
Polymerisation-induced self-assembly (PISA) as a straightforward formulation strategy for stimuli-responsive drug delivery systems and biomaterials: recent advances
Hien
Phan
 *a,
Vincenzo
Taresco
b,
Jacques
Penelle
a and
Benoit
Couturaud
*a
*a,
Vincenzo
Taresco
b,
Jacques
Penelle
a and
Benoit
Couturaud
*a
aUniv Paris Est Creteil, CNRS, Institut de Chimie et des Matériaux Paris-Est (ICMPE), UMR 7182, 2 rue Henri Dunant, 94320 Thiais, France. E-mail: Couturaud@icmpe.cnrs.fr
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 9th October 2020
Abstract
Stimuli-responsive amphiphilic block copolymers have emerged as promising nanocarriers for enhancing site-specific and on-demand drug release in response to a range of stimuli such as pH, the presence of redox agents, and temperature. The formulation of amphiphilic block copolymers into polymeric drug-loaded nanoparticles is typically achieved by various methods (e.g. oil-in-water emulsion solvent evaporation, solid dispersion, microphase separation, dialysis or microfluidic separation). Despite much progress that has been made, there remain many challenges to overcome to produce reliable polymeric systems. The main drawbacks of the above methods are that they produce very low solid contents (<1 wt%) and involve multiple-step procedures, thus limiting their scope. Recently, a new self-assembly methodology, polymerisation-induced self-assembly (PISA), has shown great promise in the production of polymer-derived particles using a straightforward one-pot approach, whilst facilitating high yield, scalability, and cost-effectiveness for pharmaceutical industry protocols. We therefore focus this review primarily on the most recent studies involved in the design and preparation of PISA-generated nano-objects which are responsive to specific stimuli, thus providing insight into how PISA may become an effective formulation strategy for the preparation of precisely tailored drug delivery systems and biomaterials, while some of the current challenges and limitations are also critically discussed.
 Hien Phan | Hien Phan earned her Master's degree from the Erasmus Mundus Joint Master Programme NANOMED in 2019. Hien joined the group of Dr Jacques Penelle and Dr Benoit Couturaud at the Institute of Chemistry and Materials Paris Est (ICMPE), France for her Ph.D. thesis in polymer therapeutics for the treatment of cancer. Her research interests lie in the design, production, and assessment of stimuli-responsive nanomedicine for advanced drug delivery systems in cancer and infectious diseases, aiming at the potential translations of nanomedicine into industrial and clinical perspectives. |
 Vincenzo Taresco | Dr Vincenzo Taresco, from 2014–2019, worked at the School of Pharmacy of the University of Nottingham as Postdoc. In this period, he has divided his research into the design of responsive polymers, their use as smart delivery carriers and their screening through high-throughput methodologies. Since May 2019, he has been Senior Research Officer in the group of Prof. Steven Howdle where he is aiming to combine the fields of green chemistry, bio-medicine and pharmaceutics. |
 Jacques Penelle | Dr Jacques Penelle is currently a Research Director with the French CNRS. His main area of expertise covers macromolecular chemistry and polymer structure–property relationships. He has held permanent faculty positions in Belgium (the Catholic University of Louvain, Department of Chemistry), in the United States (the University of Massachusetts – Amherst, Polymer Science & Engineering Department), and in France since 2003. His chief current research interests involve the development of synthetic methodologies toward unusual polymer structures, the use of high-pressure techniques to obtain polymers from poorly reactive monomers, and the design of bioactive polymer drugs. |
 Benoit Couturaud | Dr Benoit Couturaud is Assistant Professor at the Institute of Chemistry and Materials Paris-Est (ICMPE) since 2018. He got his PhD from the University of Montpellier in 2013. After a PostDoc under the direction of Prof. Patrice Woisel (UMET, Lille), he moved to the University of Nottingham working on an EPSRC programme led by Prof. Morgan and Cameron Alexander. In 2016, he was awarded a Marie Curie Individual Fellowship involving Prof. Rachel O'Reilly (the University of Birmingham) and Prof. Kristofer Thurecht (the University of Queensland, Brisbane). |
1. Introduction
The highly complex behaviour of diseases demands that drug delivery systems and biomaterials should be designed in such a way that they can respond to various pathogenic environments in an opportune and judicious manner. To improve the therapeutic reliability, it is of utmost importance for particles to exhibit high drug loading and site-specific delivery, while efficiently favouring drug release in the diseased body areas.1,2 In this regard, functionalised materials displaying such tailored features have rapidly emerged as promising candidates for the design of many advanced drug delivery systems.3,4 The functionalisation of materials can be achieved via the encoding of stimuli-sensitive functionalities into their molecular structures, thus endowing on-demand responses to endogenous factors such as pH, oxidation, and reduction, or to exogenous triggers such as photoirradiation, temperature, and ultrasonic activation.5–11 On the other hand, synthetic polymeric nanomaterials are of particular interest as their colloidal properties and surface chemistry can be readily tuned to precisely meet the demand of individual applications.12–14 Altogether, stimuli-responsive polymer-based nanocarriers have gained rapidly growing research attention in biomedical sciences as a versatile and exciting platform for materials design strategies.15–19 However, polymer synthesis and its formulation into polymeric nanoparticles (NPs) usually involve multiple-step procedures, addition of toxic surfactants, and extensive intermediate purification steps. In an attempt to minimise these tedious synthetic efforts whilst maintaining batch-to-batch consistency and clinical relevance, amphiphilic block copolymers have been adopted as nano-vehicle materials since they can inherently self-assemble in aqueous media to form stabilised core–shell nano-objects.20,21 Amphiphilic diblock copolymers have therefore been widely exploited in a range of therapeutic purposes, including cancer22,23 and gene therapy,24 and also in diagnosis.25Typically, the room temperature self-assembly of amphiphilic block copolymers with a glassy hydrophobic block is usually performed using the co-solvent or solvent switch method.26 In this technique, the amphiphilic block copolymer is initially dissolved in an organic solvent such as DMF, acetone, or THF that is compatible with both blocks, followed by slow addition of a selective solvent, e.g. water, which is a good solvent for only one block, leading to the microphase separation and then to spontaneous self-assembly into nano-objects with a solvophobic core coated with a solvophilic layer.21 This allows a relatively low polymer concentration (<1 wt%) to be used.27–30 In addition, the self-assembly usually occurs via post-polymerisation of well-defined precursor copolymers, and hence is not economically ideal for scale-up and industrial environments. While the solution-phase self-assembly can still leverage a diverse set of particle morphologies such as micelles, nanofibers, and vesicles,31,32 which is primarily dictated by the packing parameter (p),26 in practice it is tedious to obtain higher order morphologies.
To meet such technical challenges, polymerisation-induced self-assembly (PISA) has recently been proposed. PISA has tremendously redefined the concept of block copolymer self-assembly by jointly facilitating polymerisation and self-assembly in a one-pot approach.33–40 Two types of polymerisation methods are used in PISA, dispersion and emulsion. The former requires both monomers and a macromolecular precursor to be solvophilic and compatible with each other in a continuous phase, either in an organic solvent or in water-based systems. Once a critical degree of polymerisation of the core-forming monomer is reached, this block becomes insoluble and tends to precipitate. The initial particles act as the loci for polymerisation on which monomers continue to be added covalently to the stabilised particles. Meanwhile, the second method is usually implemented in water, using a soluble homopolymer precursor that is chain-extended with an immiscible monomer to a critical degree of polymerisation. This leads to the formation of monomer droplets stabilised by the initial homopolymers. Both polymerisation methods generate in situ self-assembled nano-objects with various morphologies as depicted in Fig. 1, which are a very important and valuable asset compared to the typical nanoprecipitation technique through which mostly spherical particles are produced while the same copolymers produced by PISA can be shaped in several ways.41 The morphological evolution initially starts with the early formation of spherical micelles and provides a not-kinetically-frozen structure that evolves to cylindrical or worm-like nanoparticles, and finally to vesicles over the growing course of the core-forming block. The control over particle size and shape has been thoroughly discussed in recent reviews.42–49 Generally, the formation and transformation of NP morphologies are controlled and predictable. A higher order sphere-to-vesicle transition is often promoted by an increase in the degree of polymerisation (DP) and by modification of the reaction conditions. This feature facilitates the precise control of targeted particle shapes. In general, controlling the particle shape is of substantial importance to enhance the performance of drug delivery systems as each morphology has its own effects on the in vivo fate, biodistribution and cellular uptake after administration.50–52 More specifically, oblate particles exhibit a reduced uptake by macrophages and a prolonged blood circulation, strongly enhancing the odds of reaching the targeted sites. Due to their high surface areas, elongated NPs can also form stronger ligand–receptor bindings compared to nanospheres, and thus improve the selectivity and specificity during endocytosis.53–55 On the other hand, NPs with sharp edges can disrupt the endosomal membranes, inducing less exocytosis, thereby staying longer in the cytoplasm than the spherical particles.56 By producing a wide scope of well-controlled nanostructures, PISA can provide more possibilities for achieving better cellular uptake and tracking,57 which ultimately results in an enhanced bioavailability of the NPs. NP uptakes at target cells are also directly governed by their sizes. NPs should ideally be designed with sizes between 5 and 100 nm in diameter. Sizes lower than 5 nm are known to generate particles that will be eliminated by renal filtration,53 whereas higher diameters (over 200 nm) trigger excessive accumulation in the liver and spleen.58 Particles with diameters lower than 100 nm exhibit less adsorption of serum proteins while circulating in the blood, thus enhancing half-lives in vivo.59,60 Moreover, PISA can be performed at relatively high solid contents up to 50 wt% and usually imparts very high monomer conversions within short reaction times. Hence, PISA is of high benefit to the formulation of NPs, whose shapes and sizes are tuneable even during large-scale productions. Besides, simultaneous self-assembly and drug encapsulation may enable relatively high loading efficiencies (67% for doxorubicin-encapsulated NPs,61 54% for cisplatin-loaded micelles and 24% for BSA-loaded vesicles,62 under simple working conditions), likely owing to high polymer concentrations. PISA synthesis has been carried out using several types of living polymerisation techniques, but reversible addition–fragmentation chain transfer (RAFT) is probably the most prevalent synthesis method currently available to prepare NPs by PISA approach due to its adaptability to a broad range of reaction conditions and monomer families.63–68 RAFT dispersion or emulsion PISA processes can be initiated via various approaches, most commonly by thermal initiation but also by other new alternative approaches such as photochemical, ultrasonic and microwave activations, or using enzymes.49 Moreover, a few types of polymerisation techniques that have recently emerged can be of interest to PISA for drug delivery formulations, e.g. ring opening polymerisation (ROP)69 or ring-opening metathesis polymerisation PISA (ROMPISA).70 In conclusion, with superior cellular uptakes resulting from diverse and tuneable morphologies as well as relatively high drug loading under industrially relevant approaches, PISA appears to be a versatile synthesis and formulation technique for polymer-based nanomaterials in drug delivery systems.
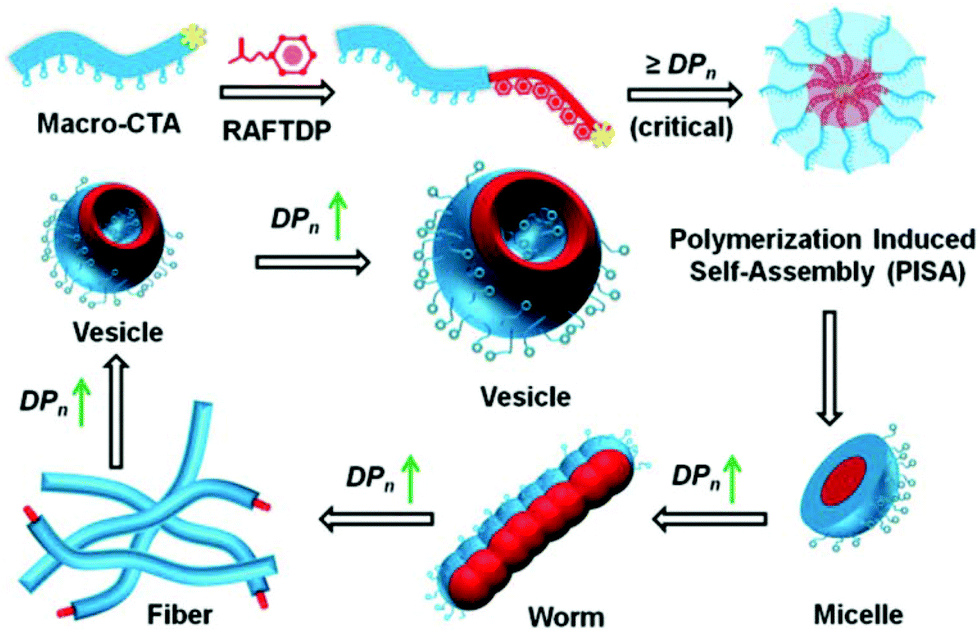 | ||
| Fig. 1 Illustrative mechanism of NP morphological transformation by RAFT-PISA. Reproduced from ref. 77 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Based on simple and accessible formulations of NPs from PISA, an extensive amount of literature has been focused on producing stimuli-sensitive NPs, using mostly RAFT and some ROP/ROMP combinations, thereby conferring an efficient programmable drug release through a timely and direct nano-formulation.
The present review therefore aims to highlight a number of the latest examples regarding the design and preparation of polymeric NPs sensitive to a variety of triggering stimuli by using mainly RAFT-PISA and a few ROP/ROMPISA methods that show remarkable applications in many precise drug delivery systems. The authors will conclude with several critical remarks on the challenges and opportunities underlying the role of PISA as a formulation strategy for polymer-based nanomedicines in the context of clinical translatability.
2. Summary of the several stimuli used for drug delivery systems obtained by PISA
2.1. pH-Responsive systems
Differences in pH profiles under pathogenic conditions compared to normal cell microenvironments is one of the most prominent features to exploit in the development of intelligent tools in nanomedicine. Many inflammation sites and solid tumours display relatively acidic pH values (<6.5) compared to normal tissues (pH 7.4).71,72 Integrating functionalities into particles that are stable under physiological pH whilst cleavable at low pH triggers drug unloading in a well-programmed way.For example, Karagoz et al. described a one-pot PISA synthetic route to asymmetric-block-copolymer NPs: poly[oligo(ethyleneglycol)methacrylate]-block-[poly(styrene)-co-poly(vinyl benzaldehyde)] (POEGMA-b-P(ST-co-VBA)) was obtained using a RAFT-mediated dispersion polymerisation in methanol and enabling self-assembly into various morphologies from spheres, worm-like and rod-like to vesicles.73 After the particle synthesis, the aldehyde groups in the core-forming blocks were conjugated with doxorubicin (DOX) via its amine functional groups to form the pH-responsive imine moieties (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) on Schiff bases. A release study demonstrated that more than 80% DOX was liberated from the NPs at pH 5 due to the acidic pH-breakable bonds, whereas only 10% were released at pH 7.4. Furthermore, while particle morphologies did not affect the DOX release, they had a significant impact on the cell internalisation in MCF-7 breast cancer cell line, whereby worm- and rod-like particles displayed an improved cell internalisation compared to spherical micelles and vesicles. The authors reasoned that cylindrical morphologies imparted greater aspect ratios or surface areas, allowing for higher adhesions to cell membranes and thus favouring cellular uptake. In addition, rod-like and worm-like DOX-particles exhibited higher cytotoxicity than vesicles and micelles, as observed from IC50 variations on MCF-7 cell line (Fig. 2). In summary, this system, among all the possible obtained morphologies, has demonstrated an effective combination of responsiveness, cellular internalisation, and cytotoxicity, a feature that is very difficult to achieve from conventional nanoprecipitation.
N–) on Schiff bases. A release study demonstrated that more than 80% DOX was liberated from the NPs at pH 5 due to the acidic pH-breakable bonds, whereas only 10% were released at pH 7.4. Furthermore, while particle morphologies did not affect the DOX release, they had a significant impact on the cell internalisation in MCF-7 breast cancer cell line, whereby worm- and rod-like particles displayed an improved cell internalisation compared to spherical micelles and vesicles. The authors reasoned that cylindrical morphologies imparted greater aspect ratios or surface areas, allowing for higher adhesions to cell membranes and thus favouring cellular uptake. In addition, rod-like and worm-like DOX-particles exhibited higher cytotoxicity than vesicles and micelles, as observed from IC50 variations on MCF-7 cell line (Fig. 2). In summary, this system, among all the possible obtained morphologies, has demonstrated an effective combination of responsiveness, cellular internalisation, and cytotoxicity, a feature that is very difficult to achieve from conventional nanoprecipitation.
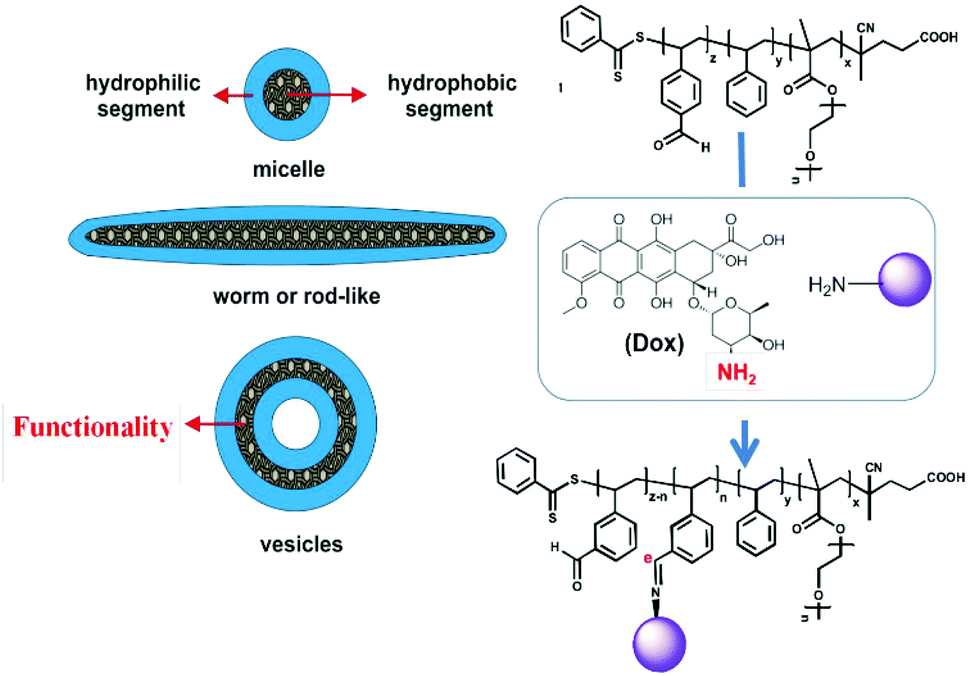 | ||
| Fig. 2 Schematic diagram of in situ self-assembly of POEGMA-b-P(ST-co-VBA) using PISA and its conjugation with DOX through a pH-cleavable bond. Reproduced from ref. 73 with permission from the Royal Society of Chemistry, copyright 2014. | ||
Similarly, (methacryloxyethoxy)benzaldehyde (MAEBA) was polymerised in the presence of poly((N,N′-dimethylamino)ethyl methacrylate) used as a macro-Chain Transfer Agent (macro-CTA) under RAFT dispersion PISA conditions, generating PDMAEMA block copolymer nano-objects as spheres, nanorods, nanowires, and vesicles.74 DOX was conjugated with the PMAEBA block via the acidic pH-breakable imine linkage (–CN–) in a similar way as in ref 73. A drug release study indicated that less than 1% of the DOX had been released at pH 7.4, while the fastest release was observed at pH 5 due to the cleavage of the aromatic imine under acidic conditions.
Lately, Armes and coworkers synthesised bio-inspired framboidal-like vesicles that simulated some Dengue virus features, with the vesicles exhibiting both moieties targeting the SR-B1 scavenger receptors that are available on triple-negative breast cancer cells, and pH-sensitive surface areas for local drug delivery.75 In particular, the two macro-CTAs poly(glycerol monomethacrylate) (PGMA) and poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC) were first chain-extended using 2-hydroxypropyl methacrylate (HPMA) and glycidyl methacrylate (GlyMA) that had been conjugated with rhodamine B piperazine (Rh). The obtained PGMA-P(HPMA-stat-GlyMARh) polymer was then chain-extended with the 2-(diisopropylamino)ethyl methacrylate (DPA) monomer via RAFT aqueous-seeded emulsion polymerisation, resulting in pH-responsive framboidal-like triblock copolymer vesicles. In this respect, the phosphorylcholine functionalities of PMPC endowed the vesicles with selective ligands to target the SR-B1 receptors on MDA-MB-231 cells, while the tertiary amines on DPA became protonated to liberate the plasmid DNA in the nucleus through intracellular acidification (Fig. 3).
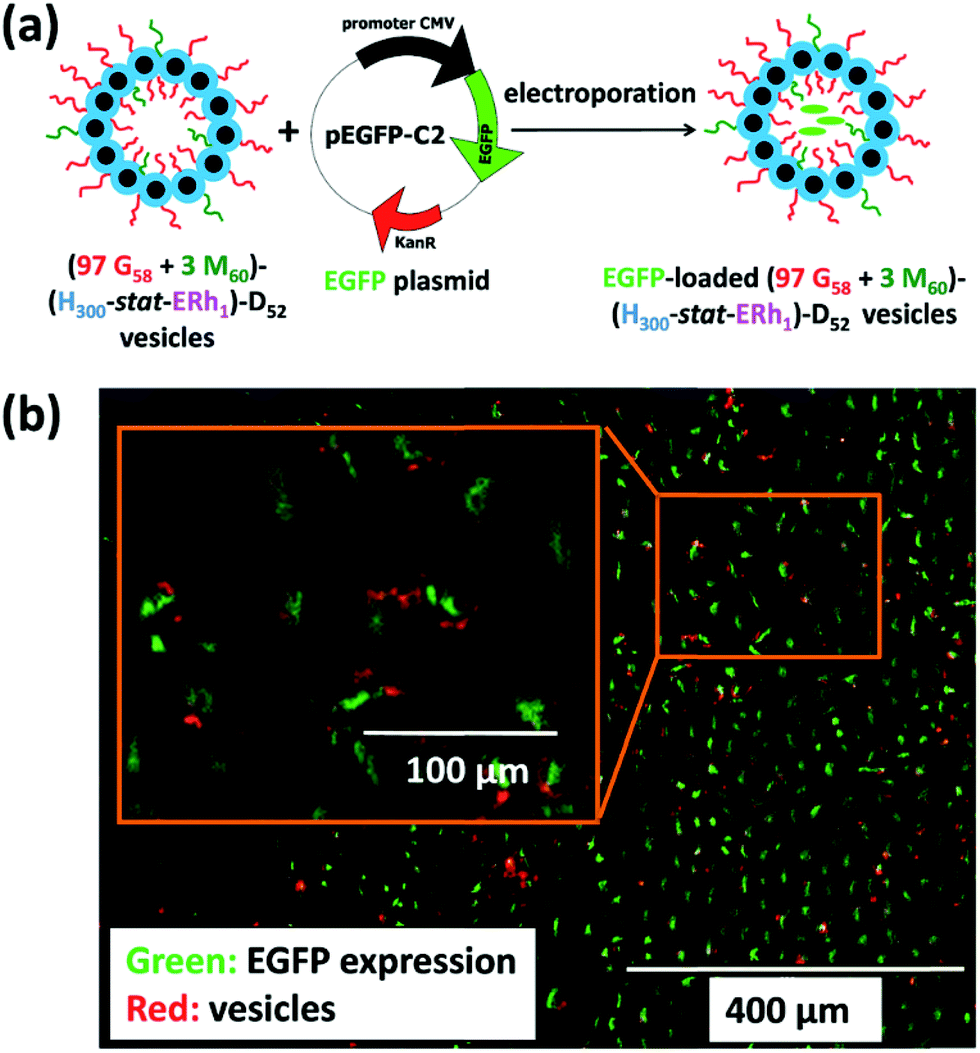 | ||
| Fig. 3 (a) Illustration of the intracellular release of a EGFP plasmid DNA within MDA-MB-231 cells using (97 PGMA58 + 3 PMPC60)–P(HPMA300-stat-GlyMARh1)–PDPA52 (97 G58 + 3 M60)–(H300-stat-ERh1)–D52 framboidal vesicles. (b) Schematic diagram of the loading of plasmid EGFP DNA in these triblock copolymer vesicles (the green colour indicates a successful EGFP expression while the red colour indicates the location of the rhodamine-labeled vesicles). Reproduced from ref. 75 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Another recent example has been reported on the synthesis of cisplatin drug-encapsulated spherical micelles using ROMPISA.76 One of the most attractive features of ROMPISA is its tolerance to a wide range of initiators and functionalities, allowing the straightforward polymerisation of monomers in one pot. Amphiphilic block copolymers were synthesised in water, particularly a poly(oligo ethylene glycol) macro CTA, which was chain-extended by comonomers of a cisplatin norbornene analogue and pH-responsive 2-(diisopropylamino)ethyl norbornene dicarboximide.
The effect of sizes and surface charges of the resulting micelles on cellular uptake and cytotoxicity was investigated, indicating that smaller sizes exhibited improved cellular uptake and cytotoxicity, whilst the positively-charged NPs were better absorbed than the neutral or negatively-charged ones. A drug loading of 20% was achieved, with a selective release at pH 6.
2.2. Reduction-responsive systems
High intracellular redox potential is another hallmark of carcinogenesis, resulting from an overproduction of reducing agents such as the glutathione tripeptide γ-glutamyl–cysteinyl–glycine (GSH), whose concentration ranges from 2 to 10 mM in the cytosol of tumour cells compared to the 1–2 mM concentration, the typical range in normal cells. It is thus widely adopted to facilitate targeted and on-demand drug releases.78–81 Reduction responsiveness is also critically important in hypoxia, a biological condition associated with oxygen deprivation in various diseases such as rheumatoid arthritis, ischemic stroke, and solid tumours. Hypoxia is associated with the presence of reductive species such as azoreductases and nitroreductases (NTR), thus nanomedicine tools can be tailored using azobenzene (Azo), nitroaromatic or quinone derivatives as hypoxia-responsive functionalities which are likely cleavable within the hypoxia-affected areas.82,83In this regard, the anticancer drug camptothecin (CPT) was integrated with a prodrug monomer (CPTM). 2-(2-(Methoxyethoxy)ethyl methacrylate (MEO2MA) and the obtained CPTM comonomer were then polymerised in the presence of poly(ethylene glycol)-4-cyano-4-((phenylcarbono-thioyl)thio) pentanoic acid (PEG113-CPDB) used as macro-CTA, via a RAFT dispersion technique, leading to the formation of a diblock prodrug copolymer PEG-b-P(MEO2MA-co-CPTM).84 This synthetic technique resulted in prodrug NPs with a three-layered structure. In particular, the reduction-sensitive core was cross-linked via copolymerisation of benzyl methacrylate (BzMA) and N,N-cystaminebismethacrylamide (CBMA); the second layer contained the CPT-copolymer with the CPT conjugated to the polymer chains via reduction-sensitive disulfide linkages; and the outer layer included stabilising hydrophilic PEG chains. This prodrug NPs facilitated the cleavage of the conjugated CPTs upon exposure to a reductive microenvironment, e.g. at high levels of GSH in the cytosol. This synthetic strategy enabled a drug delivery system to achieve (1) simultaneous polymerisation and drug encapsulation in a one-pot approach at a relatively high concentration of 100 mg mL−1, (2) a high monomer conversion, (3) simultaneous core-cross-linking during prodrug particle preparation, (4) a CPT concentration that could be precisely tuned by altering the MEO2MA/CPT ratio, and (5) efficient drug unloading upon cell internalisation due to the high concentration of GSH in the cytoplasm.
Recently, the development of dual-functional polymer NPs exhibiting a fluorescence-assisted tracking capability and reduction-responsive triggered drug release has been reported.85 Accordingly, aza-boron dipyrromethene (BODIPY), a near-infrared (NIR) fluorescence dye, was loaded in situ into particles via a RAFT aqueous dispersion PISA without specific purification steps. Firstly, a reduction-responsive polyprodrug camptothecin-monomer (pCPTM) was prepared via chain extension of a PPEGMA macro-CTA during the RAFT copolymerisation of PEGMA and CPTM. Secondly, PEGMA comonomers and pre-synthesised NIR fluorescent monomer (NIRM) were co-polymerised in the presence of the PPEGMA macro-CTA, yielding pNIRM. The last step was required to prepare core-cross-linked bifunctional NPs, wherein pCPTM and pNIRM were hydrophilic macro-CTA agents that were chain-extended with the HPMA monomer and dimethacrylate cross-linker (EGDMA) via one-pot RAFT aqueous dispersion polymerisation. The resulting spherical micelles had a three-layered structure, with a cross-linked core encapsulating the CPT drug, surrounded by the pNIRM fluorescent layer, and externally coated by the hydrophilic PPEGMA corona. The covalent attachment of the BODIPY imaging agent endowed distinctive optical properties to the spheres, enabling real-time monitoring and tracking of the loaded drugs and NPs via an NIR imaging technique. In addition, both cell internalisation and intracellular drug release induced by a high level of GSH (10 mM) was confirmed in HeLa cells in vitro. The in vivo imaging also clearly indicated a passive targeting of the spherical micelles in the tumour site over time. To summarise, PISA has successfully allowed the preparation of highly delicate spherical micelles with reduction-responsive drug release and stable optical characteristics via a direct and simple one-pot formulation strategy, thus paving a novel pathway for tracking dynamic processes and biological imaging therapies (Fig. 4).
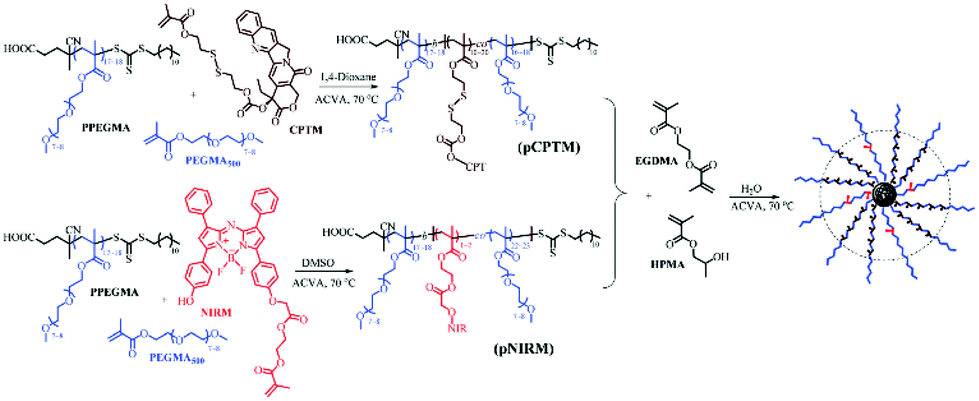 | ||
| Fig. 4 Synthetic routes for reduction-responsive, prodrug and NIR-fluorescence imaging dual-functional NPs by RAFT mediated dispersion polymerisation, producing P(CPTM-co-NIRM)-b-P(HPMA-co-EDGMA) spherical micelles in water. Reproduced from ref. 85 with permission from the Royal Society of Chemistry, copyright 2018. | ||
Couturaud et al. synthesised functionalised fluorine-based amphiphilic block copolymers using a photo-mediated RAFT dispersion polymerisation method, as promising scaffolds for drug delivery.86 As ester-activated pentafluorophenyl methacrylate (PFMA) can be readily functionalized with amino compounds, it was employed and polymerised with the poly(ethylene glycol)-4-cyano-4-(ethylthiocarbonothioylthio)pentanoic acid (PEG113-CEPA) macro-CTA via light-triggered PISA at 405 nm and 37 °C, yielding uniform spherical micelles independently of DP variations. A post-functionalisation of the PFMA-based polymer with primary diamines, e.g. ethylenediamine (EDA) or cystamine, generated cross-linking of the core-forming block, imparting a greater colloidal stability to the spherical particles. Upon exposure to increasing levels of GSH, the cystamine-bearing nanostructures dissociated due to the reduction in GSH-sensitive disulfide cross-links, whereas the diamine-cross-linked spherical micelles remained intact. The designed cross-linked polymeric NPs also had excellent biocompatibilities in in vitro human cells (Fig. 5).
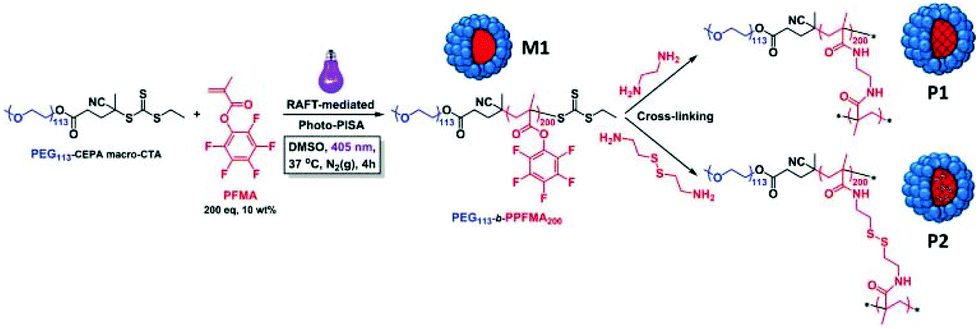 | ||
| Fig. 5 Schematic illustration of the preparation of PEG-b-PPFMA via photo-initiated PISA in DMSO at 37 °C (M1), followed by cross-linking reactions using either ethylenediamine or cystamine (P1 and P2, respectively) for transfer into aqueous media. Reproduced from ref. 86 with permission from Wiley Online Library, copyright 2018. | ||
A recent work by Hong and colleagues has demonstrated the use of PISA for dual reduction-triggered drug release and pH-responsive prodrug NP preparation.87 Poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) and poly(2-(diethylamino)ethyl methacrylate) (PDEAEMA) were used as macro CTAs and were chain-extended with CPTM to produce PHPMA/PDEAEMA-stabilized NPs. The PDEAEMA chains were protonated in acidic tumours and transitioned from a hydrophobic to a hydrophilic state, which in turn enabled an increased drug release and cancer cell uptake.
2.3. Oxidation-responsive systems
While a moderate level of reactive oxygen species (ROS) is a key necessity to maintain normal functions in cell signalling pathways and metabolism through oxygen-regulating homeostasis, a large increase in ROS is associated with a variety of pathological conditions such as cancer, diabetes, and inflammations.88,89 Hence, the overexpression of ROS has been employed in the design of smart oxidation-responsive polymer-based nanomaterials.90–94Having realised that the current stimuli-sensitive nanocarriers usually require tedious preparations, Sobotta et al. developed a one-pot synthetic pathway using RAFT-mediated dispersion PISA to produce well-defined spherical micelles of tuneable sizes that display ROS-triggered drug release.95 A series of block copolymers with varying chain ratios was prepared, whereby poly(N-acryloylmorpholine) (PNAM) was utilised as a macro-CTA for chain extension with N-acryloylthiomorpholine (NAT), a monomer carrying an oxidation-sensitive thioether moiety. This procedure readily afforded P(NAM-b-NAT) spherical particles of 25–73 nm in diameter (DLS) via the PISA process. The sphere size-range could be tailored by tuning the size or composition of the diblock copolymers. Nile Red dye was chosen as a model for drugs and was encapsulated into the spheres. In response to a H2O2 concentration of 10 mM, the hydrophobic thioether was oxidised to the hydrophilic sulfone, triggering a solubility transition of the core-forming block. As a result, the micellar structures dissociated, and the drug was released as confirmed by the fluorescence quenching of Nile Red proportional to the increased H2O2 levels. In addition to oxidation-triggered drug release, the formulated nano-vehicle showed excellent biocompatibility and no cytotoxicity, therefore providing a versatile, selective, and controlled drug delivery to the sites of interest (Fig. 6).
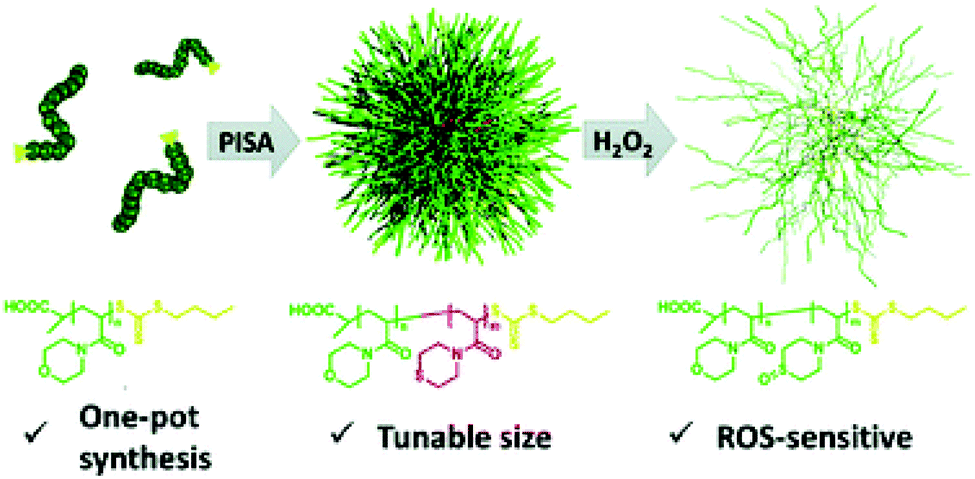 | ||
| Fig. 6 Schematic diagram of the one-pot synthesis of P(NAM-b-NAT) micelles of tuneable sizes displaying ROS-triggered drug release. Reproduced from ref. 95 with permission from the Royal Society of Chemistry, copyright 2018. | ||
2.4. Carbon dioxide (CO2)-responsive systems
Unlike other experimental models for stimuli that usually involve chemical stimulations and possibly generate by-products that can have side effects on biological cells, CO2 appears as an alternate stimulus since it is a key endogenous metabolite.Additionally, CO2 exhibits excellent biocompatibility and membrane permeability.96–100 Even though CO2 responsiveness has not been largely adopted in the context of therapeutic treatments, some of the designs available for CO2-sensitive polymeric materials have been considered as proofs-of-concepts for uses in drug delivery.
Recently, silica nanoparticles (SiO2 NPs) have been utilised as effective adhesives for hydrogels and biological tissues.101,102 It was reported that SiO2 NPs (20 nm) can be encapsulated into PISA-produced hybrid vesicles displaying an on-demand release of the internal contents due to the CO2-responsive characteristic of the core-forming block. Here, mPEG113 was used as a macro-CTA for the photo-initiated RAFT aqueous dispersion copolymerisation of 2-hydroxypropyl methacrylate (HPMA) and 2-(dimethylamino)ethyl methacrylate (DMAEMA) at room temperature. The obtained results indicated that complete monomer conversion was rapidly achieved within 15 min upon exposure to 405 nm visible-light, affording a well-defined CO2-sensitive diblock copolymer mPEG-b-P(HPMA-co-DMAEMA). The SiO2 NPs were concurrently encapsulated into the lumen of the polymeric vesicles via PISA, in a convenient one-pot synthetic strategy. The loaded cargos were released from the NPs in response to CO2 bubbling under mild conditions, due to the presence of the DMAEMA monomer in the hydrophobic block. After treatment with CO2 for 2 minutes, DMAEMA was protonated, enhancing the hydrophilicity of the core-forming block, and the vesicles therefore disintegrated to release the SiO2 NPs (Fig. 7).103 The same research group used the same concept to encapsulate fragile biomacromolecules such as bovine serum albumin (BSA) (5 wt%) into the polymeric vesicles prepared from an aqueous photo-initiated PISA under mild conditions. BSA was encapsulated in situ into the hybrid vesicles made of PEGMA-b-P(HPMA-co-DMAEMA). The addition of increased amounts of DMAEMA led to higher order morphologies during the block copolymer self-assembly, due to increased hydrogen bonding interactions. In the presence of CO2, BSA was unloaded from the vesicles due to the disassembly of copolymer chains induced by the protonation of DMAEMA.104
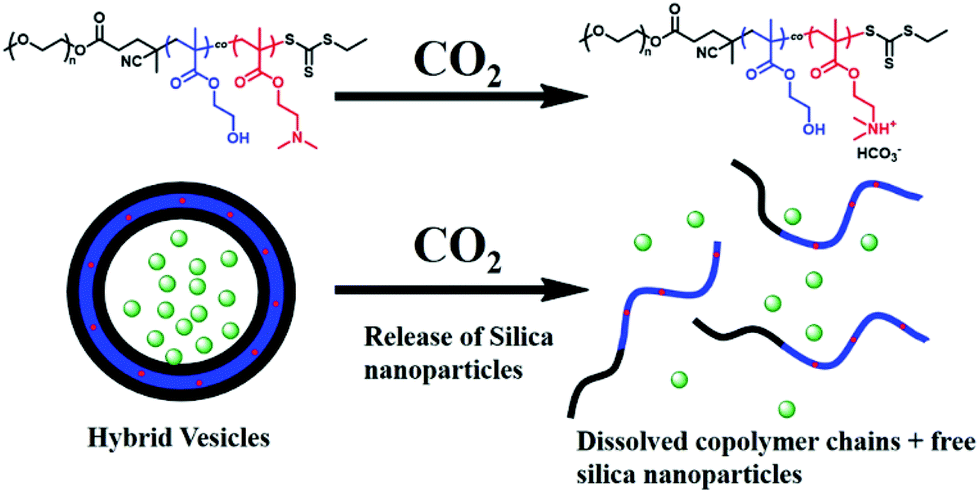 | ||
| Fig. 7 CO2-triggered release of SiO2 NPs from mPEG-b-P(HPMA-co-DMAEMA) hybrid vesicles. Reproduced from ref. 104 with permission from Wiley Online Library, copyright 2017. | ||
Nevertheless, several challenges remain to produce CO2-sensitive NPs via PISA. First, the polymerisation solution has to be degassed by purging inert gas or freeze–pump–thaw cycles, thus hampering large-scale manufacturing of those NPs. Second, the obtained particles are likely to disintegrate upon exposure to surfactants or organic solvents. Third, the switchability of CO2 with inert gas (N2 or Argon) is limited. To overcome these challenges, Yu et al. described a robust in situ cross-linking strategy that provides (1) a mild PISA process using photo-induced initiation in water; (2) an open-air polymerisation using enzyme-assisted degassing; and (3) CO2-responsive vesicles to precisely release the internal cargos. Particularly, HPMA and DMAEMA were polymerised by RAFT aqueous dispersion copolymerisation, with a 4-cyano-4-(ethylthiocarbonothioylthio)pentanoic acid (CDPA) macro-CTA, to directly produce PGMA-b-P(HPMA-co-DMAEMA-co-AMA) vesicles via photo-induced PISA.105 The reaction was deoxygenated by adding glucose oxidase and glucose. In situ crosslinking was achieved by including allyl methacrylate (AMA), an asymmetric cross-linker, during the PISA process. Under CO2 treatment, the core-contained DMAEMA was protonated and became hydrophilic, in a similar way to the aforementioned studies. However, due to the AMA cross-linked core, the vesicles were not disrupted but became prone to swelling or expansion in volume and size. On the other hand, when purging with N2, the swelling vesicles shrunk because of the deprotonation of DMAEMA under these inert conditions. This swelling–shrinking process was reversible over four CO2/N2 cycles. Methylene Blue (MB) dye was used as a drug model in a proof-of-concept release experiment and revealed enhanced release when subjected to a CO2 treatment.
In the meantime, these CO2-sensitive vesicles could carry an increased amount of SiO2 NPs after the CO2 treatment. This was explained by the interactions between SiO2 NPs and tertiary amines on DMAEMA in the hydrophobic block and the higher surface area of the vesicles favouring more SiO2 NPs to be loaded (Fig. 8).
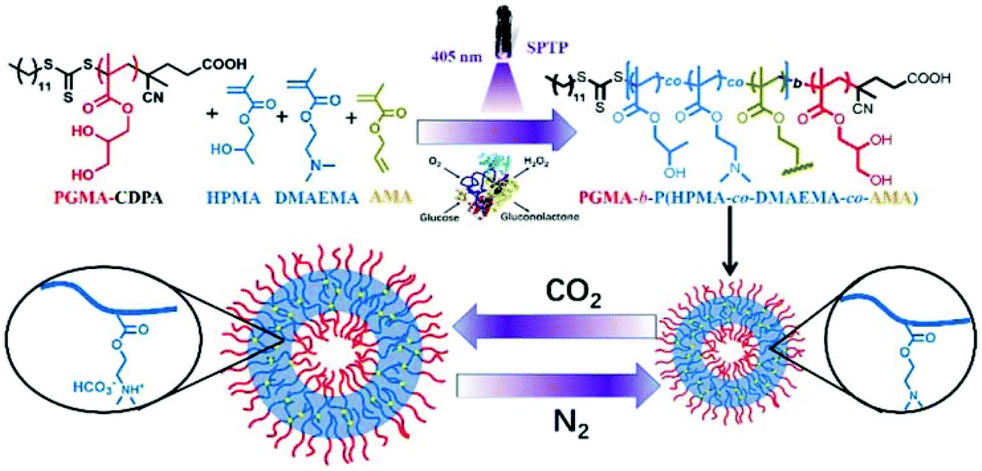 | ||
| Fig. 8 Schematic illustration of an enzyme-assisted photo-PISA for preparing cross-linked CO2-responsive vesicles. Reproduced from ref. 105 with permission from the Royal Society of Chemistry, copyright 2019. | ||
2.5. Temperature-responsive systems
Typically, a temperature-sensitive block copolymer exhibits a lower critical solution temperature (LCST) at the interface between a stabilising block and a core-forming block, below which it is soluble and above which it becomes insoluble. A temperature oscillation around the LCST triggers a particle morphological transformation, leading to the disassembly of nanostructures and to the unloading of the internal payloads. This feature is interesting in tissue engineering and regeneration as illustrated in the following examples.Lately, Mable et al. prepared a series of in situ SiO2-encapsulated amphiphilic block copolymer vesicles via a one-pot RAFT aqueous dispersion polymerisation method, in which a poly(glycerol monomethacrylate) (PGMA) macro-CTA was chain-extended with the HPMA monomer while simultaneously adding the hydrophilic 18 nm SiO2 NPs.62 SiO2 NPs were utilised as a bio-related active species due to their intrinsic self-healing effects on synthetic hydrogels or biological tissues.106
The targeted degree of polymerization was maintained relatively low to explore the thermo-responsiveness of the PHPMA core-forming block. SAXS confirmed that the release of SiO2 NPs occurs within the first 12 min upon cooling to 0 °C, as the PGMA58-b-PHPMA250 diblock copolymer underwent a morphological transformation from vesicles to a mixture of spherical and short-worm micelles, thereby triggering the release of internal cargos. Furthermore, an onset of vesicle dissociation was observed at about 10 °C, and promptly increased as the temperature decreased. The authors reported that this smart formulation and effective on-demand release of SiO2 NPs would be applied to self-healing adhesives of synthetic hydrogels and living tissues, thus enabling a possible model for encapsulating globular proteins (BSA), enzymes, and antibodies (Fig. 9).
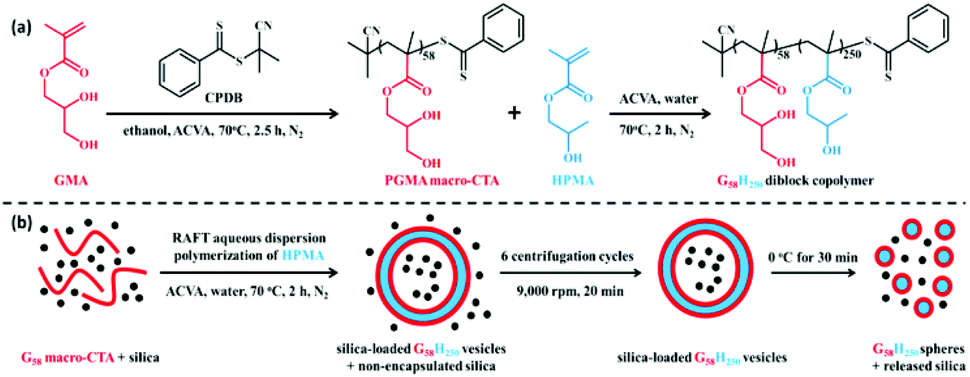 | ||
| Fig. 9 (a) Synthesis of PGMA58-b-PHPMA250 (G58H250) diblock copolymer via RAFT aqueous dispersion; (b) schematic illustration of the in situ encapsulation of SiO2 NPs via PISA and the subsequent release of SiO2 NPs upon cooling to 0 °C due to vesicle dissociation. Reproduced from ref. 62 with permission from ACS Publications, copyright 2015. | ||
On the other hand, worm-like gels have been widely adopted as potential materials in a range of biomedical applications. The group of Armes reported the preparation of biocompatible poly(glycerol monomethacrylate)-poly(2-hydroxypropyl methacrylate) (PGMA-b-PHPMA) diblock copolymer nanoworm gel that was responsive to temperature changes.107 These PGMA-b-PHPMA diblock copolymer worm-like micelles display a reversible thermal-responsive behaviour. Here, a PGMA-b-PHPMA diblock copolymer was dispersed into fluorophore-labelled Staphylococcus aureus at 4 °C, the temperature at which nanoworm degelation occurred, and led to worm-to-sphere transition, thus lowering the solution viscosity. This solution was then filtered through a 0.45 μm membrane, allowing for the smaller diblock copolymer particles to cross whereas the micrometer-sized bacteria stayed on the membrane. Brought back to ambient temperature, the obtained solution reversibly shifted from spheres to worms, favouring the gelation, which in turn served as an injectable scaffold for stem cell growth. Later, the same research group exploited the use of PGMA-b-PHPMA diblock copolymer nanoworm gel in human Pluripotent Stem Cells (hPSCs) and human embryos.108 When cooled down from 37 to 5 °C, PGMA-b-PHPMA worm gels switched from worms to spheres, allowing for a reversible gel formation-destruction via an order-order transition, on which cells can be embedded and recovered. These worm gels showed a good aptitude for mimicking natural mammalian mucins and for forming an excellent 3D matrix to maintain the viability and pluripotency of hPSCs without any passaging for 14 days, as well as for possible short-term storage of hPSCs and human embryos without a need for cryopreservation.
2.6. Enzymatically responsive systems
Enzymes are key components in cell regulation, and enzymatic dysfunction or overexpression is associated with a number of pathological conditions, particularly in cancer.109–111 These dysfunctions or overexpression can be exploited as a potential target in drug delivery designs. For instance, drugs can be leveraged when being exposed and degraded in a dysfunctional enzyme environment.Although the majority of reported examples have used the RAFT-PISA technique, other potential types of polymerisation techniques can be of interest for the production of biodegradable and responsive polymer NPs. To address the challenges of high polymerisation temperatures and strictly oxygen-free conditions, ROMPISA has been used to synthesise particles with high control over the polymerisation process under aerobic conditions.
In a recent report, Wright et al. generated peptide-based NPs sensitive to the proteolytic enzyme thermolysin in aqueous media, via a one-pot method in open air by ROMPISA at a high solid concentration (10 wt%).70 Such enzyme-responsive poly(norbornene) block copolymers are key to a series of future applications where more enzymatically sensitive biodegradable materials can be prepared via the versatile and robust ROMPISA method.
Recently, vesicles made from the ROP of N-carboxyanhydride (NCA)-induced self-assembly (NCA-PISA) have been reported. The polymerisation of L-phenylalanine NCA (L-Phe NCA) was initiated by methoxypolyethylene glycol amine(PEG45-NH2) at 10 °C without the need for degassing, using 20% solid contents.69 The obtained vesicles were then degraded by trypsin over 96 h.
2.7. Dual photo- and pH-responsive systems
The following formulations have a dual responsiveness, endowing particles with the UV-induced cross-linking for enhanced colloidal stability under physiological conditions, while conferring pH-triggered drug unloading in acidic microenvironments. In this contribution, the two photosensitive monomers 2-nitrobenzyl methacrylate (NBMA) and 7-(2-methacryloyloxy-ethoxy)-4-methyl-coumarin (CMA) were employed as comonomers in RAFT aqueous dispersion polymerisation with poly(N-(2-hydroxypropyl) methacrylamide (PHPMA) used as a macro-CTA. This approach readily afforded well-defined polymeric nano-structures with four types of morphologies: spheres, worms, lamellae, and vesicles.112 Upon UV irradiation (365 nm), the NBMA moieties were cleaved and replaced with methacrylic acid (MAA), inducing the hydrophobic-to-hydrophilic transition of the core-forming block; at the same time, the coumarin segments were dimerised to generate a core cross-linking, thus preventing the NP disassembly. DOX was loaded into the PHPMA-b-P(MAA-co-CMA) nano-objects via an electrostatic interaction between the –NH3+ ammonium cations on DOX and the –COO− carboxylate anions on P(MAA-co-CMA) in water, enabling a relatively high drug loading of 24.9 wt%. In vitro release revealed that DOX rapidly escaped from the nano-objects at pH 5.4 in intracellular endosomes/lysosomes due to the protonation of the –COO− groups on PMAA, thus disrupting the electrostatic interactions between DOX and the hydrophobic P(MAA-co-CMA) core. In summary, the above formulation method has enabled the production of polymeric NPs with powerful dual responsive behaviours, exhibiting a robust photo-induced cross-linked core for greater stabilisation and on-demand DOX release in acidic intracellular microenvironment upon cellular uptake.In a similar work, Pan et al. described the RAFT dispersion copolymerisation of DIPEMA and CMA in the presence of PHPMA as a macro-RAFT agent to produce in situ vesicles.113 Upon irradiation with UV light at 365 nm, the coumarin moieties anchored in the vesicle membrane underwent dimerisation to crosslink together, thus bringing an improved structural stabilisation to the polymeric vesicles. Furthermore, the presence of tertiary amino groups on the DIPEMA segments of the membrane triggered a swelling–shrinking transition upon tuning the solution pH from acidic to basic. At pH 4, the DIPEMA was protonated and shifted to a hydrophilic state, inducing the swelling of the core-forming block, thus enabling the pores of the membrane vesicles to open and facilitating the transmembrane traffic of substances. In acidic solutions, both the membrane (DIPEMA) and DOX were protonated, and the escape of DOX was inhibited due to electrostatic repulsion, although the membrane pores were large enough for DOX to get through. In contrast to DOX, fluorescein was effectively released after loading through the vesicle membrane as the size of fluorescein is smaller than the pores and due to the absence of electrostatic repulsion.
2.8. Dual reduction- and pH-responsive systems
To enhance the chemical versatility and responsiveness of the amphiphilic block copolymer vesicles, pH-responsive and reduction-cleavable bonds were both incorporated into their membrane-forming blocks during PISA-mediated copolymerisation.114 First, PEG-CPDB was chain-extended with comonomers including 2-(diisopropylamino)-ethyl methacrylate (DIPEMA), and either methacrylamide multivinyl comonomer cystaminebismethacrylamide (CBMA) or bis(2-methacryloyloxyethyl) disulfide (DSDMA) using RAFT-mediated dispersion polymerisation. It was indicated that the PISA of DIPEMA with the CBMA cross-linker imparted more robust crosslinked membranes. Rhodamine B was used as a drug model and encapsulated into the polymeric vesicles. Owing to the dual stimuli-responsive character of the vesicles, the dye was rapidly unloaded by the synergistic effect of DL-dithiothreitol (DTT) (10 mM) and an acidic medium at pH 4 mimicking intracellular conditions. At acidic pH values, DIPEMA was protonated and the system was switched to a hydrophilic state, vesicles were thus swollen, and the dye was conclusively liberated. Furthermore, drug release was also augmented due to the DTT-induced cleavage of disulfide cross-links in the acidic aqueous medium. This study demonstrated a straightforward one-pot strategy combining concomitantly crosslinked membrane, self-assembly, and drug encapsulation, endowing enhanced structural integrity and stabilisation to the vesicle membrane as well as an improved intracellular drug release (Fig. 10).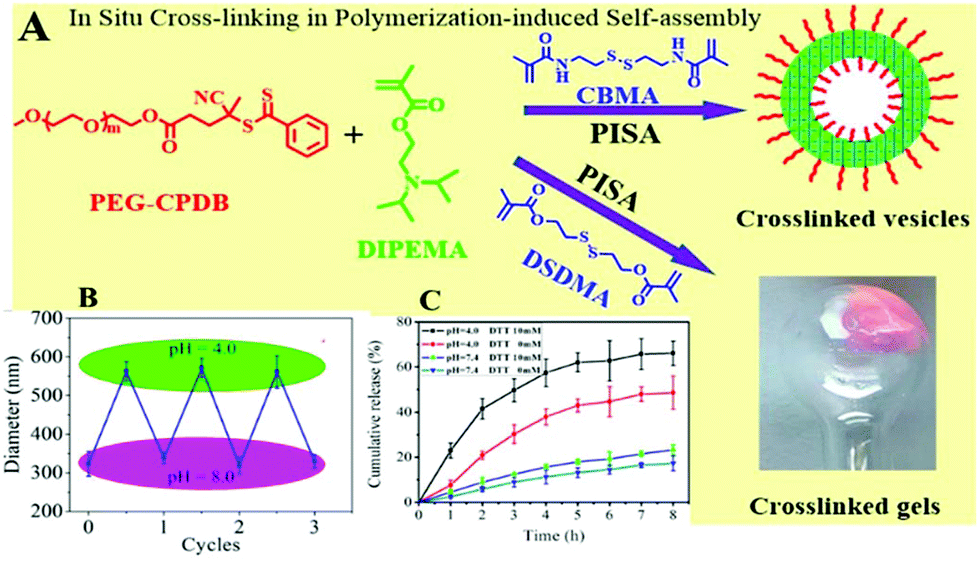 | ||
| Fig. 10 (A) In situ crosslinking in PISA to prepare crosslinked PEG-CPDB-b-P(DIPEMA90-co-CBMA10) membrane vesicles (D90C10); (B) reversible changes in the diameter of D90C10 vesicles upon switching solution pH between 4.0 and 8.0. (C) Cumulative release profiles of rhodamine B from D90C10 vesicles under different conditions. Reproduced from ref. 114 with permission from ACS Publications, copyright 2019. | ||
3. Outlooks and future challenges
The present review discussed the latest exciting studies describing the design and formulation of in situ stimuli-responsive NPs via mostly RAFT-PISA and a few ROP/ROMP-PISA methods, in order to achieve on-demand drug release while favouring a simple and versatile synthetic approach. The heterogeneity of diseased areas and their associated pathogenic conditions such as a higher reduction potential, the presence of ROS and/or low pH values have long been potential targets for the development of drug delivery systems that are responsive to these specific factors and enable drug unloading at the appropriate sites of action. The use of PISA is of particular interest and widely recognised as a powerful technique to prepare self-assembled NPs in a one-pot synthetic pathway, with limited intermediate purification steps, while facilitating scalability and industrial demands as opposed to traditional methods. A range of PISA-prepared self-assemblies incorporating functional moieties has been highlighted that respond to pathogenically mimicking stimuli. For example, NP pH-responsiveness to release drugs could be accomplished by a hydrophobic-to-hydrophilic solubility transition of the core-forming block.This transition resulted from the incorporation of ionisable groups that, at acidic pH values associated with the cell pathology, induced protonation and hydrophilicity of the internal fragments, leading to nano-objects disassembling and drugs being released.
The nano-structural dissociation arising from solubility transition was also observed under ROS-abundant conditions, whereby the hydrophobic thioether moieties in the core were oxidised to hydrophilic sulfones, disintegrating the core–shell structures. Micelles cross-linked via disulfide bonds displayed an enhanced colloidal stability under normal cell conditions; however, in the presence of reducing agents, they disintegrated to liberate the internal payloads owing to the cleavage of reduction-sensitive disulfide bonds. On the other hand, thermal responsiveness of spherical micelles triggered drug release in response to temperature changes because of the morphological evolutions. Dual triggers such as pH-and-reduction or pH-and-light stimuli combinations synergistically increased the sensitivity of particles for an enhanced drug release efficiency.
Although PISA research has vigorously been growing and showing great promise to overcome the drawbacks of traditional solution self-assembly techniques, many challenges remain to be addressed. One of the major bottlenecks preventing the expansion of PISA into more biomaterial applications is the use of relatively high temperatures and absolutely oxygen-free environments required for radical polymerisation methods, which can be challenging to achieve and hamper the final desired reaction yields. Most of the existing (meth)acrylate-based polymers are also difficult to degrade in biological media resulting in some increased accumulation and toxicity. Fortunately, some of these issues can be addressed using ROMP or ROP methods, involving the use of new and biodegradable monomers to synthesise self-assembled objects under mild conditions, or by using UV initiation to avoid the need for high reaction temperatures. These approaches are also of great benefit to the encapsulation of fragile therapeutic drugs or biomolecules such as peptides, antibodies, or DNA/RNA, thus avoiding their denaturation or degradation. Currently the majority of PISA formulations for drug delivery and biomaterials have been carried out based on RAFT polymerisation methods, some on ROP or ROMP. Hence there is a need to exploit other types of living radical polymerisation methods such as nitroxide-mediated polymerisation (NMP),115 iodine-mediated polymerisation (ITP),116 and sulfur-free RAFT117,118 for potentially preparing less toxic polymers. Though atom transfer radical polymerization (ATRP) has been extensively adopted to synthesise PISA-derived nano-objects,119,120 its application in drug delivery is very limited due to the use of cytotoxic copper catalysts for polymerisation.
It is also of importance to be able to design multifunctional PISA-made particles that can load multiple therapeutic agents and/or diagnostic probes while being conjugated with targeting ligands for achieving enhanced site-specific drug delivery as well as precisely controlled drug release on demand. Additionally, more attempts are required to investigate the behaviour of these PISA-prepared NPs in complex 3D cell culture models as well as in vivo to rapidly accelerate translations to clinical studies. In principle, a formulation method of future interest should also display reasonable drug loading and increased cellular uptake or trafficking, and PISA has proven itself to be an outstanding candidate. Attempts have been made to apply external stimuli to impart higher control over sizes and morphologies of PISA-prepared NPs. Interestingly, light has been used to enable spatiotemporal control of initiation,121 cross-linking,112 or modification of the hydrophobic/hydrophilic block ratio.122
Nevertheless, while it is critical to carry out PISA in aqueous media for medical purposes, it can limit the ability of encapsulating hydrophobic drugs, especially via physical entrapment. Probably, the use of prodrug monomers as aforementioned could potentially provide a solution by which drugs can be conjugated to monomers or macro-CTAs prior to or during the PISA process via stimuli-responsive linkages, thus both increasing the loading efficiency and achieving local release. The question has also been raised about whether the particle shapes may have an impact on the resulting loading efficiency and cell internalisation since there have not been many reports addressing this issue. Hence more attention should be drawn to this critical aspect. Meanwhile, hydrophilic drugs can be easily compartmentalised into PISA-generated NPs, particularly in vesicles, imparting the protection of internal cargos from degradation by biological media. This aspect could be very useful for the preparation of less invasive oral formulations which can protect drugs from the extreme conditions of the digestive and intestinal tracks before reaching the disease areas. Finally, the majority of PISA-related work up to now has been focusing on anticancer drug delivery while it is very likely that the same methodology could be applied to other therapeutic treatments, such as diabetes, and cardiovascular, or infectious diseases (Table 1).
| Block copolymer | Stimulus/stimuli | Drug/payload | Morphology | Ref. |
|---|---|---|---|---|
| POEGMA-b-P(ST-co-VBA) | Acidic pH | Doxorubicin (DOX) | Spheres, worms, rods, vesicles | 73 |
| PDMAEMA | Acidic pH | DOX | Spheres, rods, wires, vesicles | 74 |
| P(HPMA300-stat-GlyMARh1)–PDPA52 | Acidic pH | Rhodamine B | Vesicles | 75 |
| PEG-b-P(MEO2MA-co-CPTM) | GSH 10 mM | Camptothecin (CPT) | Spheres | 84 |
| P(CPTM-co-NIRM)-b-P(HPMA-co-EDGMA) | GSH 10 mM | BODIPY | Spheres | 85 |
| PEG113-b-PPFMA | GSH 10 mM | None | Spheres | 86 |
| P(NAM-b-NAT) | H2O2 10 mM | Nile red | Spheres | 95 |
| mPEG-b-P(HPMA-co-DMAEMA) | CO2 | SiO2 NPs | Vesicles | 103 |
| PEGMA-b-P(HPMA-co-DMAEMA) | CO2 | BSA | Vesicles | 104 |
| PGMA-b-P(HPMA-co-DMAEMA-co-AMA) | CO2 | Methylene blue (MB)/SiO2 NPs | Vesicles | 105 |
| PGMA-b-PHPMA | Temperature | SiO2 NPs | Vesicles | 62 |
| PHPMA-b-P(NBMA-co-CMA) | UV and acidic pH | DOX | Spheres, nanorods, lamellae, vesicles | 112 |
| PHPMA-b-P(DIPEMA-co-CMA) | UV and acidic pH | AuNPs/DOX/fluorescein | Vesicles | 113 |
| PEG-CPDB-b-P(DIPEMA-co-CBMA) | Reductive (DTT) and acidic pH | Rhodamine B | Vesicles | 114 |
Beyond these challenges, it is widely believed that PISA will continue to thrive and rapidly expand, building on our growing understanding of synthetic parameters and formulation strategies. To date, PISA has successfully enabled the self-assembly, drug loading, and incorporation of stimuli-responsive functionalities in a one-pot synthetic approach. In the near future, one believes the rapid maturation of PISA can instil more important aspects of precise drug delivery into a self-assembled nano-object, thus furthering a range of therapeutic values to the ultimate translation from the bench to the bedside.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Ministère de l'Enseignement supérieur, de la Recherche et de l'Innovation (MESRI) and Université Paris-Est Créteil.References
- A. K. Pearce and R. K. O'Reilly, Bioconjugate Chem., 2019, 30, 2300–2311 CrossRef CAS.
- V. Taresco, C. Alexander, N. Singh and A. K. Pearce, Adv. Ther., 2018, 1, 1800030 CrossRef.
- P. Couvreur and C. Vauthier, Pharm. Res., 2006, 23, 1417–1450 CrossRef CAS.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. Masoud Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- Z. S. Al-Ahmady, W. T. Al-Jamal, J. V. Bossche, T. T. Bui, A. F. Drake, A. J. Mason and K. Kostarelos, ACS Nano, 2012, 6, 9335–9346 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- J. V. John, C. W. Chung, R. P. Johnson, Y.-I. Jeong, K.-D. Chung, D. H. Kang, H. Suh, H. Chen and I. Kim, Biomacromolecules, 2016, 17, 20–31 CrossRef CAS.
- M. Liu, H. Du, W. Zhang and G. Zhai, Mater. Sci. Eng., C, 2017, 71, 1267–1280 CrossRef CAS.
- Z. Li, N. Song and Y. W. Yang, Matter, 2019, 1, 345–368 CrossRef.
- G. Battistelli, A. Cantelli, G. Guidetti, J. Manzi and M. Montalti, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 139–150 CAS.
- D. Shenoy, S. Little, R. Langer and M. Amiji, Pharm. Res., 2005, 22, 2107–2114 CrossRef CAS.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Chem. – Eur. J., 2015, 21, 13164–13174 CrossRef CAS.
- N. Song, W. Liu, Q. Tu, R. Liu, Y. Zhang and J. Wang, Colloids Surf., B, 2011, 87, 454–463 CrossRef CAS.
- T. Thambi, V. G. Deepagan, H. Y. Yoon, H. S. Han, S. H. Kim, S. Son, D. G. Jo, C. H. Ahn, Y. D. Suh, K. Kim, I. C. Kwon, D. S. Lee and J. H. Park, Biomaterials, 2014, 35, 1735–1743 CrossRef CAS.
- C. I. C. Crucho, ChemMedChem, 2015, 10, 24–38 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Polym. Chem., 2013, 4, 31–45 RSC.
- H. Phan, R. I. Minut, P. McCrorie, C. Vasey, R. R. Larder, E. Krumins, M. Marlow, R. Rahman, C. Alexander, V. Taresco and A. K. Pearce, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1801–1810 CrossRef CAS.
- P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers, Elsevier, Amsterdam, 2000 Search PubMed.
- E. Batrakova, S. Lee, S. Li, A. Venne, V. Alakhov and A. Kabanov, Pharm. Res., 1999, 16, 1373–1379 CrossRef CAS.
- Y. Yang, D. Pan, K. Luo, L. Li and Z. Gu, Biomaterials, 2013, 34, 8430–8443 CrossRef CAS.
- X.-B. Xiong, A. Falamarzian, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248–261 CrossRef CAS.
- V. S. Trubetskoy, Adv. Drug Delivery Rev., 1999, 37, 81–88 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- R. Raveendran, K. M. Mullen, R. M. Wellard, C. P. Sharma, R. Hoogenboom and T. R. Dargaville, Eur. Polym. J., 2017, 93, 682–694 CrossRef CAS.
- S. J. T. Rezaei, M. R. Nabid, H. Niknejad and A. A. Entezami, Int. J. Pharm., 2012, 437, 70–79 CrossRef CAS.
- F. Xu, H. Li, Y. L. Luo and W. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 5181–5192 CrossRef CAS.
- L. Zhang, H. Shen and A. Eisenberg, Macromolecules, 1997, 30, 1001–1011 CrossRef CAS.
- S. Yadav, A. K. Sharma and P. Kumar, Front. Bioeng. Biotechnol., 2020, 8, 127 CrossRef.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- N. P. Truong, J. F. Quinn, M. V. Dussert, N. B. T. Sousa, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2015, 4, 381–386 CrossRef CAS.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS.
- M. Semsarilar, I. Canton and V. Ladmiral, Methods in Molecular Biology, Humana Press Inc., 2016, vol. 1367, pp. 89–108 Search PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- D. Ikkene, A. A. Arteni, M. Ouldali, J.-L. Six and K. Ferji, Polym. Chem., 2020, 11, 4729–4740 RSC.
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid Commun., 2017, 38, 1–14 CrossRef.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1–22 CrossRef.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1–21 CrossRef.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2019, 40, 1–10 Search PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- Y. Zhao, Y. Wang, F. Ran, Y. Cui, C. Liu, Q. Zhao, Y. Gao, D. Wang and S. Wang, Sci. Rep., 2017, 7, 1–11 CrossRef.
- J. Zhao, H. Lu, S. Wong, M. Lu, P. Xiao and M. H. Stenzel, Polym. Chem., 2017, 8, 3317–3326 RSC.
- A. B. Jindal, Int. J. Pharm., 2017, 532, 450–465 CrossRef CAS.
- S. Salatin, S. M. Dizaj and A. Y. Khosroushahi, Cell Biol. Int., 2015, 39, 881–890 CrossRef CAS.
- R. Toy, P. M. Peiris, K. B. Ghaghada and E. Karathanasis, Nanomedicine, 2014, 9, 121–134 CrossRef CAS.
- R. Toy, E. Hayden, C. Shoup, H. Baskaran and E. Karathanasis, Nanotechnology, 2011, 22, 115101 CrossRef.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C. Y. Fang, I. Rehor, P. Cigler, H. C. Chang, G. Lin, R. Liu and Q. Li, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- W. Zhao, H. T. Ta, C. Zhang and A. K. Whittaker, Biomacromolecules, 2017, 18, 1145–1156 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J.-M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef.
- C. Fang, B. Shi, Y. Y. Pei, M. H. Hong, J. Wu and H. Z. Chen, Eur. J. Pharm. Sci., 2006, 27, 27–36 CrossRef CAS.
- J. Engström, H. Asem, H. Brismar, Y. Zhang, M. Malkoch and E. Malmström, Macromol. Chem. Phys., 2020, 221, 1900443 CrossRef.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- B. Karagoz, C. Boyer and T. P. Davis, Macromol. Rapid Commun., 2014, 35, 417–421 CrossRef CAS.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- J. Tan, Y. Bai, X. Zhang, C. Huang, D. Liu and L. Zhang, Macromol. Rapid Commun., 2016, 37, 1434–1440 CrossRef CAS.
- J. Siirilä, S. Häkkinen and H. Tenhu, Polym. Chem., 2019, 10, 766–775 RSC.
- J. Lesagedelahaye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS.
- J. Jiang, X. Zhang, Z. Fan and J. Du, ACS Macro Lett., 2019, 8, 1216–1221 CrossRef CAS.
- D. B. Wright, M. P. Thompson, M. A. Touve, A. S. Carlini and N. C. Gianneschi, Macromol. Rapid Commun., 2019, 40, 1800467 CrossRef.
- A. Punnia-Moorthy, J. Oral Pathol. Med., 1987, 16, 36–44 CrossRef CAS.
- G. Hao, Z. P. Xu and L. Li, RSC Adv., 2018, 8, 22182–22192 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- L. Qiu, C. Xu, F. Zhong, C. Hong and C. Pan, ACS Appl. Mater. Interfaces, 2016, 8, 18347–18359 CrossRef CAS.
- C. J. Mable, I. Canton, O. O. Mykhaylyk, B. Ustbas Gul, P. Chambon, E. Themistou and S. P. Armes, Chem. Sci., 2019, 10, 4811–4821 RSC.
- D. B. Wright, M. T. Proetto, M. A. Touve and N. C. Gianneschi, Polym. Chem., 2019, 10, 2996–3000 RSC.
- K. Bauri, A. Narayanan, U. Haldar and P. De, Polym. Chem., 2015, 6, 6152–6162 RSC.
- M. P. Gamcsik, M. S. Kasibhatla, S. D. Teeter and O. M. Colvin, Biomarkers, 2012, 17, 671–691 Search PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS.
- J. M. Estrela, A. Ortega and E. Obrador, Crit. Rev. Clin. Lab. Sci., 2006, 43, 143–181 CrossRef CAS.
- R. Bakalova, Z. Zhelev, I. Aoki and T. Saga, Clin. Cancer Res., 2013, 19, 2503–2517 CrossRef CAS.
- Z. Li, M. Wu, H. Bai, X. Liu and G. Tang, Chem. Commun., 2018, 54, 13127–13130 RSC.
- J. Zhen, S. Tian, Q. Liu, C. Zheng, Z. Zhang, Y. Ding, Y. An, Y. Liu and L. Shi, Biomater. Sci., 2019, 7, 2986–2995 RSC.
- W. Zhang, C. Hong and C. Pan, Biomacromolecules, 2016, 17, 2992–2999 CrossRef CAS.
- C. Tian, J. Niu, X. Wei, Y. Xu, L. Zhang, Z. Cheng and X. Zhu, Nanoscale, 2018, 10, 10277–10287 RSC.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1–6 CrossRef.
- X. Zhao, M. Chen, W. G. Zhang, C. H. Wang, F. Wang, Y. Z. You, W. J. Zhang and C. Y. Hong, Macromol. Rapid Commun., 2020, 41, 2000260 CrossRef CAS.
- P. Jia, C. Dai, P. Cao, D. Sun, R. Ouyang and Y. Miao, RSC Adv., 2020, 10, 7740–7750 RSC.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS.
- L. Lin, H. Gong, R. Li, J. Huang, M. Cai, T. Lan, W. Huang, Y. Guo, Z. Zhou, Y. An, Z. Chen, L. Liang, Y. Wang, X. Shuai and K. Zhu, Adv. Sci., 2020, 7, 1903138 CrossRef CAS.
- X. Xia, X. Yang, P. Huang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 18301–18308 CrossRef CAS.
- J. Shi, Z. Chen, B. Wang, L. Wang, T. Lu and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28554–28565 CrossRef CAS.
- Y. Yao, H. Zhang, Z. Wang, J. Ding, S. Wang, B. Huang, S. Ke and C. Gao, J. Mater. Chem. B, 2019, 7, 5019–5037 RSC.
- Y. Na, J. S. Lee, J. Woo, S. Ahn, E. Lee, W. Il Choi and D. Sung, J. Mater. Chem. B, 2020, 8, 1906–1913 RSC.
- F. H. Sobotta, F. Hausig, D. O. Harz, S. Hoeppener, U. S. Schubert and J. C. Brendel, Polym. Chem., 2018, 9, 1593–1602 RSC.
- A. Darabi, P. G. Jessop and M. F. Cunningham, Chem. Soc. Rev., 2016, 45, 4391–4436 RSC.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS.
- J. Gutknecht, M. A. Bisson and F. C. Tosteson, J. Gen. Physiol., 1977, 69, 779–794 CrossRef CAS.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS.
- Q. Yan and Y. Zhao, Chem. Commun., 2014, 50, 11631–11641 RSC.
- H. Wu, F. Li, S. Wang, J. Lu, J. Li, Y. Du, X. Sun, X. Chen, J. Gao and D. Ling, Biomaterials, 2018, 151, 66–77 CrossRef CAS.
- J. H. Kim, H. Kim, Y. Choi, D. S. Lee, J. Kim and G. R. Yi, ACS Appl. Mater. Interfaces, 2017, 9, 31469–31477 CrossRef CAS.
- J. Tan, D. Liu, X. Zhang, C. Huang, J. He, Q. Xu, X. Li and L. Zhang, RSC Adv., 2017, 7, 23114–23121 RSC.
- J. Tan, X. Zhang, D. Liu, Y. Bai, C. Huang, X. Li and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1–8 Search PubMed.
- L. Yu, Y. Zhang, X. Dai, Q. Xu, L. Zhang and J. Tan, Chem. Commun., 2019, 55, 11920–11923 RSC.
- S. Rose, A. Prevoteau, P. Elzière, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382–385 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS.
- C. E. Callmann, C. V. Barback, M. P. Thompson, D. J. Hall, R. F. Mattrey and N. C. Gianneschi, Adv. Mater., 2015, 27, 4611–4615 CrossRef CAS.
- J. E. Ghadiali and M. M. Stevens, Adv. Mater., 2008, 20, 4359–4363 CrossRef CAS.
- M. Li, G. Zhao, W.-K. Su and Q. Shuai, Front. Chem., 2020, 8, 647 CrossRef CAS.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Biomacromolecules, 2017, 18, 1210–1217 CrossRef CAS.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15086–15095 CrossRef CAS.
- M. Chen, J. W. Li, W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromolecules, 2019, 52, 1140–1149 CrossRef.
- E. Groison, S. Brusseau, F. D'Agosto, S. Magnet, R. Inoubli, L. Couvreur and B. Charleux, ACS Macro Lett., 2012, 1, 47–51 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS.
- A. Lotierzo, R. M. Schofield and S. A. F. Bon, ACS Macro Lett., 2017, 6, 1438–1443 CrossRef CAS.
- J. Sarkar, L. Xiao, A. W. Jackson, A. M. Van Herk and A. Goto, Polym. Chem., 2018, 9, 4900–4907 RSC.
- J. Wang, Z. Wu, G. Wang and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800332 CrossRef.
- Y. Wang, G. Han, W. Duan and W. Zhang, Macromol. Rapid Commun., 2019, 40, 1800140 CrossRef.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308–3314 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |