
Simultaneous extraction and analysis of clozapine and lorazepam from human plasma using dual solvent-stir bar microextraction with different acceptor phases followed by high-performance liquid chromatography ultra-violet detection
Mahsa
Sheikh
 ,
Mohammad Reza
Hadjmohammadi
and
Mohammad Hossein
Fatemi
*
,
Mohammad Reza
Hadjmohammadi
and
Mohammad Hossein
Fatemi
*
Department of Analytical Chemistry, Faculty of Chemistry, University of Mazandaran, NirooHavayii Boulevard, 47416-95447 Babolsar, Iran. E-mail: mhfatemi@umz.ac.ir; Fax: +98 1125242002; Tel: +98 1125242031
First published on 25th November 2020
Abstract
A new design of dual solvent stir bar microextraction (DSSBME) was developed and combined with HPLC-UV for the simultaneous extraction of clozapine (CLZ) and lorazepam (LRP) from human plasma with different acceptor phases. Two short hollow fibers immobilized with an organic extraction solvent were used as the solvent bars for microextraction of CLZ and LRP from the sample solution. The solvent bars were fixed with a staple pin which served as the stirrer. The target analytes were simultaneously and selectively extracted from the sample solution into their corresponding solvent bar. Extraction parameters such as organic solvent type, pH of the sample solution, the acceptor phase concentration, salt incorporation into the solution, stirring rate, and extraction time were optimized to achieve the best extraction results. Under the optimum conditions (1-undecanol as extraction solvent, pH of sample solution = 9.0, 10% w/v NaCl, concentration of HCl = 10 mM, concentration of NaOH = 100 mM, stirring rate of 1400 rpm and extraction time of 30 min at ambient temperature) the limit of detection for CLZ was 0.4 ng mL−1 and for LRP it was 1.1 ng mL−1. The linear range for CLZ was 1.3–1000.0 ng mL−1 (R2 = 0.9991) and for LRP it was 3.6–800.0 ng mL−1 (R2 = 0.9993). Extraction recovery and the enrichment factor for CLZ were 95.4% and 343 and for LRP they were 74.3% and 263, respectively. Finally, the method developed was successfully applied for the simultaneous determination of CLZ and LRP in human plasma samples.
1. Introduction
CLZ (Fig. 1) is the first “atypical antipsychotic” drug, effectively used in the treatment of both the negative and positive symptoms of schizophrenia. It causes lower extrapyramidal side effects and hyperprolactinemia as compared with traditional antipsychotics. However, it may result in severe agranulocytosis and granulocytopenia. The other side effects of CLZ are sedation, hypersalivation, orthostatic hypotension, and seizure.1 Schizophrenia patients are commonly treated with several simultaneous medications. These agents may be psychotropic drugs such as LRP (benzodiazepine (Fig. 1)) which are adjunctively employed to treat psychosis and antipsychotic-induced side-effects, or pharmacotherapies targeted at symptoms associated with concomitant mood disorders or anxiety. Adverse drug reactions experienced with concomitant administration of CLZ and LRP include delirium, ataxia, hypersalivation, sedation and slurred speech. Nevertheless, LRP should be cautiously administered (in low dosages) in combination with CLZ and administered for a short time if necessary. Therefore, their concentrations should be quantized in biological samples such as plasma.2,3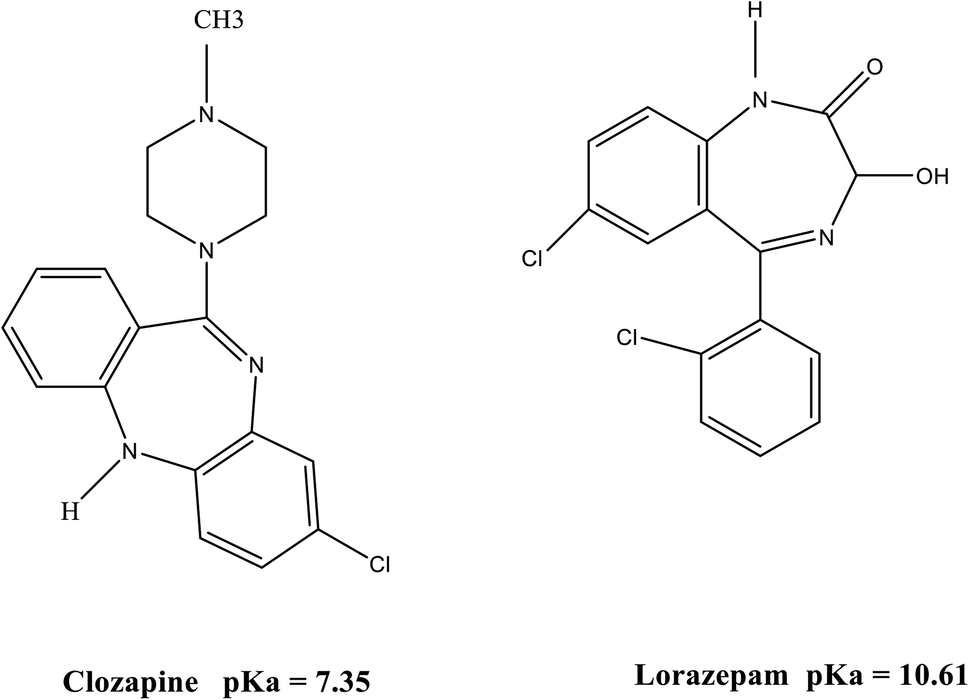 | ||
| Fig. 1 Molecular structure of CLZ and LRP. | ||
Regarding the incompatibility of biological samples with analytical instruments, the sample preparation step has become an inseparable part of almost all bioanalytical methods. Moreover, biological samples are too complex for direct injection and they are usually present at a low concentration.4
Liquid–liquid extraction (LLE) and solid phase extraction (SPE) are traditional sample preparation methods commonly used for the preconcentration of analytes in biological samples. Despite their effective and precise results, they are time-consuming and involve tedious procedures requiring large amounts of expensive and toxic organic solvents. To overcome these problems, several new microextraction techniques including solid phase microextraction (SPME),5–7 single drop microextraction (SDME),8 cloud point extraction (CPE),9 stir-bar sorptive extraction (SBSE),10 dispersive liquid–liquid microextraction (DLLME),11,12 and hollow fiber–liquid-phase microextraction (HF-LPME)13,14 have been developed. These environmentally friendly, feasible, and faster extraction techniques have been extensively used for different types of samples.
HF-LPME was first introduced by Pedersen-Bjergaard and Rasmussen in 1999 to improve the stability and reliability of LPME.15 In HF-LPME the extraction solvent is immobilized as a thin supported liquid membrane (SLM) in the HF pores protected with a porous membrane. Target analytes are extracted from the donor phase (DP) into the organic solvent inside the pores of the HF, and further into the lumen of the hollow fiber containing the acceptor phase (AP) (organic or aqueous). HF-LPME can be performed in both two-phase and three-phase modes. The two-phase mode is mainly utilized for hydrophobic compounds and involves target analyte extraction from the sample into the solvent located both inside the lumen and pores of the hollow fiber, based on the classical partition. In the other mode, the acceptor phase can be an aqueous solution, providing a three-phase extraction system which is preferable for the extraction of compounds with ionizable functionalities. HF-LPME benefits from several advantages, such as ease of operation, low cost, negligible consumption of organic solvent, no memory effect, tolerance to wide pH ranges, excellent sample clean-up, and high ratio of the sample volume to acceptor volume (high enrichment factor).16,17
Solvent bar microextraction (SBME) is a modification of HF-LPME with no use of the microsyringe.18 In this method, the organic solvent is trapped into a short piece of HF (sealed both ends) placed in a stirred DP. The mass transfer of analytes will be increased due to the random free movement of the solvent bar in the sample solution giving rise to decreased extraction time and improved extraction efficiency in comparison with HF-LPME.19
In this paper, a new design of the dual solvent stir bar microextraction technique was developed based on SBME combined with HPLC-UV for the simultaneous extraction of CLZ and LRP in human plasma with different APs. In the proposed device, two pieces of immobilized hollow fibers (solvent bars) are connected on a steel staple pine site and the HF moves along the staple pin in the DP. The target analytes are simultaneously and selectively extracted from the sample solution into their corresponding solvent bar. The proposed device is cost-effective and easy to operate. Moreover, this device is capable of self-stirring and can resolve the problem of solvent bar floatation on the sample solution. Effective parameters on the DSSBME of CLZ and LRP such as the type of organic solvent, pH of the sample solution, the concentration of acceptor phases (hydrochloric acid and sodium hydroxide), salt addition to the sample solution, stirring rate, and extraction time were optimized for the simultaneous extraction of CLZ and LRP. To the best of our knowledge, such a dual solvent-stir bar method has not been applied for the simultaneous extraction of two drugs with various acceptor phases from human plasma.
2. Experimental
2.1. Chemicals and materials
CLZ and LRP were donated by Sobhan Darou Company (Tehran, Iran). The Accurel Q3/2 polypropylene hollow fiber (0.2 μm pore size, 600 μm internal diameter and 200 μm wall thickness) was from Membrana GmbH (Wuppertal, Germany). HPLC-grade methanol, acetone, and acetonitrile, sodium chloride, hydrochloric acid, phosphoric acid, potassium dihydrogen phosphate, sodium hydroxide, 1-octanol, 5-nonanol, benzyl alcohol, 1-undecanol, and dodecane were from Merck (Darmstadt, Germany). Tetrahydrofuran (THF) was obtained from Sigma-Aldrich (Steinheim, Germany). Double distilled deionized water was used for the preparation of the mobile phase. Water filtration was done through a 0.45 μm filter (Millipore membranes, Bedford MA, USA). Stock standard solutions of CLZ and LRP (500 μg mL−1) were prepared in HPLC-grade methanol and stored at 4 °C in a dark place.2.2. Instrumentation and operating conditions
Chromatographic determination was performed using a Waters HPLC system equipped with an LC pump (model 515 Waters Assoc. Milford, MA, USA), a manual injector (model 7725i) with a 20 μL sample loop, and a UV detector (model LC-95) set at 235 nm (PerkinElmer, Norwalk, CT, USA). The column used C18 (250 × 4.6 mm, 10 μm) from Dr Ing. H. Knauer GmbH (Berlin, Germany). The applied mobile phase was a mixture of 25 mM phosphate buffer (pH 3.5)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : acetonitrile: THF (59:40:1 v/v/v) at a flow rate of 1.0 mL min−1 at ambient temperature. The pH of the solutions was measured using a pH meter (3030 Jenway Leeds, UK). A magnetic stirrer plate Heidolph (Kelheim, Germany) was used to stir the sample solutions.
: acetonitrile: THF (59:40:1 v/v/v) at a flow rate of 1.0 mL min−1 at ambient temperature. The pH of the solutions was measured using a pH meter (3030 Jenway Leeds, UK). A magnetic stirrer plate Heidolph (Kelheim, Germany) was used to stir the sample solutions.
2.3. Plasma sample
Blank plasma samples were obtained from the blood transfusion center of Babol, Mazandaran Province, Iran. For protein elimination, 4 mL acetone (HPLC-grade) was added to 1 mL of plasma sample and the mixture was vortexed for 1 min. The resulting mixture was stored at −20 °C for 10 min. The cold plasma samples were thawed at room temperature and centrifuged at 5000 rpm for 15 min. The supernatant was then decanted into another clean tube and filtered through a 0.45 μm filter. Spiked plasma samples were prepared by adding the standard solution of CLZ and LRP to drug-free dilute plasma samples for optimization and method validation.2.4. DSSBME procedure
To prepare dual solvent-stir bars, the hollow fiber was first cut into 3.5 cm segments and cleaned with HPLC grade acetone in an ultrasonic bath to remove all possible impurities followed by drying at ambient temperature. Then, 9 μL of the AP was loaded into a 100 microliter microsyringe. The needle tip was then inserted into one open end of the hollow fiber, while the other open end of the hollow fiber segment was pressure-sealed with the flat tip of conventional tweezers. This hollow fiber was immersed in the organic solvent for 30 s. It was then washed with double distilled deionized water to clean the fiber surface. In the next step, the aqueous acceptor phase (10 mM HCl solution) was injected into the lumen of the fiber. The needle of the syringe was removed from the fiber and the open end of the fiber was sealed with one side of the staple pin. The mentioned steps were repeated to fix the other segment of the HF on the other side of the staple pin, using a different acceptor phase (100 mM NaOH solution). The whole assembly was placed into 9 mL of the sample solution of CLZ and LRP and stirred at 1400 rpm with a magnetic stirrer. After 30 min of extraction, the device was taken out from the sample solution and the staple pin was removed from the HFs. The sealed sides of the HFs were cut with a scissor and the acceptor phases were withdrawn from both HF segments and collected in a microtube. Finally, the microtube containing (both drugs) was diluted with 5 μL of the mobile phase and injected into the HPLC-UV system. To avoid any possible memory effects, a fresh hollow fiber was used for each extraction. A schematic diagram of the proposed DSSBME method is shown in Fig. 2.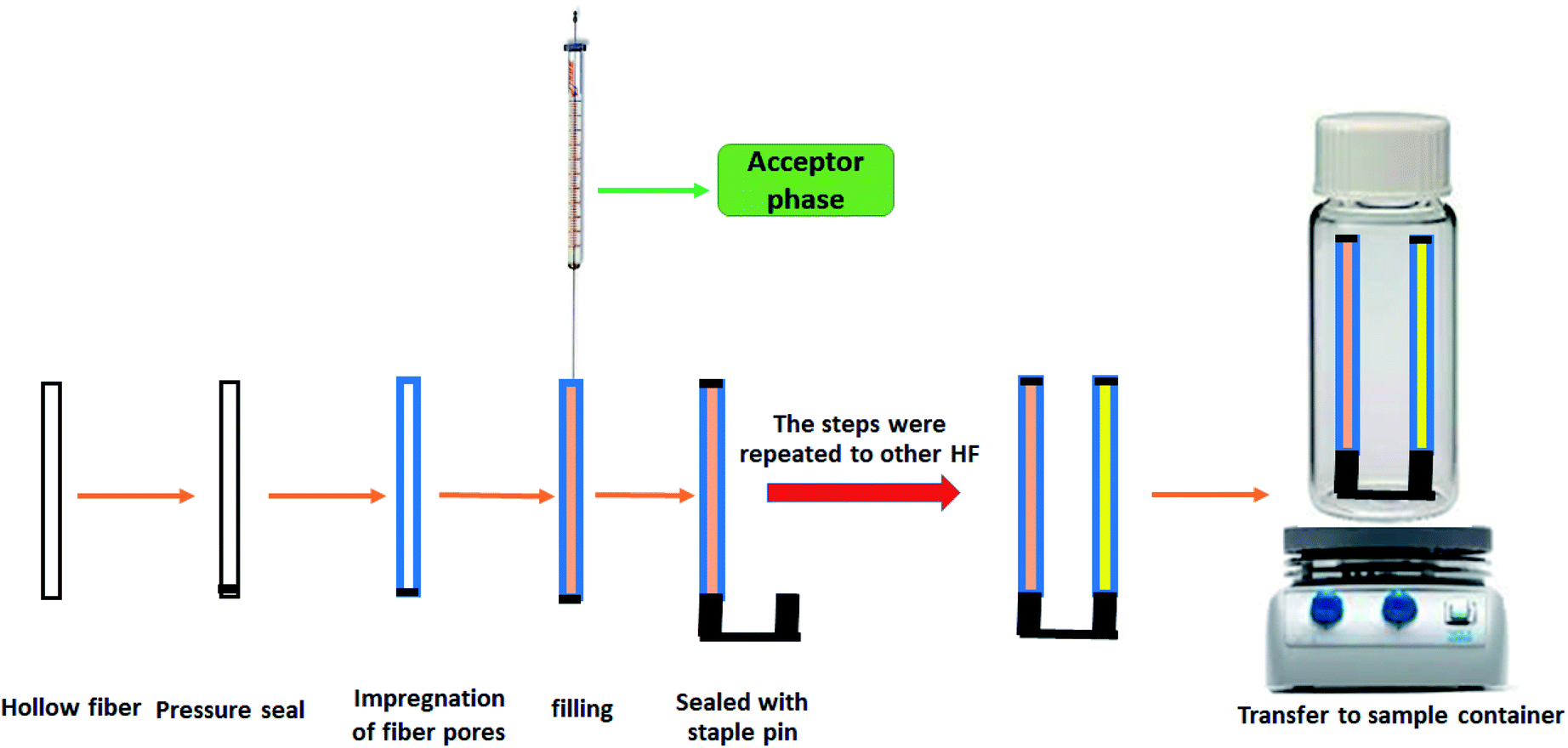 | ||
| Fig. 2 Schematic diagram of the setups of the proposed DSSBME method. | ||
3. Results and discussion
3.1. Optimization of DSSBME conditions
To achieve the highest recovery in the DSSBME procedure for the simultaneous extraction of CLZ and LRP, parameters affecting the extraction recovery including, type of organic solvent, pH in the donor phase, the concentration of acceptor phases, salt incorporation into the sample solution, stirring rate, and extraction time were optimized. The extraction recovery (ER) of the analytes was used to evaluate the extraction performance and is obtained from the following equation:20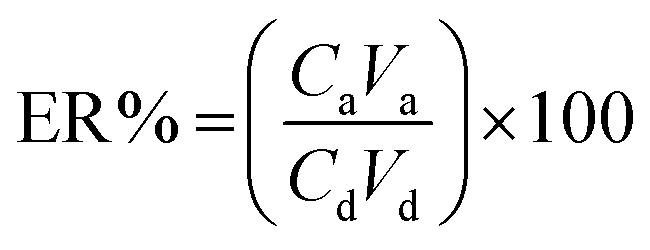 | (1) |
Another parameter in the extraction method is the enrichment factor (EF), and is defined as the ratio of the final concentration of the analyte in the AP (Ca) to its initial concentration in the DP (Cd). So, the EF and ER% could be shown as follows.20
 | (2) |
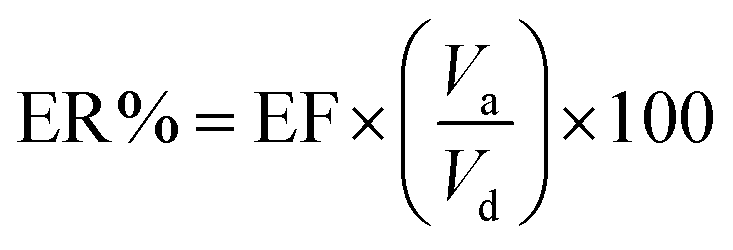 | (3) |
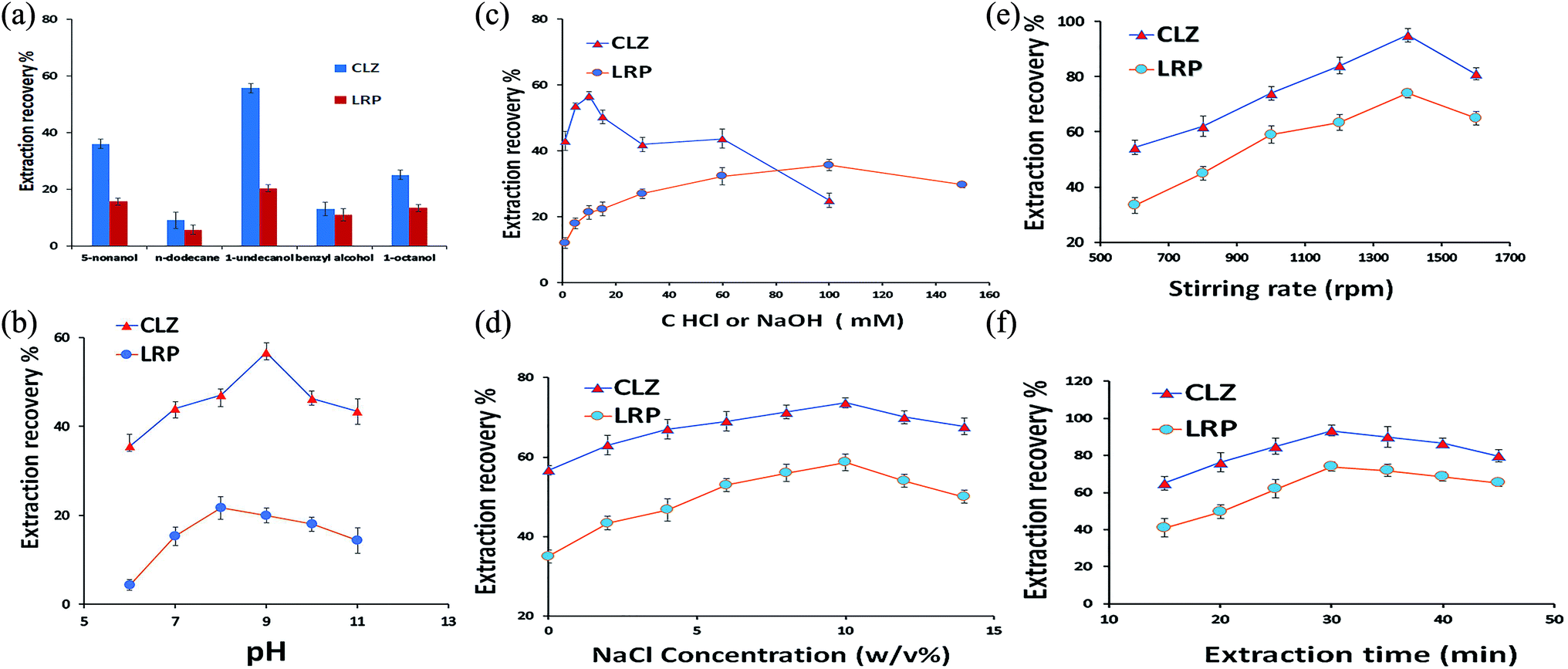 | ||
| Fig. 3 Influence of the experimental parameters including the effect of type of extraction solvents (a), effect of sample solution pH (b), effect of concentrations of HCl and NaOH as the APs (c), effect of salt addition to the sample solution (d), effect of stirring rate (e), and effect of extraction time (f), on the extraction recovery of CLZ and LRP. DSSBME conditions: organic solvent: 1-undecanol (for (b)–(f)), pH of the sample solution: 9 (for (c)–(f)), AP for CLZ = 10 mM HCl and for LRP = NaOH 100 mM (for (a), (b) and (d)–(f)), concentration of NaCl = 0% w/v (for (a)–(c), (e)–(f)), extraction time: 30 min ((a)–(e)) and stirring rate = 1000 rpm (for (a)–(d) and (f)) at room temperature. | ||
3.1.2. Effect of the sample solution pH
Another important parameter affecting the extraction recovery in the three-phase DSSBME process is the pH of the DP and AP.23 The pH of the donor phase should be adjusted to keep the analytes in the neutral form, while the pH of the acceptor phase should be altered for analyte ionization. The pH difference between the DP and AP is the driving force for mass transfer from the sample solution to the immobilized organic solvent and subsequently, to the acceptor solution in the lumen of the HF. In the present work, the sample solution consisted of CLZ and LRP with different functional groups. The pH of the sample solution should be adjusted at least 1.5 units lower than the pKa of acidic compounds and 1.5 units above the pKa of alkaline compounds to possess the highest molecular form of both drugs in the sample solution.24 The influence of sample solution pH was explored by varying pH in the range of 6.0–11.0. As shown in Fig. 3b, the maximum extraction recovery of CLZ was achieved in pH = 9. Since there are no significant differences between the ER of LRP in pH = 9 and 8, therefore pH = 9 was selected as the optimum pH for the DSSBME procedure.Since the DSSBME method is an equilibrium process, the mass transfer of target analytes is time-dependent. Accordingly, the extraction recovery is expected to improve with extraction time to establish an equilibrium state between the sample and acceptor solution. As shown in Fig. 3f, the extraction recovery for both drugs was significantly increased by raising the extraction time to 30 min, beyond which the extraction efficiency declined due to the possibility of organic solvent loss at long extraction runs.
3.2. Method validation
Analytical characteristics of the proposed DSSBME method such as extraction recovery (ER%), enrichment factor (EF), limit of detection (LOD), the linear range (LR), determination coefficient (R2), and intra- and inter-day relative standard deviation (RSD%) were determined by spiking known amounts of each drug into blank plasma samples under optimized extraction conditions. The calibration curve was obtained by spiking different concentrations of standard solutions into plasma samples diluted with double-distilled and deionized water (1:1). The calibration curves were linear in the range of 1.3–1000.0 ng mL−1 for CLZ and 3.6–800.0 ng mL−1 for LRP. The LOD is defined as 3Sb/m; where Sb is the standard deviation of the blank with 6 replications and m shows the slope of a calibration curve. The LOD values of CLZ and LRP were 0.4 and 1.1 ng mL−1, respectively. The ER and EF were calculated for the sample spiked with 100 ng mL−1 of analytes as 95.4% and 343 for CLZ and 74.3 and 263 for LRP, respectively. The repeatability and reproducibility of the proposed method were addressed by the inner- and inter-day RSD. The inner-day RSD was assessed by three determinations of analytes at two spiked levels (75 and 350 ng mL−1) during a day, while intra-day RSD was evaluated by three determinations in three subsequent days. The obtained results are summarized in Table 1.
| Compounds | LODb | LRb | R 2 | Addedb | Intra-day (n = 3) RSD% | Inter-day (n = 3) RSD% | % ER | EF |
|---|---|---|---|---|---|---|---|---|
|
a Conditions: sample volume = 9.0 mL, pH of the sample solution = 9, AP for CLZ = 10 mM HCl, AP for LRP = 100 mM NaOH, concentration of NaCl = 10% w/v, extraction time = 30 min, and stirring rate = 1400 rpm at room temperature. Mobile phase: 25 mM phosphate buffer (pH 3.5):acetonitrile:THF (59:40:1 v/v/v), column, C18 (250 × 4.6 mm, 10 μm), flow rate = 1.0 mL min−1 at room temperature, and wavelength detection at 235 nm.
b ng ml−1.
|
||||||||
| CLZ | 0.4 | 1.3–1000.0 | 0.9991 | 75 | 2.2 | 5.3 | 95.4 | 343 |
| 350 | 1.8 | 4.4 | ||||||
| LRP | 1.1 | 3.6–800.0 | 0.9993 | 75 | 2.7 | 5.8 | 74.3 | 267 |
| 350 | 2.1 | 4.6 | ||||||
The proposed DSSBME method showed high extraction recovery, appropriate LOD, and precision values for analysis of trace amounts of drugs, and thus it met the criteria for analyzing plasma samples of patients who simultaneously used the mentioned drugs.
3.3. Real sample analysis
To investigate the applicability of the DSSBME method, extraction and determination of CLZ and LZP in spiked plasma samples were carried out at various concentrations. The experimental results are presented in Table 2. Relative Recovery (RR) was determined as follows:27 | (4) |
| Compounds | Addedb | Foundb | Intra-day precision (n = 3) | RR% |
|---|---|---|---|---|
|
a Conditions: sample volume = 9.0 mL, pH of the sample solution = 9, AP for CLZ = 10 mM HCl, AP for LRP = 100 mM NaOH, concentration of NaCl = 10% w/v, extraction time = 30 min, and stirring rate = 1400 rpm at room temperature. Mobile phase: 25 mM phosphate buffer (pH 3.5):acetonitrile:THF (59:40:1 v/v/v), column, C18 (250 × 4.6 mm, 10 μm), flow rate = 1.0 mL min−1 at room temperature, and wavelength detection at 235 nm.
b ng mL−1.
|
||||
| CLZ | 30 | 28.4 | 3.1 | 94.8 |
| 80 | 77.28 | 2.3 | 96.6 | |
| LRP | 30 | 21.39 | 2.8 | 71.3 |
| 80 | 58.96 | 2.5 | 73.7 | |
Chromatograms of the drug-free and spiked plasma sample are presented in Fig. 4 for CLZ and LRP (150 ng mL−1 of each drug). The chromatogram of the spiked plasma sample showed no interference for the simultaneous determination of CLO and LZP.
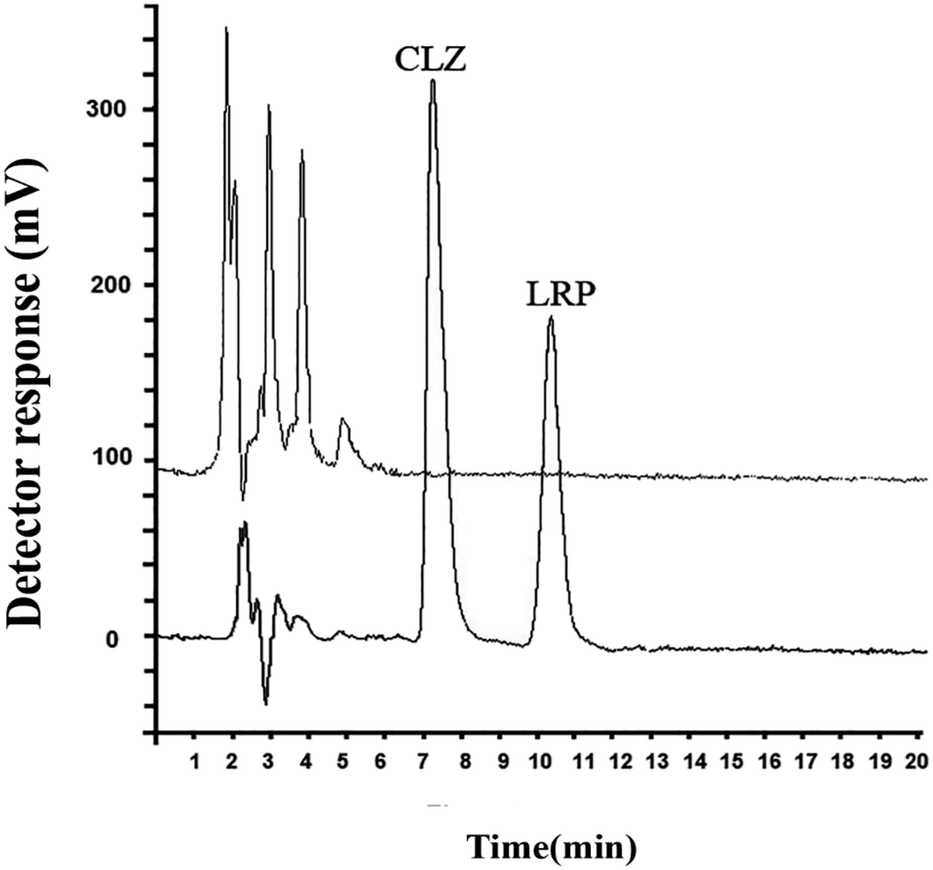 | ||
| Fig. 4 Chromatograms of the blank plasma sample (A) and spiked blank plasma sample (B) at 150 ng mL−1 of CLZ and LRP Under the optimum conditions. Conditions: sample volume = 9.0 mL, pH of the sample solution = 9.0, AP for CLZ = 10 mM HCl, AP phase for LRP = 100 mM NaOH, concentration of NaCl = 10% w/v, extraction time = 30 min, and stirring rate = 1400 rpm at room temperature. | ||
3.4. Comparative studies
To investigate the applicability of DSSBME to quantitative analysis of CLZ and LZP in the plasma sample, the performance of this method was compared with that of the conventional SBME techniques under the same extraction conditions. Diluted plasma samples (1:1) were spiked with the standard solution of drugs (150 ng mL−1) and applied for the comparative extraction. For the conventional SBME, the preparation of solvent bars was the same as that of DSSBME, except that a magnet stirrer was used instead of a staple pin and both ends of the HFs were pressure-sealed. Also, CLZ and LZP were extracted separately. The obtained results (Fig. 5) showed that the proposed DSSBME provides a high extraction recovery with higher precision in comparison with the SBME method. In the conventional SBME method, the HF membrane structure is porous and the density of the organic solvents is less than 1.0. Thus, the solvent bar will float on the DP at low stirring rates during the extraction process. In the proposed DSSBME, the HF-solvent bars are located at a fixed position on the staple pine, and as a result, the solvent bars are always submerged in the DP. Therefore, the proposed set-up improved the repeatability and extraction recovery of drugs by providing a reproducible and high contact area between the stirred sample and the solvent bars.
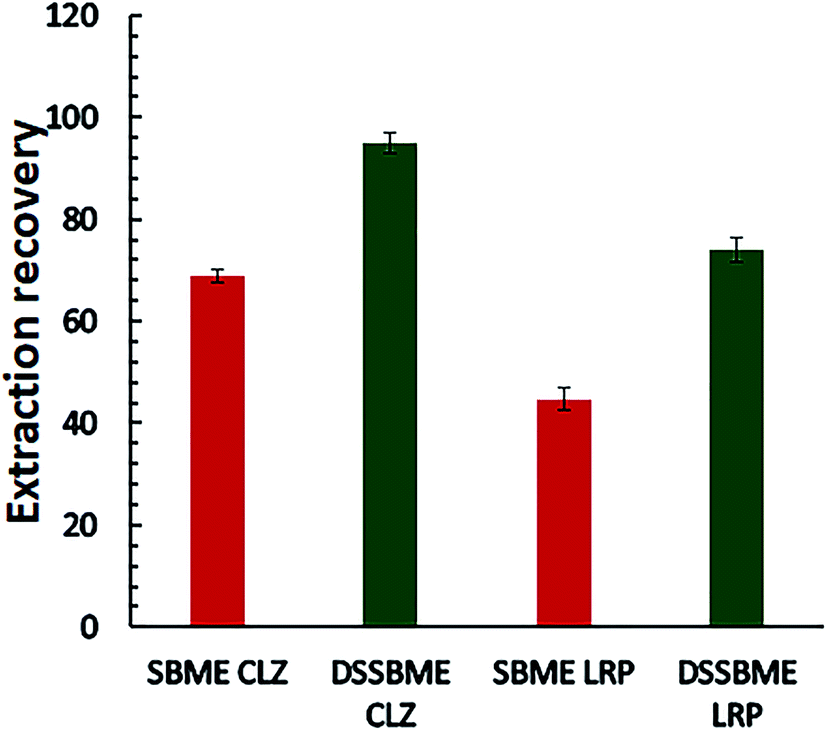 | ||
| Fig. 5 Comparison of the performance of DSSBME and conventional SBME for the extraction of CLZ and LRP in the plasma sample. Conditions of DSSBME as Fig. 4 and SBEM: stirring rate = 800 rpm and other conditions as Fig. 4. | ||
The analytical characteristics of the proposed method were compared with those of other methods used for the extraction and determination of CLZ and LRP. The results are summarized in Table 3. As can be observed, the proposed method has an adequately low LOD and good RSD with a wide linear range. Although LOD values of the microextraction in packed sorbent-gas chromatography-tandem mass spectrometry and HF-LPME-LC-MS methods are lower, these methods were used. In comparison with HPLC-UV, LC-MS and GC-MS are more sensitive and expensive. The combination of the proposed microextraction method with a more sensitive instrument can highly enhance the sensitivity of the method. Another highlighted feature of the proposed method is that it did not require volatile and chlorinated organic solvents. In general, the developed microextraction method is cost-effective and feasible for routine trace analysis of CLZ and LRP in plasma samples.
| Method | Analyte | LODa | LDRa | RSD (%) (inner-day) | RSD (%) (inter-day) | References |
|---|---|---|---|---|---|---|
| a ng mL−1. | ||||||
| In-tube SPME/LC-MS | CLZ | 2.95 | 20–500 | 4.9 | 11.4 | 28 |
| TDLLME-HPLC-UV | CLZ | 1.5 | 5–3000 | 5.1 | 6.8 | 29 |
| UA-LDS-DLLME | CLZ | — | 5–500 | 2.8 | 4 | 30 |
| DLLME-HPLC-UV | CLZ | 0.92 | 5–500 | 4.3 | 5.6 | 1 |
| SPME-HPLC-UV | CLZ | 7 | 20–2500 | 1.8 | 4.9 | 31 |
| HF-LPME-GC-MS | LRP | 15 | — | <9.7 | <11.9 | 32 |
| UA-DLLME-HPLC-PDA | LRP | 3.2 | 10–5000 | — | — | 33 |
| HF-LPME-LC-MS | LRP | 0.4 | 1.2–200 | 4.6 | 4.8 | 34 |
| SDME-HPLC-DAD | LRP | 2.1 | 10–1000 | 8.9 | — | 35 |
| DSSBME-HPLC-UV | CLZ | 0.40 | 1–1000 | 1.83 | 4.42 | This work |
| DSSBME-HPLC-UV | LRP | 1.11 | 10–800 | 2.12 | 4.66 | This work |
4. Conclusion
In this work, a new DSSBME method was introduced and successfully applied for the simultaneous determination of trace amounts of CLZ and LRP in the plasma sample with acceptable precision. This device could be easily assembled using inexpensive tools. It can be selectively applied in two-phase and three-phase modes for each solvent bar. The proposed device can be self-stirred and selectively extract each drug to its corresponding HF. Also, the problem of the floating bar was overcome. Thanks to its high stirring rate, this procedure offers high mass transfer and consequently fast extraction. The protective effect of the hollow fiber also allows extraction and preconcentration in a single step not requiring additional cleanup procedures. Since fibers are disposable no memory effect could be seen during the extraction procedure. Finally, the method proposed allows a good analytical performance, retrieving a low LOD, wide linear range, good RSD, and high extraction recovery and enrichment factor. It is therefore suitable for the analysis of plasmatic CLZ and LRP.Conflicts of interest
The authors declare no conflict of interest.References
- S. M. Majidi and M. R. Hadjmohammadi, J. Iran. Chem. Soc., 2019, 16, 2307–2314 CrossRef CAS.
- S. C. Edge, J. S. Markowitz and C. L. Devane, Hum. Psychopharmacol., 1997, 12, 5–20 CrossRef CAS.
- N. Wahid, G. Chin, A. H. Turner and A. Seegan, Mental Illness, 2017, 9, 57–58 CrossRef.
- K. D. Clark, C. Zhang and J. L. Anderson, Anal. Chem., 2016, 88, 11262–11270 CrossRef CAS.
- N. Reyes-Garcés, E. Gionfriddo, G. A. Gómez-Ríos, M. N. Alam, E. Boyacı, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2017, 90, 302–360 CrossRef.
- A. Roszkowska, N. Miękus and T. Bączek, J. Sep. Sci., 2019, 42, 285–302 CrossRef CAS.
- V. Jalili, A. Barkhordari and A. Ghiasvand, Microchem. J., 2020, 152, 104319 CrossRef CAS.
- S. Tang, T. Qi, P. D. Ansah, J. C. N. Fouemina, W. Shen, C. Basheer and H. K. Lee, TrAC, Trends Anal. Chem., 2018, 108, 306–313 CrossRef CAS.
- G. Kojro and P. Wroczyński, J. Chromatogr. Sci., 2020, 58, 151–162 CAS.
- F. David, N. Ochiai and P. Sandra, TrAC, Trends Anal. Chem., 2019, 112, 102–111 CrossRef CAS.
- S. M. Majidi and M. R. Hadjmohammadi, Biomed. Chromatogr., 2020, e4877 CAS.
- S. Dmitrienko, V. Apyari, V. Tolmacheva and M. Gorbunova, J. Anal. Chem., 2020, 75, 1237–1251 CrossRef CAS.
- Z. c. Liu, S. m. Zhang, R. q. Wang, X. Chen, S. Hu and X. h. Bai, J. Sep. Sci., 2019, 42, 2977–2984 CrossRef CAS.
- A. Gjelstad, TrAC, Trends Anal. Chem., 2019, 113, 25–31 CrossRef CAS.
- S. Pedersen-Bjergaard and K. E. Rasmussen, Anal. Chem., 1999, 71, 2650–2656 CrossRef CAS.
- S. R. Banforuzi and M. R. Hadjmohammadi, Talanta, 2017, 173, 14–21 CrossRef.
- S. R. Bandforuzi and M. R. Hadjmohammadi, J. Chromatogr. A, 2018, 1561, 39–47 CrossRef CAS.
- X. Jiang and H. K. Lee, Anal. Chem., 2004, 76, 5591–5596 CrossRef CAS.
- M. R. Hadjmohammadi, J. Chromatogr. A, 2017, 1496, 1–8 CrossRef.
- S. M. Majidi and M. R. Hadjmohammadi, J. Chromatogr. A, 2020, 461030 CrossRef.
- A. Fashi, A. A. Salarian and A. Zamani, Talanta, 2018, 182, 299–305 CrossRef CAS.
- S. Maddadi, M. Qomi and M. Rajabi, J. Liq. Chromatogr. Relat. Technol., 2017, 40, 806–812 CrossRef CAS.
- N. Y. Mlunguza, S. Ncube, P. N. Mahlambi, L. Chimuka and L. M. Madikizela, Environ. Monit. Assess., 2020, 192, 1–14 CrossRef.
- E. Carasek and J. Merib, Anal. Chim. Acta, 2015, 880, 8–25 CrossRef CAS.
- R. Venson, A.-S. Korb and G. Cooper, J. Chromatogr. B, 2019, 1108, 32–53 CrossRef CAS.
- N. N. Nia and M. R. Hadjmohammadi, Anal. Methods, 2019, 11, 5134–5141 RSC.
- S. R. Bandforuzi and M. R. Hadjmohammadi, Anal. Chim. Acta, 2019, 1078, 90–100 CrossRef.
- M.-M. Zheng, S.-T. Wang, W.-K. Hu and Y.-Q. Feng, J. Chromatogr. A, 2010, 1217, 7493–7501 CrossRef CAS.
- A. Asghari, E. Fahimi, M. Bazregar, M. Rajabi and L. Boutorabi, J. Chromatogr. B, 2017, 1052, 51–59 CrossRef CAS.
- X. Chen, S. Zheng, J. Le, Z. Qian, R. Zhang, Z. Hong and Y. Chai, J. Pharm. Biomed. Anal., 2017, 142, 19–27 CrossRef CAS.
- L. Mercolini, F. Bugamelli, E. Kenndler, G. Boncompagni, L. Franchini and M. A. Raggi, J. Chromatogr. B, 2007, 846, 273–280 CrossRef CAS.
- A. V. de Bairros, R. M. de Almeida, L. Pantaleão, T. Barcellos, S. M. e Silva and M. Yonamine, J. Chromatogr. B, 2015, 975, 24–33 CrossRef CAS.
- M. A. Farajzadeh and M. R. A. Mogaddam, Anal. Chim. Acta, 2012, 728, 31–38 CrossRef CAS.
- Z. Es'haghi, A. Nezhadali, S. Bahar, S. Bohlooli and A. Banaei, J. Chromatogr. B, 2015, 980, 55–64 CrossRef.
- K. Molaei, A. A. Asgharinezhad, H. Ebrahimzadeh, N. Shekari, N. Jalilian and Z. Dehghani, J. Sep. Sci., 2015, 38, 3905–3913 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |