K. Parker, Biomaterials, 2014, 35, 5462–5471 CrossRef CAS.
- D. A. Soscia, D. Lam, A. C. Tooker, H. A. Enright, M. Triplett, P. Karande, S. K. G. Peters, A. P. Sales, E. K. Wheeler and N. O. Fischer, Lab Chip, 2020, 20, 901–911 RSC.
- D. Lam, H. A. Enright, S. K. G. Peters, M. L. Moya, D. A. Soscia, J. Cadena, J. A. Alvarado, K. S. Kulp, E. K. Wheeler and N. O. Fischer, J. Neurosci. Methods, 2020, 329, 108460 CrossRef.
- S. Lee, D. Sasaki, D. Kim, M. Mori, T. Yokota, H. Lee, S. Park, K. Fukuda, M. Sekino, K. Matsuura, T. Shimizu and T. Someya, Nat. Nanotechnol., 2019, 14, 156–160 CrossRef CAS.
- V. J. Kujala, F. S. Pasqualini, J. A. Goss, J. C. Nawroth and K. K. Parker, J. Mater. Chem. B, 2016, 4, 3534–3543 RSC.
- J. Yan, Y. Sun, H. Zhu, L. Marcu and A. Revzin, Biosens. Bioelectron., 2009, 24, 2604–2610 CrossRef CAS.
- M. Lian, X. Chen, Y. Lu and W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 25036–25042 CrossRef CAS.
- M. Kitsara, D. Kontziampasis, O. Agbulut and Y. Chen, Microelectron. Eng., 2019, 203–204, 44–62 CrossRef CAS.
- P. Fattahi, G. Yang, G. Kim and M. R. Abidian, Adv. Mater., 2014, 26, 1846–1885 CrossRef CAS.
- J. L. Bourke, A. F. Quigley, S. Duchi, C. D. O'Connell, J. M. Crook, G. G. Wallace, M. J. Cook and R. M. I. Kapsa, J. Tissue Eng. Regener. Med., 2018, 12, 490–493 CrossRef CAS.
- S. P. Paşca, Nature, 2018, 553, 437–445 CrossRef.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS.
- A. Kalmykov, C. Huang, J. Bliley, D. Shiwarski, J. Tashman, A. Abdullah, S. K. Rastogi, S. Shukla, E. Mataev, A. W. Feinberg, K. J. Hsia and T. Cohen-Karni, Sci. Adv., 2019, 5, eaax0729 CrossRef CAS.
- A. P. Napolitano, D. M. Dean, A. J. Man, J. Youssef, D. N. Ho, A. P. Rago, M. P. Lech and J. R. Morgan, BioTechniques, 2007, 43, 494–500 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- P. M. Misun, J. Rothe, Y. R. F. Schmid, A. Hierlemann and O. Frey, Microsyst. Nanoeng., 2016, 2, 1–9 Search PubMed.
- D. Phan, L. Y. Siong, P. N. Pathirana and A. Seneviratne, in 2015 International Symposium on Bioelectronics and Bioinformatics (ISBB), 2015, pp. 144–147 Search PubMed.
- Z. Lou, L. Wang and G. Shen, Adv. Mater. Technol., 2018, 3, 1800444 CrossRef.
- S. Pan, F. Zhang, P. Cai, M. Wang, K. He, Y. Luo, Z. Li, G. Chen, S. Ji, Z. Liu, X. J. Loh and X. Chen, Adv. Funct. Mater., 2020, 26, 1909540 CrossRef.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS.
- Y. Zhao, Z. Li, S. Song, K. Yang, H. Liu, Z. Yang, J. Wang, B. Yang and Q. Lin, Adv. Funct. Mater., 2019, 29, 1901474 CrossRef.
- Y. Liu, T. Yang, Y. Zhang, G. Qu, S. Wei, Z. Liu and T. Kong, Adv. Mater., 2019, 31, 1902783 CrossRef.
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- G. E. Dunbar, B. Y. Shen and A. A. Aref, Clin. Ophthalmol., 2017, 11, 875–882 CrossRef CAS.
- M. Senior, Nat. Biotechnol., 2014, 32, 856–856 CrossRef CAS.
- D. H. Keum, S.-K. Kim, J. Koo, G.-H. Lee, C. Jeon, J. W. Mok, B. H. Mun, K. J. Lee, E. Kamrani, C.-K. Joo, S. Shin, J.-Y. Sim, D. Myung, S. H. Yun, Z. Bao and S. K. Hahn, Sci. Adv., 2020, 6, eaba3252 CrossRef.
- P. Mostafalu, A. Tamayol, R. Rahimi, M. Ochoa, A. Khalilpour, G. Kiaee, I. K. Yazdi, S. Bagherifard, M. R. Dokmeci, B. Ziaie, S. R. Sonkusale and A. Khademhosseini, Small, 2018, 14, 1703509 CrossRef.
- Z. Lou, L. Wang, K. Jiang, Z. Wei and G. Shen, Mater. Sci. Eng., R, 2020, 140, 100523 CrossRef.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS.
- L. Hsu and X. Jiang, Trends Biotechnol., 2019, 37, 795–796 CrossRef CAS.
- H. Liu, M. Li, C. Ouyang, T. J. Lu, F. Li and F. Xu, Small, 2018, 14, 1801711 CrossRef.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Chem. Mater., 2011, 23, 1254–1262 CrossRef CAS.
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 12, 1224–1246 RSC.
- H. Chopra, S. Kumar and I. Singh, in Bioadhesives in Drug Delivery, John Wiley & Sons, Ltd, 2020, ch. 6, pp. 147–170 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |