
Presence of anthraquinone in coffee and tea samples. An improved methodology based on mass spectrometry and a pilot monitoring programme†
Francisco José
Díaz-Galiano
 ,
Maria
Murcia-Morales
,
María del Mar
Gómez-Ramos
,
Carmen
Ferrer
and
Amadeo R.
Fernández-Alba
*
,
Maria
Murcia-Morales
,
María del Mar
Gómez-Ramos
,
Carmen
Ferrer
and
Amadeo R.
Fernández-Alba
*
Agrifood Campus of International Excellence ceiA3 (ceiA3), European Union Reference Laboratory for Pesticide Residues in Fruits and Vegetables, Department of Chemistry and Physics, University of Almería, La Cañada de San Urbano, 04120, Almería, Spain. E-mail: amadeo@ual.es
First published on 26th November 2020
Abstract
Anthraquinone has been linked to potential adverse effects on human health and the environment. The most commonly employed methods for the analysis of coffee and tea cause the extraction of matrix interferents such as the methylxanthines caffeine and theobromine, which hinder the analysis of anthraquinone. A new manual extraction method – using ethyl acetate as the extraction solvent with a dispersive solid-phase extraction clean-up step based on primary–secondary amines – has been developed. The new developed method allows for the quantitation of anthraquinone at 5 μg kg−1 concentration levels, four times lower than the current maximum residue limit for coffee and tea in the European Union (20 μg kg−1). Alongside, a new automated extraction method has also been developed. Finally, a pilot monitoring programme of 90 coffee and tea samples from several countries within the European Union has been performed, in which anthraquinone has been detected in a concentration range of 5.1–18.8 μg kg−1 in 32% of the monitored samples, below the current 20 μg kg−1 maximum residue limit, and in 48% of the monitored tea samples, revealing the need for including anthraquinone in a more extensive monitoring programme of tea.
1. Introduction
The incomplete burning of wood, coal, and other fuel sources is known to be the origin of persistent organic compounds such as polycyclic aromatic hydrocarbons (PAHs). These organic contaminants can be carried by winds and adsorbed on plants over considerable distances.1 Anthraquinone (9,10-dioxoanthracene) (AQ) is one such aromatic organic compound of risk to human health and to the environment, belonging to the oxygenated PAH (oxy-PAH) subgroup. AQ is a pervasive PAH that can be found in most of the samples contaminated with PAHs, such as soil and sediment samples.2 The toxicity of AQ and its derivatives has been thoroughly studied in the literature,3,4 and recently, it has been linked to a potential increase of cancer risk in humans.5 It can be found on food due to its formation and adsorption during the aforementioned combustion processes, some of which are used to dry plant products such as tea, chamomile or coffee.6 Contamination of tea with AQ can also take place due to its application as a biopesticide or bird repellent.7 An alternative contamination source of tea and coffee is the proximity of the drying patios, where leaves and coffee beans are left to dry, to sources of burning, from which AQ can be deposited.2,6 The European Commission recommends its control at low concentrations, since the consumption of these plant products is high.8 The first notification by the European Food Safety Agency (EFSA) on AQ in tea was issued in 2012, and ever since, a total of 52 notices have been issued for this commodity, the last one (at the time of writing) on January 10, 2020. AQ has also been found in herbs, spices and other food products such as chili peppers.9 The concentration mentioned in these notifications ranged between 15 μg kg−1 and 360 μg kg−1, although only one case of concentration below 20 μg kg−1 was reported in this eight-year period, in 2019.The analysis of dry commodities such as tea, dry herbs or spices can present certain difficulties. The SANTE document recommends the addition of water prior to their extraction to improve the extraction efficiency.10 However, Chen et al. studied the effect of a hydration step prior to the extraction of tea and found it to be counterproductive.11 In their study, the authors found that only very polar pesticides – which were a minority – benefited from sample hydration. Higher baselines (by two orders of magnitude) were obtained, which means worse method performance for the vast majority of pesticides; darker extracts were also obtained, and flocculent precipitation was observed when tea samples were hydrated prior to their extraction. Due to the increased co-extraction of matrix components because of sample hydration, more matrix interferences in the analysis of pesticides are to be expected. In this work, some of these interferences arising from co-extracted matrix components have been determined.
Nevertheless, as previously mentioned, avoiding sample hydration means lower extraction efficiency of polar pesticides. If sample hydration is to be avoided, and only organic solvent is applied for the extraction, energetic extraction conditions must be employed, which are generally outside the capabilities of standard extraction techniques present in routine laboratories. These higher energy conditions can be achieved using an automated sample extraction instrument, such as one based on pressurized liquid extraction and sample heating.
Automated extraction is attracting more interest from laboratories due to its increased robustness and reproducibility compared to manual sample treatment methodologies. In this regard, Wang et al. demonstrated the automation of the QuEChERS extraction procedure using a vortex vibration-centrifuge device with promising results,12 but no further developments can be found in the literature. Axial shakers are already being employed to partially automate extraction procedures which offer a reproducible shaking step, such as the AGYTAX®13,14 – which also allows for concurrent sample heating15 – or the SPEX Geno/Grinder®16 – which also allows for sample milling17. In recent years, new devices are being brought into the market, which offer the possibility of completely automating the whole extraction process, with only sample weighing needed from the user. Automated extraction devices based on pressurized liquid extraction allow the extraction of traditionally difficult commodities without the need for performing sample hydration or the need for costly clean-up steps, and within a fraction of the time a manual sample extraction would require. Indeed, pressurized liquid extraction has been successfully employed to extract veterinary drugs from feeding stuffs using acidified formic acid and a simple espresso machine,18 semi-volatile organic compounds with an accelerated solvent extractor (ASE) and dichloromethane,19 and other organic contaminants such as polycyclic aromatic hydrocarbons (PAHs) using a homemade pressurized liquid extraction device and a combination of different solvents.20
This study's objectives are (i) to evaluate the difficulties associated with the currently used multiresidue methods for food within the European Union – in particular due to the inclusion of hydration steps; (ii) to develop manual and automated methods that allow the control of AQ at very low concentrations – below the current maximum residue limit (MRL) of 20 μg kg−1 for tea, coffee, herbal infusions and cocoa;21 and (iii) to perform a monitoring study of potentially contaminated dry commodities. A sub-objective was also to test if deuterated anthraquinone (AQ-D8) could be used to quantitate AQ and, if so, be included in multiresidue methods with a hydration step as a screening strategy for the detection of AQ.
2. Materials and methods
2.1. Reagents and materials
LC-MS grade acetonitrile (AcN), pesticide residue analysis grade acetone (Ac) and ethyl acetate (AcOEt), and ultra-gradient HPLC-grade AcN were obtained from Sigma-Aldrich (Steinheim, Germany). Anhydrous magnesium sulphate, sodium chloride, sodium hydrogencitrate sesquihydrate, calcium chloride and sodium citrate tribasic dihydrate were purchased from Sigma-Aldrich. Primary–secondary amine-bonded silica (PSA) and Carbopack X 120/400 mesh (GCB) bulk sorbents and laboratory grade sand were supplied by Supelco (Bellefonte, PA, USA), and Bondesil-C18 (C18) bulk sorbent was supplied by Agilent Technologies (Santa Clara, CA, USA). Pre-weighed MgSO4, PSA and ChloroFiltr® in 15 mL PTFE centrifuge tubes were acquired from United Chemical Technologies (UCT) (Bristol, PA, USA).All high-purity pesticide standards were purchased from Sigma-Aldrich, LGC (Teddington, United Kingdom) or Riedel-de-Haën (Seelze, Germany) and were stored at a temperature of −30 °C. Individual stock solutions with concentrations between 1000 and 2000 mg L−1 were prepared in acetonitrile and were stored in amber screw-capped glass vials in the dark at −20 °C. In the case of AQ and AQ-D8, the solvent used was a mixture of Ac![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcN (8:92, v/v) and the concentration was 600 mg L−1.
:AcN (8:92, v/v) and the concentration was 600 mg L−1.
A Sonopuls HD 3100 ultrasonic system supplied by Bandelin Electronic GmbH & Co. KG (Berlin, Germany) was employed. The apparatus was equipped with a GM3100 high-intensity generator (100 W), a UW 3100 ultrasonic converter, an SH 70 G standard horn, and a 3 mm-diameter titanium MS73 probe for 2–50 mL volumes. An AGYTAX® automatic axial extractor supplied by Cirta Lab. S.L. (Spain) was also used. Finally, an EDGE instrument supplied by CEM Corporation (Charlotte, North Carolina, United States of America) was used in the development of an automated dry food commodity extraction method. Q-Cups and Q-Discs® (G1 and C9 varieties) were also provided by CEM Corporation.
2.2. Manual sample treatment
The extraction procedure for AQ is as follows: 4 g of tea, dry herbs or coffee are weighed in a 50 mL PTFE centrifuge tube, followed by the addition of 10 mL of AcOEt. Next, the sample plus the solvent is sonicated at 75% amplitude for 42 s (six 5 s extraction cycles plus one 2 s pause in between). Afterwards, the centrifuge tubes are centrifuged for 5 min at 4000 rpm at room temperature. A 3 mL aliquot of the supernatant is transferred into a 15 mL PTFE centrifuge tube containing 180 mg of PSA to perform a dispersive Solid Phase Extraction (dSPE) clean-up step. Finally, the 15 mL centrifuge tubes are centrifuged for 5 min at 4000 pm, the extracts transferred into 4 mL clear glass vials, and the PSA is neutralized with 30 μL of formic acid (FA) in AcN (5:95, v/v).
All analyses were carried out using gas chromatography coupled to tandem mass spectrometry (GC-QqQ-MS/MS), for which 50 μL of the final extract in AcOEt were directly injected into the GC system.
2.3. Automated sample treatment
The automated sample treatment procedure is as follows: first, a layered Q-Disc® setup is placed in the Q-Cup in the following way: first, a C9 (cellulose) Q-Disc®, then a G1 (glass fibre) Q-Disc® and finally a second C9 Q-Disc® on top of the G1 one. Following this step, 4 g of tea are weighed into the Q-Cup and the tea is covered by approximately 5 g of fine laboratory grade sand. The Q-Cup is placed into the EDGE instrument alongside a 50 mL PTFE falcon tube, which is used to collect the sample extract.The method optimized for the EDGE extraction is the following: the solvent used is AcN in two consecutive cycles. During the first extraction cycle, the Q-Cup containing the sample is loaded within the instrument, sealed, and 10 mL of AcN added. The sample and the solvent are heated up to 40 °C, pressurized at 2 bar and these conditions are held for 150 s. Afterwards, the 10 mL of AcN extract are transferred into the collection PTFE tube. In the second extraction cycle i.e. the rinse cycle, 5 mL of AcN are added to the Q-Cup and then immediately transferred into the collection PTFE tube. The total extraction volume is 15 mL. Finally, a wash step is performed by passing 10 mL of AcN at 40 °C to prepare the EDGE instrument for the next sample extraction.
2.4. GC-EI-QqQ-MS/MS analysis
The analyses by gas chromatography were performed in an Agilent Intuvo 9000 GC system equipped with an Agilent 7693 autosampler and an Agilent 7010 GC MS/MS triple quadrupole. The instrumental parameters can be found elsewhere.13For the optimization of the MS parameters, AQ and AQ-D8 were monitored in full scan mode in the 50–250 m/z range. The first step was the selection of the precursor ion/s for each analyte and the retention time, injecting individual solutions for each pesticide at 1 mg kg−1 in full-scan mode. The ion with the highest intensity and m/z relationship was selected as the precursor ion. Precursor ion fragmentation was performed by collision-induced dissociation with nitrogen, from which the best fragment ions were chosen. Once the fragment ions were selected, the adequate CE for each transition was assayed in the 3–40 eV range.
For AQ, the selected transitions were 208.0 > 180.0, 208.0 > 152.0 and 180.0 > 152.0. The corresponding transitions for AQ-D8 were 216.0 > 188.0, 216.0 > 160.0 and 188.0 > 160.0.
2.5. Method performance
The identification and quantitation of compounds and method validation were performed on the basis of the SANTE document (SANTE/12682/2019) criteria.10 The method development was performed using spiked tea samples. The positive findings showed two fully overlapping transitions with the same retention time as that of the standard (±0.1 min time tolerance) (three overlapping transitions in the case of AQ and AQ-D8). The ion ratio of these ions was within the ±30% deviation range with regard to the calibration standards. The signal-to-noise ratio was higher than 3 for all the reported positives. Linearity was checked from 2 to 200 μg L−1 using six calibration points. Lindane-D6 was added to all injection vials at a concentration of 50 μg L−1 as a form of quality control. Method validation parameters were also checked for coffee and cocoa.2.6. Analysis by GC-EI-TOF-MS
The separation of the organic compounds from the extracts was carried out using an Agilent 7890A gas chromatograph (Agilent Technologies, Palo Alto, CA, USA). The samples were injected using a multimode injector equipped with an ultra-inert inlet liner, with glass wool obtained from Agilent. The multimode injector operated in solvent vent mode with a temperature program of 70 °C (0.1 min), 800 °C min−1 to 325 °C. The injection volume was 5 μL and helium (99.999% purity) was used as the carrier gas. The GC separation was performed using two fused silica HP-5MS UI capillary columns of 15 m × 0.250 μm inner diameter and a film thickness of 0.25 μm (from Agilent) connected by a capillary flow technology (CFT) union. The oven temperature was programmed as follows: 60 °C for 1 min, 40 °C min−1 to 170 °C and finally up to 310 °C at 10 °C min−1. The total run time was 20.75 min with three additional minutes for backflushing at 310 °C. Backflushing was used to shorten the analysis time and reduce system maintenance avoiding the arrival of undesirable compounds of the matrix to the detector. The end of the chromatographic column is connected to the second column through a CFT union (used as a purged capillary flow device), which allows system backflushing. The instrument worked at a constant flow. During the run time, the flow was set at 0.75 mL min−1 in the first column and 0.95 mL min−1 in the second column (with a difference of 0.2 mL min−1 over the flow in the first column). Retention time locked (RTL) setting was used to eliminate the need for adjusting retention times of the compounds, for further analysis when needed. Chlorpyrifos methyl was used as a locking compound at a retention time of 9.14 min.The gas chromatography system was connected to a quadrupole time-of-flight (QTOF) mass spectrometer, Agilent 7250 (Agilent Technologies, Santa Clara, USA), operating in electron impact ionization (EI) mode (70 eV). The ion source and transfer line were set at 280 °C and the quadrupole temperature was set at 150 °C. A solvent delay of 3 min was selected in order to prevent damage in the ion source filament. The TOF-MS was operated in full-scan mode from m/z 60 to 500 and an acquisition rate of 3 spectra per s with a resolution of 40000 FWHM (m/z 263). Perfluorotributylamine (PFTBA) was used for MS calibration. The mass accuracy of the generated ions was controlled using an internal mass calibration performed before every two injections.
3. Results and discussion
3.1. Manual method development
Following these results, it was evident that the method required adjustments to correctly extract and analyse AQ. Each method tested involved the extraction of a blank aliquot of Earl Grey tea plus three aliquots spiked at 5, 10 and 20 μg kg−1 each, which were the standard spiking levels chosen for all remaining experiments.
The first step in the method described by Lozano et al. is a hydration step involving the addition of 4 mL of Milli-Q water (0.5 g of sample per 1 mL of water), mixing it and allowing it to stand for 30 min prior to the extraction procedure.22 The clean-up step involving dSPE uses 50 mg mL−1 PSA and 50 mg mL−1 CaCl2. Several different strategies were then tested in order to improve AQ's analysis (Table 1).
| Method (MXX) | Hydration step | Solvent used | Dilution factor (V m−1) | Salts used | Extraction method | Clean-up (dispersive solid phase extraction) | LOQ (μg kg−1) | Rec. (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Extract olume (mL) | MgSO4 (mg) | PSA (mg) | GCB (mg) | C18 (mg) | ChloroFiltr® (mg) | FA (5%) | ||||||||
|
a Hydration step: addition of twice the sample mass as water mass, shake, wait 30 min; dilution factor: extraction volume to sample mass ratio; Ac:AcOEt: 1:1 (v/v); salts used: 8 g MgSO4 plus 1.5 g NaCl; shaking: 7 min AGYTAX® axial shaker; ultrasound: 42 s Sonolpuls HD 3100; FA (5%): 10 μL of 5% formic acid in AcN (v/v) per mL of clean-up extract.
|
||||||||||||||
| M01 | Yes | AcN | 5.0 | Yes | Shaking | 3 | 450 | 150 | — | 150 | — | Yes | >20 | 296 |
| M02 | Yes | AcN | 5.0 | Yes | Shaking | 3 | 450 | 150 | 22.5 | 150 | — | Yes | >20 | 253 |
| M03 | Yes | AcN | 5.0 | Yes | Shaking | 3 | 450 | 150 | 7.5 | — | — | Yes | >20 | 342 |
| M04 | Yes | AcN | 5.0 | Yes | Shaking | 3 | 450 | 75 | 22.5 | 75 | — | Yes | >20 | 350 |
| M05 | No | AcN | 5.0 | No | Shaking | 3 | — | 75 | 22.5 | — | — | Yes | — | — |
| M06 | No | AcN | 5.0 | No | Shaking | 3 | — | 75 | 22.5 | 75 | — | Yes | — | — |
| M07 | No | AcN | 5.0 | No | Shaking | 3 | — | 75 | — | — | — | Yes | 10 | 77 |
| M08 | No | AcN | 5.0 | No | Shaking | 3 | — | 75 | 75 | — | — | Yes | — | — |
| M09 | No | AcN | 5.0 | No | Shaking | 5 | 900 | 300 | — | — | 150 | Yes | 10 | 54 |
| M10 | No | AcN | 5.0 | No | Shaking | 5 | 900 | 300 | 40 | — | 150 | Yes | — | — |
| M11 | Yes | AcN | 5.0 | Yes | Shaking | 5 | 900 | 300 | — | — | 150 | Yes | >20 | 166 |
| M12 | Yes | AcN | 5.0 | Yes | Shaking | 5 | 900 | 300 | 40 | — | 150 | Yes | >20 | 573 |
| M13 | No | AcOEt | 5.0 | No | Ultrasound | 3 | — | 180 | — | — | — | Yes | 10 | 101 |
| M14 | No | AcOEt | 5.0 | No | Ultrasound | 3 | — | 360 | — | — | — | Yes | 10 | 119 |
| M15 | No | AcOEt | 5.0 | No | Ultrasound | 5 | 900 | 300 | — | — | 150 | Yes | 10 | 100 |
| M16 | No | AcOEt | 5.0 | No | Shaking (Δ) | 3 | — | 180 | — | — | — | Yes | 10 | 101 |
| M17 | No | AcOEt | 5.0 | No | Shaking (Δ) | 3 | — | 360 | — | — | — | Yes | 10 | 92 |
| M18 | No | AcOEt | 5.0 | No | Shaking (Δ) | 5 | 900 | 300 | — | — | 150 | Yes | 10 | 102 |
| M19 | No | Ac:AcOEt | 5.0 | No | Ultrasound | 3 | — | 800 | 25 | — | — | Yes | >20 | 10 |
| M20 | No | Ac:AcOEt | 5.0 | No | Ultrasound | 3 | — | 180 | — | — | — | No | 10 | 93 |
| M21 | No | Ac:AcOEt | 5.0 | No | Ultrasound | 3 | — | 180 | — | — | — | Yes | 10 | 90 |
| M22 | No | AcOEt | 5.0 | No | Shaking | 3 | — | 180 | — | — | — | Yes | 10 | 96 |
| M23 | No | AcOEt | 2.5 | No | Shaking | 3 | — | 180 | — | — | — | Yes | 10 | 84 |
| M24 | No | AcOEt | 2.5 | No | Ultrasound | 3 | — | 180 | — | — | — | Yes | 5 | 98 |
| M25 | Yes | AcOEt | 2.5 | Yes | Ultrasound | 3 | — | 180 | — | — | — | Yes | >20 | 207 |
First, four different clean-up strategies were investigated (M01–M04) based on the original method. Following this, 2 g of Earl Grey tea were weighed into 50 mL PTFE centrifuge tubes, hydrated with 4 mL of Milli-Q water and left to stand for 30 min. Next, 10 mL of AcN were added and the tubes were shaken automatically in the AGYTAX® axial shaker for 7 min. QuEChERS-based extraction salts were added to the centrifuge tube (4 g MgSO4, 1 g NaCl, 1 g sodium citrate tribasic dihydrate and 0.5 g sodium hydrogencitrate sesquihydrate) and the tubes were again shaken automatically for 7 min. The samples were centrifuged at 4000 rpm for 5 min at the maximum rotational speed, and the supernatants were subjected to different clean-up procedures. For methods M01–M04, 3 mL of the supernatant were transferred into 15 mL PTFE centrifuge tubes containing varying combinations of MgSO4 and PSA, GCB and C18 sorbents, vortexed for 30 s and centrifuged at 4000 rpm for 5 min at the maximum rotational speed. Finally, about 2 mL of the extracts were transferred into 4 mL vials and 30 μL of formic acid (FA) solution in AcN (5:95, v/v) were added. The extracts were then directly injected into the GC-QqQ-MS/MS instrument for analysis.
The interfering co-extracted co-eluting matrix components were still present, and thus, AQ identification was still not possible. When studying the behaviour of AQ-D8, it was observed that its area was halved for M02 and M04 when compared to M01 and M03. It was suspected that AQ was being adsorbed onto GCB in M02 and M04.
Afterwards, in order to evaluate the effect of sample hydration on the co-extraction of matrix components, M05 to M08 in which the hydration step was removed were tested. Hence, the same extraction procedure described for M01 to M04 was followed, but the hydration step, the addition of extraction salts and one of the shaking steps were skipped. Then, again, 3 mL of the supernatant were subjected to four different clean-up strategies with PSA, GCB, C18 and combinations of these. For the first time in this study, AQ was properly identified using M07, whose clean-up step comprised 25 mg mL−1 PSA only. Hence, the preliminary data suggested that avoiding sample hydration might be key to properly extracting and analysing AQ. Avoiding sample hydration has been previously discussed by other authors,11 who in a large multiscale experiment determined that for most analytes besides the most polar of them, sample hydration posed more disadvantages than benefits. AQ, with a logP of 3.39,6 appears to be one of these compounds adversely affected by tea hydration. Furthermore, due to the elimination of the hydration step, the effect of matrix interferences was greatly reduced (Fig. 1).
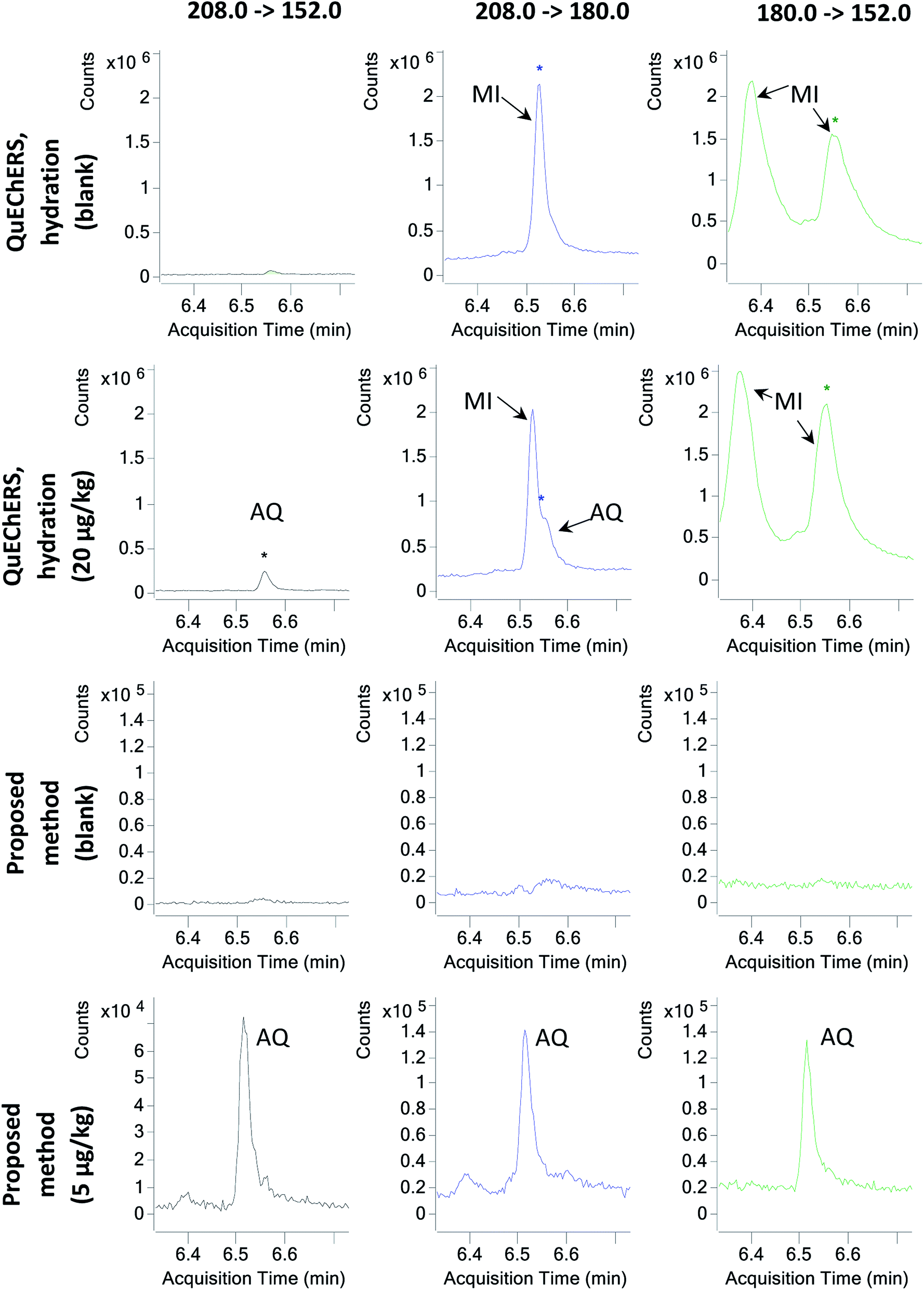 | ||
| Fig. 1 Chromatograms of the MRM transitions of AQ in a QuEChERS Earl Grey tea extract with a hydration step (first row, blank sample; second row, sample spiked with 20 μg kg−1 AQ) and the same sample extracted with the proposed manual method (third row, blank sample; fourth row, sample spiked with 5 μg kg−1 AQ). MI = matrix interference(s) per s; AQ = anthraquinone. | ||
To check whether hydration ought to be removed, or if using a different clean-up strategy would suffice, four more method modifications were tested (M09–M12). M09 and M10 would skip sample hydration, whereas M11 and M12 would not. The extraction procedure was equivalent to methods M05–M08 for M09 and M10, while M11 and M12's extraction procedure was the same as that of M01–M04. The clean-up step, however, was modified to use pre-weighed UCT 15 mL PTFE centrifuge tubes containing 900 mg MgSO4, 300 mg PSA and 150 mg ChloroFiltr®. To M10 and M12 clean-up tubes, 40 mg GCB were also added. The volume of extract required for these tubes was 5 mL, as stated by the manufacturer. Matrix interferences were still present in M11 and M12, but M09 allowed the identification and quantitation of AQ, although with a low recovery value (54%). AQ was not detected with M08, indicating adsorption of AQ onto GCB. It was concluded best not to use GCB as a dSPE sorbent in the analysis of AQ using AcN as the solvent. However, the intensity of the co-extracted matrix interferences in AQ's three MRM transitions was consistently lower in methods with GCB, which pointed at GCB partially removing some of the co-extracted matrix interferences.
With the intent of increasing recovery values and lowering the LOQ, AcOEt was tested as the extraction solvent in combination with two different extraction methods. In one set of experiments (M13 to M15), a Sonopuls HD 3100 instrument was employed to perform an ultrasound assisted extraction (UAE), which consisted of six 5 s extraction cycles with 2 s pauses between them. The second set of experiments (M16 to M18) used the same AGYTAX® automatic shaker, but with the aid of a different 50 mL centrifuge tube holder, and the samples were heated up to 35 °C. M13 and M16's clean-up step consisted of 60 mg PSA per mL extract, M14 and M17's step consisted of 120 mg PSA per mL extract, and M15 and M18's step used the same pre-weighed 15 mL PTFE centrifuge tubes as for M09 to M12. The amount of PSA did not affect AQ's analysis, and there were not any differences when compared to ChloroFiltr® centrifuge tubes. Recovery values were successfully increased to close to 100% in all cases except for M14. When taking into account these results in conjunction with the information provided by M07, it was decided that 60 mg PSA per mL extract was the adequate clean-up solvent. Identification and quantitation of AQ at 5 μg kg−1 was still not possible, so further method modifications were required.
The solubility of AQ is poor in most solvents except in acetone or sulphuric acid in high concentration,23,24 so in order to check whether the extraction solvent was the limiting factor in lowering the LOQ, an Ac:AcOEt (1:1, v/v) mixture was tested as the extraction solvent in M19 to M21. One source claimed to use PSA alongside GCB as a dSPE clean-up step obtaining a good recovery and LOQ,25 so it was decided to test it again in M19 to confirm our previous results. However, recoveries were again negligible and the LOQ was over 20 μg kg−1, which further advised against the use of this sorbent for the analysis of AQ (see Section 3.1.2.). M20 and M21 were based on M13, which had provided good results and a short sample treatment time. Neutralization of the final extract was checked to determine if it had any effect on AQ, but no differences were found between methods. Recovery values for M20 and M21 were similar to those in previous experiments, but the LOQ could not be lowered.
Since M20 and M21 did not yield better results than previous methods, for M22 to M25 the choice of solvent was reverted to AcOEt, which is less volatile than acetone and poses less trouble in the extraction procedure. In M22 and M23, increasing the sample mass to solvent volume ratio was compared; M23 and M24 compared the extraction method, i.e. automatic shaking versus ultrasound extraction. The recovery and LOQ of M22 were very similar to those of M13, so no significant differences were found between the use of the ultrasound probe or the AGYTAX® shaker in these two methods. While AQ could not be correctly identified in M23 at 5 μg kg−1 due to a non-complying ion ratio, the peak shape at this concentration level showed a substantial improvement.
Finally, with M24, an LOQ of 5 μg kg−1 could be achieved. Quantitation and identification of AQ and AQ-D8 were possible at the 5 μg kg−1 spiking level (Fig. 1). The reason why M24 performs better than M23 is probably the extraction methodology: the ultrasound probe is capable of a better extraction than shaking for such a relatively high sample amount, which seems to be critical at very low concentrations for this method.
3.2. Automated method development
Although the proposed manual method successfully recovers AQ and AQ-D8, no other pesticides have been studied in this work. It is widely accepted that water hydration is still necessary to achieve good analytical performance regarding more polar and incurred pesticides.11,28 To overcome the lack of a hydration step, a more energetic extraction method was developed based on the automated pressurized liquid extraction of dry matrixes, developed on the basis of the results of the proposed manual method (M24). Method validation for the automated sample extraction using the EDGE instrument was carried out in several phases. Earl Grey tea samples were spiked with AQ and AQ-D8 at 5, 10 and 20 μg kg−1. First, both solvent choice (AcN, AcOEt) and clean-up steps were optimized (AM01–AM06) (Table 2). For all these methods, 4 g of tea were added to a Q-Cup containing a Q-Disc® setup as described in Section 2.3., and then the sample was covered with 5 g of fine laboratory grade sand. This was done to prevent tea, which is a lightweight matrix, from floating on top of the solvent, which prevents the sample from being completely in contact with the extraction solvent. For the extraction, the instrument added 10 mL of AcN or AcOEt on top of the sample, the system was heated up to 40 °C and pressurized at 2 bar, and then held for 120 s. The extraction solvent was transferred into the collection tube, and the system was washed with 10 mL of the appropriate solvent prior to the extraction of the following sample. Recovery values were almost twice for methods using AcN compared to those employing AcOEt as the extraction solvent. Then, the effect of a dSPE clean-up step using PSA, analogous to M24, was checked. No significant improvement in the recoveries was observed, nor in the TIC obtained with the GC-EI-TOF-MS instrument. Thus, the use of a dSPE step with PSA was discarded going onwards.| Method (AMXX) | Solvent | Volume (mL) | Bubbling time (s) | Hold time (s) | Temp. (°C) | Rinse step | Rinse solvent volume (mL) | Total solvent (mL) | Dilution factor (V m−1) | Clean-up (dSPE) | LOQ (μg kg−1) | Rec. (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Dilution factor: extraction volume to sample mass ratio; FA (5%): 20 μL of 5% formic acid in AcN (v/v) per mL of clean-up extract. | ||||||||||||
| AM01 | AcN | 10 | — | 120 | 40 | No | — | 10 | 2.50 | — | 20 | 46 |
| AM02 | AcN | 10 | — | 120 | 40 | No | — | 10 | 2.50 | PSA | 20 | 49 |
| AM03 | AcN | 10 | — | 120 | 40 | No | — | 10 | 2.50 | PSA, FA | 20 | 54 |
| AM04 | AcOEt | 10 | — | 120 | 40 | No | — | 10 | 2.50 | — | 20 | 27 |
| AM05 | AcOEt | 10 | — | 120 | 40 | No | — | 10 | 2.50 | PSA | 20 | 29 |
| AM06 | AcOEt | 10 | — | 120 | 40 | No | — | 10 | 2.50 | PSA, FA | 20 | 28 |
| AM07 | AcN | 10 | 60 | 60 | 40 | No | — | 10 | 2.50 | — | >20 | — |
| AM08 | AcN | 10 | 90 | 60 | 40 | No | — | 10 | 2.50 | — | 20 | 31 |
| AM09 | AcN | 5 | 60 | 60 | 40 | Yes | 5 | 10 | 2.50 | — | 20 | 41 |
| AM10 | AcN | 10 | — | 90 | 40 | Yes | 5 | 15 | 3.75 | — | 10 | 78 |
| AM11 | AcN | 10 | 30 | 90 | 40 | Yes | 5 | 15 | 3.75 | — | 20 | 57 |
| AM12 | AcN | 10 | — | 150 | 40 | Yes | 5 | 15 | 3.75 | — | 7.5 | 101 |
Afterwards, a method setting called ‘bubbling’ was tested. During bubbling, air is passed through the sample–solvent mixture at ambient pressure prior to the pressurized extraction step. Several combinations of bubbling and hold time were tested in methods AM07 to AM09 (Table 2). Recovery values for AM07 and AM08 were lower than for AM01. In the case of lightweight matrixes such as tea, coffee or dry herbs, bubbling might impact negatively the extraction step, causing the sample to float on top of the solvent and resulting in part of the sample not being in contact with the solvent. The recovery value was higher for AM08 likely due to the sample being longer in contact with the solvent (150 s total) than in AM07 (120 s total), even if bubbling is counterproductive. For AM09, the total solvent volume was kept at 10 mL but added in two different cycles: a regular extraction cycle and a ‘rinse only’ cycle. The recovery for AQ was 41% in AM09 compared to no recovery in AM07 and 31% in AM08.
AM10 and AM11 were tested, which comprised two cycles: first, a 90 s hold of 10 mL AcN at 40 °C, and then a 5 mL AcN rinse. The first cycle in AM11 included a 30 s bubbling step. The recovery value was significantly higher in AM10 than in AM11 (78% vs. 57%). However, the LOQ for AM10, which did not employ bubbling, reached 10 μg kg−1.
A final method (AM12) based on AM10 was tested. In this case, bubbling was completely removed from the first cycle and the hold time was significantly increased from 90 s to 150 s. The recovery for AQ in this final method was 103% at 10 μg kg−1.
Initially, the lowest spiking level was 5 μg kg−1. However, taking into consideration the instrumental quantitation limit of 2 μg kg−1 and the dilution factor of 3.75 for AM12, the LOQ for the developed automatic extraction method AM12 was 7.5 μg kg−1, which was experimentally confirmed, with a recovery value of 101%.
The total run time per sample, including Q-Cup placement by the robotic arm within the extraction chamber, sample extraction and the wash step, is 7 min. Sample throughput using the automated method AM12 is approximately 70 samples extracted in an 8 h period, or about 206 samples in a 24 h period,per EDGE instrument employed.
AQ-D8 was also studied alongside AQ, showing similar results to those presented in Table 2. For the optimized method, AM12, the recovery value for this deuterated compound was 96%.
3.3. Analysis of AQ using AQ-D8 as a surrogate standard
The analysis of AQ using sample treatment methods with a hydration step means that AQ cannot be correctly identified using the MRM transitions mentioned herein. In particular, the affected transitions by co-extracted matrix interferences are 208.0 > 180.0 and 180.0 > 152.0. While not being the most sensitive transition for AQ, 208.0 > 152.0 presents the advantage of not being affected by co-extracted matrix interferents. For methods without a hydration step, none of the transitions are affected by any co-eluting matrix component (Fig. 1). In the case of AQ-D8, however, none of the equivalent transitions to those of AQ, i.e. 216.0 > 188.0, 180.0 > 160.0 and 216.0 > 160.0, exhibit any type of interference (Fig. S1†).When comparing the slopes of calibration curves constructed using AQ and AQ-D8 equivalent transitions 208.0 > 152.0 and 216.0 > 160.0, respectively, it can be observed that the slope of the AQ calibration curve is 90.1% of the calibration curve of AQ-D8 (Fig. S2†). In addition, at 5 μg kg−1, the instrumental response of AQ is 89.8% of the analytical response of AQ-D8. The significance of these values is that AQ-D8 can be used to quantitate AQ with high accuracy. Furthermore, since the transition 208.0 > 152.0 for AQ is unaffected by extraction methods with a hydration step, and this transition is equivalent to 216.0 > 160.0 for AQ-D8, this deuterated compound can be included in multiresidue methods with a hydration step as a non-confirmatory, quantitative screening tool for the detection of AQ, whose presence and exact concentration can be later determined with the aid of a tailored extraction method, such as M24.
3.4. Determination of the co-extracted matrix interferents using GC-EI-TOF-MS
An Agilent 7250 GC-Q-TOF high-resolution accurate mass spectrometer was employed in the determination of the co-extracted matrix interferents hindering the analysis of AQ. First, the total ion chromatograms (TIC) of Earl Grey tea extracts using M01 and AM12 were compared (Fig. 2). The baseline is approximately five times higher in the case of M01 than for AM12. Beginning at 8.8 min, a very intense and broad peak can be observed, which in the case of M01 presents a tail which extends well into 10.5 min. Since AQ elutes at 10 min under these conditions, and these matrix interferences are much more intense in the case of M01, to the point of saturation, it was hypothesized that it might consist in a compound interfering with the analysis of AQ.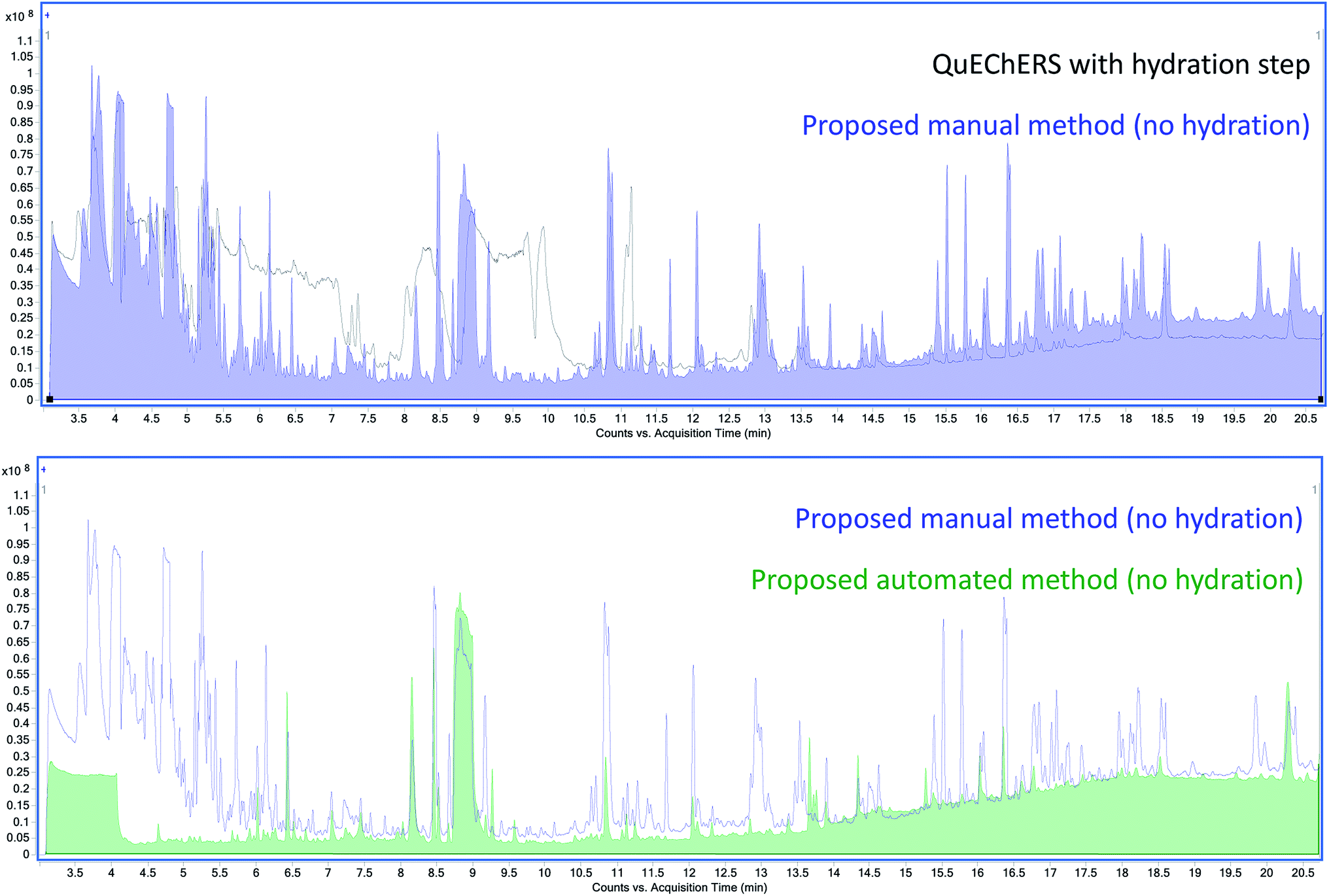 | ||
| Fig. 2 GC-TOF-MS total ion chromatogram (TIC) of Earl Grey tea extracted using the QuEChERS method (black, top) and the proposed manual method (blue, top). The wide peak that includes caffeine can be observed at 8.8 min, whose tail in the case of the QuEChERS method TIC can be observed to prolong until almost 11 min. The automated extraction of tea (bottom, green) provides an even cleaner extract than the proposed manual method (bottom, blue). | ||
The co-extracted natural compounds which affect AQ's transition 180.0 > 152.0 in the QqQ analysis have been tentatively identified as the methylxanthines caffeine and theobromine. First, the extracted-ion chromatogram (XIC) of 180 ± 1 m/z was obtained and the mass spectra studied. The molecular ion 194.0806 m/z was found. Then, caffeine was tentatively identified by comparison against the NIST library. Finally, caffeine was confirmed using accurate mass data, molecular ion and its fragments, with a mass error ≤ ±5 ppm and by comparison with an analytical standard. Theobromine was found in the search for metabolites, with a 180.0642 m/z and tentatively identified by comparison against the NIST library and accurate mass data of the molecular ion and mass fragments with a mass error ≤ ±5 ppm.
Although caffeine, theobromine and anthraquinone do not share a retention time neither in the GC-EI-QqQ-MS/MS nor in the GC-EI-TOF-MS instruments, when using hydration methods, the abundance of these methylxanthines is so high that these peaks tail well into the retention time of AQ (Fig. 3). This tailing gives rise to the interferences observed in the MRM transition 180.0 > 152.0 of AQ in the GC-EI-QqQ-MS/MS instrument. These methylxanthines might still be extracted when using methods without a hydration step; however, their extraction is not as efficient without the addition of water and they were not observed to interfere with the analysis of AQ.
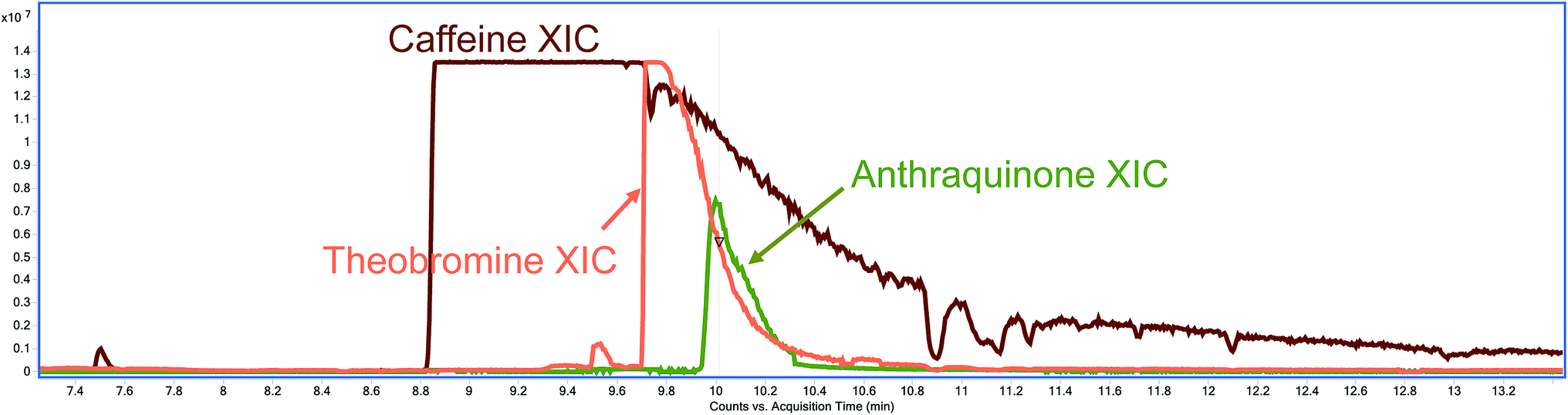 | ||
| Fig. 3 XIC of caffeine (brown), theobromine (orange) and anthraquinone (green) from a QuEChERS Earl Grey tea extract with a hydration step. | ||
3.5. Method performance
The performance of methods M24 and AM12 was tested in terms of LOQ, accuracy, precision and linearity. The LOQ was successfully set at the instrumental quantitation method (2 μg kg−1) for M24 and AM12, 5 μg kg−1 and 7.5 μg kg−1, respectively. Method performance data are summarized in Table 3.| Method | Matrix | Solvent | Dilution factor (V m−1) | Instrumental limit of quantitation (μg kg−1) | LOQ (μg kg−1) | Linear range (μg kg−1) | R 2 (%) | Recovery (n = 5) (%) | Repeatability (n = 5) (%) | Reproducibility (n = 5, 5 days) (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Dilution factor: extraction volume to sample mass ratio. | ||||||||||
| M24 | Tea | AcOEt | 2.50 | 2.0 | 5.0 | 5.0–200 | 1.000 | 98 | 8 | 12 |
| M24 | Coffee | AcOEt | 2.50 | 2.0 | 5.0 | 5.0–200 | 0.999 | 105 | 10 | 14 |
| M24 | Cocoa | AcOEt | 2.50 | 2.0 | 5.0 | 5.0–200 | 1.000 | 95 | 8 | 12 |
| AM12 | Tea | AcN | 3.75 | 2.0 | 7.5 | 7.5–200 | 1.000 | 101 | 3 | 7 |
| AM12 | Coffee | AcN | 3.75 | 2.0 | 7.5 | 7.5–200 | 0.999 | 108 | 4 | 8 |
| AM12 | Cocoa | AcN | 3.75 | 2.0 | 7.5 | 7.5–200 | 0.999 | 104 | 4 | 6 |
3.6. Pilot monitoring programme of real samples for the detection of AQ
A total of 90 samples purchased in three different countries within the European Union (Denmark, Portugal and Spain) were analysed for this work using both M24 and AM12. The matrixes analysed were tea – several varieties, including chamomile – coffee (ready-to-use capsules and coffee grains) and other dry herbs, such as oregano or parsley. None of the samples contained AQ at concentration levels higher than the current MRL of 20 μg kg−1; however, 29 samples contained AQ at concentration levels between the LOQ and 18.8 μg kg−1. The number of samples positive for AQ below 7.5 μg kg−1 was only 4; thus, the performance of the automated extraction was very close to the manual one, although with a lower consumption of chemicals, shorter extraction times and cleaner extracts, which means less instrumental maintenance required. The number of samples positive for AQ was 29 out of the 90 samples studied, meaning 32% of the monitored samples contained AQ at concentration levels above the LOQ (28% if concentrations above the LOQ for AM12 only are considered). Among tea samples, the number of positive AQ findings was 48% (29 out of 61) (Table 4), or 41% if only positives reported using the automated extraction method (AM12) are considered. Detailed information about the samples and AQ concentrations can be found in Table S1.†| Number of samples | Number of positive samples | Concentration range (μg kg−1) | Percentage of positive samples (%) |
|---|---|---|---|
| 61 | 29 | 5.1–18.8 | 48 |
4. Conclusions
Caffeine and theobromine have been tentatively identified as the main co-extracted interferents in methods with a hydration step. The presence of these two methylxanthines in sample extracts prevents the correct identification and determination of anthraquinone in coffee and tea. Avoiding sample hydration has been determined to eliminate interferences in the analysis of anthraquinone in coffee and tea. The new method's quantitation limit is 5 μg kg−1, four times lower than the current maximum residue level of 20 μg kg−1 for coffee, tea, and herbal infusions established by the European Commission. The automated extraction method provides a quantitation limit of 7.5 μg kg−1, which is also below the current maximum residue level. Detection of anthraquinone using the automated method allowed the detection and quantitation of all the monitored real samples at levels above 7.5 μg kg−1. Additionally, the automated method based on pressurized liquid extraction offers several advantages: there is no need for a dispersive solid phase extraction step, performance in terms of repeatability and reproducibility is better, and a potential sample throughput of 200 in a 24 h period per instrument. Sample treatment automation provides comparable results with a reduced fungible use and faster sample treatment time. A monitoring study of dry commodities has detected anthraquinone in 32% of the samples at levels far below the current maximum residue limit, showcasing its better performance than the current multiresidue methods. Anthraquinone has been detected in 48% of the monitored tea samples. This high percentage of positive tea samples in anthraquinone suggests the convenience of including anthraquinone in a more extensive monitoring programme. Deuterated anthraquinone is unaffected by co-extracted natural products in dry commodities. A screening strategy involves using this compound to detect the possible presence of anthraquinone.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the European Commission, DG SANTE (Document SANTE/12682/2019), and the European Union Reference Laboratory for Fruits and Vegetables (EURL-FV). Francisco José Díaz-Galiano acknowledges the pre-doctoral Training University Lecturers (FPU) fellowship program (grant FPU18/05113) awarded by the Spanish Ministry of Science, Innovation and Universities.References
- A. M. Abd El-Aty, J. H. Choi, M. M. Rahman, S. W. Kim, A. Tosun and J. H. Shim, Food Addit. Contam., 2014, 31, 1794–1804 CrossRef CAS.
- E. A. Rodgers-Vieira, Z. Zhang, A. C. Adrion, A. Gold and M. D. Aitken, Appl. Environ. Microbiol., 2015, 81, 3775–3781 CrossRef CAS.
- L. E. Sendelbach, Toxicology, 1989, 57, 227–240 CrossRef CAS.
- V. Shukla, S. Asthana, P. Gupta, P. D. Dwivedi, A. Tripathi and M. Das, Adv. Mol. Toxicol., 2017, 11, 1–50 Search PubMed.
- IARC- International Agency for Research on Cancer, IARC Monogr. Eval. Carcinog. Risks Hum., 2012, 101, 41–66 Search PubMed.
- X. Wang, L. Zhou, F. Luo, X. Zhang, H. Sun, M. Yang, Z. Lou and Z. Chen, Food Chem., 2018, 244, 254–259 CrossRef CAS.
- S. J. Werner, S. T. DeLiberto, A. M. Mangan, S. E. Pettit, J. W. Ellis and J. C. Carlson, Crop Prot., 2015, 72, 158–162 CrossRef CAS.
- European Food Safety Authority, EFSA J., 2018, 16(7), 5348 Search PubMed.
- RASFF Portal, https://webgate.ec.europa.eu/rasff-window/portal/, accessed 6 February 2020.
- European Commission, Eur. Comm. Dir. Heal. Food Saf., 2019, 12682, 1–53 Search PubMed.
- X. Chen, Y. Li, Q.-Y. Chang, X.-Y. Hu, G.-F. Pang and C.-L. Fan, J. AOAC Int., 2015, 98, 149–159 CrossRef CAS.
- J. Wang, Z. He, L. Wang, Y. Xu, Y. Peng and X. Liu, J. Chromatogr. A, 2017, 1521, 10–18 CrossRef CAS.
- M. Murcia Morales, M. J. Gómez Ramos, P. Parrilla Vázquez, F. J. Díaz-Galiano, M. García Valverde, V. Gámiz López, J. Manuel Flores and A. R. Fernández-Alba, Sci. Total Environ., 2020, 710, 136288 CrossRef CAS.
- B. Reichert, I. R. Pizzutti, I. H. Costabeber, A. Uclés, S. Herrera and A. R. Fernández-Alba, Talanta, 2015, 134, 415–424 CrossRef CAS.
- L. Pareja, F. Jesús, H. Heinzen, M. D. Hernando, Ł. Rajski and A. R. Fernández-Alba, Anal. Methods, 2019, 11, 2123–2128 RSC.
- D. Chepyala, I. L. Tsai, H. W. Liao, G. Y. Chen, H. C. Chao and C. H. Kuo, J. Chromatogr. A, 2017, 1491, 57–66 CrossRef CAS.
- M. J. Martínez Bueno, F. J. Díaz-Galiano, Ł. Rajski, V. Cutillas and A. R. Fernández-Alba, J. Chromatogr. A, 2018, 1546, 66–76 CrossRef.
- R. B. Hoff, L. Molognoni, C. T. P. Deolindo, M. O. Vargas, C. R. Kleemann and H. Daguer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1152, 122232 CrossRef CAS.
- F. Mercier, E. Gilles, G. Saramito, P. Glorennec and B. Le Bot, J. Chromatogr. A, 2014, 1336, 101–111 CrossRef CAS.
- J. S. S. Pinto, L. M. Assis, M. A. Andrade and F. M. Lancas, J. Chromatogr. Sci., 2018, 56, 761–769 CAS.
- The European Commission, Off. J. Eur. Communities: Legis., 2014, 57, 3–60 Search PubMed.
- A. Lozano, Ł. Rajski, N. Belmonte-Valles, A. Uclés, S. Uclés, M. Mezcua and A. R. Fernández-Alba, J. Chromatogr. A, 2012, 1268, 109–122 CrossRef CAS.
- Handbook of Chemistry and Physics, ed. R. C. Weast, CRC Press Inc., Boca Raton, Florida, USA, 60th edn, 1979, vol. 8 Search PubMed.
- R. J. S. Leweis, Hawley's Condensed Chemical Dictionary, John Wiley & Sons, Inc., New York, USA, 14th edn, 2001 Search PubMed.
- C. Sivanandha Reddy, S. Kulavardhana Reddy and G. V. S. Reddy, Rasayan J. Chem., 2018, 11, 582–588 CrossRef.
- D. G. Hayward, J. W. Wong and H. Y. Park, J. Agric. Food Chem., 2015, 63, 8116–8124 CrossRef CAS.
- G.-F. Pang, C.-L. Fan, Q.-Y. Chang, Y. Li, J. Kang, W.-W. Wang, J. Cao, Y.-B. Zhao, N. Li, Z.-Y. Li, Z.-M. Chen, F.-J. Luo and Z.-Y. Lou, J. AOAC Int., 2013, 96, 887–896 CrossRef CAS.
- T. Cajka, C. Sandy, V. Bachanova, L. Drabova, K. Kalachova, J. Pulkrabova and J. Hajslova, Anal. Chim. Acta, 2012, 743, 51–60 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay01962c |
| This journal is © The Royal Society of Chemistry 2021 |