
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of porphyrin glycoconjugates varying in linker length: preliminary effects on the photodynamic inactivation of Mycobacterium smegmatis†
Charlie F. Dixon‡
a,
Ana N. Nottingham‡ b,
Ana Frances Lozanoa,
J. Alexander Sizemoreb,
Logan A. Russellb,
Chelsea Valitona,
Kimberly L. Newella,
Dominique Babinb,
William T. Bridgesa,
Matthew R. Parrisa,
David V. Shchirova,
Nicole L. Snyder*b and
Joshua V. Ruppel*a
b,
Ana Frances Lozanoa,
J. Alexander Sizemoreb,
Logan A. Russellb,
Chelsea Valitona,
Kimberly L. Newella,
Dominique Babinb,
William T. Bridgesa,
Matthew R. Parrisa,
David V. Shchirova,
Nicole L. Snyder*b and
Joshua V. Ruppel*a
aUSC Upstate, 800 University Way, Spartanburg, SC, USA. E-mail: RuppelJ@uscupstate.edu
bDavidson College, Department of Chemistry, Box 7120, Davidson, NC 28035, USA. E-mail: nisnyder@davidson.edu
First published on 10th February 2021
Abstract
Porphyrins have served as common photosensitizing agents in photomedicine due to their unique properties and broad therapeutic potential. While photodynamic therapy (PDT) offers a promising avenue for novel drug development, limitations in application due to selectivity, and the inherent hydrophobicity and poor solubility of porphyrins and other organic photosensitizers has been noted. Porphyrin glycoconjugates have recently gained attention for their potential to overcome these limitations. However, little has been done to explore the effects of the linker between the carbohydrate and porphyrin analog. Here we report the synthesis of over 30 new carbohydrate–porphyrin conjugates which vary in the nature of the sugar (Gal, Glc, GalNAc, GlcNAc, Lac and Tre) and the distance between the porphyrin macrocycle and the carbohydrate. Porphyrin glycoconjugates were synthesized in three steps from a readily available meso-brominated diphenylporphyrin analog by (i) C–O coupling of an appropriate TMS-protected alkynol consisting of two to six carbon spacers (ii) removal of the TMS protecting group, and (iii) CuAAC conjugation with an appropriate glycosyl azide. First studies with trehalose-based glycoporphyrins and M. smeg were used to determine the effects of the linker in photodynamic inactivation (PDI) studies. Preliminary results demonstrated an increase in photodynamic inactivation with a decrease in linker length. Investigations are underway to determine the mechanism for these results.
Recently, porphyrins have gained traction as common photosensitizing agents in photomedicine due to their unique electronic structures, photochemical properties, and broad therapeutic potential.1 Specifically, porphyrins have found use in photodynamic therapy (PDT) for the treatment of certain cancers, dermatological disorders, microbial diseases, and other localized infections.2,3 While PDT offers a promising avenue for novel drug development in governing highly-selective methods of treatment, limitations in application due to the inherent hydrophobicity and poor solubility of porphyrins and other organic photosensitizers has been noted.4–6 To overcome solubility issues, zinc insertion has been shown to increase the lipophilicity of compounds, resulting in reduced aggregation, increased vesicle and cell uptake, more effective membrane binding and photooxidation, as well as higher specificity in target site delivery.7 Glycosylation of various porphyrin systems has also proved to be an effective strategy in expanding both water-solubility and specificity in organic photosensitizers,8 and has been shown to facilitate carbohydrate–protein interactions in cellular recognition.9,10 Additionally, the distance between the sugar and the porphyrin macrocycle, which can be modulated by linker length, is another feature with the potential to factor into the application and effectiveness of glycoporphyrins within biological systems.
Carbohydrate–protein interactions are ubiquitous in nature and play fundamental roles in diverse biological processes including inflammation, cellular adhesion, molecular recognition, catalysis, pathogenic infections, and signal transduction events.11–13 Because glycoproteins and glycolipids represent major components on the outer surface of most mammalian cells,11,13 carbohydrate–protein interactions are often involved in the initial point of contact for adhesion, recognition, attachment, and colonization for microorganisms, toxins, lectins, antibodies, and pathogens.14 Thus, the presentation and expression of glycosylated structures on cell surfaces plays an important role in promoting certain carbohydrate–protein binding events towards specific target structures in biological systems. Understanding the molecular principles that contribute to the underlying features of these crucial and widespread carbohydrate–protein interactions is an area of great interest, especially in the development of novel diagnostics and therapeutics, which often focus on targeting important biological processes involved in or mediated by carbohydrate–protein binding events.
Unlike other biological polymers such as proteins and nucleic acids, carbohydrates offer the unique potential for diverse structural architectures in natural and synthetic chemistry.13 This is important, since linker morphology and length can have a significant impact on the ability of carbohydrates to bind to specific proteins. For example, Engel and co-workers demonstrated that linker size could be tuned in order to increase the photo efficiency of liposome carriers utilized in PDT.15 Freichel and co-workers additionally reported the significance of linker diversity on glycomacromolecular systems,16 highlighting the potential impact of both linker length and morphology on carbohydrate–lectin binding affinities.17–19 Temme et al. further examined features contributing to carbohydrate–lectin binding profiles, noting that linker length as well as linker composition, in addition to differences in glycan densities, significantly contributed to variability factors across microarray binding profiles.20 Similarly, Kilcoyne and co-workers analyzed the impact of linker profiles on carbohydrate–lectin microarray binding, reporting higher yields of carbohydrate recognition among lectins of interest when using a more flexible linker.21 Interestingly, Pertici et al. presented a rigid spacer with flexible ends as an extremely potent lectin inhibitor,22 while Reynolds and co-workers described the importance of both linker rigidity and flexibility from a thermodynamic perspective in terms of maximizing binding avidity in multivalent carbohydrate–protein binding systems.23 Although a variety of factors can impact carbohydrate–protein binding interactions, these findings demonstrate both the utility and ubiquity of spacers to optimize carbohydrate-mediated binding interactions in biological systems, and highlight the vast potential for the development of novel therapeutics within rational drug design and synthetic chemistry.
As part of our on-going program to develop biologically relevant carbohydrate–porphyrin conjugates, we are interested in exploring new methods in the synthesis of various photosensitizer analogs with different linker lengths. Herein, we report the synthesis of over 30 new carbohydrate–porphyrin conjugates with different linkers and sugar moieties including glucose (Glc), galactose (Gal), N-acteylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), and the disaccharides lactose (Lac) and trehalose (Tre) to further study single carbohydrate–protein interactions.
Expanding on our previous work in which we utilized copper-promoted azide–alkyne cycloaddition to create carbohydrate conjugates of porphyrins, bacteriochlorins, and phthalocyanines,8,24,25 we sought to vary the distance between the carbohydrate moiety and the photosensitizer. We found that utilizing a Pd-catalyzed cross-coupling strategy allowed for the development of a series of alkynylated porphyrins made from a common brominated porphyrin precursor (Scheme 1).26–28 C–O cross-coupling of brominated diphenylporphyrin (1) and a series of protected alkynylated aliphatic alcohols (2a–e) followed by deprotection provided the desired alkynylated porphyrins in respectable yields for the two-step process (Scheme 1). It is worth noting that attempts to produce the alkynylated porphyrin analog from the n = 1 protected alkynylated alcohol resulted in only trace amounts of the desired product.
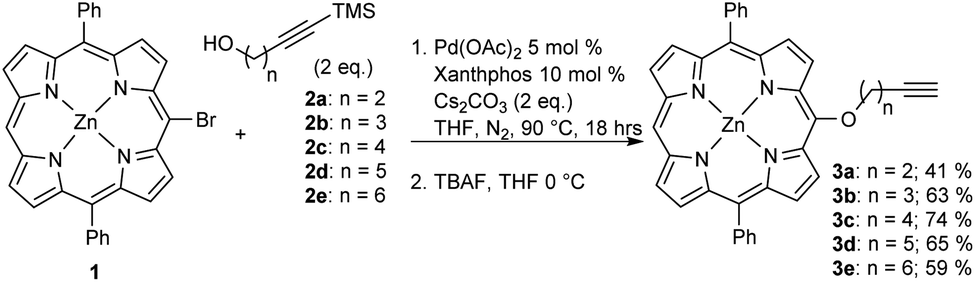 | ||
| Scheme 1 Pd-catalyzed C–O cross-coupling of 1 with a series of protected alkynylated alcohols (2a–2e). | ||
Once the series of alkyl-linked alkynylated porphyrins was obtained, our goal was to create a library of glycoconjugates using a copper-promoted azide–alkyne cycloaddition (CuAAC) protocol with a small library of azido mono- and di-saccharides (Scheme 2). In our previous work, we found that tuning the acidic or basic conditions of the CuAAC protocol was important to improve yields.24 Although our previous study seemed to indicate that the acidic conditions would prove to generate high yields, we were surprised to initially find that using the acidic CuAAC conditions led to lower than expected yields (40%). Utilizing a base-promoted variant, specifically using excess base,24 we found that yields were dramatically improved.
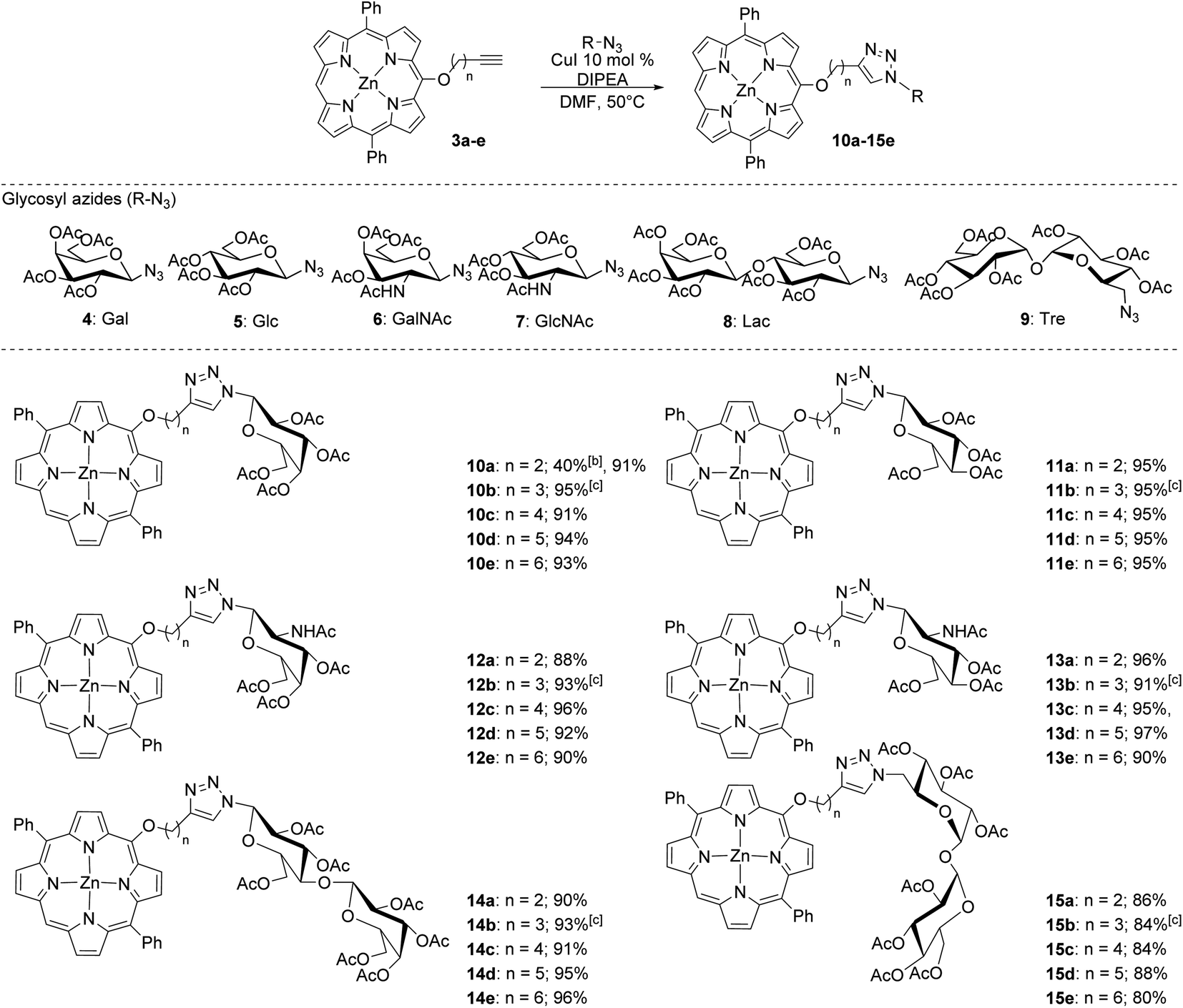 | ||
| Scheme 2 Glycoconjugation of a series of alkynylated DPP derivatives via copper-promoted azide–alkyne 1,3-dipolar cycloaddition (CuAAC).a aReactions were carried out at 50 °C for 48 h under N2 in DMF with 10 mol% of CuI and 100 eq. of DIPEA. bReaction carried out with 10 mol% of CuSO4·5H2O and 50 mol% of ascorbic acid. cAverage of 2 trials. | ||
As shown in Scheme 2, the base-promoted CuAAC glycoconjugation of the alkynylated porphyrins with a series of acetylated-azido carbohydrates produced 30 unique porphyrin glycoconjugates in high to excellent yields. The monosaccharides (4–7) utilized in this study were especially effective, providing yields of greater than 90% in most cases. Likewise, the disaccharides lactose (8) and trehalose (9) were well tolerated and produced in high yields, although the trehalose derivatives were obtained in slightly lower yields (80–88%). We found that this system was highly reproducible, finding only a slight variation in the obtained yield (±2%) across multiple trials (Scheme 2). The base-promoted CuAAC glycoconjugation strategy did not seem to be sensitive to the length of the linker (n = 2–6) and the slight variations found in isolated yields was likely due to product loss during purification.
Upon synthesis, we chose to further analyze trehalose diphenylporphyrin (DPP) derivatives in first studies to demonstrate the potential utility of such compounds within biological settings, especially in targeting localized bacterial infections with PDT. Because trehalose is an important metabolite for many types of mycobacteria, compounds containing trehalose have been used effectively for both diagnostic and therapeutic studies.8,24 Such applications rely on the nondiscriminatory enzyme, antigen 85 (Ag85A, Ag85B, Ag85C), to incorporate units of trehalose esters into the mycomembrane of mycobacteria via transesterification reactions between units of free trehalose (Tre), trehalose monomycolate (TMM), and trehalose dimycolate (TDM).24 Given the diverse substrate tolerance of Ag85, exploitation of this enzyme could present as an effective and highly selective pathway for drug delivery to treat a wide variety of mycobacterial infections. Indeed, Bertozzi and co-workers demonstrated that 4-N,N-dimethylamino-1,8-naphthalimide-trehalose, or DMN-Tre, could be metabolically incorporated into Mycobacterium tuberculosis (Mtb) and used as a solvochromic diagnostic.29 In addition, Dutta and co-workers recently synthesized a series of trehalose-based photosensitizers, including porphyrin and BODIPY-based systems; the two BODIPY dyes were able to adequately kill Mycobacterium smegmatis (M. smeg) in the μM range, while the porphyrin systems showed limited cytotoxicity.30
Since linker morphology and length have been shown to impact specific carbohydrate–protein binding interactions, we were interested in exploring the functional applications of our compounds within this context. As an example, compounds 16a–e and 17 (Fig. 1) were biologically evaluated in vitro against M. smeg in photodynamic inactivation (PDI) studies.
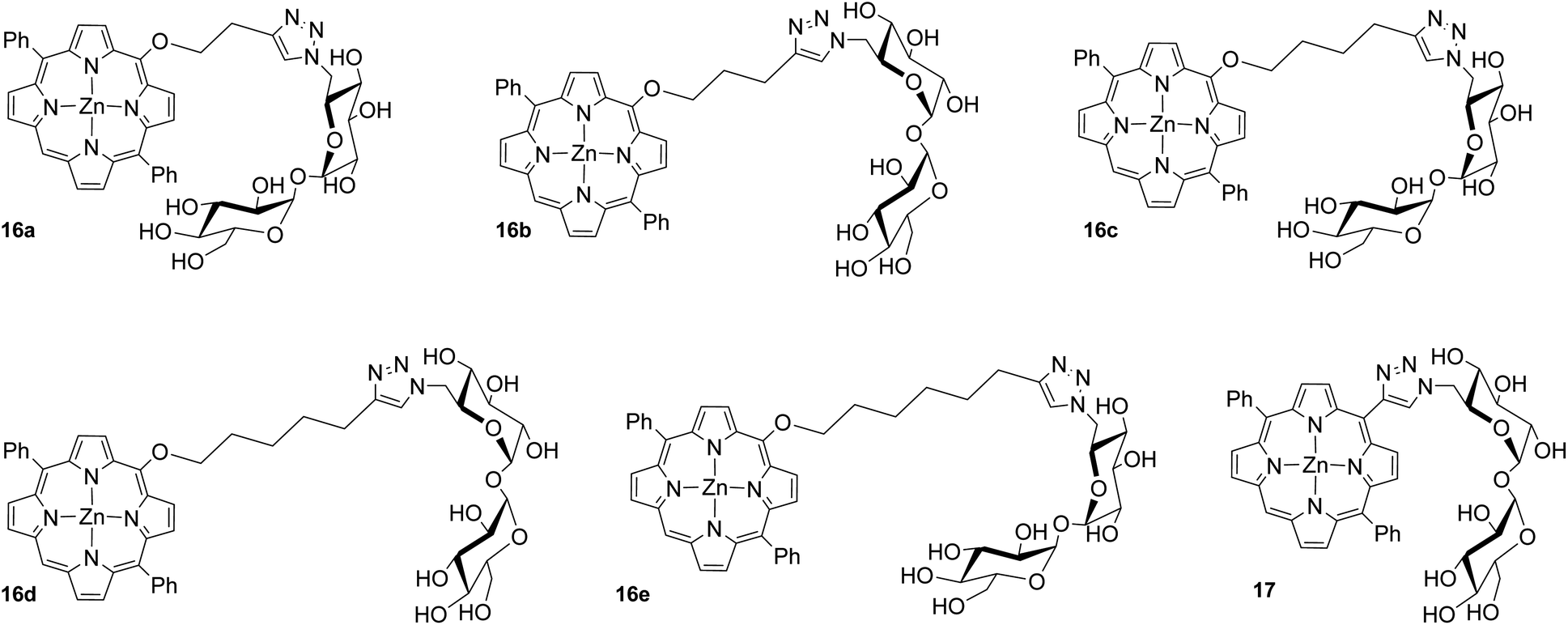 | ||
| Fig. 1 Trehalose diphenylporphyrin glycoconjugates used in preliminary cell viability studies. | ||
To investigate the relative biological activities between similar compounds with different linker lengths, a resazurin assay was performed,30 yielding both qualitative and quantitative results for analysis. As oxidation reactions occur within the metabolism of growing cells, the blue resazurin dye is reduced and converted into resorufin, a pink fluorescent dye, providing a qualitative visual indicator of relative cell viability. We first chose to examine the shortest and longest analogs, 16a and 16e, respectively, at 10 μM. This value was selected due to our previous findings using a meso analog with zero carbon spacer units (17).31 Upon irradiation, visual examination revealed that the trehalose conjugate with the two carbon linker, 16a, exhibited higher levels of cytotoxicity against M. smeg compared to 16e with the six carbon linker (Fig. 2A). To quantitatively determine the relative cytotoxic effects of 16a and 16e, the percentage of reduced resazurin was calculated from raw data, and standardized to allow for relative comparison (Fig. 2B). Compounds 16a performed similar to 17 and was superior to 16e in cell killing.
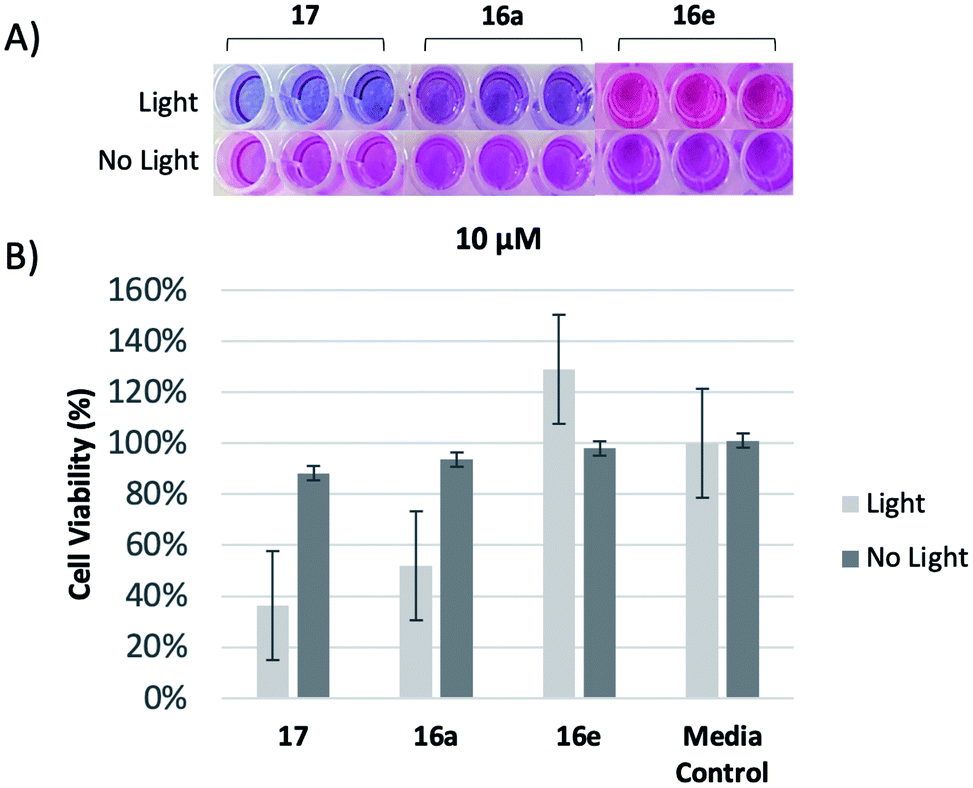 | ||
| Fig. 2 (A) Qualitative visual indicator of relative cell viability of compounds 16a, 16e, and 17 at 10 μM, performed in triplicate. (B) Cell viability of compounds 16a, 16e, and 17 against M. smeg relative to control. | ||
In order to assess the intermediate length compounds 16b–d we chose to double the concentration to 20 μM to increase cell killing. The results are shown in Fig. 3. As the length of the linker increased from three carbons, 16b, to five carbons, 16d, cell viability increased, further demonstrating conjugates with shorter linkers are more effective at killing M. smeg (Fig. 3). Notably, all conjugates (16a–e) showed limited dark toxicity. Previous research has demonstrated that it is likely these compounds have the potential to be metabolically incorporated into the mycomembrane.30 We hypothesize in this case that the longer linker most likely interferes with metabolic incorporation. Elucidation of the mode of action for cell killing is currently underway.
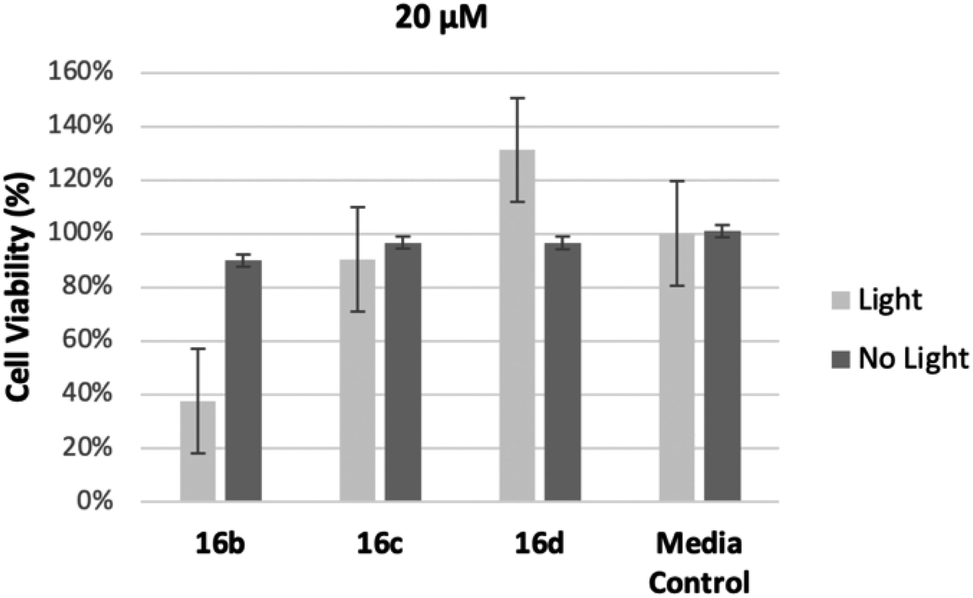 | ||
| Fig. 3 Cell viability of compounds 16b–d at 20 μM against M. smeg relative to control. | ||
Conclusion
Herein, we report on the synthesis of a series of new porphyrin glycoconjugates. These compounds, which vary in the distance between the porphyrin macrocycle and the carbohydrate, were synthesized first by C–O coupling of an appropriate TMS-protected alkynol consisting of two to six carbon spacers to a meso-brominated diphenyl porphyrin analog. After removal of the TMS protecting group, CuAAC conjugation with the appropriate glycosyl azide was performed. The final conjugates were obtained in high yields and with high purity. A series of trehalose-based porphyrin glycoconjugates were then applied in first studies against M. smeg to ascertain whether the linker length has an impact on cell killing in PDI studies. Preliminary results demonstrated an increase in cell killing with a decrease in linker length. Investigations are underway to determine the mechanism for these results.Experimental section
General considerations
All starting materials were synthesized following published literature procedures from commercially available reagents and solvents.32–36 1H NMR and 13C NMR were obtained using a Bruker Advance III 400 with Sample Xpress Lite auto sampler. UV/Vis spectra were obtained in DMSO using an Agilent Technologies Cary 8454 UV/Vis spectrometer. High-resolution mass spectra were obtained on an Agilent Technologies 6520B Accurate-Mass Q-TOF MS with a Dual ESI ion source interfaced to an Agilent Technologies 1260 Infinity II LC. MALDI measurements were made with a MassTech AP-MALDI(ng) HR ion source attached to the Agilent 6520B Q-TOF MS using a CHCA matrix.General procedure for the Pd-catalyzed C–O cross coupling of 1 (Scheme 1)
ZnBrDPP (1) (194 mg, 0.32 mmol), Pd(OAc)2 (3.6 mg, 0.016 mmol), xantphos (18.5 mg, 0.032 mmol), and Cs2CO3 (209 mg, 0.64 mmol) were added to a dry Schlenk tube and evacuated with stirring for 45 minutes. The protected alkynyl alcohol (2a–e, 0.64 mmol) and toluene (8 mL) was added under nitrogen atmosphere and the Schlenk tube was capped. The mixture was placed in an oil bath at 90 °C for 18 hours. The crude reaction mixture was concentrated under vacuum and purified using flash chromatography (silica gel, hexanes![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM, 50:50). After concentrating under vacuum, the residue was dissolved in a minimum amount of THF and allowed to cool to 0 °C in an ice bath. TBAF (1 M solution in THF, 3 molar equivalents) was added to the solution. The reaction mixture was allowed to warm to room temperature and stirred for an additional 15 minutes. Upon complete deprotection as evidenced by TLC (hexanes:DCM, 20:80), the reaction mixture was extracted with DCM, washed with water, dried over Na2SO4, filtered, and concentrated under vacuum. The reaction mixture was purified by automated chromatography (Teledyne ISCO Combiflash Rf+) using a Redisep Rf 12 gram, 16.8 mL column with a flow rate of 32 mL min−1 (hexanes/methylene chloride gradient, 100:0 to 0:100) to remove impurities. Characterization data and copies of 1H and 13C NMR spectra for compounds 3a–3e can be found in the ESI.†
:DCM, 50:50). After concentrating under vacuum, the residue was dissolved in a minimum amount of THF and allowed to cool to 0 °C in an ice bath. TBAF (1 M solution in THF, 3 molar equivalents) was added to the solution. The reaction mixture was allowed to warm to room temperature and stirred for an additional 15 minutes. Upon complete deprotection as evidenced by TLC (hexanes:DCM, 20:80), the reaction mixture was extracted with DCM, washed with water, dried over Na2SO4, filtered, and concentrated under vacuum. The reaction mixture was purified by automated chromatography (Teledyne ISCO Combiflash Rf+) using a Redisep Rf 12 gram, 16.8 mL column with a flow rate of 32 mL min−1 (hexanes/methylene chloride gradient, 100:0 to 0:100) to remove impurities. Characterization data and copies of 1H and 13C NMR spectra for compounds 3a–3e can be found in the ESI.†
General procedure for carbohydrate conjugation
Alkyne-Cn-ZnDPP (0.027 mmol), the azido-carbohydrate (0.025 mmol), and copper iodine (10 mol%) were added to a dry Schlenk tube and evacuated with stirring for 45 minutes. DMF (2.5 mL) and DIPEA (0.4 mL) were added under nitrogen atmosphere and the Schlenk tube was capped. The mixture was placed in an oil bath at 50 °C for 48 hours. The mixture was extracted with methylene chloride (3×), washed with water (3×), dried over Na2SO4, filtered, and concentrated under vacuum. The reaction mixture was purified by automated chromatography (Teledyne ISCO Combiflash Rf+) using a Rf 12 gram, 16.8 mL column with a flow rate of 32 mL min−1 (hexanes/acetone gradient, 100:0 to 0:100) to remove impurities. Characterization data and copies of 1H and 13C NMR spectra for the glycoconjugated products can be found in the ESI.†
General procedure for deacetylation of trehalose porphyrin glycoconjugates
Acetate protecting groups were removed in a 15 mL 0.25% wt NaOCH3 in MeOH solution that was left to stir for 1 hour. The reaction was monitored by TLC in 1:1 EtOAc:hexanes until full conversion was observed. The solution was subsequently neutralized with methanol-washed Amberlite™ IR120 H form ion-exchange resin, filtered, dried with Na2SO4, concentrated, and dried in vacuo to afford the deprotected product, confirmed by HRMS.
Optical density studies
Optical density studies were modeled off of a protocol published by Dutta and co-workers.30 Liquid M. smeg cell cultures were prepared by culturing freeze dried M. smeg (ATCC® 19980™) in 5 mL Middlebrook 7H9 media vials with 10% w/v TWEEN 80 growth medium. Vials were loosely capped and incubated with shaking at 37 °C for 48–72 hours. The liquid cell cultures were stored at 4 °C and resuspended by gentle vortexing before use in cell assays. Cell stock was diluted into Middlebrook 7H9 media with 10% w/v TWEEN 80 growth medium to generate cell solutions with OD600 = 0.01 at 600 nm (Varian Cary 100 Bio UV-Visible Spectrophotometer).General procedure for irradiation studies
Two identical 96-microwell plates (Thermo Scientific, Nunclon Delta Surface, Cat. No. 167008, Lot No. 137647) were prepared using stock solutions ranging from 0.44–1.2 mg mL−1 of compounds16a–e and 17 suspended in DMSO. 17 μM wells of each trehalose diphenylporphyrin compound were plated in triplicate to yield final volumes of 100 μL. Blank control wells were set up in triplicate by adding 100 μL Middlebrook 7H9 media vials with 10% w/v TWEEN 80 growth medium into each well. 100 μL 1:10 DMSO/media control wells were also set up in triplicate. An additional 50 μL of the OD600 = 0.01 M. smeg cells was subsequently added to each well on both plates, to yield final volumes of 150 μL in each well, with compound well concentrations at 10–20 μM. The plates were wrapped to block ambient light and incubated at 37 °C with shaking. After 24 hours, the “Light” plate was irradiated for 80 minutes to deliver 80 J cm−2 of 415 nm light, while the “No Light” plate was kept in the dark for the same 80 minutes. 20 μL of 0.1 mg mL−1 resazurin solution (R&D Systems, Cat. No. AR002, Lot No. 1549220) was subsequently added to each well on both plates. The plates were again wrapped and incubated at 37 °C with shaking for an additional 24 hours. Absorbance values were read on a microplate reader (BioTek Synergy H1) at 570 nm (resorufin) and 602 nm (resazurin) to yield raw data.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. V. R. and N. L. S. would like to thank the National Institutes of Health National Institute of General Medical Sciences for partial support of this work through an Academic Research Enhancement Award (1R15GM119067-01). J. V. R. would like to thank USC Upstate and the Office of Sponsored Awards and Research Support for partial funding of this work. N. L. S. would like to thank the National Science Foundation for an Major Research Instrumentation award to Davidson College (#1624377) to support this work. N. L. S. would also like to thank Davidson College for partial support through a Faculty Study and Research Grant. C. F. D. and D. S. would like to thank the University of South Carolina and the Office of Undergraduate Research for a Magellan Scholarship. A. N. N., J. A. S. and L. A. R. would like to thank the Davidson Research Initiative for support. The authors would also like to thank Morgan A. Burch, Rebecca Vance and David N. Blauch at Davidson College for their support with synthesis, biological assays and analytics, respectively.References
- M. Shakiba, J. Chen and G. Zheng, in Applications of Nanoscience in Photomedicine, ed. M. R. Hamblin and P. Avci, Chandos Publishing, Oxford, 2015. pp. 511–526 Search PubMed.
- T. Dai, Y.-Y. Huang and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2009, 6(3), 170–188 CrossRef CAS.
- S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115(18), 10261–10306 CrossRef CAS.
- E. Feese, H. S. Gracz, P. D. Boyle and R. A. Ghiladi, J. Porphyrins Phthalocyanines, 2019, 23(11n12), 1414–1439 CrossRef CAS.
- K. B. Guterres, G. G. Rossi, L. B. Menezes, M. M. Anraku de Campos and B. A. Iglesias, Tuberculosis, 2019, 117, 45–51 CrossRef CAS.
- T. G. St Denis and M. R. Hamblin, in Applications of Nanoscience in Photomedicine, ed. M. R. Hamblin and P. Avci, Chandos Publishing, Oxford, 2015, 22, pp. 465–485 Search PubMed.
- C. Pavani, A. F. Uchoa, C. S. Oliveira, Y. Iamamoto and M. S. Baptista, Photochem. Photobiol. Sci., 2009, 8(2), 233–240 RSC.
- S. A. L. Cooper, K. W. Graepel, R. C. Steffens, D. G. Dennis, G. A. Cambronero, R. Q. Wiggins, J. V. Ruppel and N. L. Snyder, J. Porphyrins Phthalocyanines, 2019, 23(07n08), 850–855 CrossRef CAS.
- X. Chen, L. Hui, D. A. Foster and C. M. Drain, Biochemistry, 2004, 43(34), 10918–10929 CrossRef CAS.
- K. Fujimoto, T. Miyata and Y. Aoyama, J. Am. Chem. Soc., 2000, 122(14), 3558–3559 CrossRef CAS.
- Y. M. Chabre and R. Roy, Adv. Carbohydr. Chem. Biochem., 2010, 63, 165–393 CrossRef CAS.
- L. L. Kiessling, T. Young, T. D. Gruber and K. H. Mortell, in Glycoscience: Chemistry and Chemical Biology, ed. B. O. Fraser-Reid, K. Tatsuta and J. Thiem, Springer, Berlin, Heidelberg, 2008, pp. 2483–2523 Search PubMed.
- R. A. Dwek, Chem. Rev., 1996, 96(2), 683–720 CrossRef CAS.
- C. A. Bewley, in Protein-Carbohydrate Interactions in Infectious Diseases, RSC Biomolecular Sciences, Royal Society of Chemistry, Cambridge, 2006, pp. 6–29 Search PubMed.
- A. Engel, S. K. Chatterjee, A. Al-Arifi and P. Nuhn, J. Pharm. Sci., 2003, 92(11), 2229–2235 CrossRef CAS.
- T. Freichel, D. Laaf, M. Hoffmann, P. B. Konietzny, V. Heine, R. Wawrzinek, C. Rademacher, N. L. Snyder, L. Elling and L. Hartmann, RSC Adv., 2019, 9(41), 23484–23497 RSC.
- M. F. Marchiori, D. E. Pires Souto, L. O. Bortot, J. Francisco Pereira, L. T. Kubota, R. D. Cummings, M. Dias-Baruffi, I. Carvalho and V. L. Campo, Bioorg. Med. Chem., 2015, 23(13), 3414–3425 CrossRef CAS.
- V. Denavit, D. Lainé, T. Tremblay, J. St-Gelais and D. Giguère, Trends Glycosci. Glycotechnol., 2018, 30(172), SE21–SE40 CrossRef.
- M. van Scherpenzeel, E. E. Moret, L. Ballell, R. M. J. Liskamp, U. J. Nilsson, H. Leffler and R. J. Pieters, ChemBioChem, 2009, 10(10), 1724–1733 CrossRef CAS.
- J. S. Temme, C. T. Campbell and J. C. Gildersleeve, Faraday Discuss., 2019, 219, 90–111 RSC.
- M. Kilcoyne, J. Q. Gerlach, M. Kane and L. Joshi, Anal. Methods, 2012, 4(9), 2721–2728 RSC.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48(33), 4008–4010 RSC.
- M. Reynolds and S. Pérez, C. R. Chim., 2011, 14(1), 74–95 CrossRef CAS.
- M. C. Bennion, M. A. Burch, D. G. Dennis, M. E. Lech, K. Neuhaus, N. L. Fendler, M. R. Parris, J. E. Cuadra, C. F. Dixon, G. T. Mukosera, D. N. Blauch, L. Hartmann, N. L. Snyder and J. V. Ruppel, Eur. J. Org. Chem., 2019, 6496–6503 CrossRef CAS.
- G. T. Mukosera, T. P. Adams, R. F. Rothbarth, H. Langat, S. Akanda, R. G. Barkley, R. Dolewski, J. V. Ruppel and N. L. Snyder, Tetrahedron Lett., 2015, 56, 73–77 CrossRef CAS.
- G.-Y. Gao, A. J. Colvin, Y. Chen and X. P. Zhang, Org. Lett., 2002, 5, 3261–3264 CrossRef.
- G.-Y. Gao, J. V. Ruppel, D. B. Allen, Y. Chen and X. P. Zhang, J. Org. Chem., 2007, 72, 9060–9066 CrossRef CAS.
- G.-Y. Gao, J. V. Ruppel, K. B. Fields, X. Xu, Y. Chen and X. P. Zhang, J. Org. Chem., 2008, 73, 4855–4858 CrossRef CAS.
- M. Kamariza, P. Shieh, C. S. Ealand, J. S. Peters, B. Chu, F. P. Rodriguez-Rivera, M. R. B. Sait, W. V. Treuren, N. Martinson, R. Kalscheuer, B. D. Kana and C. R. Bertozzi, Sci. Transl. Med., 2018, 10, eaam6310 CrossRef.
- A. K. Dutta, E. Choudhary, X. Wang, M. Záhorszka, M. Forbak, P. Lohner, H. J. Jessen, N. Agarwal, J. Korduláková and C. Jessen-Trefzer, ACS Cent. Sci., 2019, 5(4), 644–650 CrossRef CAS.
- L. A. Russel, unpublished work.
- B. Littler, M. Miller, C. H. Hung, R. Wagner, D. O'Shea, P. Boyle and J. Lindsey, J. Org. Chem., 1999, 64, 1391–1396 CrossRef CAS.
- S. DiMagno, V. Lin and M. Therien, J. Org. Chem., 1993, 58, 5983–5993 CrossRef CAS.
- F. Gou, X. Jiang, R. Fang, H. Jing and Z. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 6697–6703 CrossRef CAS.
- R. Frei, M. Wodrich, D. Hari, P. A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563–16573 CrossRef CAS.
- H. Y. Lin, J. Haegele, M. Disare, Q. Lin and Y. Aye, J. Am. Chem. Soc., 2015, 137, 6232–6244 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10793j |
| ‡ Authors made equal contributions. |
| This journal is © The Royal Society of Chemistry 2021 |