
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in glycoside and oligosaccharide synthesis
Conor J.
Crawford
 a and
Peter H.
Seeberger
*ab
a and
Peter H.
Seeberger
*ab
aDepartment of Biomolecular Systems, Max Planck Institute for Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: peter.seeberger@mpikg.mpg.de
bInstitute for Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 13th October 2023
Abstract
The structural complexity of glycans poses a serious challenge in the chemical synthesis of glycosides, oligosaccharides and glycoconjugates. Glycan complexity, determined by composition, connectivity, and configuration far exceeds what nature achieves with nucleic acids and proteins. Consequently, glycoside synthesis ranks among the most complex tasks in organic synthesis, despite involving only a simple type of bond-forming reaction. Here, we introduce the fundamental principles of glycoside bond formation and summarize recent advances in glycoside bond formation and oligosaccharide synthesis.
 Conor J. Crawford | Conor J. Crawford completed his bachelor and doctoral studies at University College Dublin, Ireland, during this time, he was a visiting scholar at the Johns Hopkins Bloomberg School of Public Health. Since 2021, he has been a Marie Skłodowska-Curie fellow at the Max Planck Institute of Colloids and Interfaces, and in 2023, was a visiting scientist at the Max Planck Institute for Marine Microbiology. |
 Peter H. Seeberger | Peter Seeberger completed his bachelor studies in chemistry at the University of Erlangen Nuremberg, Germany, and earned a PhD in biochemistry in Boulder, USA. He carried out postdoctoral research at the Sloan-Kettering Cancer Centre Research in New York. Subsequently, he held positions as an Associate Professor at MIT and a Full Professor at ETH Zürich. Since 2009, he has been a Director at the Max Planck Institute of Colloids and Interfaces in Potsdam and a Professor at Freie Universität Berlin. In 2023, he founded the Center for the Transformation of Chemistry (CTC). |
1. Introduction
Glycans are the most abundant biomolecules in nature and constitute the majority of earth's biomass. The chemical bonds that connect glycans and link them to other biomolecules are called glycosidic bonds. Over the past century, the biological importance of glycans and glycoconjugates is becoming increasingly clear, as the field of glycobiology continues to develop.1–3 The complexity of glycans, has rendered insights into the structure and function of these biomolecules more difficult to gain than for proteins and nucleic acids. Consequently, glycans have been referred to as the ‘dark matter of the biological universe’.3 The lack of template-encoded expression, homogeneous biological samples and access to defined standards have made it difficult to establish structure–function relationship on the molecular level (Fig. 1).4–11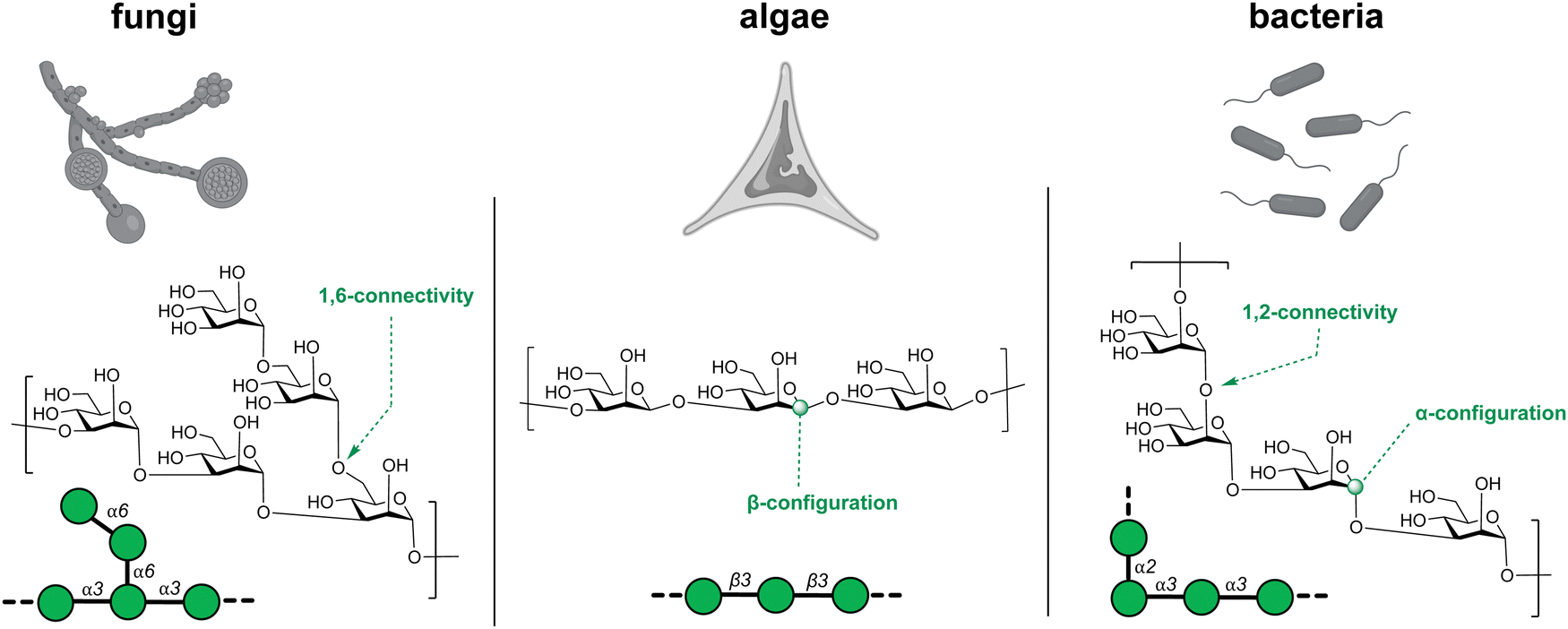 | ||
| Fig. 1 The glycan structural diversity barrier. Glycans with identical monosaccharide compositions but different connectivity and configurations perform diverse functions in nature. | ||
Pure, well-defined glycans are essential tools to advance the glycosciences as they enable molecular investigations into the interactions between glycans and proteins, such as antibodies11,12 and carbohydrate-active enzymes (CAZy);13,14 facilitate the creation of well-defined microarrays;10,15,16 allow for the examination of glycan secondary structure;17–20 and contribute to the development of vaccines21–23 and chemical biology tools.24,25 Synthetic glycans can play many other roles in environmental, material and health research.
Proteins and nucleic acids can be obtained in homogeneous form using biological methods, such as recombinant expression or the polymerase chain reaction (PCR). On the other hand, glycans produced in biological systems are inherently heterogeneous and chemical synthesis is the primary method for obtaining homogeneous glycans. Enzyme-based approaches exist and can be used in combination with chemical methods to produce well-defined glycans.26–34 Enzymatic syntheses are limited by access to well-defined enzymes, such that most enzymatic syntheses target mammalian glycans.35–44 Given the importance of chemical synthesis to procure oligosaccharides, this review will focus on the chemical methods for glycosidic bond formation.
The molecular complexity of glycans has been a challenge to generations of chemists. Glycan complexity can be described by: (i) composition, (ii) connectivity and (iii) configuration (Fig. 1).
(i) The composition of monosaccharides is frequently that of stereoisomers, e.g., glucose and mannose. They differ in the orientation of a single hydroxyl group (epimer), which complicates structural analysis and potential sequencing efforts. An important consequence of this structural similarity is that many monosaccharides are indistinguishable by classical mass spectrometry.45–51
(ii) Connectivity describes how monosaccharides are connected to each other. Each monosaccharide contains multiple hydroxyl groups, each of which can serve as a point of attachment to others glycosides, including branches, as well as other non-glycosidic groups such as sulfates or acetates. In the context of chemical synthesis, this means that chemists often use protecting groups to assemble complex glycosides and oligosaccharides. Protecting group manipulations frequently represent the majority of the chemical steps.
(iii) Configuration describes the information embedded in the glycosidic linkage that is essential for the function of a given glycan. For instance, cellulose and amylose are both polymers connected polymers by 1–4 glucose linkages, yet they serve completely different functions in nature. Cellulose is β-linked (1,2-trans) and provides strength to plant cell walls,52 while amylose is α-linked (1,2-cis) and functions as an energy storage molecule in plants.53 As chemists connect glycans, they must control the configuration of this stereogenic centre (Fig. 2).
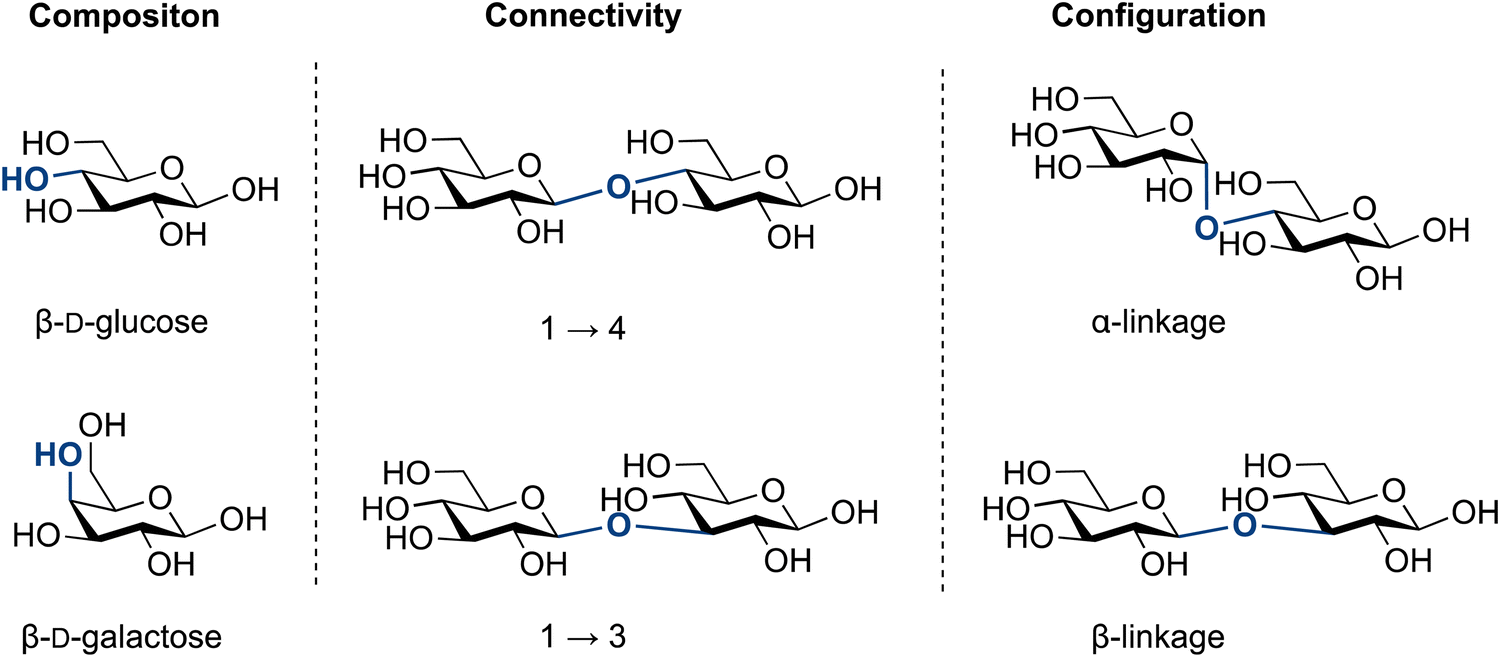 | ||
| Fig. 2 Levels of complexity in glycans: composition, connectivity and configuration. | ||
Collectively, composition, connectivity and configuration combinatorially multiply the molecular diversity of glycans by orders of magnitude beyond what nature achieves with nucleic acids and proteins. This complexity renders the synthesis of glycosides as one the major challenges in organic chemistry.54–60
Below, the following topics will be discussed: (i) general mechanistic aspects of the glycosylation reaction, (ii) variables in the glycosylation reaction, (iii) robust methods for the synthesis of glycosides, and (iv) recent examples highlighting advances in the synthesis of complex glycosides and oligosaccharides.
2. Glycan composition
Glycans possess diverse chemical architectures, exhibiting molecular complexity and diversity beyond other biomolecules.61 Particularly, microbial, plant and algal glycans, utilize many different monosaccharides as building blocks.62–64 Bacteria employ hundreds of monosaccharides including heptose, 3-deoxy-D-manno-2-octulosonic acid (Kdo), legionaminic acid, bacillosamine and fucoseamine (Fig. 3). Currently, these monosaccharide building blocks are not commercially available such as D-glucose or D-mannose. Therefore, chemical and enzymatic processes are being developed to provide better access to these monosaccharides.65–73 | ||
| Fig. 3 Examples of rare monosaccharides. Abbreviations: 2-keto-3-deoxy-D-glycero-D-galacto-nononic acid (Kdn) and 3-deoxy-D-manno-2-octulosonic acid (Kdo). | ||
The synthesis of ‘rare’ sugars can be accomplished via chemical and enzymatic approaches. Chemical methods encompass epimerization,74 deoxygenation,75 and chain elongation,76 which have been employed in the chemical synthesis of a variety of glycans. These include N-acetylgalactosamine,77,78 6-deoxy-L-gulose,79 di-N-acetyl D-bacillosamine,80–82N-acetyl L-pneumosamine,83 Kdo,72,76,84 2-keto-3-deoxy-D-glycero-D-galactononulosonic acid (Kdn),85–87 legionaminic acid88,89 and L-fucosamine (Fig. 3).80,90
Epimerisation of biomass-derived carbohydrates can produce rare sugar isomers using either chemical or enzymatic methods.91,92 Photochemical conditions to site-selectively epimerize a variety of minimally protected and non-protected carbohydrates (Scheme 1) employed catalytic quantities of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4-CzIPN), quinuclidine, adamantane thiol and tetrabutylammonium p-chlorobenzoate in acetonitrile and DMSO at room temperature under blue light irradiation. The kinetic products of epimerization are formed through two sequential steps: (i) hydrogen-atom abstraction and (ii) hydrogen-atom donation. Thereby, several valuable monosaccharide building blocks were prepared on gram scale as demonstrated for the conversion of glucose to methy α-D-allopyranoside (Scheme 1).
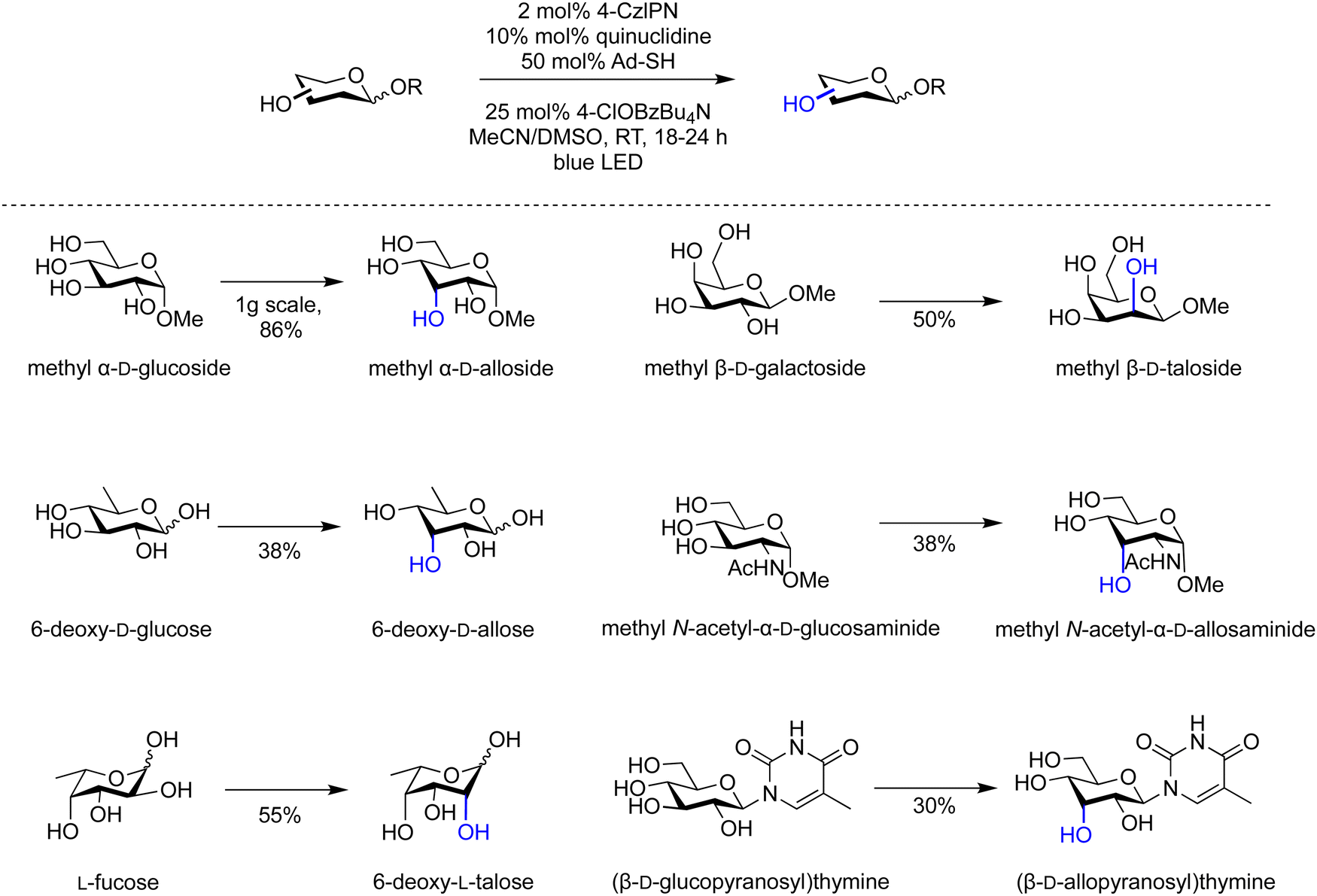 | ||
| Scheme 1 Site-selective epimerisation of glycans.91 | ||
Improved synthetic routes to rare monosaccharides provided sufficient material to establish their inherent chemical properties as glycosylating agents and nucleophiles.71,88 These insights were applied in the synthesis of complex glycosides that incorporate these rare monosaccharides to study their structure and function.66,93–96
3. Glycan connectivity
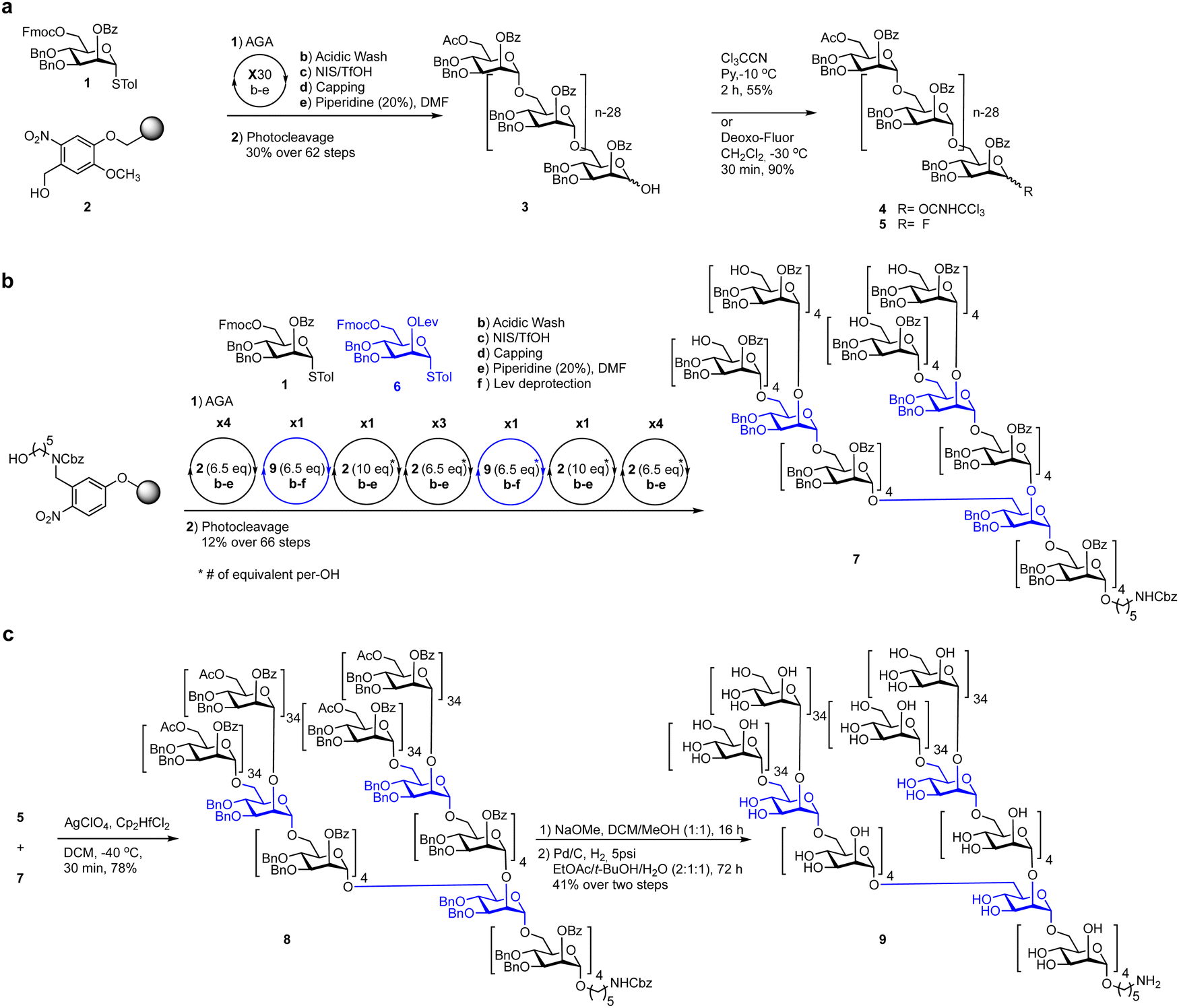
A challenge in glycan synthesis is the necessity to selectively modify one specific hydroxyl group at a time in the presence of many others. Therefore, chemical glycan synthesis demands meticulous planning to achieve the desired protecting group pattern on both mono- and oligosaccharides. These protecting groups serve to ‘mask’ the inherent reactivity of a hydroxyl group. These chemical modifications enable the precise assembly of molecules in a controlled manner.58,97 Ideally, hydroxyl-protecting groups are selectively added and removed from a glycan to manipulate the exposed hydroxyl group. Particularly valuable are protecting groups and methods that permit the selective modification of carbohydrate polyols such as benzylidene acetals that can be regioselectively opened under reductive conditions.98–102 Some one-pot protocols allow for the preparation of advanced synthetic intermediates.103–105 Dibutyltin oxide and diarylborinic acid catalysts enable regioselective acylation or alkylation in the presence of multiple hydroxyl groups.106–114 In a typical synthetic scheme, the selectively exposed hydroxyl group serves as a nucleophile for the regioselective addition of another saccharide unit.59 Glycosides with amine groups are found in animals, plants and microbes115–119 and a host of nitrogen protecting groups have been developed (Fig. 4).120 Selecting the best nitrogen protecting group for a particular target involves often trial and error experimentation (see Section 6.4, Scheme 3).115,117,121–124 | ||
| Fig. 4 Examples of common protecting groups masking hydroxyl and nitrogen atoms. | ||
Similarly, the choice of hydroxyl protecting groups impacts the synthesis and the order of manipulation is essential for a successful synthesis.125–130 Permanent protecting groups,131,132 such as benzyl ethers (Fig. 4) stay in place throughout the synthesis and are removed after assembling the target structure. Temporary protecting groups,97,132,133 such as chloroacetyl esters,134,135 are selectively removed during the synthesis reveal the desired hydroxyl group for selective functionalization (Fig. 4).
4. Glycoside configuration
A glycosidic linkage is formed through the activation of a glycosylating agent (donor) to create a reactive electrophilic species that couples with the nucleophile (glycosyl acceptor) (Fig. 5). This coupling reaction results in the formation of α- or β-stereoisomers. The stereospecific formation of glycosidic bonds is a major synthetic challenge in glycan synthesis. A variety of methods are available to generate stereospecific glycosidic linkages whereby the yield and the stereochemical outcome of these reactions depend on steric and electronic effects of both glycosylating agent and nucleophile.136–138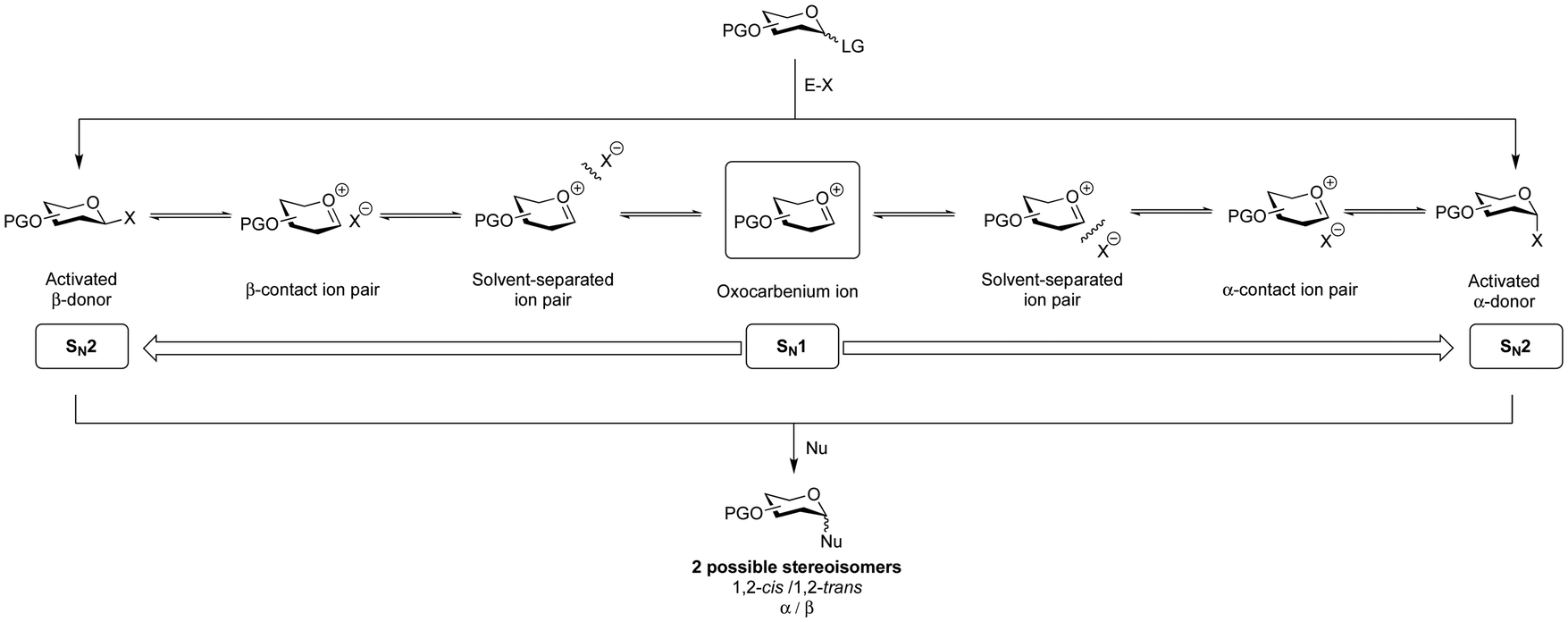 | ||
| Fig. 5 Possible reaction mechanisms of glycosylation reactions. Glycosylation reactions can take place on a spectrum ranging from SN1 to SN2 mechanisms. PG, protecting group; E-X, promoter system; and Nu, nucleophile. | ||
1,2-trans stereochemistry at the anomeric centre can be ensured by the use of participating protecting groups, such as an ester, on the 2-hydroxyl group. Neighbouring protecting group participation,139–142 in the glycosylation reaction means that ester protecting groups form a cyclic oxonium ion intermediate, that shields one face of the glycosylating agent intermediate from reacting with the nucleophile that approaches from the opposite face and results in the formation of the 1,2-trans glycosidic linkage (Section 6.5, Fig. 9a).
The construction of 1,2-cis glycosides is more difficult126,143–148 as a general method has yet to be established. Often mixtures of anomers are formed and separated by column chromatography. Recent developments in this area are discussed in Section 6.
5. Glycosylation reaction mechanism
An improved understanding of the glycosylation reaction mechanism informs the design and execution of more efficient protocols for the selective synthesis of glycosides. Much effort has been directed towards understanding this complex chemical event.149–152Different reaction pathways, on a spectrum from SN2 to SN1,153,154 are possible for glycoside formation, resulting in distinct diastereomeric products (α/β) (Fig. 5). The first step involves activating a glycosylating agent, which leads to the formation of an array of electrophilic intermediates, including contact ion-pairs that contain the activator-derived counterion (X) (Fig. 5).
In the absence of a participating group on C2, the reaction mechanism is more complex. Covalent reactive intermediates and the reactive oxocarbenium ion species are responsible for product formation. Low-temperature NMR techniques have detected covalent intermediates on the SN2 end of the reaction mechanism spectrum, including reactive species such as glycosyl triflates and imidates.151,155 Subsequent substitution of these reactive species with nucleophiles defines the SN2 side of the reaction mechanism spectrum.
In contrast, the SN1 side of the spectrum is less well understood and is a focus of current research. The substitution pattern on the glycosylating agent impacts the stability and reactivity of the various reactive intermediates. Efforts to establish structure–reactivity–stereoselectivity relationships of these SN1 species have been reported.154
6. Variables in glycoside bond formation
The stability and reactivity of glycosylating agents, and the factors that control their diastereoselective substitution with nucleophiles, are affected by a multitude of variables (Fig. 6). Predicting the outcome of a particular glycosylation reaction in a complex oligosaccharide is difficult. The order of assembly, protecting group patterns, temperatures and solvents all contribute the success of glycoside formation.138,149,156 This inherent complexity often requires experimental trial and error to identify a viable synthetic route. | ||
| Fig. 6 Variables in glycoside synthesis. | ||
For less complex substrates the following levels of difficulty are broadly accepted. The formation of α-manno-type structures is favoured by the anomeric effect, and neighbouring group participation can be enlisted (Fig. 7).157–159 β-Glucosides are readily accessible when neighbouring group participation is possible. However, the formation of α-glucosides structures depends strongly on thermodynamic control or SN2-type reactions without neighbouring group participation. The synthesis of β-mannosides does not benefit from either the thermodynamic anomeric effect or neighbouring group participation but relies strictly dependent on an SN2-type reaction, which starts with the readily accessible α-mannosylating agents. β-Mannosides pose one of the greatest challenges in glycoside to preparation.160
 | ||
| Fig. 7 Scale of difficulty in glycoside synthesis. | ||
6.1. Influence of the promoter on activation of glycosylating agent
The choice of promoter significantly influences the outcome of glycosylation reactions. Often, iterative testing is required to identify the most suitable promoter system for a specific glycosylation as different types of anomeric leaving groups are explored. The possibility to interconvert different types of glycosylating agents is important.Thioglycosides are very stable, can be converted into most commonly used glycosylating agents and are stable to many protecting group manipulations (Fig. 8).161
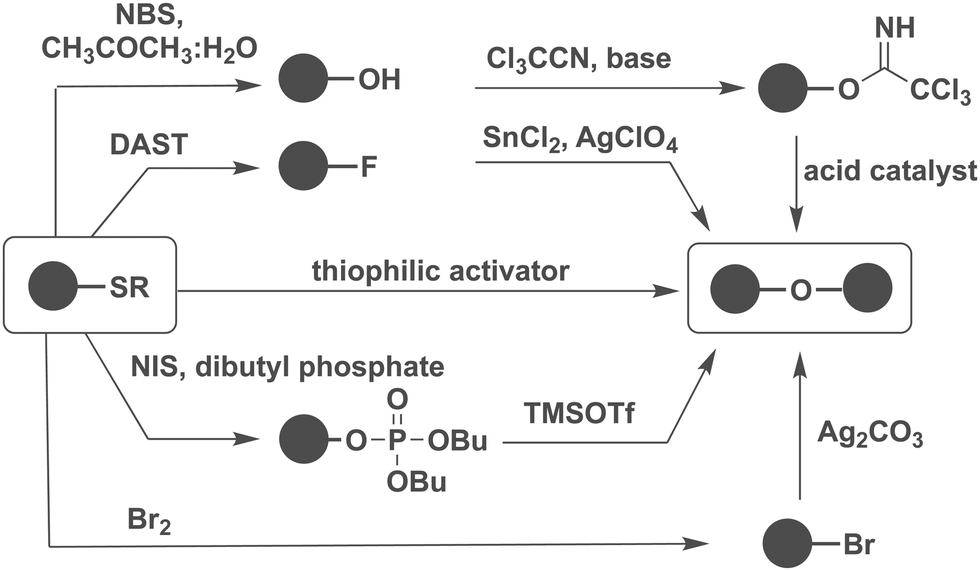 | ||
| Fig. 8 Conversion of thioglycosides into other types of glycosylating agents. | ||
6.2. Solvent
The choice of solvent has a major impact on glycosylation reactions, as it affects the abundance of the various covalent and ion pair species present during the reaction. Solvents with poor electron-donating properties, such as dichloromethane and toluene, are commonly employed in glycoside synthesis because they promote SN2-type reactions. Solvents capable of donating electron lone pairs, such as ethers (e.g. diethyl ether, dioxane) and acetonitrile, lead to distinct changes in the reaction course due to their varying roles in stabilizing cationic reaction intermediates (Table 1).Ethereal solvents have a tendency to drive glycosylations in an α-selective fashion, while nitrile solvents give higher ratios of β-products.167–170 These results are rationalized as follows: ethereal solvents, such as diethyl ether or dioxane lead to higher levels of equatorial intermediates,144,167,169–176 resulting in an SN2 type mechanism giving rise to higher levels of α-product. Solvents with higher dielectric constants often lead to lower diastereocontrol in glycoside synthesis. These higher polarity solvents are thought to better stabilize ion pairs and individual ions to a greater extent, thus promoting dissociative mechanisms and the resulting lower stereoselectivity (Fig. 5).
Solvent additives can alter the course and outcome of glycosylations. DMF as solvent or co-solvent facilitates the formation of 1,2-cis glycosides,155 similar to the selectivity of halide ion catalysed glycosylation reactions.177,178 Glycosylations in the presence of DMF produce the axial glycosyl imidate, as observed by NMR. The reaction may proceed via a minor but more reactive equatorial imidate, leading to high selectivity (Scheme 2).155
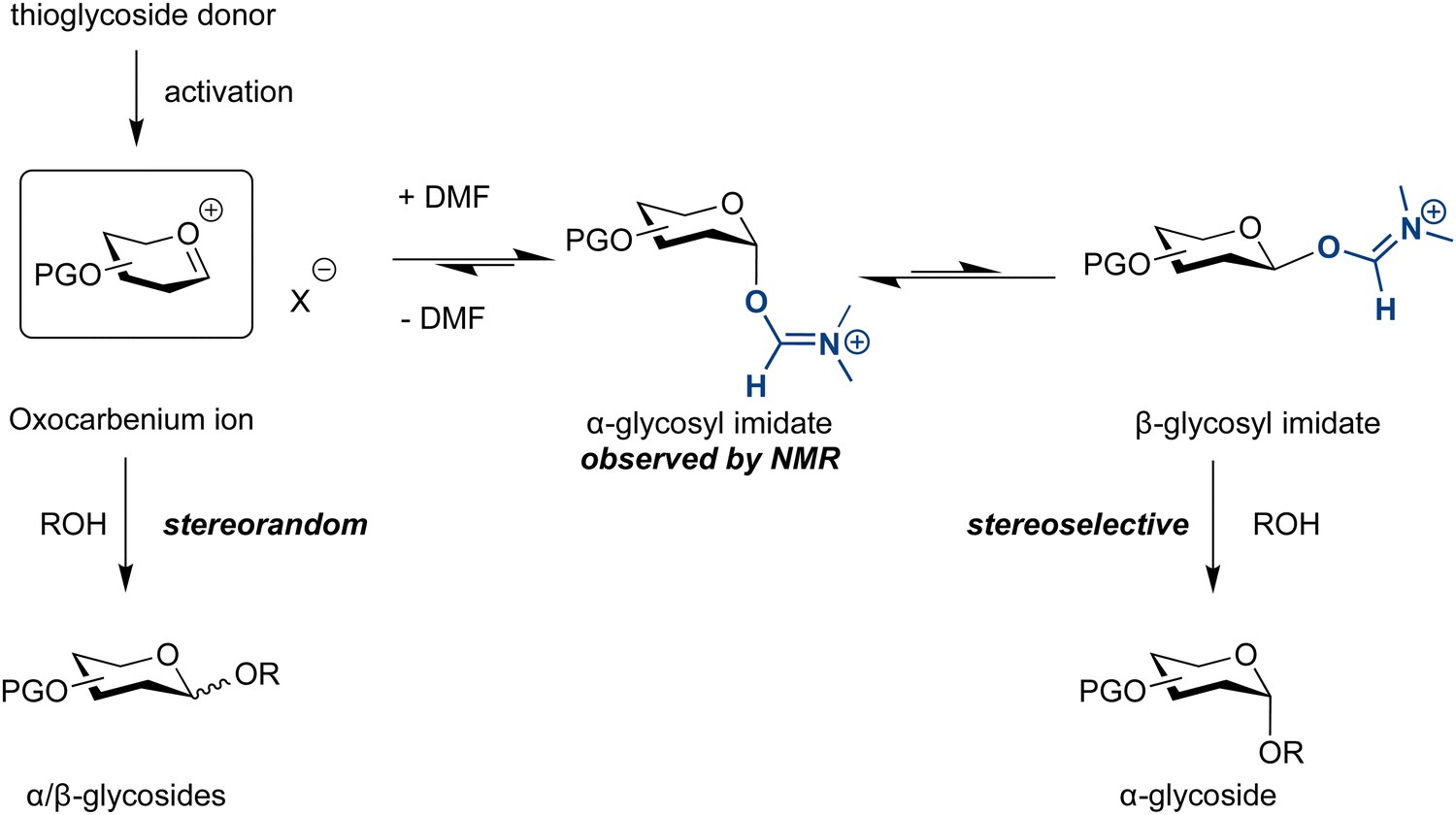 | ||
| Scheme 2 Proposed mechanism of the DMF-modulated glycosylation.155 | ||
Glycosylations in acetonitrile produce the axial nitrilium cation in situ, that in turn results in higher stereoselectivity in the synthesis of equatorially substituted α-sialosides or β-glucosides.162 Thereby, 1,2-trans glucosides are formed even with glycosylating agents bearing a non-participating protecting group.
6.3. Temperature
Many glycosylating agents are very temperature sensitive and reaction temperatures at or above room temperature are uncommon. However, exceptions do exist e.g., synthesis of Shigella flexneri oligosaccharides, (Section 7.4, Scheme 12), where temperatures of 75 °C were required for effective glycosylations.In addition, low reaction temperatures facilitate stereoelectronic control via SN2-type reactions. Systematic studies of glycosylations reactions in flow reactors have identified temperature as the strongest influence on α/β selectivity.149 However, solubility and reactivity are significantly affected by the reaction temperature selection. Classically, solution-phase glycosylations are initiated at low temperature, to lower the chance of unwanted side-reactions and allowed to warm slowly to ambient temperatures, in hopes of coupling with minimal side-product formation.
A more systematic temperature selection for glycosylations aims at quantifying the differing thermal stability of thioglycosides to guide temperature selection. To date, this approach does not account for differing acceptor nucleophilicity150 but this shortcoming is being addressed right now. This approach is now being united with the relative reactivity values (RRV) to create a path forward towards models for chemists to more effectively synthesize glycosides.179–183
Quantifying the reactivity of different thioglycosides will help to rationalise chemoselective one-pot protocols varying the of thioglycoside aglycon with different electron withdrawing groups.184–188 Thioglycoside aglycon modifications enable reactivity tuning of building block reactivity without the need to alter protecting group patterns.184 An orthogonal approach is discussed in Section 6.5.189,190 The utility of quantifying donor temperature stability was demonstrated in the context of an automated assembly of a β-1–4 glucan tetrasaccharide, where the crude HPLC trace improved while the amount of building block used was reduced.150
6.4. Influence of protecting groups on the nucleophile
The reactivity of the nucleophile can vary widely due to a combination of steric, stereoelectronic and inductive effects.137,138,190–197 The nature of the glycosyl acceptor can influence the outcome of a glycosylation reaction, both in terms of isolated yield and stereoselectivity.198 When all groups are equatorial, as is the case of a glucose acceptor, in the 1C4-conformation, the following reactivity order is observed: 6-OH > 3-OH > 2-OH > 4-OH. These considerations can be used to estimate and tune the reactivity of a given carbohydrate alcohol: (i) axial alcohols are less reactive than equatorial alcohols. This observation has been applied in regioselective saccharide syntheses with partially O-protected glycosyl acceptors (Scheme 14).16,115,199 (ii) Glycosyl acceptors with a neighbouring alcohol in an equatorial position, are more reactive than acceptors where one of the adjoining alcohol is axial, e.g. the 3-OH of mannose vs. glucose acceptor (iii) ester protecting (electron withdrawing group) groups beside the acceptor decrease the reactivity of the acceptor in comparison to electron donating groups, e.g. benzyl ethers (iv) the nucleophilicity of the acceptor affects glycosylation stereoselectivity, with reactive nucleophiles being β-selective and weaker nucleophiles having higher α-selectivity.136,200–202 Robust structure–reactivity relationships for nucleophiles are currently being established,198,203 in order to prevent poor stereoselectivities and yields as a result of a lack of insight into acceptor reactivity.204–217The C4 hydroxyl of glucosamine is well-studied and generally considered to be a poor nucleophile.218–220 Different nitrogen protecting groups and the bulk of neighbouring hydroxyl protecting groups have been shown to affect the 4-OH acceptor nucleophilicity. It is hypothesised that intermolecular hydrogen-bonding networks involving the N-acetyl group play a key role. The N-acetyl-2N,3O-oxazolidinone-protected system renders the alcohol more reactive as it apparently reduces the steric hindrance around the C4 hydroxyl group.123,221 This linkage posed a challenge in the automated glycan assembly (AGA) of chitin and chitosan oligosaccharides (Scheme 3).222 Glycosyl phosphates were found to be more effective than thioglycosides with final yields of 34% vs. 8%, respectively. The choice of nitrogen protecting group influences the synthesis, with incorporation of any N-benzyloxycarbonyl (Cbz) bearing glycosides anywhere in the growing oligosaccharide chain decreasing the overall yield when compared to oligosaccharides containing exclusively N-trichloroacetyl protecting groups (Scheme 3).
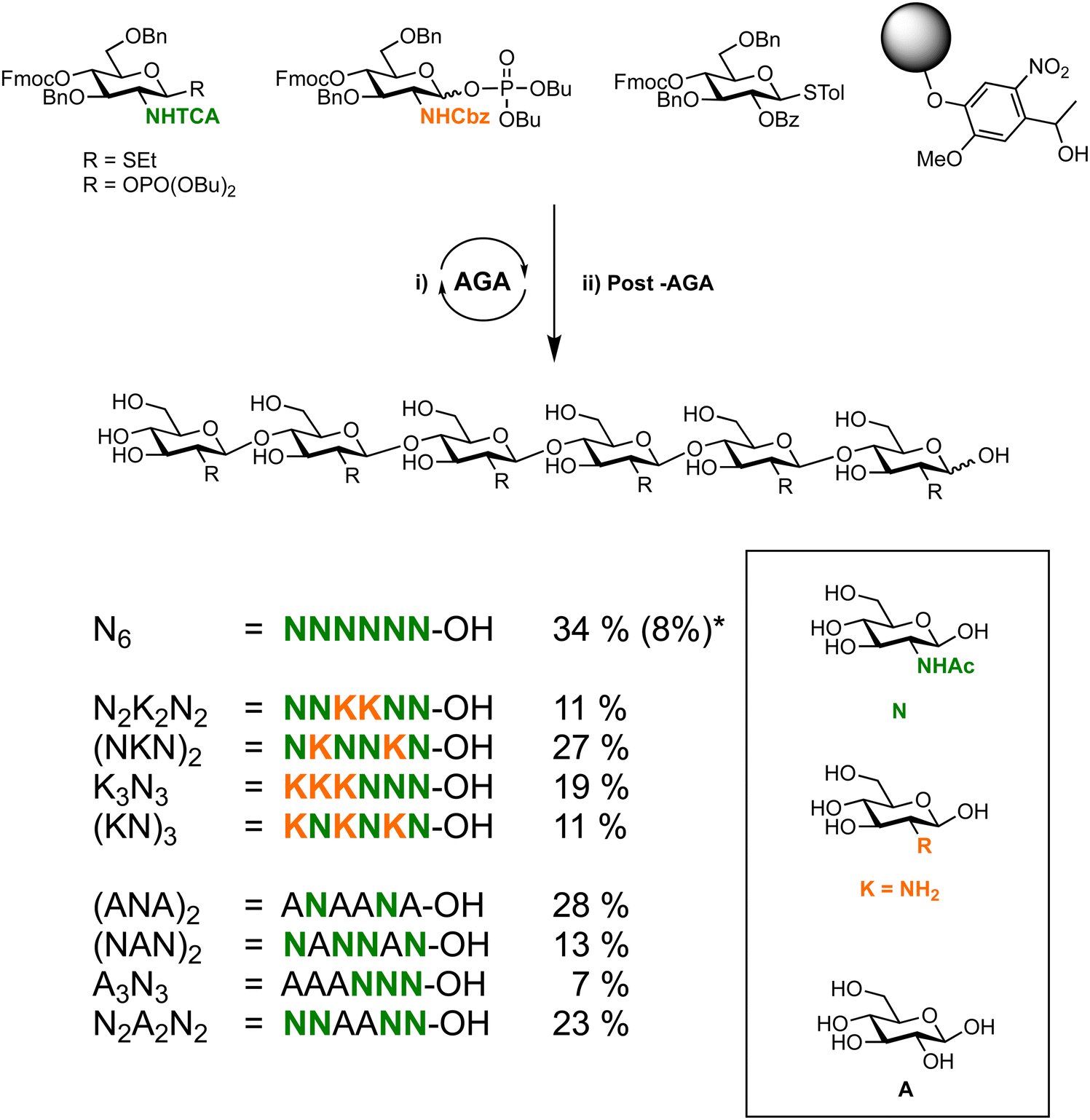 | ||
Scheme 3 Automated glycan assembly of a library of chitin oligosaccharides.222 *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yield obtained when synthesis was complete using BB1a. Yield obtained when synthesis was complete using BB1a. | ||
The role of the protecting group pattern on acceptor nucleophilicity can be observed in the synthesis of the trisaccharide repeating unit of Pseudomonas aeruginosa O11 antigen (Scheme 4).126 Coupling disaccharide acceptors revealed that results depended upon the protecting group on the C3 hydroxyl group of the fucosamine nucleophile. Acetate, allyl and 2-naphthylmethyl ether protecting groups were compared with the 2-naphthylmethyl ether protecting group performing best. Lowering the temperature from 0 °C (25% yield) and to −60 °C to −10 °C drastically improved the yield to 84% (Scheme 4).
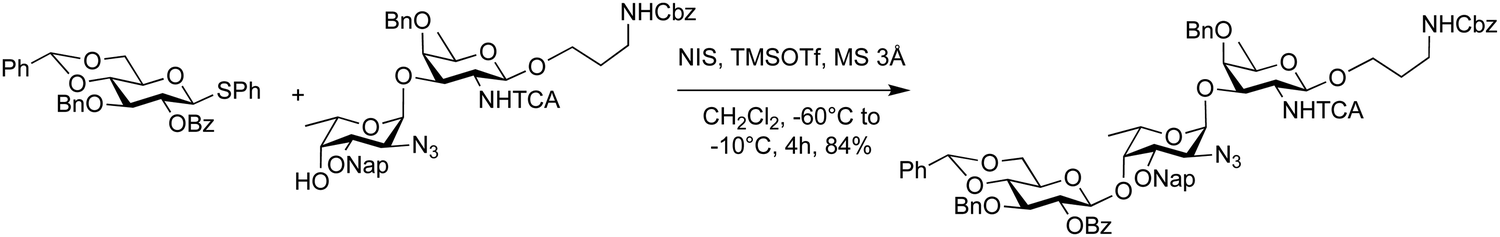 | ||
| Scheme 4 Synthesis of the trisaccharide repeating unit of Pseudomonas aeruginosa O11 antigen.223 | ||
The optimization of glycosylation reactions typically focuses on the glycosylating agent and external factors such as solvent and reaction temperature. However, there is growing recognition that fine-tuning the reactivity of the nucleophile can improve both yield and stereoselectivity in glycosylations. Unlike glycosylating agents, where relative reactivity values have been assigned in a number of cases,224 glycosyl acceptors remain less thoroughly understood. Establishing a robust numerical method to compare the reactivity of various acceptors will enable synthetic chemists to make informed decisions and develop of more efficient and selective processes for synthesizing complex glycans.225
6.5. Influence of protecting groups on the glycosylating agent
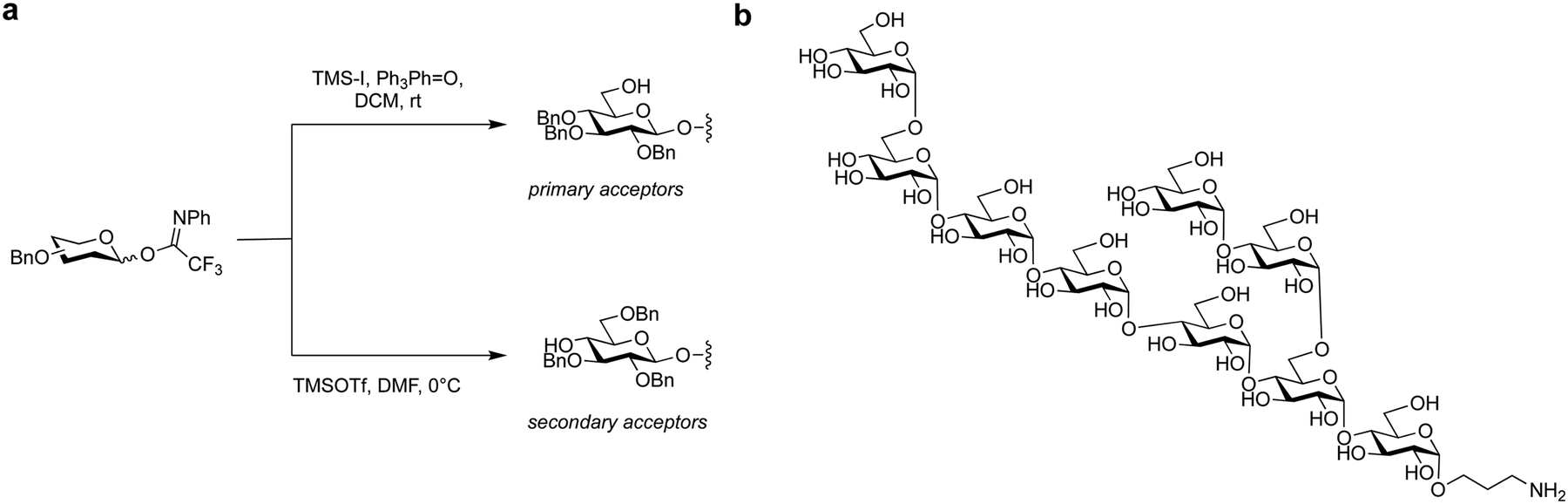
Protecting groups influence the reactivity of the glycosylating agent and the stereochemical outcome of glycosylations.57 Inductively donating protecting groups, such as benzyl ethers, stabilize electron-deficient transition states of SN2-type reactions and SN1-type reaction intermediates. These protecting groups accelerate glycosylation reactions, as they enhance the overall reactivity of the glycosyl donor. Electron-withdrawing protecting groups, such as benzoyl esters, have the opposite effect as they reduce the reactivity of the glycosyl donor and make glycosylation reactions slower and less efficient.226 By judiciously selecting the appropriate protecting groups, chemists can finely control the reactivity and selectivity of glycosylation reactions, thus influencing the formation of specific glycosidic linkages with the desired stereochemistry.Electronic effects of protecting groups on the reactivity of glycosylating agents have been labelled as ‘arming’ and ‘disarming’, as glycosylating agents bearing benzyl groups on pentenyl glycosides can be selectively activated in the presence of ‘disarmed’ ones.226 Subsequent efforts to quantify this effect enabled one-pot syntheses of oligosaccharides.227–231 The systematic quantification of donor reactivity established an extensive series of relative reactivity values (RRVs) for thioglycosides as the foundation for the ‘programmable’ one-pot synthesis of complex oligosaccharides. The sequential addition and activation of mono-hydroxylated donors in a reaction flask resulted in a growing oligomeric chain.179,181,182,232
Controlling stereoselectivity in glycosylations remains a major challenge in the synthesis of complex oligosaccharides, especially for 1,2-cis linkages. While no general solution for the construction of 1,2-cis linkages exists, remote participation by distal acyl groups has shown promise in improving stereoselectivity (Fig. 9b).144,233–235 However, the role and strength of this effect, primarily due to the transient nature of the dioxolenium ion intermediate, is still under discussion.234,236–238 An in-depth study of distal acyl groups using cryogenic infrared spectroscopy, DFT computations, and a series of systematic glycosylations, has revealed that certain ester groups can play a decisive role in shaping the stereochemical outcome of glycosylations.234,237,238 For example, remote participation of 3-OH ester groups on mannosides is stronger than 3-OH ester groups on galactosides, while remote participation of 3-OH ester groups in glucosyl and galactosyl donors provide less assistance in guiding the stereochemical course of glycosylations. Reports on the role of 6-OH ester groups are mixed.144,237,238 The remote participation of 4-OH ester groups in galactose can lead to covalent bonds between the carbonyl oxygen and the anomeric carbon, to promote α-selective galactosylations.234,238 Although the strength of remote participation requires further clarification for different types of glycosides, it offers a viable option for improving stereocontrol in glycoside synthesis.
 | ||
| Fig. 9 (a) Neighbouring protecting group participation. (b) Long-range participation. E-X promoter system, Nu nucleophile. | ||
Cyclic protecting groups such as 4,6-O-benzylidene can significantly impact the stereochemical outcome of glycosylations as shown most prominently for the synthesis of challenging β-mannosidic linkages.239,240 Initially, this methodology relied on a pre-activation promoter system involving sulfoxides and thioglycosides, resulting in superior β-selectivity compared to glycosylating agents lacking this protecting group. The 4,6-O-benzylidene protecting group effect also applies to other glycosylating agents such as trichloroacetimidates and phosphates.241,242 Anomeric tethers influence the stereochemical outcome of glycosylations as first applied to the β-selective synthesis of fructofuranosides and the first stereospecific synthesis of sucrose.243 The selectivity was attributed to the tetraisopropyldisiloxane tethering group blocking the α-face, ensuring that the acceptor approaches only from the β-face. The stereoselective synthesis of β-xylulofuranoses used a siloxane tether244 on a thioglycoside to yield the desired β-linked xylofuranose. Similarly, a macrocyclic anomeric tether for α-sialylation, relied on the tethering group blocking the β-face for the nucleophile to approach from the α-face.245 The cyclic di-tert-butylsilylene (DTBS) protecting group can improve selectivity246 in particular for α-selective galactosylation. This reaction remains α-selective even in the presence of a C-2 participating benzoate ester protecting group (Scheme 5).247,248
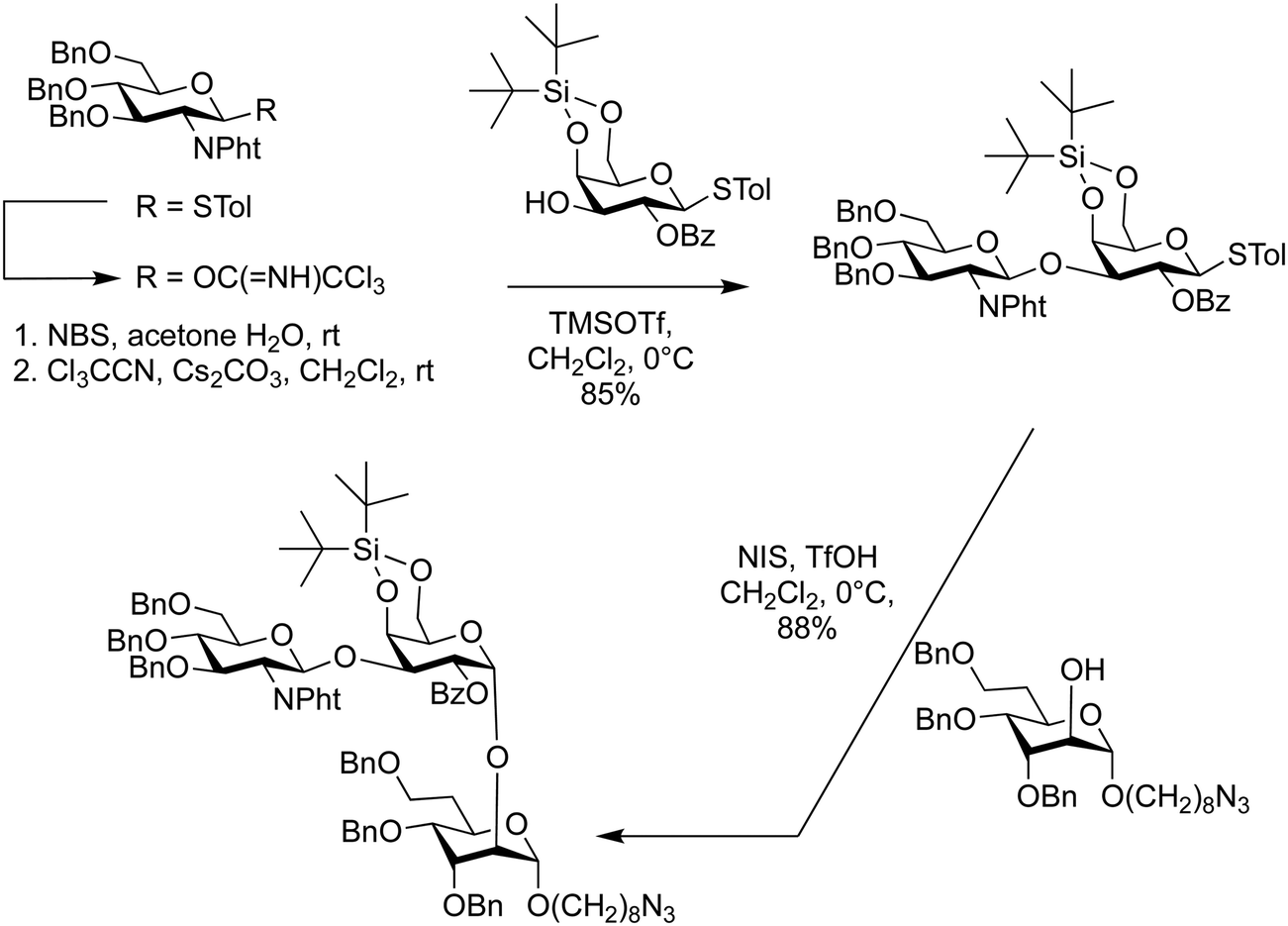 | ||
| Scheme 5 Combination of conformational restriction and glycosyl imidates for the synthesis of Campylobacter jejuni capsular polysaccharide repeating unit.249 | ||
Protecting group control of glycosylating agents was enlisted in the synthesis of the capsular polysaccharide repeating unit of Campylobacter jejuni 81–176 to determine the absolute configuration of its O-methyl phosphoramidate motif.249 This synthesis required addressing several challenges, including the stereoselective installation of the α-galactoside, the preparation of the 6-deoxyheptose building block with correct stereochemistry, and the introduction of the O-methyl phosphoramidate motif. A key in the construction was the use of a glycosyl trichloroacetimidate, that was prepared from the corresponding thioglycoside (Scheme 5). This glycosyl imidate was coupled with the C3 hydroxyl group of galactose using trimethylsilyltrifluoromethanesulfonate (TMSOTf) as the catalyst, yielding the desired β-linked disaccharide in 85% yield. The galactose portion of this disaccharide bore a di-tert-butylsilyl acetal on O-4 and O-6, enabling α-selective galactosylation, even in the presence of a neighbouring participating group (Scheme 5).247,248 This strategy was used to stereoselectively couple a disaccharide with the 6-deoxy-altro-heptoside acceptor by activating the thioglycoside with N-iodosuccimide and triflic acid in 88% yield.
7. Commonly used glycosylating agents
The pursuit of developing broadly applicable, stereocontrolled glycosylation reactions remains an ongoing challenge in chemical synthesis due to the numerous variables involved in the process, including anomeric leaving groups, protecting groups, promoters, acceptor nucleophilicity and solvents. Much effort has focused on the anomeric leaving group, leading to the development of a range of glycosylating agents.250–256 Here, six major approaches for glycoside synthesis are discussed:(1) the Fischer–Helferich method; (2) anomeric halides; (3) thioglycosides; (4) glycosyl imidates and (5) glycosyl phosphates; and (6) organic and transition-metal catalysis.
Each of these methods possesses its own advantages and limitations. The synthesis of oligosaccharides often requires a pragmatic approach that combines multiple methods to achieve the desired outcomes effectively. Despite the challenges, continuous research and innovation are paving the way for advancements in glycoside synthesis, thereby expanding the possibilities in chemical synthesis.
7.1. The Fischer–Helferich method
In the classical Fischer–Helferich method, O-glycosides are formed via an acid-catalyzed reaction between hemiacetals and alcohols (Fig. 10).257 This reaction is reversible and not applicable to the synthesis of oligosaccharides.54 | ||
| Fig. 10 Fischer–Helferich mediated synthesis of glycosides. | ||
Diastereocontrol at the anomeric position is based on thermodynamic energy differences between the corresponding α- and β-anomers. The axial position for the anomeric OR group is disfavoured by 1,3-diaxial interactions and favoured by the anomeric effect (stereoelectronic interactions between the ring oxygen and the anomeric carbon–oxygen bond).176 These two opposing effects often have similar magnitudes, to yield mixtures of products that, due to a slight dominance of the anomeric effect, contain more of the α-anomer. The α:β ratios vary with individual sugars and the reaction conditions.
The method produces a mixture of α- and β-furanosides and pyranosides, through a competing equilibrium that likely proceeds via open-chain intermediates, α,β anomerizations, and oxocarbenium ions. Furanosides are first formed and the equilibrium subsequently shifts towards pyranosides (Fig. 11).258,259 Chromatography is often required to isolate pure anomers from such reaction mixtures.55
 | ||
| Fig. 11 Equilibrium present in Fischer glycosylation. | ||
Combining this simple acid-catalysed glycoside synthesis with additional acetal formation in the presence of a ketone such as acetone can allow access advanced synthetic intermediates in one pot (Table 2). Therefore, many such compounds are now commercially available. Diacetone sorbose is obtained from acetone and L-sorbose under acidic conditions as an intermediate in the synthesis of vitamin C.260 Today, the Fischer–Helferich method is used in the synthesis of simple glycosides, chiral synthons for total synthesis or in the preparation of chemical biology probes.260–267
| Starting material | Product | |
|---|---|---|
| a References are in brackets. | ||
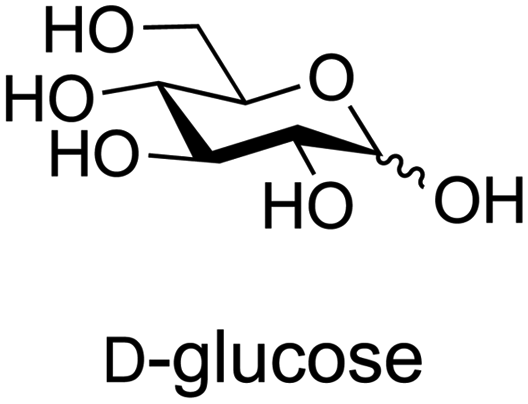
|
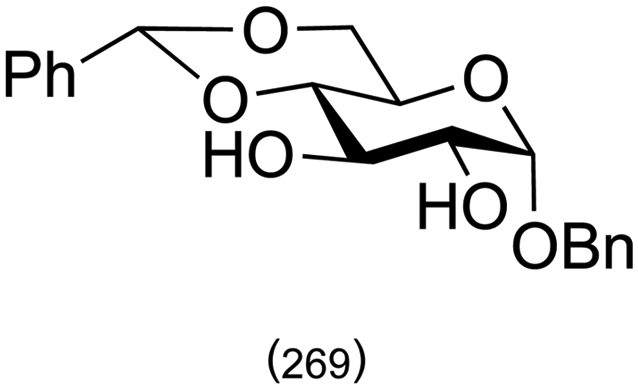
|
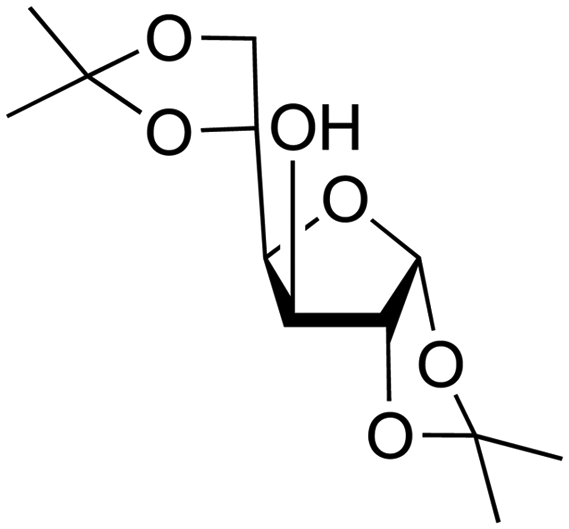
|
|

|
|
|
|

|
|

|

|
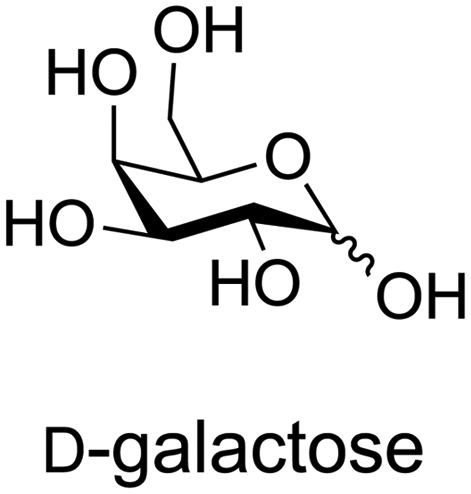
A Fischer glycosylation was used in the synthesis of UDP-galactofuranose (Scheme 6), whereby, D-galactose was converted to an anomeric mixture of methyl furanosides and methyl α-D-galactopyranoside under ferric chloride-catalyzed conditions.275 Thereafter, several protecting group manipulations and a glycosylation with dibenzyl phosphate yielded the desired phosphorylated furanoside. This 1-phosphorylated monosaccharide was then coupled with an activated 5′-N-methyl phosphorylimidazolide nucleoside to yield UDP-Galf (Scheme 6).
 | ||
| Scheme 6 Utility of the Fischer–Helferich method in the synthesis of UDP-galactofuranose.265 | ||
7.2. Glycosyl halides
Anomeric halides are a classical means to synthesise glycosides dating back to 1901 (Fig. 12) (Table 3).276 The Koenigs–Knorr method and its subsequently developed variants277–281 utilize glycosyl halides (bromide, chloride, iodide and fluoride) activated by heavy metal salts such as silver or tin to facilitate coupling with a glycosyl acceptor.276–278,282–284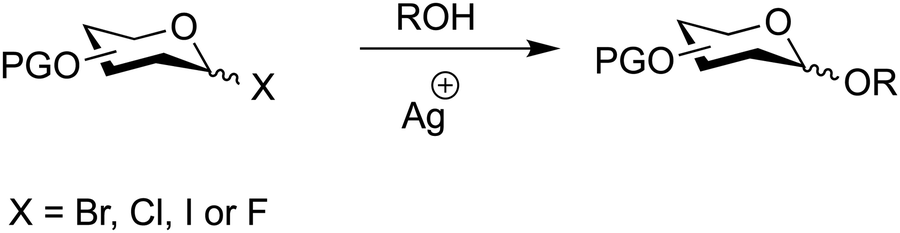 | ||
| Fig. 12 Anomeric halide mediated synthesis of glycosides. | ||
The exchange of the anomeric hydroxyl group for bromine, chlorine, iodine or fluorine is typically carried out with halogenating agents with all other hydroxy groups O-protected.285–292 Glycosyl bromides, chlorides and iodides mainly lead to the product with the halogen atom in an axial position, whereas fluoride glycosyl donors can occur in α/β mixtures with the ratio depending on the solvent.292
| Leaving group | Promoter | Lead author(s) |
|---|---|---|
| Bromide and chloride | Ag2CO3 | Koenigs and Knorr293 |
| Bromide and chloride | Hg(CN)2 | Helferich294 |
| Bromide and chloride | AgOTf | Lemieux295 |
| Fluoride | SnCl2/AgClO4 | Mukaiyama,296 |
| Fluoride | BF3·Et2O | Nicolaou297 and Kunz298 |
| Fluoride | Cp2ZrCl2 (or Cp2HfCl2)/AgClO4 (or AgOTf) | Suzuki299,300 |
| Iodide | No promoter | Helferich301 |
| Iodide | TBAI | Gervay-Hague302,303 |
The stability and reactivity of glycosyl halides is greatly dependent on the halogen, the sugar and the protecting groups.304 The thermal stability of these glycosyl donors increases from iodide, to bromide, to chloride, to fluoride. The protective groups electron-donating or electron-withdrawing character also affects the stability of glycosyl halides.304
For instance, 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide can be prepared and stored at 0 °C, whereas 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl bromide is thermally unstable at as low as −78 °C. Glycosyl iodides are usually very unstable and are prepared in situ.146,177,305
In general, many glycosyl halides are: (1) prepared in situ; (2) used with minimal further purification and (3) are highly sensitive to hydrolysis. These stability issues can sometimes necessitate the use of non-homogeneous/impure compounds, with the exception of glycosyl fluorides, that are more stable to heat, less sensitive to hydrolysis and can often be purified by chromatography.306–309 Initially, glycosyl fluorides were thought to be too stable for glycosylation reactions due to the large bond-dissociation energy of the C–F bond (552 kJ mol−1) but this has been found not to be the case.296
The reactivity trend of the glycosyl halides is the inverse to their stability (I > Br > Cl > F) with protecting groups exerting remote electronic effects. The relative influence of the protecting group is not only dependent on its distance from the anomeric carbon atom but in general, electron-withdrawing O-protective groups can lower the reactivity of a glycosylating agent if too unstable.
Anomeric halides have been employed in the synthesis of a wide range of complex glycans and glycoconjugates307,310–317 as well as AGA of mannan polysaccharides (Scheme 7), rhamnogalacturonan-II (Scheme 8) and Mycobacterium tuberculosis cell wall α-glucans (Fig. 13).146,306,318,319
 | ||
| Scheme 7 Glycosyl fluorides enable the late-stage assembly of 151-mer mannan polysaccharide.306 | ||
 | ||
| Scheme 8 Synthesis of rhamnogalacturonan-II structures using glycosyl fluorides. | ||
 | ||
| Fig. 13 Reagent controlled stereoselective synthesis of α-glucans. (a) Reagent-controlled methodology and (b) target α-glucan nonasaccharide.146 | ||
Glycosyl fluorides were key to the assembly of a 151-mer mannan polysaccharide, via block coupling of four linear 30-mer α-(1–6) polymannosides with a branched 31-mer polymannoside tetraol acceptor (Schemes 7a and b).306,320,321 While a glycosyl trichloroacetimidate failed to give the desired product in the block coupling strategy glycosyl fluorides furnished the desired 151-mer via a 31 + 30 + 30 + 30 + 30 block coupling to (Scheme 7c). Silver perchlorate performed better than silver triflate–hafnocene dichloride as it resulted in complete glycosylation in 30 minutes with only minimal levels of by-products formed.299 Purification by HPLC gave the desired 151-mer that was subsequently deprotected to yield the 151-mer.
Glycosyl fluorides have been used in the synthesis of portions of rhamnogalacturonan-II that contain 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) branches.307 Different Kdo-glycosyl fluoride were iteratively tested with the glycosyl acceptor at different temperatures, promoters and glycosyl donor protecting group patterns. Addition of one equivalent of BF3·Et2O every three hours at −40 °C proved be best to suppress donor elimination and minimize acetal protecting group cleavage. Thereby, the target pentasaccharide was obtained in 31% yield with complete α-stereoselectivity without the 7,8-O-isopropylidene acetal protecting group. Following hydrolysis of the 4,5-O-isopropylidene acetal with acetic acid and hydrogenolysis of the benzyl ethers the desired RG-II fragment was obtained (Scheme 8).
For the synthesis of α-glucans from the cell wall of Mycobacterium tuberculosis a combination of glycosyl imidates, anomeric halides and additives was employed to stereoselectively construct cis-glucosidic linkages with both primary and secondary alcohols (Fig. 13). The higher reactivity of the primary compared to the secondary alcohol meant the use of trimethylsilyl iodide and triphenylphosphine oxide was ideal for the stereoselective cis-glucosylation with primary alcohols. For secondary alcohols, a combination of trimethylsilyltriflate (TMSOTf) and DMF was selected. Using these two glycosylation conditions in combination with selective protecting group manipulations allowed for the assembly of a nonasaccharide α-glucan following global deprotection.146
In summary, the reactivity of the glycosyl halides can be varied over a wide range of conditions and can be modulated the choice of the halogen, the protecting group pattern, the promoter, the solvent and the temperature.322 In addition, the reactivity of the nucleophile can be adjusted.138 The diastereoselectivity during the glycoside formation is attained by participation of neighbouring groups for 1,2-trans glycosides and for 1,2-cis glycosides by the in situ anomerization method.177 The anomeric halide method has some inherent disadvantages that are difficult to overcome, including the low thermal stability of the glycosylating agent and its sensitivity to hydrolysis (with the exception of glycosyl fluorides) as well as the the use of excess often toxic heavy metal salts that make them unsuitable for large scale synthesis.
7.3. Thioglycosides
Thioglycosides are versatile glycosylation agents that were first explored in the early 1970s,323 but their full potential was realized in the 1980s when several new promoters emerged.175,324,325 Today, thioglycosides are popular glycosylating agents, especially for the synthesis of oligosaccharides (Fig. 14).223,326–331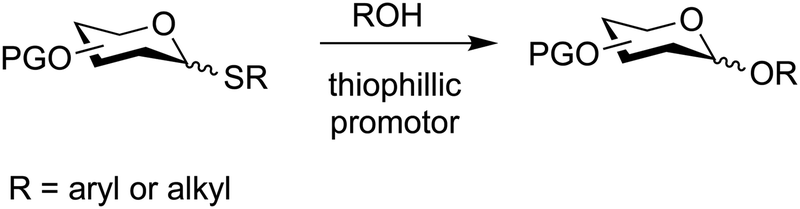 | ||
| Fig. 14 Glycosylation based on thioglycosides. | ||
Thioglycosides can be used directly in the synthesis of O-glycosides by coupling with a nucleophile in the presence of a thiophilic promoter and they can be converted into most other glycosylating agents (Fig. 8). Thereby, thioglycosides provide chemists with the flexibility required for the construction of oligosaccharides and glycoconjugates that often requires trial and error.
Most thioglycosides are simple alkyl and aryl glycosides and can be synthesized on a large scale. The products are often crystalline and can be purified without chromatography. Many paths to prepare thioglycosides have been described, but frequently peracetylated saccharides are reacted with a thiol in the presence of a Lewis acid such as BF3·Et2O. Alternatively, thioglycosides can be synthesized from glycosyl halides or phosphates.150,161
Common protecting groups such as esters (benzoates, levulinates), ethers (benzyl, allyl, silyl), acetals (isopropylidene, benzylidene) and orthoesters can be introduced, manipulated and removed on thioglycosides rendering them distinct among other types of glycosylating agents. The removal of benzyl groups using hydrogenolysis is difficult, as the sulfur atom can poison the catalyst but the chemoselective removal of 2-naphthylmethyl ether protecting groups in the presence of benzyl ethers on thioglycosides has been demonstrated.332 A new oxidative photocatalytic method for benzyl ether removal can overcome this challenge.333 Oxidation reactions, e.g. to obtain glycan uronic acids, using thioglycoside building blocks requires vigilance as oxidation of the sulfur group to a sulfoxide is possible but often DMSO, TEMPO-BAIB and PDC-mediated oxidations can be performed successfully.334 Aryl thioglycosides are less prone to this oxidation when compared to alkyl thioglycosides.
The first report glycosylations involving thioglycosides used mercuric acetate that was soon replaced by other promoters.175 Using methyl triflate as activator meant thioglycosides having a neighbouring participating substituent at O-2 gave the expected 1,2-trans glycosides, whereas those with a non-participating O-2 substituent gave anomeric mixtures. Subsequently, several other, high-yielding thiophilic promoters have been developed (Table 4).
Thioglycosides can be effective in the construction of 1,2-trans and 1,2-cis linkages, including a recent AGA of starch and glycogen α-glucan oligosaccharides (Scheme 9).144 Key to the assembly of these complex structures was the synergistic use of remote participation, solvent effects and optimal building blocks. The ideal building blocks for the repeated synthesis of α-(1 → 4) and α-(1 → 6) glycosides were identified by a series of systematic glycosylations. Two building blocks from a set of twenty-one were found to be ideal for the stereoselective synthesis of these glycans (Fig. 14). AGA of the α-(1 → 4) glucan backbone worked best when a 3,6-di-O-benzoylated thioglycoside with a 4-O-fluorenylmethyloxycarbonyl (Fmoc) protecting group was used. The building block for α-(1 → 6) linkages, contained a 3-O-benzoate ester, a 4-O-Fmoc and a 6-O-levulinoyl ester. Using AGA these two thioglycoside building blocks were combined with the help of N-iodosuccimide and triflic acid for the construction of a collection of linear and branched α-glucans, including a 20-mer amylopectin polysaccharide, one of the largest synthetic 1,2-cis-linked glycan to date.
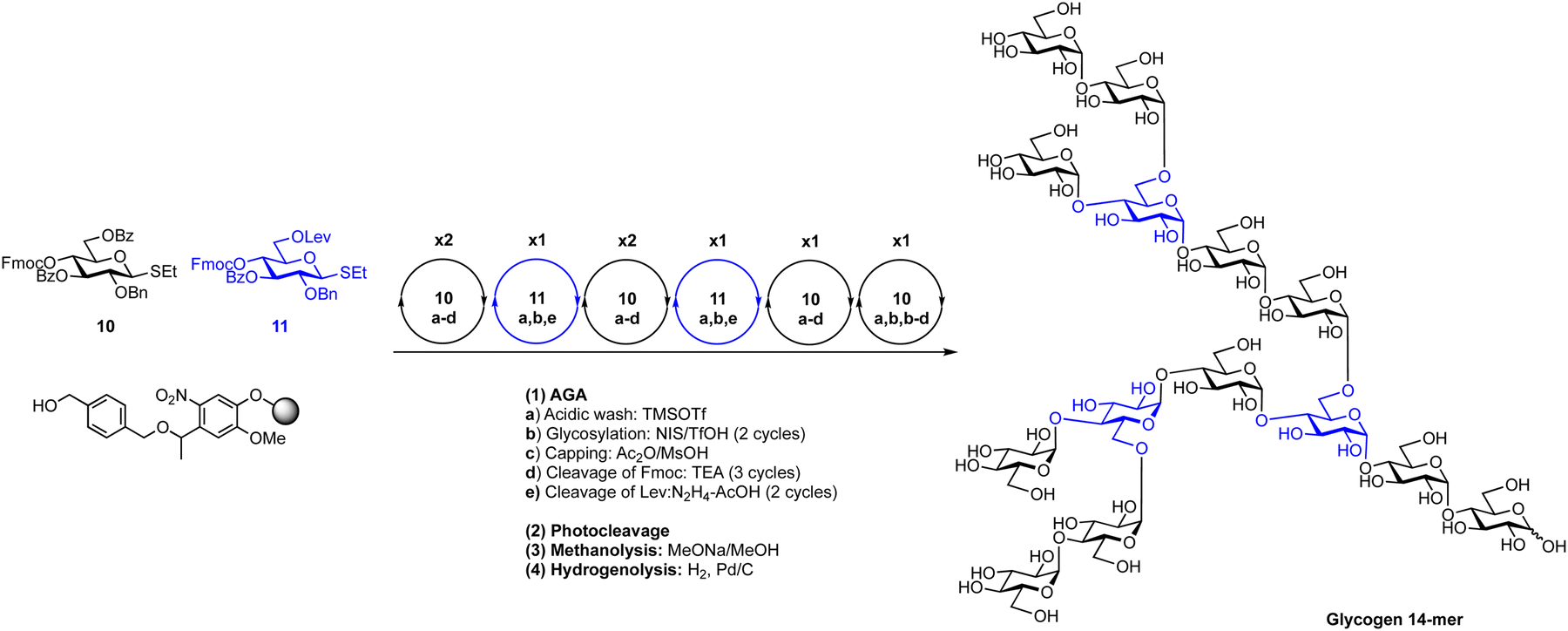 | ||
| Scheme 9 Combination of building block optimization, solvent effects and remote participation for the automated assembly of 1,2-cis starch and glycogen oligosaccharides.144 | ||
Thioglycosides can be used in block syntheses of oligosaccharides,341–344 such as the convergent synthesis of a library of glucuronoxylomannans from Cryptococcus neoformans (Scheme 10).345 The β-xylose and β-glucuronic acid branches were installed at the disaccharide stage using neighbouring protecting group participation before multi-step protecting group manipulations were required to install the 6-O-acetylation pattern of the α-mannan backbone. Tetra- and hexasaccharide thioglycoside building blocks were assembled from glycosyl imidates. These thioglycoside building blocks were assembled using dimethyl(methylthio)sulfonium trifluoromethanesulfonate (DMTST) as a promoter, with diethyl ether as a solvent to enhance α-selectivity. Hexasaccharide thioglycoside building blocks allowed for the assembly of glycans up to an 18-mer with yields ranging from 78–86%.
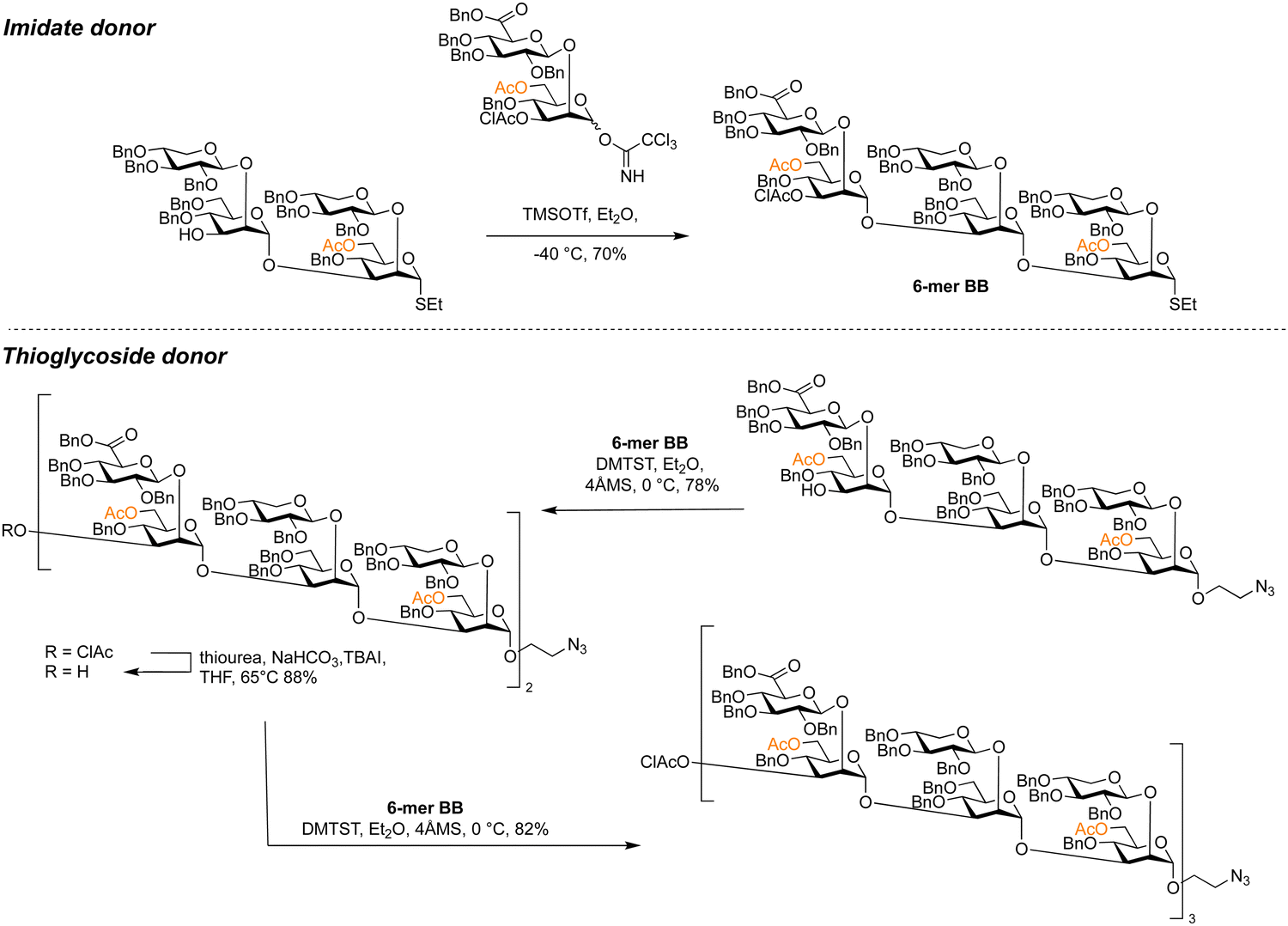 | ||
| Scheme 10 Glycosyl imidates and thioglycosides used in the assembly of a library of fungal glucuronoxylomannans.345 | ||
Many shelf-stable thioglycosides with diverse protecting group patterns can be prepared and are commercially available as reagents for oligosaccharide synthesis.346 A wide range of promoter systems for thioglycoside activation, offer significant flexibility for glycosylations. The ability to readily convert thioglycosides into other glycosylating agents makes them one of the most reliable and robust methods for synthesizing complex glycans.
7.4. Glycosyl imidates
O-Glycosyl imidates are useful glycosylating agents under acidic conditions (Fig. 15).54,347–352N-Methylacetimidate donors, initially developed, did not gain broad application due to their low reactivity.353,354 Subsequently, electron deficient trichloroacetimidates proved more robust and reliable in the synthesis of glycosides.348 Today, reactions with nucleophiles are performed with catalytic amounts of Brønsted or Lewis acids such as TMSOTf and BF3·OEt2. Typically, TMSOTf is the catalyst of first choice and is used in 0.1 equivalents based on the glycosylating agent. The glycosyl imidate is prepared by reacting the free reducing end of pyranose or furanose with an electron deficient nitrile, e.g. trichloroacetonitrile, under basic conditions to furnish the desired glycosyl imidate. This glycosyl imidate can then be used in a subsequent glycosylation under acid catalysis.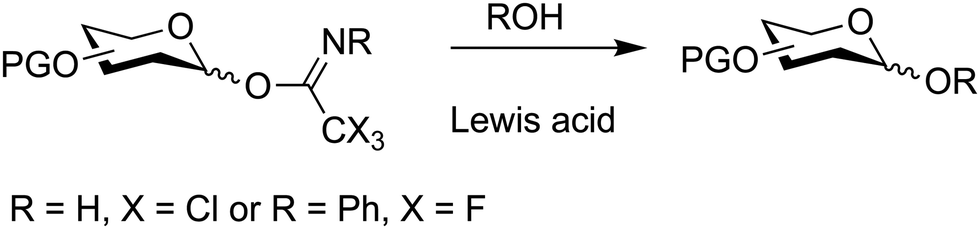 | ||
| Fig. 15 Glycoside synthesis based on glycosyl imidates. | ||
Controlling the anomeric configuration of the glycosylating agent is possible by careful choice of base, for instance K2CO3 can be used to prepare the β-anomer, while DBU, can be used to prepare the α-anomer (Scheme 11).355 This selectivity is a result of the greater nucleophilicity of the β-oxide, that is attributed to kinetic effects, while the use of DBU leads to the thermodynamically more stable α-product.356 Glycosyl trichloroacetimidates bearing multiple electron donating groups are sensitive to hydrolysis. Chromatographic purification can require pre-neutralisation of silica gel with a base, e.g. 1% solution of triethylamine, and pure building blocks should be stored frozen. An alternative to glycosyl trichloroacetimidates is the use of the less reactive glycosyl N-(phenyl)trifluoroacetimidate that is more stable than the corresponding trichloroacetimidates but also less reactive.357–360
 | ||
| Scheme 11 Reagent-controlled synthesis of different glycosyl imidate anomers. | ||
In contrast to most other glycosylation methods that require stoichiometric amounts of promoters O-glycosyl imidates are acid catalyzed and have been used to synthesise a variety of complex glycosides.
Activation of glycosyl imidates using boron trifluoride etherate starting at −40 °C and raising slowly to room temperature in dichloromethane is a popular set of conditions shown to be suitable to synthesise a range of glycosides.307 When neighbouring groups such as 2-O-benzoyl esters are present, the expected 1,2-trans products are obtained. With the non-participating protecting groups, e.g. a benzyl ether, the inversion of configuration at the anomeric centre is preferred. The amount of 1,2-cis-glycosides can be further enhanced by using diethyl ether as the solvent and trimethylsilyl triflate or tert-butyldimethylsilyl trifluoromethanesulfonate as the catalyst.361
Glycosyl trichloroacetimidates can rearrange to the corresponding stable N-trichloroacetylglycosyl amine via acid catalysis. This reaction can be minimized by lower temperatures,362 altering the solvent363–365 and changing the quantity and nature of the promoter.366,367 Using “inverse glycosylation conditions” can also improve the outcome of the synthesis. N-Phenyl based glycosyl imidate donors are less prone to the rearrangement reaction.368,369
Glycosyl imidates method have been used for block couplings of complex oligosaccharides128,345,350,370 such as the assembly of di-, tri- and tetrasaccharides for the synthesis of lipopolysaccharide O-antigens of Shigella flexneri vaccines (Scheme 12).371–373 The coupling of a trisaccharide trichloroacetimidate donor and a glucosamine acceptor had to be optimized. Initial experiments at low and room temperature failed to give satisfactory yields of the hexasaccharide, possibly due to poor acceptor solubility. However, when the coupling was carried out at 75 °C in 1,2-dichloroethane with a catalytic amount of triflic acid, 76% of the coupled product were obtained.371,374 Following selective removal of the temporary O-acetyl group, the 3-OH glucosamine acceptor was coupled under similar conditions but notably a different temperature regime of −35 °C to 10 °C, to furnish target decasaccharide in 82% yield. (Scheme 12).
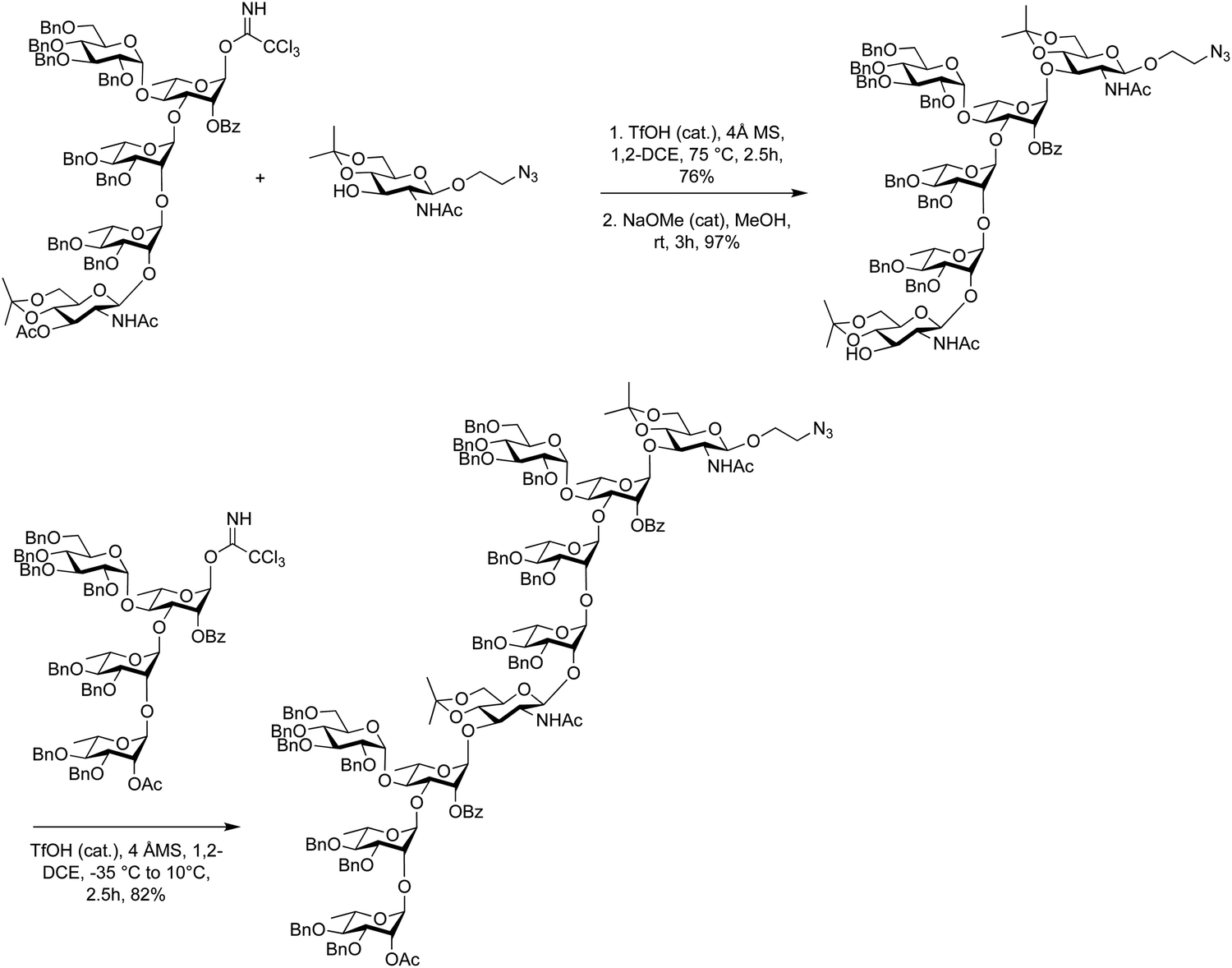 | ||
| Scheme 12 Using glycosyl imidates in the assembly of Shigella flexneri Serotype 2a oligosaccharides. | ||
Imidates were used in the synthesis of glycosylated peptides that bear a phosphonate group (Scheme 13).375 This phosphonate group mimics naturally occurring mannose-6-phosphate that causes glycoproteins to be trafficked to the lysosome.376–379 The trichloroacetimidate donor was prepared using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to afford exclusively the α-anomer and reacted with a L-serine benzyl ester at −78 °C using TMSOTf as a catalyst. After the successful coupling, the amino acid was polymerized and conjugated to antibodies creating chimaeras that can cause proteins to be degraded catalytically.375
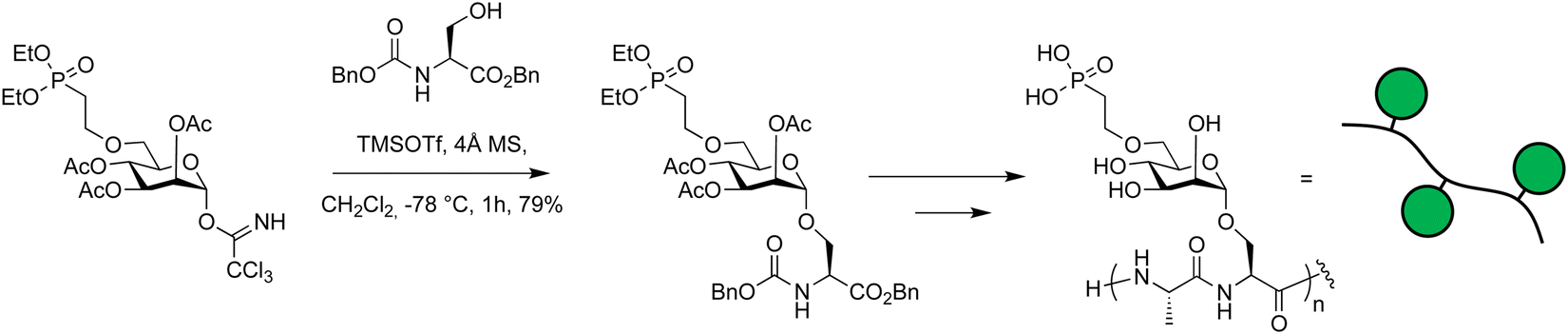 | ||
| Scheme 13 Glycosyl imidate building blocks used in the synthesis of glycopeptides.375 | ||
7.5. Glycosyl phosphates and phosphites
Glycosyl phosphates are key intermediates in the biosynthesis of glycans, and have found application in chemical synthesis as anomeric leaving groups.380,381 Early reports included diphenylphosphate and glycosyl phosphites for effective chemical glycosylations.199,382–388 Subsequently several variants have been synthesized by varying the substituents on the phosphorus atom (Fig. 16).385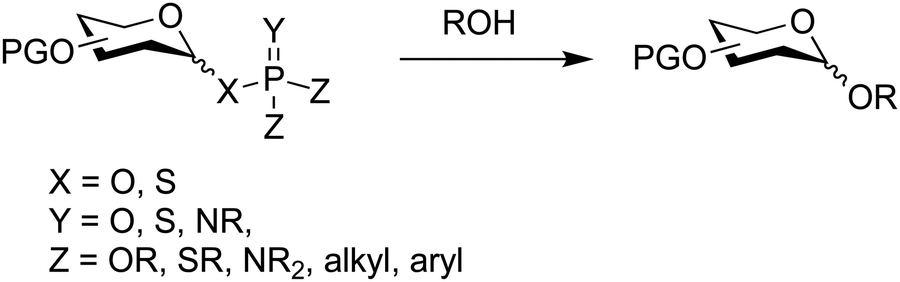 | ||
| Fig. 16 Anomeric phosphates as glycosylating agents. | ||
Diphenyl, dialkyl and propane-1,3-diyl glycosyl phosphates can be introduced at the anomeric carbon and utilized as leaving groups for the glycosylation reactions.199,382–384,386,388,389 Mostly, either the starting material is a free reducing sugar (1-OH free) or the glycosyl phosphate is prepared by converting one type of glycosylating agent, such as a thioglycosides, into a glycosyl phosphate. The stability of glycosyl phosphates depends on the chemical substituents that decorate the phosphorus atom but dibutyl phosphate donors can be purified by flash chromatography and stored at sub-zero temperatures for months. Under acidic conditions, β-phosphates anomerize to the thermodynamically more stable α-phosphates, making it theoretically possible that during the glycoside forming reaction, anomerization of the glycosyl phosphate can occur.
Dibutyl phosphates are a frequently used anomeric leaving group on glycosylating agents.389 The ratio of the resulting α- and β-glycosyl phosphates is solvent dependent, toluene and dichloromethane gave β-phosphates and the use of THF produced almost exclusively α-phosphates. Generally, in glycosylations where neighbouring protecting groups are present, the expected 1,2-trans glycoside is formed.390 In the synthesis of 1,2-cis glycosides, anomeric phosphates have shown good utility, e.g. in the formation of challenging α-sialosides.16,199 A dibutyl phosphate donor was used in the synthesis of O-acetylated sialosides to probe virus receptor binding preferences (Scheme 14).16 Dibutyl glycosyl phosphates have been used to construct a wide range of glycans by AGA.116,235,390 such as a collection of β-(1 → 3) glucans (Scheme 15). Constructing these branched structures required two building blocks, one for the β-1,3 backbone and one for the β-1,6 branching points. The backbone building block was a di-4,6-O-benzylated building block with a 3-O-Fmoc temporary protecting group and a 2-O-benzoate ester ensured the formation of the desired 1,2-trans selectivity. The branching building block contained a 6-O-levulinoyl ester protecting group for orthogonal removal and the subsequent installation of branches. These two building blocks were combined together via TMSOTf promoted glycosylations to construct a collection of β-1,3 glucans. These synthetic glucans were printed on a microarray and scanned with human sera to explore aspects of fungal immunology.
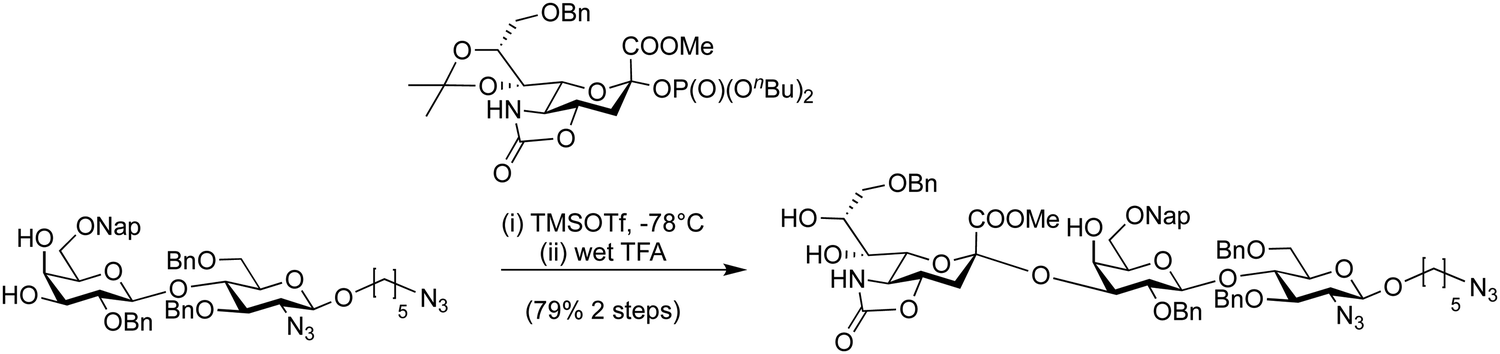 | ||
| Scheme 14 Regioselective glycosylation with sialic acid dibutyl phosphate.16 | ||
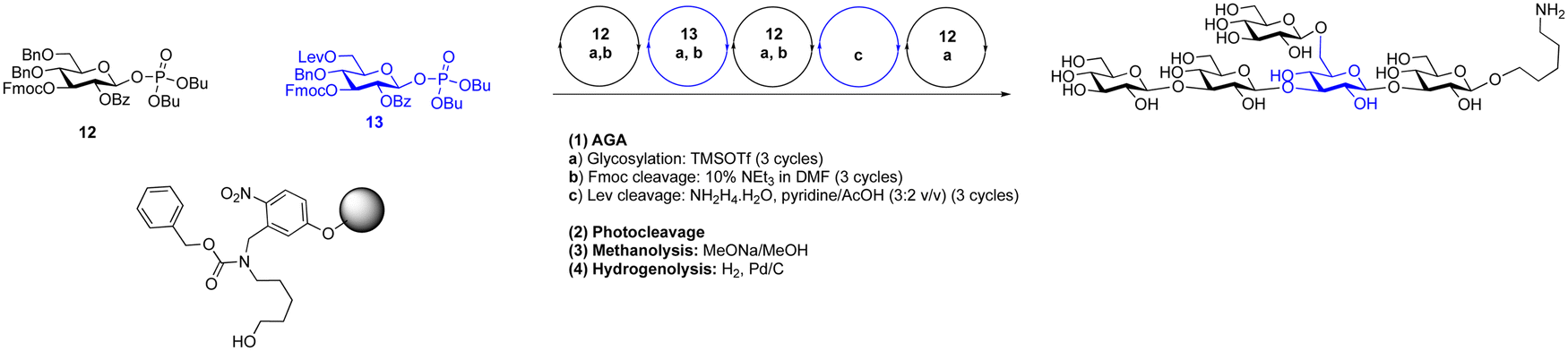 | ||
| Scheme 15 AGA of a β-glucan pentasaccharide using glycosyl phosphates.390 | ||
Phosphorus-based glycosylating agents have gained prominence as a viable option for glycoside synthesis. By altering the substituents on the phosphorus atom, it becomes possible to fine-tune the glycosyl donor properties. An advantage of this method, is its operational simplicity, requiring the use of stoichiometric amounts of Lewis acid promoters e.g., TMSOTf. The glycosylations are high yielding and highly stereoselective. Dibutyl phosphate donors, in particular, have proven valuable in AGA of a wide range of glycans.391,392
7.6. Organic and transition-metal catalysis
New approaches to construct glycosides via organocatalytic and transition metal-mediated methodologies have been developed. Among the catalytic methods, the glycosyl imidate approach, discussed in Section 7.4, has seen by far the most wide-spread use.Both thiourea and organoboron-based catalysts have been used for selective glycosylations of unprotected or partially protected glycosyl acceptors.74,408 Selective glycosylations of molecules containing multiple hydroxyl groups with similar reactivity is possible by discriminating nucleophiles based on complex stereochemical environments.409,423,424 Regioselective glycosylations of carbohydrate polyols have been reported using dibutyltin oxide424–427 and diarylborinic acid catalysts.106–113 Both organoboron and tin-based reagents offer the same regioselectivity and can be used in building block synthesis to enable regioselective acylation and alkylation of polyols (discussed in Section 3).
A recent example of regioselective organocatalysis in glycoside synthesis was reported using a bis-thiourea catalyst to synthesise 1,2-cis glycosides with partially and unprotected glycosyl acceptors (Scheme 16).74 Stereo- and regio-selective galactosylations and mannosylations were reported using an assortment of polyfunctional nucleophiles. The anomeric configuration of glycosyl acceptors was found to impact the regioselectivity with α-acceptors affording lower regioselectivity. Eight catalysts were screened with different aryl groups and more electron-rich arenes resulted in higher selectivity for (1,2)-galactosylations. Computational studies suggested that catalyst selectivity arose in part from stabilizing C–H/π interactions between the catalyst and the acceptor, a phenomenon well established for carbohydrate–protein interactions.
 | ||
| Scheme 16 Regioselective organocatalysis using a bis-thiourea catalyst for site-selective mannosylation.74 High stereoselectivities (>20:1 β:α) were observed in every case. aReaction performed at 40 °C. bReaction performed at 23 °C. | ||
Gold-mediated glycosylations are distinct from other transition-metal-mediated glycoside syntheses as novel leaving groups have been introduced.453–456 Glycosyl ortho-alkynyl benzoates and propargyl 1,2-orthoesters share a similar mechanism that exploits the alkynophilic nature of gold.452,457 Glycosyl ortho-alkynyl benzoates were used to synthesise a 128-mer of the O-antigen from Bacteroides vulgatus (Scheme 17).458 The challenging 1,2-cis-mannopyranoside linkages were completed early in the synthesis and neighbouring group participation was enlisted to stereoselectivity assembly the 1,2-trans-rhamnopyranoside linkages. This transition-metal-mediated approach allowed for the decagram-scale synthesis of a tetrasaccharide building block that was then used in iterative 2n+1-mer cycles, to assemble an 8-mer, 16-mer and a 32-mer on gram-scale. Two 64-mers were combined under gold(I)-mediated catalysis to yield 74% of protected 128-mer.
 | ||
| Scheme 17 Iterative gold-mediated catalysis for the assembly of a 128-mer O-antigen of Bacteroides vulgatus. | ||
Transition metal and organo-catalysis methods have been used to synthesize a variety of N-, C- and S-linked glycosides. Overall, they provide complementary for efficiently synthesizing of complex glycosides.
8. Global deprotection of synthetic glycosides
After completing a synthesis, permanent protecting groups are removed to furnish glycosides found in nature. This “global deprotection” at the final stage is often the least optimised part of the synthesis.458–462 High-quality reagents are essential to maximise the yields of target glycans, especially in the case of hydrogenolysis reactions, where the quality of palladium on carbon catalysts varies widely among commercial suppliers.463 Additional consideration should be given to the glycan structure, the role of solvent(s) (and solvent combinations),464,465 temperature(s) and pressure(s), that can play a decisive role in determining the success of the hydrogenolysis reaction.462,463 Hydrogenation of trichloroacetamide or hydrogenolysis of benzyl ethers can be difficult to remove or convert. Saturation of aromatic protecting groups such as benzyl and 2-naphthylmethyl ethers, has been reported on some glycosides and may be an overlooked side-reaction (Fig. 17).459,462,466–468 | ||
| Fig. 17 Hydrogenation of aromatic protecting groups in global deprotection reactions. | ||
Difficulties in removing ester protecting groups can arise during global deprotection as pivalate and benzoate esters can be difficult to remove from oligosaccharides at times.125,469 Adjusting temperature, base, solvent and protecting group patterns can be used to optimise a synthesis.
The challenge of global deprotection is exemplified by the synthesis of a 128-mer polysaccharide resembling the repeating unit of Bacteroides vulgatus via gold-promoted glycosylations (Scheme 17). The global deprotection involved five steps: (1) cleavage of one TBS ether with tetrabutylammonium (TBAF); (2) removal of the anomeric p-methoxyphenyl (MP) with ammonium cerium(IV) nitrate (CAN); (3) benzoylation of the resulting hemiacetal; (4) hydrogenolysis of the 64 benzyl ethers and 64 benzylidene acetals and (5) methanolysis of the 128 benzoyl groups with sodium methoxide solution. A capping step (3) was important for hydrogenolysis to avoid glycan degradation. Hydrogenolysis of glycans larger than 16-mers were challenging due to solubility issues that are not uncommon when transforming a fully protected, hydrophobic structure into a hydrophilic. The solubility of a range of amphiphilic intermediates that form over the course of a hydrogenolysis reaction, as benzyl ethers are cleaved has to be considered. In this case, a combination of 10% Pd/C and 10% Pd(OH)2 was used to deprotect the 128-mer, before chromatography using a Sephadex column yielded 15% of the 128-mer over five steps.
The global deprotection of oligosaccharides is not yet routine. Enhancing the efficiency of this chemical step(s) holds the potential to significantly advance oligosaccharide synthesis.
9. Summary and outlook
There is no universal glycosylation method to synthesize all possible glycosides. However, methodological advancements are enabling the synthesis of increasingly complex glycans and automated glycan synthesis. Glycoside synthesis remains more complex than the synthesis of both nucleic acids and oligopeptides. The chemical synthesis of well-defined glycosides and oligosaccharides remains essential to explore the many roles glycans play in nature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. J. C. was funded by MSCA grant: MARINEGLYCAN. Funding from the Max Planck Society is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed
.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed
-
A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor, NY, 2022, p. 823
- J. Boutet, P. Blasco, C. Guerreiro, F. Thouron, S. Dartevelle, F. Nato, F. J. Cañada, A. Ardá, A. Phalipon, J. Jiménez-Barbero and L. A. Mulard, Chem. – Eur. J., 2016, 22, 10892–10911 CrossRef CAS PubMed
- A. Phalipon, C. Costachel, C. Grandjean, A. Thuizat, C. Guerreiro, M. Tanguy, F. Nato, B. Vulliez-Le Normand, F. Bélot, K. Wright, V. Marcel-Peyre, P. J. Sansonetti and L. A. Mulard, J. Immunol., 2006, 176, 1686–1694 CrossRef CAS PubMed
- Y. Yu, S. Gim, D. Kim, Z. A. Arnon, E. Gazit, P. H. Seeberger and M. Delbianco, J. Am. Chem. Soc., 2019, 141, 4833–4838 CrossRef CAS PubMed
- X. Wu, M. Delbianco, K. Anggara, T. Michnowicz, A. Pardo-Vargas, P. Bharate, S. Sen, M. Pristl, S. Rauschenbach, U. Schlickum, S. Abb, P. H. Seeberger and K. Kern, Nature, 2020, 582, 375–378 CrossRef CAS PubMed
- M. Delbianco, A. Kononov, A. Poveda, Y. Yu, T. Diercks, J. Jiménez-Barbero and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 5421–5426 CrossRef CAS PubMed
- W. Weis, J. H. Brown, S. Cusack, J. C. Paulson, J. J. Skehel and D. C. Wiley, Nature, 1988, 333, 426–431 CrossRef CAS PubMed
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. Van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C. H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17033–17038 CrossRef CAS PubMed
- J. Stevens, O. Blixt, T. M. Tumpey, J. K. Taubenberger, J. C. Paulson and I. A. Wilson, Science, 2006, 312, 404–410 CrossRef CAS PubMed
- B. Vulliez-Le Normand, F. A. Saul, A. Phalipon, F. Bélot, C. Guerreiro, L. A. Mulard and G. A. Bentley, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9976–9981 CrossRef CAS PubMed
- V. Solanki, K. Krüger, C. J. Crawford, A. Pardo-Vargas, J. Danglad-Flores, K. L. M. Hoang, L. Klassen, D. W. Abbott, P. H. Seeberger, R. I. Amann, H. Teeling and J.-H. Hehemann, ISME J., 2022, 16, 1818–1830 CrossRef CAS PubMed
- L. Doyle, O. G. Ovchinnikova, B. S. Huang, T. J. B. Forrester, T. L. Lowary, M. S. Kimber and C. Whitfield, J. Biol. Chem., 2023, 299, 104609 CrossRef CAS PubMed
- A. K. Pröbstel, X. Zhou, R. Baumann, S. Wischnewski, M. Kutza, O. L. Rojas, K. Sellrie, A. Bischof, K. Kim, A. Ramesh, R. Dandekar, A. L. Greenfield, R. D. Schubert, J. E. Bisanz, S. Vistnes, K. Khaleghi, J. Landefeld, G. Kirkish, F. Liesche-Starnecker, V. Ramaglia, S. Singh, E. B. Tran, P. Barba, K. Zorn, J. Oechtering, K. Forsberg, L. R. Shiow, R. G. Henry, J. Graves, B. A. C. Cree, S. L. Hauser, J. Kuhle, J. M. Gelfand, P. M. Andersen, J. Schlegel, P. J. Turnbaugh, P. H. Seeberger, J. L. Gommerman, M. R. Wilson, L. Schirmer and S. E. Baranzini, Sci. Immunol., 2020, 5, eabc7191 CrossRef PubMed
- Z. Li, Y. Lang, L. Liu, M. I. Bunyatov, A. I. Sarmiento, R. J. de Groot and G. J. Boons, Nat. Chem., 2021, 13, 496–503 CrossRef CAS PubMed
- M. Delbianco, A. Kononov, A. Poveda, Y. Yu, T. Diercks, J. Jiménez-Barbero and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 5421–5426 CrossRef CAS PubMed
- J. Kwon, A. Ruda, H. F. Azurmendi, J. Zarb, M. D. Battistel, L. Liao, A. Asnani, F. I. Auzanneau, G. Widmalm and D. I. Freedberg, J. Am. Chem. Soc., 2023, 145, 10022–10034 CrossRef CAS PubMed
- G. Fittolani, T. Tyrikos-Ergas, A. Poveda, Y. Yu, N. Yadav, P. H. Seeberger, J. Jiménez-Barbero and M. Delbianco, Nat. Chem., 2023, 2023, 1–9 Search PubMed
- A. Canales, A. Mallagaray, J. Pérez-Castells, I. Boos, C. Unverzagt, S. André, H. J. Gabius, F. J. Cañada and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2013, 52, 13789–13793 CrossRef CAS PubMed
- R. M. F. Van Der Put, C. Smitsman, A. De Haan, M. Hamzink, H. Timmermans, J. Uittenbogaard, J. Westdijk, M. Stork, O. Ophorst, F. Thouron, C. Guerreiro, P. J. Sansonetti, A. Phalipon and L. A. Mulard, ACS Cent. Sci., 2022, 8, 449–460 CrossRef CAS PubMed
- P. H. Seeberger, Chem. Rev., 2021, 121, 3598–3626 CrossRef CAS PubMed
- V. Verez-Bencomo, V. Fernández-Santana, E. Hardy, M. E. Toledo, M. C. Rodríguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera, M. Izquierdo, A. Villar, Y. Valdés, K. Cosme, M. L. Deler, M. Montane, E. Garcia, A. Ramos, A. Aguilar, E. Medina, G. Toraño, I. Sosa, I. Hernandez, R. Martínez, A. Muzachio, A. Carmenates, L. Costa, F. Cardoso, C. Campa, M. Diaz and R. Roy, Science, 2004, 305, 522–525 CrossRef CAS PubMed
- C. J. Crawford, M. P. Wear, D. F. Q. Smith, C. D’Errico, S. A. McConnell, A. Casadevall and S. Oscarson, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2016198118 CrossRef CAS PubMed
- S. Cecioni, R. A. Ashmus, P. A. Gilormini, S. Zhu, X. Chen, X. Shan, C. Gros, M. C. Deen, Y. Wang, R. Britton and D. J. Vocadlo, Nat. Chem. Biol., 2022, 18, 332–341 CrossRef CAS PubMed
- H. Kunz, J. C. Paulson and C. Unverzagt, J. Am. Chem. Soc., 1990, 112, 9308–9309 CrossRef
- T. Li, L. Liu, N. Wei, J.-Y. Yang, D. G. Chapla, K. W. Moremen and G.-J. Boons, Nat. Chem., 2019, 11, 229–236 CrossRef CAS PubMed
- P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11, 1784–1794 CrossRef CAS PubMed
- O. Blixt and T. Norberg, J. Org. Chem., 1998, 63, 2705–2710 CrossRef CAS PubMed
- S. Hanashima, S. Manabe and Y. Ito, Angew. Chem., Int. Ed., 2005, 44, 4218–4224 CrossRef CAS PubMed
- P. Sears and C. H. Wong, Science, 2001, 291, 2344–2350 CrossRef CAS PubMed
- A. R. Prudden, L. Liu, C. J. Capicciotti, M. A. Wolfert, S. Wang, Z. Gao, L. Meng, K. W. Moremen and G. J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6954–6959 CrossRef CAS PubMed
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. De Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379–383 CrossRef CAS PubMed
- J. P. Dolan, S. C. Cosgrove and G. J. Miller, JACS Au, 2023, 3, 47–61 CrossRef CAS PubMed
- A. D. Srivastava, L. Unione, M. Bunyatov, I. A. Gagarinov, S. Delgado, N. G. A. Abrescia, A. Ardá and G. J. Boons, Angew. Chem., Int. Ed., 2021, 60, 19287–19296 CrossRef CAS PubMed
- I. A. Gagarinov, T. Li, N. Wei, J. Sastre Toraño, R. P. de Vries, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2019, 58, 10547–10552 CrossRef CAS PubMed
- T. Li, L. Liu, N. Wei, J.-Y. Yang, D. G. Chapla, K. W. Moremen and G.-J. Boons, Nat. Chem., 2019, 11, 229–236 CrossRef CAS PubMed
- L. Liu, A. R. Prudden, C. J. Capicciotti, G. P. Bosman, J.-Y. Yang, D. G. Chapla, K. W. Moremen and G.-J. Boons, Nat. Chem., 2019, 11, 161–169 CrossRef CAS PubMed
- R. Karlsson, P. Chopra, A. Joshi, Z. Yang, S. Y. Vakhrushev, T. M. Clausen, C. D. Painter, G. P. Szekeres, Y.-H. Chen, D. R. Sandoval, L. Hansen, J. D. Esko, K. Pagel, D. P. Dyer, J. E. Turnbull, H. Clausen, G.-J. Boons and R. L. Miller, Sci. Adv., 2021, 7, 6026 CrossRef PubMed
- R. J. Fair, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2015, 51, 6183–6185 RSC
- H. Kondo, Y. Ichikawa and C. H. Wong, J. Am. Chem. Soc., 1992, 114, 8748–8750 CrossRef CAS
- C. H. Wong, S. L. Haynie and G. M. Whitesides, J. Org. Chem., 1982, 47, 5416–5418 CrossRef CAS
- D. G. Drueckhammer and C. H. Wong, J. Org. Chem., 1985, 50, 5912–5913 CrossRef CAS
- C. H. Wong, M. Schuster, P. Wang and P. Sears, J. Am. Chem. Soc., 1993, 115, 5893–5901 CrossRef CAS
- E. Mucha, A. Isabel Gonzμlez Flórez, M. Marianski, D. A. Thomas, W. Hoffmann, W. B. Struwe, H. S. Hahm, S. Gewinner, W. Schçllkopf, P. H. Seeberger, G. von Helden, K. Pagel, E. Mucha, D. Flórez, D. arianski, D. AThomas, W. Hoffmann, S. Gewinner, W. Schçllkopf, D. Helden, D. SHahm, D. HSeeberger and D. BStruwe, Angew. Chem., Int. Ed., 2017, 56, 11248–11251 CrossRef CAS PubMed
- K. Pagel and D. J. Harvey, Anal. Chem., 2013, 85, 5138–5145 CrossRef CAS PubMed
- J. Hofmann and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 8342–8349 CrossRef CAS PubMed
- C. J. Gray, L. G. Migas, P. E. Barran, K. Pagel, P. H. Seeberger, C. E. Eyers, G. J. Boons, N. L. B. Pohl, I. Compagnon, G. Widmalm and S. L. Flitsch, J. Am. Chem. Soc., 2019, 141, 14463–14479 CrossRef CAS PubMed
- C. J. Gray, I. Compagnon and S. L. Flitsch, Curr. Opin. Struct. Biol., 2020, 62, 121–131 CrossRef CAS PubMed
- B. Schindler, L. Barnes, G. Renois, C. Gray, S. Chambert, S. Fort, S. Flitsch, C. Loison, A. R. Allouche and I. Compagnon, Nat. Commun., 2017, 8, 1–7 CrossRef CAS PubMed
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed
-
T. P. Nevell and H. S. Zeronian, Cellulose chemistry and its applications, Ellis Horwood Limited, Chichester, 1st edn, 1985 Search PubMed
- S. G. Ball, M. H. B. J. Van De Wal and R. G. F. Visser, Trends Plant Sci., 1998, 3, 462–467 CrossRef
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1982, 21, 155–173 CrossRef
- P. J. Garegg, Adv. Carbohydr. Chem. Biochem., 1997, 52, 179–205 CrossRef CAS PubMed
-
P. H. Seeberger and H. S. Overkleeft, Chemical Synthesis of Glycans and Glycoconjugates, Cold Spring Harbor Laboratory Press, 2022 Search PubMed
- Sébastien Vidal, Protecting Groups, Wiley, 2019.
-
S. Oscarson and A. N. Cheallaigh, Comprehensive Glycoscience, Elsevier, 2021, vol. 2, pp. 1–48 Search PubMed
-
T. K. Lindhorst, Essentials of Carbohydrate Chemistry and Biochemistry, 2007, pp. 143–146 Search PubMed
-
P. H. Seeberger, Essentials of Glycobiology, Chapter 2, Monosaccharide Diversity, Cold Spring Harbor Laboratory Press, 4th edn, 2022 Search PubMed
- W. S. York, A. G. Darvill, M. McNeil and P. Albersheim, Carbohydr. Res., 1985, 138, 109–126 CrossRef CAS
- M. Bligh, N. Nguyen, H. Buck-Wiese, S. Vidal-Melgosa and J.-H. Hehemann, Curr. Opin. Chem. Biol., 2022, 71, 102204 CrossRef CAS PubMed
- P. Luong and D. H. Dube, Bioorg. Med. Chem., 2021, 42, 116268 CrossRef CAS PubMed
- T. Sugai, G. J. Shen, Y. Ichikawa and C. H. Wong, J. Am. Chem. Soc., 1993, 115, 413–421 CrossRef CAS
- H. B. Pfister and L. A. Mulard, Org. Lett., 2014, 16, 4892–4895 CrossRef CAS PubMed
- P. Wang, C. Huo, S. Lang, K. Caution, S. T. Nick, P. Dubey, R. Deora and X. Huang, Angew. Chem., Int. Ed., 2020, 59, 6451–6458 CrossRef CAS PubMed
- M. Emmadi and S. S. Kulkarni, J. Org. Chem., 2018, 83, 14323–14337 CrossRef CAS PubMed
- M. Brasholz and H. U. Reissig, Angew. Chem., Int. Ed., 2007, 46, 1634–1637 CrossRef CAS PubMed
- C. Bernlind and S. Oscarson, J. Org. Chem., 1998, 63, 7780–7788 CrossRef CAS
- P. Kosma, Tetrahedron Lett., 2016, 57, 2133–2142 CrossRef CAS PubMed
- D. Crich and E. Onobun, J. Org. Chem., 2020, 85, 16035–16042 CrossRef PubMed
- S. Yang, C. J. Chu and T. L. Lowary, Org. Lett., 2022, 24, 5614–5618 CrossRef CAS PubMed
- Q. Li, S. M. Levi, C. C. Wagen, A. E. Wendlandt and E. N. Jacobsen, Nature, 2022, 608, 74–79 CrossRef CAS PubMed
- H. M. Carder, Y. Wang and A. E. Wendlandt, J. Am. Chem. Soc., 2022, 144, 11870–11877 CrossRef CAS PubMed
- Y. Feng, J. Dong, F. Xu, A. Liu, L. Wang, Q. Zhang and Y. Chai, Org. Lett., 2015, 17, 2388–2391 CrossRef CAS PubMed
- E. Glibstrup and C. M. Pedersen, Org. Lett., 2016, 18, 4424–4427 CrossRef CAS PubMed
- M. Guberman, B. Pieber and P. H. Seeberger, Org. Process Res. Dev., 2019, 23, 2764–2770 CrossRef CAS
- T. G. Frihed, C. M. Pedersen and M. Bols, Eur. J. Org. Chem., 2014, 7924–7939 CrossRef CAS
- M. Emmadi and S. S. Kulkarni, Nat. Protoc., 2013, 8, 1870–1889 CrossRef CAS PubMed
- D. Leonori and P. H. Seeberger, Org. Lett., 2012, 14, 4954–4957 CrossRef CAS PubMed
- J. Van Mechelen, J. Voorneveld, H. S. Overkleeft, D. V. Filippov, G. A. Van Der Marel and J. D. C. Codée, Org. Biomol. Chem., 2020, 18, 2834–2837 RSC
- S. R. Sanapala and S. S. Kulkarni, J. Am. Chem. Soc., 2016, 138, 4938–4947 CrossRef CAS PubMed
- P. Ngoje and D. Crich, J. Am. Chem. Soc., 2020, 142, 7760–7764 CrossRef CAS PubMed
- D. Crich and C. Navuluri, Org. Lett., 2011, 13, 6288–6291 CrossRef CAS PubMed
- E. A. Voight, C. Rein and S. D. Burke, J. Org. Chem., 2002, 67, 8489–8499 CrossRef CAS PubMed
- M. G. Banwell, N. L. Hungerford and K. A. Jolliffe, Org. Lett., 2004, 6, 2737–2740 CrossRef CAS PubMed
- S. Siyabalapitiya Arachchige and D. Crich, Org. Lett., 2022, 24, 2998–3002 CrossRef CAS PubMed
- S. Matthies, P. Stallforth and P. H. Seeberger, J. Am. Chem. Soc., 2015, 137, 2848–2851 CrossRef CAS PubMed
- D. Leonori and P. H. Seeberger, Beilstein J. Org. Chem., 2013, 9, 332–341 CrossRef CAS PubMed
- Y. Wang, H. M. Carder and A. E. Wendlandt, Nature, 2020, 578, 403–408 CrossRef CAS
- B. T. Menavuvu, W. Poonperm, K. Leang, N. Noguchi, H. Okada, K. Morimoto, T. B. Granström, G. Takada and K. Izumori, J. Biosci. Bioeng., 2006, 101, 340–345 CrossRef CAS PubMed
- R. K. Singh, J. Sianturi and P. H. Seeberger, Org. Lett., 2022, 24, 2371–2375 CrossRef CAS PubMed
- S. Zhang, M. Sella, J. Sianturi, P. Priegue, D. Shen and P. H. Seeberger, Angew. Chem., Int. Ed., 2021, 60, 14679–14692 CrossRef CAS PubMed
- G. C. Sati, V. A. Sarpe, T. Furukawa, S. Mondal, M. Mantovani, S. N. Hobbie, A. Vasella, E. C. Böttger and D. Crich, ACS Infect. Dis., 2019, 5, 1718–1730 CrossRef CAS PubMed
- M. M. Bharali and A. Santra, J. Org. Chem., 2023, 88, 8770–8780 CrossRef CAS PubMed
-
P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2006 Search PubMed
- P. J. Garegg, H. Hultberg and S. Wallin, Carbohydr. Res., 1982, 108, 97–101 CrossRef
- M. Sakagami and H. Hamana, Tetrahedron Lett., 2000, 41, 5547–5551 CrossRef CAS
- A. Borbás, Z. B. Szabó, L. Szilágyi, A. Bényei and A. Lipták, Tetrahedron, 2002, 58, 5723–5732 CrossRef
-
P. G. Goekjian and S. Vidal, Protecting Groups, Wiley, 2019, pp. 109–143 Search PubMed
- C. R. Shie, Z. H. Tzeng, S. S. Kulkarni, B. J. Uang, C. Y. Hsu and S. C. Hung, Angew. Chem., Int. Ed., 2005, 44, 1665–1668 CrossRef CAS PubMed
- C. C. Wang, S. S. Kulkarni, J. C. Lee, S. Y. Luo and S. C. Hung, Nat. Protoc., 2008, 3, 97–113 CrossRef CAS PubMed
- A. A. Joseph, V. P. Verma, X. Y. Liu, C. H. Wu, V. M. Dhurandhare and C. C. Wang, Eur. J. Org. Chem., 2012, 744–753 CrossRef CAS
- C. C. Wang, J. C. Lee, S. Y. Luo, S. S. Kulkarni, Y. W. Huang, C. C. Lee, K. L. Chang and S. C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS
- K. A. D’Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058–11066 CrossRef PubMed
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed
- D. Lee, C. L. Williamson, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260–8267 CrossRef CAS PubMed
- C. Gouliaras, D. Lee, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 13926–13929 CrossRef CAS PubMed
- D. Lee and M. S. Taylor, Synthesis, 2012, 3421–3431 CAS
- D. Lee and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 3724–3727 CrossRef CAS PubMed
- M. S. Taylor, Acc. Chem. Res., 2015, 48, 295–305 CrossRef CAS PubMed
- S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS
- Y. Zhou, K.-S. Liao, T.-Y. Chen, Y. S. Y. Hsieh and C.-H. Wong, J. Org. Chem., 2023, 88, 7141–7151 CrossRef CAS PubMed
- C. Romanò and S. Oscarson, Org. Biomol. Chem., 2019, 17, 2265–2278 RSC
- M. Guberman, M. Bräutigam and P. H. Seeberger, Chem. Sci., 2019, 10, 5634–5640 RSC
- J. Sianturi, P. Priegue, J. Hu, J. Yin and P. H. Seeberger, Angew. Chem., Int. Ed., 2022, 61, e202209556 CrossRef CAS
- I. A. Gagarinov, T. Fang, L. Liu, A. D. Srivastava and G. J. Boons, Org. Lett., 2015, 17, 928–931 CrossRef CAS PubMed
- L. Sun, P. Chopra and G. J. Boons, J. Org. Chem., 2020, 85, 16082–16098 CrossRef CAS PubMed
-
S. Vidal, Protecting Groups, Wiley, 2019, pp. 169–200 Search PubMed
- M. Tsutsui, J. Sianturi, S. Masui, K. Tokunaga, Y. Manabe and K. Fukase, Eur. J. Org. Chem., 2020, 1802–1810 CrossRef CAS
- R. K. Singh, J. Sianturi and P. H. Seeberger, Org. Lett., 2022, 24, 2371–2375 CrossRef CAS PubMed
- J. D. M. Olsson, L. Eriksson, M. Lahmann and S. Oscarson, J. Org. Chem., 2008, 73, 7181–7188 CrossRef CAS PubMed
- C. de Meo, A. V. Demchenko and G. J. Boons, J. Org. Chem., 2001, 66, 5490–5497 CrossRef CAS PubMed
- M. Reihill, V. Fournière, A. N. Cheallaigh, J. O. Edlund, G. J. Miller, T. Borén, M. Lahmann and S. Oscarson, Chem. – Eur. J., 2023, 29, e202203672 CrossRef CAS
- A. Behera, D. Rai and S. S. Kulkarni, J. Am. Chem. Soc., 2020, 142, 456–467 CrossRef CAS PubMed
- C. Qin, L. Li, G. Tian, M. Ding, S. Zhu, W. Song, J. Hu, P. H. Seeberger and J. Yin, J. Am. Chem. Soc., 2022, 144, 21068–21079 CrossRef CAS PubMed
- F. Bélot, C. Guerreiro, F. Baleux and L. A. Mulard, Chem. – Eur. J., 2005, 11, 1625–1635 CrossRef PubMed
- H. D. Premathilake, C. P. Gobble, P. Pornsuriyasak, T. Hardimon, A. V. Demchenko and C. De Meo, Org. Lett., 2012, 14, 1126–1129 CrossRef CAS PubMed
- C. De Meo, C. E. Wallace and S. A. Geringer, Org. Lett., 2014, 16, 2676–2679 CrossRef CAS PubMed
- M. J. Gaunt, J. Yu and J. B. Spencer, J. Org. Chem., 1998, 63, 4172–4173 CrossRef CAS
- D. Lloyd, M. Bylsma, D. K. Bright, X. Chen and C. S. Bennett, J. Org. Chem., 2017, 82, 3926–3934 CrossRef CAS PubMed
- A. Geert Volbeda, N. R. M. Reintjens, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Eur. J. Org. Chem., 2016, 5282–5293 CrossRef
- H. Paulsen, H. Biinsch, H. Pauisen and H. Bunsch, Chem. Ber., 1981, 114, 3126–3145 CrossRef CAS
- C. A. A. van Boeckel and T. Beetz, Tetrahedron Lett., 1983, 24, 3775–3778 CrossRef CAS
- J. M. A. Van Hengst, R. J. C. Hellemons, W. A. Remmerswaal, K. N. A. Van De Vrande, T. Hansen, S. Van Der Vorm, H. S. Overkleeft, G. A. Van Der Marel, J. D. C. Codée and C. Codée, Chem. Sci., 2023, 14, 1532–1542 RSC
- S. Van Der Vorm, T. Hansen, J. M. A. Van Hengst, H. S. Overkleeft, G. A. Van Der Marel and J. D. C. Codeé, Chem. Soc. Rev., 2019, 48, 4688–4706 RSC
- J. M. A. Van Hengst, R. J. C. Hellemons, W. A. Remmerswaal, K. N. A. Van De Vrande, T. Hansen, S. Van Der Vorm, H. S. Overkleeft, G. A. Van Der Marel, J. D. C. Codée and C. Codée, Chem. Sci., 2023, 14, 1532–1542 RSC
- R. U. Lemieux, Adv. Carbohydr. Chem., 1954, 9, 1–57 CAS
- S. Winstein, E. Grunwald and L. L. Ingraham, J. Am. Chem. Soc., 1948, 70, 821–828 CrossRef CAS
- S. Winstein, E. Grunwald, R. E. Buckles and C. Hanson, J. Am. Chem. Soc., 1948, 70, 816–821 CrossRef CAS
- L. Goodman, Adv. Carbohydr. Chem., 1967, 22, 109–175 CAS
- Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Angew. Chem., Int. Ed., 2021, 60, 12597–12606 CrossRef CAS PubMed
- Y. Zhu, M. Delbianco and P. H. Seeberger, J. Am. Chem. Soc., 2021, 143, 9758–9768 CrossRef CAS PubMed
- D. Z. Vinnitskiy, V. B. Krylov, N. E. Ustyuzhanina, A. S. Dmitrenok and N. E. Nifantiev, Org. Biomol. Chem., 2016, 14, 598–611 RSC
- L. Wang, H. S. Overkleeft, G. A. Van Der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed
- E. D. Kazakova, D. V. Yashunsky, V. B. Krylov, J. P. Bouchara, M. Cornet, I. Valsecchi, T. Fontaine, J. P. Latgé and N. E. Nifantiev, J. Am. Chem. Soc., 2020, 142, 1175–1179 CrossRef CAS PubMed
- K. Le Mai Hoang and X. W. Liu, Nat. Commun., 2014, 5, 1–10 Search PubMed
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 11942–11953 CrossRef CAS PubMed
- O. T. Tuck, E. T. Sletten, J. Danglad-Flores and P. H. Seeberger, Angew. Chem., Int. Ed., 2022, 61, e202115433 CrossRef CAS PubMed
- D. Crich and S. Sun, J. Am. Chem. Soc., 1997, 119, 11217–11223 CrossRef CAS
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed
-
L. Bohé and D. Crich, Comprehensive Organic Synthesis, 2nd edn, 2014, vol. 6, pp. 1–33 Search PubMed
- T. Hansen, L. Lebedel, W. A. Remmerswaal, S. Van Der Vorm, D. P. A. Wander, M. Somers, H. S. Overkleeft, D. V. Filippov, J. Désiré, A. Mingot, Y. Bleriot, G. A. Van Der Marel, S. Thibaudeau and J. D. C. Codée, ACS Cent. Sci., 2019, 5, 781–788 CrossRef CAS PubMed
- S. R. Lu, Y. H. Lai, J. H. Chen, C. Y. Liu and K. K. T. Mong, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed
- L. O. Kononov, N. N. Malysheva, A. V. Orlova, A. I. Zinin, T. V. Laptinskaya, E. G. Kononova and N. G. Kolotyrkina, Eur. J. Org. Chem., 2012, 1926–1934 CrossRef CAS
- H. Thøgersen, R. U. Lemieux, K. Bock and B. Meyer, Can. J. Chem., 1982, 60, 44–57 CrossRef
- K. Bock and R. U. Lemieux, Carbohydr. Res., 1982, 100, 63–74 CrossRef CAS
- I. V. Alabugin, L. Kuhn, N. V. Krivoshchapov, P. Mehaffy and M. G. Medvedev, Chem. Soc. Rev., 2021, 50, 10212–10252 RSC
- D. Crich and S. Sun, Tetrahedron, 1998, 54, 8321–8348 CrossRef CAS
- P. J. Garegg, Adv. Carbohydr. Chem. Biochem., 1997, 52, 179–205 CrossRef CAS PubMed
- A. Kuboki, T. Sekiguchi, T. Sugai and H. Ohta, Synlett, 1998, 479–482 CrossRef CAS
- J. R. Pougny and P. Sinaÿ, Tetrahedron Lett., 1976, 17, 4073–4076 CrossRef
- I. Braccini, C. Derouet, J. Esnault, C. H. de Penhoat, J. M. Mallet, V. Michon and P. Sinaÿ, Carbohydr. Res., 1993, 246, 23–41 CrossRef CAS
- G. Wulff and G. Röhle, Angew. Chem., Int. Ed. Engl., 1974, 13, 157–170 CrossRef CAS PubMed
- A. Demchenko, T. Stauch and G. J. Boons, Synlett, 1997, 818–820 CrossRef CAS
- G. Wulff and G. Röhle, Angew. Chem., Int. Ed. Engl., 1974, 13, 157–170 CrossRef CAS PubMed
- H. Lönn, J. Carbohydr. Chem., 1987, 6, 301–306 CrossRef
- A. Ishiwata, Y. Munemura and Y. Ito, Tetrahedron, 2008, 64, 92–102 CrossRef CAS
- R. Eby and C. Schuerch, Carbohydr. Res., 1974, 34, 79–90 CrossRef CAS
- P. J. Garegg, L. Olsson and S. Oscarson, J. Carbohydr. Chem., 1993, 12, 955–967 CrossRef CAS
- M. Alpe, S. Oscarson and P. Svahnberg, J. Carbohydr. Chem., 2004, 23, 403–416 CrossRef CAS
- L. Guazzelli, R. Ulc, L. Rydner and S. Oscarson, Org. Biomol. Chem., 2015, 13, 6598–6610 RSC
- L. Guazzelli, R. Ulc and S. Oscarson, ChemistryOpen, 2015, 4, 729–739 CrossRef CAS PubMed
- H. Lönn, J. Carbohydr. Chem., 1987, 6, 301–306 CrossRef
-
S. Gerber-Lemaire and P. Vogel, Carbohydrate Chemistry: Chemical and Biological Approaches, eds. A. Pilar Rauter and T. Lindhorst, Royal Society of Chemistry, Cambridge, 2009, vol. 35, pp. 13–32 Search PubMed
- R. U. Lemieux, K. B. Hendriks, R. V. Stick and K. James, J. Am. Chem. Soc., 1975, 97, 4056–4062 CrossRef CAS
- J. C. Hu, A. F. W. Feng, B. Y. Chang, C. H. Lin and K. K. T. Mong, Org. Biomol. Chem., 2017, 15, 5345–5356 RSC
- T. Polat and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 12795–12800 CrossRef CAS PubMed
- X. Huang, L. Huang, H. Wang and X. S. Ye, Angew. Chem., Int. Ed., 2004, 43, 5221–5224 CrossRef CAS PubMed
- F. Burkhart, Z. Zhang, S. Wacowich-Sgarbi and C.-H. Wong, Angew. Chem., Int. Ed., 2001, 40, 1274–1277 CrossRef CAS PubMed
- X. S. Ye and C. H. Wong, J. Org. Chem., 2000, 65, 2410–2431 CrossRef CAS PubMed
- Z. Wang, L. Zhou, K. El-Boubbou, X. S. Ye and X. Huang, J. Org. Chem., 2007, 72, 6409–6420 CrossRef CAS PubMed
- M. Lahmann and S. Oscarson, Can. J. Chem., 2002, 80, 889–893 CrossRef CAS
- J. C. Lee, C. Y. Wu, J. V. Apon, G. Siuzdak and C. H. Wong, Angew. Chem., Int. Ed., 2006, 45, 2753–2757 CrossRef CAS PubMed
- C. W. Cheng, Y. Zhou, W. H. Pan, S. Dey, C. Y. Wu, W. L. Hsu and C. H. Wong, Nat. Commun., 2018, 9, 5202 CrossRef CAS PubMed
- H. K. Lee, C. N. Scanlan, C. Y. Huang, A. Y. Chang, D. A. Calarese, R. A. Dwek, P. M. Rudd, D. R. Burton, I. A. Wilson and C. H. Wong, Angew. Chem., Int. Ed., 2004, 43, 1000–1003 CrossRef CAS PubMed
- T. K. Ritter, K. K. T. Mong, H. Liu, T. Nakatani and C. H. Wong, Angew. Chem., Int. Ed., 2003, 42, 4657–4660 CrossRef CAS PubMed
- Z. Zhang, I. R. Ollmann, X. S. Ye, R. Wischnat, T. Baasov and C. H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS
- C. Chang, M. Lin, C. Chan, K. Su, C. Wu, W. Lo, S. Lam, Y. Cheng, P. Liao, C. Wong and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 12413–12423 CrossRef CAS PubMed
- S. Van Der Vorm, T. Hansen, J. M. A. Van Hengst, H. S. Overkleeft, G. A. Van Der Marel and J. D. C. Codeé, Chem. Soc. Rev., 2019, 48, 4688–4706 RSC
- M. Heuckendorff, C. M. Pedersen and M. Bols, J. Org. Chem., 2013, 78, 7234–7248 CrossRef CAS PubMed
- M. B. Cid, F. Alfonso and M. Martín-Lomas, Chem. – Eur. J., 2005, 11, 928–938 CrossRef CAS PubMed
- M. Islam, G. Gayatri and S. Hotha, J. Org. Chem., 2015, 80, 7937–7945 CrossRef CAS PubMed
- J. Y. Baek, B. Y. Lee, M. G. Jo and K. S. Kim, J. Am. Chem. Soc., 2009, 131, 17705–17713 CrossRef CAS PubMed
- B. S. Komarova, M. V. Orekhova, Y. E. Tsvetkov and N. E. Nifantiev, Carbohydr. Res., 2014, 384, 70–86 CrossRef CAS PubMed
- A. Joosten, M. Boultadakis-Arapinis, V. Gandon, L. Micouin and T. Lecourt, J. Org. Chem., 2017, 82, 3291–3297 CrossRef CAS PubMed
- S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2018, 130, 8372–8376 CrossRef
- T. J. Martin and R. R. Schmidt, Tetrahedron Lett., 1992, 33, 6123–6126 CrossRef CAS
- S. Xiang, K. L. M. Hoang, J. He, Y. J. Tan and X.-W. Liu, Angew. Chem., Int. Ed., 2015, 54, 604–607 CrossRef CAS PubMed
- P. J. Garegg, P. Konradsson, I. Kvarnström, T. Norberg, S. C. T. Svensson and B. Wigilius, Acta Chem. Scand., 1985, 39, 569–577 CrossRef
- B. Schumann, S. G. Parameswarappa, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira, P. H. Seeberger, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira, P. H. Seeberger and B. Schumann, Angew. Chem., Int. Ed., 2016, 55, 14431–14434 CrossRef CAS PubMed
- J. M. A. van Hengst, R. J. C. Hellemons, W. A. Remmerswaal, K. N. A. van de Vrande, T. Hansen, S. van der Vorm, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Chem. Sci., 2023, 14, 1532–1542 RSC
- N. Teumelsan and X. Huang, J. Org. Chem., 2007, 72, 8976–8979 CrossRef CAS PubMed
- J. L. Yong, K. Lee, H. J. Eun, B. J. Heung and S. K. Kwan, Org. Lett., 2005, 7, 3263–3266 CrossRef PubMed
- L. Guazzelli, O. McCabe and S. Oscarson, Carbohydr. Res., 2016, 433, 5–13 CrossRef CAS PubMed
- S. Buda, P. Gołębiowska and J. Mlynarski, Eur. J. Org. Chem., 2013, 3988–3991 CrossRef CAS
- Y. Hu, K. Yu, L. L. Shi, L. Liu, J. J. Sui, D. Y. Liu, B. Xiong and J. S. Sun, J. Am. Chem. Soc., 2017, 139, 12736–12744 CrossRef CAS PubMed
- S. Medina, M. J. Harper, E. I. Balmond, S. Miranda, G. E. M. Crisenza, D. M. Coe, E. M. McGarrigle and M. C. Galan, Org. Lett., 2016, 18, 4222–4225 CrossRef CAS PubMed
- R. Castelli, S. Schindler, S. M. Walter, F. Kniep, H. S. Overkleeft, G. A. Van Der Marel, S. M. Huber and J. D. C. Codée, Chem. – Asian J., 2014, 9, 2095–2098 CrossRef CAS PubMed
- P. Grice, S. V. Ley, J. Pietruszka, H. W. M. Priepke and E. P. E. Walther, Synlett, 1995, 781–784 CrossRef CAS
- S. Buda, M. Nawój, P. Gołębiowska, K. Dyduch, A. Michalak and J. Mlynarski, J. Org. Chem., 2015, 80, 770–780 CrossRef CAS PubMed
- T. H. Schmidt and R. Madsen, Eur. J. Org. Chem., 2007, 3935–3941 CrossRef CAS
- T. Hashihayata, H. Mandai and T. Mukaiyama, Chem. Lett., 2003, 32, 442–443 CrossRef CAS
- P. I. Abronina, K. G. Fedina, N. M. Podvalnyy, A. I. Zinin, A. O. Chizhov, N. N. Kondakov, V. I. Torgov and L. O. Kononov, Carbohydr. Res., 2014, 396, 25–36 CrossRef CAS PubMed
- K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee and J. Park, J. Am. Chem. Soc., 2001, 123, 8477–8481 CrossRef CAS PubMed
- S. K. Kwan, D. B. Fulse, Y. J. Baek, B. Y. Lee and B. J. Jeon, J. Am. Chem. Soc., 2008, 130, 8537–8547 CrossRef PubMed
- T. Tyrikos-Ergas, V. Bordoni, G. Fittolani, M. A. Chaube, A. Grafmüller, P. H. Seeberger and M. Delbianco, Chem. – Eur. J., 2021, 27, 2321–2325 CrossRef CAS PubMed
- D. Crich and V. Dudkin, J. Am. Chem. Soc., 2001, 123, 6819–6825 CrossRef CAS PubMed
- M. H. S. Marqvorsen, M. J. Pedersen, M. R. Rasmussen, S. K. Kristensen, R. Dahl-Lassen and H. H. Jensen, J. Org. Chem., 2017, 82, 143–156 CrossRef CAS PubMed
- D. Crich and A. U. Vinod, Org. Lett., 2003, 5, 1297–1300 CrossRef CAS PubMed
- T. Tyrikos-Ergas, V. Bordoni, G. Fittolani, M. A. Chaube, A. Grafmüller, P. H. Seeberger and M. Delbianco, Chem. – Eur. J., 2021, 27, 2321–2325 CrossRef CAS PubMed
- S. R. Sanapala and S. S. Kulkarni, J. Am. Chem. Soc., 2016, 138, 4938–4947 CrossRef CAS PubMed
- J. C. Lee, W. A. Greenberg and C. H. Wong, Nat. Protoc., 2007, 1, 3143–3152 CrossRef PubMed
- C. Chang, M. Lin, C. Chan, K. Su, C. Wu, W. Lo, S. Lam, Y. Cheng, P. Liao, C. Wong and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 12413–12423 CrossRef CAS PubMed
- D. R. Mootoo, P. Konradsson, U. Udodong and B. Fraser-Reid, J. Am. Chem. Soc., 1988, 110, 5583–5584 CrossRef CAS
- N. L. Douglas, S. V. Ley, U. Lücking and S. L. Warriner, J. Chem. Soc., Perkin Trans. 1, 1998, 51–66 RSC
- M. Heuckendorff, C. M. Pedersen and M. Bols, Chem. – Eur. J., 2010, 16, 13982–13994 CrossRef CAS PubMed
- C. M. Pedersen, L. G. Marinescu and M. Bols, Chem. Commun., 2008, 2465–2467 RSC
- C. M. Pedersen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2007, 129, 9222–9235 CrossRef CAS PubMed
- M. Jæger Pedersen and C. M. Pedersen, Angew. Chem., Int. Ed., 2021, 60, 2689–2693 CrossRef PubMed
- Z. Zhang, I. R. Ollmann, X. S. Ye, R. Wischnat, T. Baasov and C. H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS
- D. Z. Vinnitskiy, V. B. Krylov, N. E. Ustyuzhanina, A. S. Dmitrenok and N. E. Nifantiev, Org. Biomol. Chem., 2016, 14, 598–611 RSC
- K. Greis, S. Leichnitz, C. Kirschbaum, C. W. Chang, M. H. Lin, G. Meijer, G. Von Helden, P. H. Seeberger and K. Pagel, J. Am. Chem. Soc., 2022, 144, 20258–20266 CrossRef CAS PubMed
- H. S. Hahm, M. Hurevich and P. H. Seeberger, Nat. Commun., 2016, 7, 1–8 Search PubMed
- D. Crich, T. Hu and F. Cai, J. Org. Chem., 2008, 73, 8942–8953 CrossRef CAS PubMed
- T. Hansen, H. Elferink, J. M. A. van Hengst, K. J. Houthuijs, W. A. Remmerswaal, A. Kromm, G. Berden, S. van der Vorm, A. M. Rijs, H. S. Overkleeft, D. V. Filippov, F. P. J. T. Rutjes, G. A. van der Marel, J. Martens, J. Oomens, J. D. C. Codée and T. J. Boltje, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed
- M. Marianski, E. Mucha, K. Greis, S. Moon, A. Pardo, C. Kirschbaum, D. A. Thomas, G. Meijer, G. von Helden, K. Gilmore, P. H. Seeberger and K. Pagel, Angew. Chem., Int. Ed., 2020, 59, 6166–6171 CrossRef CAS PubMed
- D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506–4507 CrossRef CAS PubMed
- D. Crich and S. Sun, J. Am. Chem. Soc., 1998, 120, 435–436 CrossRef CAS
- T. Tsuda, R. Arihara, S. Sato, M. Koshiba, S. Nakamura and S. Hashimoto, Tetrahedron, 2005, 61, 10719–10733 CrossRef CAS
- R. Weingart and R. R. Schmidt, Tetrahedron Lett., 2000, 41, 8753–8758 CrossRef CAS
- S. Oscarson and F. W. Sehgelmeble, J. Am. Chem. Soc., 2000, 122, 8869–8872 CrossRef CAS
- T. L. Lowary and B. S. Huang, J. Org. Chem., 2020, 85, 15895–15907 CrossRef PubMed
- N. Komura, K. Kato, T. Udagawa, S. Asano, H. N. Tanaka, A. Imamura, H. Ishida, M. Kiso and H. Ando, Science, 2019, 364, 677–680 CrossRef CAS PubMed
- M. Heuckendorff, J. Bendix, C. M. Pedersen and M. Bols, Org. Lett., 2014, 16, 1116–1119 CrossRef CAS PubMed
- A. Imamura, H. Ando, S. Korogi, G. Tanabe, O. Muraoka, H. Ishida and M. Kiso, Tetrahedron Lett., 2003, 44, 6725–6728 CrossRef CAS
- A. Imamura, H. Ando, H. Ishida and M. Kiso, Org. Lett., 2005, 7, 4415–4418 CrossRef CAS PubMed
- V. N. Thota, M. J. Ferguson, R. P. Sweeney and T. L. Lowary, Angew. Chem., Int. Ed., 2018, 57, 15592–15596 CrossRef CAS PubMed
- L. Morelli, F. Compostella, L. Panza and D. Imperio, Carbohydr. Res., 2022, 519, 108625 CrossRef CAS PubMed
- M. M. Mukherjee, R. Ghosh and J. A. Hanover, Front. Mol. Biosci., 2022, 9, 896187 CrossRef CAS PubMed
- C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed
- Y. Singh, S. A. Geringer and A. V. Demchenko, Chem. Rev., 2022, 122, 11701–11758 CrossRef CAS PubMed
- S. S. Kulkarni, C. C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P. H. Liao and S. C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed
- M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS PubMed
- G. Wulff and G. Röhle, Angew. Chem., Int. Ed. Engl., 1974, 13, 157–170 CrossRef CAS PubMed
- C. T. Bishop and F. P. Cooper, Can. J. Chem., 1963, 41, 2743–2758 CrossRef
-
P. J. Garegg, in Advances in Carbohydrate Chemistry and Biochemistry, Academic Press, 2004, vol. 59, pp. 69–134 Search PubMed
- T. Reichstein, A. Grüssner and R. Oppenauer, Helv. Chim. Acta, 1933, 16, 561–565 CrossRef CAS
- D. Arosio, I. Vrasidas, P. Valentini, R. M. J. Liskamp, R. J. Pieters and A. Bernardi, Org. Biomol. Chem., 2004, 2, 2113–2124 RSC
- S. Sattin, A. Daghett, M. Thépaut, A. Berzi, M. Sánchez-Navarro, G. Tabarani, J. Rojo, F. Fieschi, M. Clerici and A. Bernardi, ACS Chem. Biol., 2010, 5, 301–312 CrossRef CAS PubMed
- C. H. Lai, C. Y. Lin, H. T. Wu, H. S. Chan, Y. J. Chuang, C. T. Chen and C. C. Lin, Adv. Funct. Mater., 2010, 20, 3948–3958 CrossRef CAS
- R. T. C. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393–1398 CrossRef CAS PubMed
- A. L. Marlow and L. L. Kiessling, Org. Lett., 2001, 3, 2517–2519 CrossRef CAS PubMed
- C. d’Errico, J. O. Jørgensen, K. B. R. M. Krogh, N. Spodsberg, R. Madsen and R. N. Monrad, Biotechnol. Bioeng., 2015, 112, 914–922 CrossRef PubMed
- V. Benessere, R. Del Litto, A. De Roma and F. Ruffo, Coord. Chem. Rev., 2010, 254, 390–401 CrossRef CAS
- T. D. Inch and G. J. Lewis, Carbohydr. Res., 1972, 22, 91–101 CrossRef CAS
- F. Valentin, Collect. Czech. Chem. Commun., 1932, 4, 364–375 CrossRef CAS
-
S. Tipson, Methods Carbohydr. Chem., 1963, vol. 2 Search PubMed
- P. A. J. Gorin and A. S. Perlin, Can. J. Chem., 1961, 39, 2474–2485 CrossRef CAS
-
O. Th. Schmidt, Angew. Chem., Int. Ed. Engl., 1963, 75, 351–352 Search PubMed
- N. J. Leonard and K. L. Carraway, J. Heterocycl. Chem., 1966, 3, 485–489 CrossRef CAS
- P. A. Levene and E. T. Stiller, J. Biol. Chem., 1934, 104, 299–306 CrossRef CAS
- A. Lubineau and J.-C. Fischer, Synth. Commun., 1991, 21, 815–818 CrossRef CAS
- W. Koenigs and E. Knorr, Ber. Dtsch. Chem. Ges., 1901, 34, 957–981 CrossRef
- G. Zemplén and Z. S. Nagy, Ber. Dtsch. Chem. Ges., 1930, 63, 368 CrossRef
- B. Helferich and K.-F. Wedemeyer, Liebigs Ann. Chem., 1949, 563, 139–145 CrossRef CAS
-
E. Fischer, Untersuchungen Über Kohlenhydrate und Fermente II (1908–1919), 1922, pp. 251–257 Search PubMed
-
E. Fischer and H. Fischer, Untersuchungen Über Kohlenhydrate und Fermente II (1908–1919), 1922, pp. 218–233 Search PubMed
-
E. Fischer and L. v Mechel, Untersuchungen Über Kohlenhydrate und Fermente II (1908–1919), 1922, pp. 48–55 Search PubMed
-
R. U. Lemieux, T. Takeda and B. Y. Chung, Synthetic Methods for Carbohydrates, 1977, pp. 90–115 Search PubMed
- J. Gervay and M. J. Hadd, J. Org. Chem., 1997, 62, 6961–6967 CrossRef CAS
- A. Michael, Am. Chem. J., 1879, 1, 305–312 Search PubMed
- S. Koto, T. Yoshida, K. Takenaka and S. Zen, Bull. Chem. Soc. Jpn., 1982, 55, 3667–3668 CrossRef CAS
- S. Koto, N. Morishima, S. Shichi, H. Haigoh, M. Hirooka, M. Okamoto, T. Higuchi, K. Shimizu, Y. Hashimoto, T. Irisawa, H. Kawasaki, Y. Takahashi, M. Yamazaki, Y. Mori, K. Kudo, T. Ikegaki, S. Suzuki and S. Zen, Bull. Chem. Soc. Jpn., 1992, 65, 3257–3274 CrossRef CAS
- K. P. R. Kartha and H. J. Jennings, J. Carbohydr. Chem., 1990, 9, 777–781 CrossRef CAS
- E. Fischer and B. Helferich, Ann. Chem., 1911, 383, 68–91 CrossRef CAS
- I. Pongener, K. Nikitin and E. M. McGarrigle, Org. Biomol. Chem., 2019, 17, 7531–7535 RSC
- J. Thiem and B. Meyer, Chem. Ber., 1980, 113, 3075–3085 CrossRef CAS
- D. H. Brauns, J. Am. Chem.
Soc., 1923, 45, 833–835 CrossRef CAS
- W. Rosenbrook, D. A. Riley and P. A. Lartey, Tetrahedron Lett., 1985, 26, 3–4 CrossRef CAS
- W. Koenigs and E. Knorr, Ber. Dtsch. Chem. Ges., 1901, 34, 957–981 CrossRef
- B. Helferich and J. Zirner, Chem. Ber., 1962, 95, 2604–2611 CrossRef CAS
- R. U. Lemieux., T. Takeda. and B. Y. Chung, ACS Symp. Ser., 1976, 39, 90–115 CrossRef CAS
- T. Mukaiyama, Y. Murai and S. Shoda, Chem. Lett., 1981, 431–432 CrossRef CAS
- K. C. Nicolaou, A. Chucholowski, R. E. Dolle and J. L. Randall, J. Chem. Soc., Chem. Commun., 1984, 1155–1156 RSC
- H. Kunz and W. Sager, Helv. Chim. Acta, 1985, 68, 283–287 CrossRef CAS
- T. Matsumoto, H. Maeta, K. Suzuki and L. G. I. Tsuchihashi, Tetrahedron Lett., 1988, 29, 3567–3570 CrossRef CAS
- K. Suzuki, H. Maeta, T. Matsumoto and L. G. Ichi Tsuchihashi, Tetrahedron Lett., 1988, 29, 3571–3574 CrossRef CAS
- B. Helferich and R. Gootz, Ber. Dtsch. Chem. Ges., 1929, 62, 2788–2792 CrossRef
- S. N. Lam and J. Gervay-Hague, Carbohydr. Res., 2002, 337, 1953–1965 CrossRef CAS PubMed
- M. J. Hadd and J. Gervay, Carbohydr. Res., 1999, 320, 61–69 CrossRef CAS
- C. A. A. Van Boeckel, T. Beetz and S. F. Van Aelst, Tetrahedron, 1984, 40, 4097–4107 CrossRef CAS
- I. Pongener, D. A. Pepe, J. J. Ruddy and E. M. McGarrigle, Chem. Sci., 2021, 12, 10070–10075 RSC
- A. A. Joseph, A. Pardo-Vargas and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 8561–8564 CrossRef CAS PubMed
- E. Mancuso, C. Romanò, N. Trattnig, P. Gritsch, P. Kosma and M. H. Clausen, Chem. – Eur. J., 2021, 27, 7099–7102 CrossRef CAS PubMed
- B. Pokorny and P. Kosma, Carbohydr. Res., 2016, 422, 5–12 CrossRef CAS PubMed
- B. Pokorny and P. Kosma, ChemistryOpen, 2015, 4, 722 CrossRef CAS PubMed
- D. Ott, J. Seifert, I. Prahl, M. Niemietz, J. Hoffman, J. Guder, M. Mönnich and C. Unverzagt, Eur. J. Org. Chem., 2012, 5054–5068 CrossRef CAS
- G. Minzer and R. Hevey, ChemistryOpen, 2023, 12, e202200134 CrossRef CAS PubMed
- S. I. Van Kasteren, S. J. Campbell, S. Serres, D. C. Anthony, N. R. Sibson and B. G. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18–23 CrossRef CAS PubMed
- A. W. McDonagh, M. F. Mahon and P. V. Murphy, Org. Lett., 2016, 18, 552–555 CrossRef CAS PubMed
- P. Kosma, G. Schulz, F. M. Unger and H. Brade, Carbohydr. Res., 1989, 190, 191–201 CrossRef CAS PubMed
- P. Kosma, G. Schulz and H. Brade, Carbohydr. Res., 1988, 183, 183–199 CrossRef CAS PubMed
- I. Matsuo, K. Totani, A. Tatami and Y. Ito, Tetrahedron, 2006, 62, 8262–8277 CrossRef CAS
- M. Emmadi and S. S. Kulkarni, Org. Biomol. Chem., 2013, 11, 3098–3102 RSC
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Eur. J. Org. Chem., 2019, 1994–2003 CrossRef CAS PubMed
- L. Wang, Y. Zhang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Org. Chem., 2020, 85, 15872–15884 CrossRef CAS PubMed
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed
- H. S. Hahm, M. K. Schlegel, M. Hurevich, S. Eller, F. Schuhmacher, J. Hofmann, K. Pagel and P. H. Seeberger, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3385–E3389 CrossRef CAS PubMed
- K. Igarashi, Adv. Carbohydr. Chem. Biochem., 1977, 34, 243–283 CrossRef CAS
- R. J. Ferrier, R. W. Hay and N. Vethaviyasar, Carbohydr. Res., 1973, 27, 55–61 CrossRef CAS
- H. Lönn, Carbohydr. Res., 1985, 139, 105–113 Search PubMed
- S. Nilsson, H. Lönn and T. Norberg, Glycoconjugate J., 1989, 6, 21–34 CrossRef CAS PubMed
- D. J. Keith and S. D. Townsend, J. Am. Chem. Soc., 2019, 141, 12939–12945 CrossRef CAS PubMed
- C. J. Crawford and S. Oscarson, Carbohydr. Res., 2020, 497, 108150 CrossRef CAS PubMed
- D. Schmidt, F. Schuhmacher, A. Geissner, P. H. Seeberger and F. Pfrengle, Chem. – Eur. J., 2015, 21, 5709–5713 CrossRef CAS PubMed
- S. Lin and T. L. Lowary, Chem. – Eur. J., 2018, 24, 16992–16996 CrossRef CAS PubMed
- K. J. Doores and B. G. Davis, Org. Biomol. Chem., 2008, 6, 2692–2696 RSC
- E. Dimitriou and G. J. Miller, Org. Biomol. Chem., 2019, 17, 9321–9335 RSC
- P. O. Adero, D. R. Jarois and D. Crich, Carbohydr. Res., 2017, 449, 11–16 CrossRef CAS PubMed
- C. Cavedon, E. T. Sletten, A. Madani, O. Niemeyer, P. H. Seeberger and B. Pieber, Org. Lett., 2021, 23, 514–518 CrossRef CAS PubMed
- L. Guazzelli, R. Ulc and S. Oscarson, Carbohydr. Res., 2014, 389, 57–65 CrossRef CAS PubMed
- P. Fügedi and P. J. Garegg, Carbohydr. Res., 1986, 149, 9–12 CrossRef
- G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef CAS
- D. Crich and M. Smith, J. Am. Chem. Soc., 2001, 123, 9015–9020 CrossRef CAS PubMed
- Y. Ito and T. Ogawa, Tetrahedron Lett., 1988, 29, 1061–1064 CrossRef CAS
- K. P. R. Kartha, M. Aloui and R. A. Field, Tetrahedron Lett., 1996, 37, 5175–5178 CrossRef CAS
- P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS
- G. J. Miller, S. U. Hansen, M. Baráth, C. Johannessen, E. W. Blanch, G. C. Jayson and J. M. Gardiner, Carbohydr. Res., 2014, 400, 44–53 CrossRef CAS PubMed
- G. J. Miller, K. R. Broberg, C. Rudd, M. R. Helliwell, G. C. Jayson and J. M. Gardiner, Org. Biomol. Chem., 2015, 13, 11208–11219 RSC
- S. U. Hansen, G. J. Miller, C. Cole, G. Rushton, E. Avizienyte, G. C. Jayson and J. M. Gardiner, Nat. Commun., 2013, 4, 1–9 Search PubMed
- G. J. Miller, S. U. Hansen, E. Avizienyte, G. Rushton, C. Cole, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2013, 4, 3218–3222 RSC
- L. Guazzelli, C. J. Crawford, R. Ulc, A. Bowen, O. McCabe, A. J. Jedlicka, M. P. Wear, A. Casadevall and S. Oscarson, Chem. Sci., 2020, 11, 9209–9217 RSC
- H. S. Hahm, M. K. Schlegel, M. Hurevich, S. Eller, F. Schuhmacher, J. Hofmann, K. Pagel and P. H. Seeberger, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3385–3389 CrossRef PubMed
- R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21–123 CrossRef CAS PubMed
- R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed. Engl., 1980, 19, 731–732 CrossRef
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J. L. Reymond, Chem. Sci., 2015, 7, 166–182 RSC
- M. Mönnich, S. Eller, T. Karagiannis, L. Perkams, T. Luber, D. Ott, M. Niemietz, J. Hoffman, J. Walcher, L. Berger, M. Pischl, M. Weishaupt, C. Wirkner, R. G. Lichtenstein and C. Unverzagt, Angew. Chem., Int. Ed., 2016, 55, 10487–10492 CrossRef PubMed
- T. Luber, M. Niemietz, T. Karagiannis, M. Mönnich, D. Ott, L. Perkams, J. Walcher, L. Berger, M. Pischl, M. Weishaupt, S. Eller, J. Hoffman and C. Unverzagt, Angew. Chem., Int. Ed., 2018, 57, 14543–14549 CrossRef CAS PubMed
- P. Zhang, R. Hevey and C. C. Ling, J. Org. Chem., 2017, 82, 9662–9674 CrossRef CAS PubMed
- J. R. Pougny and P. Sinaÿ, Tetrahedron Lett., 1976, 17, 4073–4076 CrossRef
- P. Sinaÿ, Pure Appl. Chem., 1978, 50, 1437–1452 Search PubMed
- I. Ohashi, M. J. Lear, F. Yoshimura and M. Hirama, Org. Lett., 2004, 6, 719–722 CrossRef CAS PubMed
-
R. R. Schmidt and X. Zhu, Glycoscience, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 451–524 Search PubMed
- B. Yu and H. Tao, J. Org. Chem., 2002, 67, 9099–9102 CrossRef CAS PubMed
- B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS
- M. Adinolfi, G. Barone, A. Iadonisi and M. Schiattarella, Synlett, 2002, 269–270 CrossRef CAS
- M. Adinolfi, G. Barone, A. Iadonisi and M. Schiattarella, Tetrahedron Lett., 2002, 43, 5573–5577 CrossRef CAS
- B. Wegmann and R. R. Schmidt, J. Carbohydr. Chem., 1987, 6, 357–375 CrossRef CAS
- M. G. Hoffmann and R. R. Schmidt, Liebigs Ann. Chem., 1985, 2403–2419 CrossRef CAS
- P. Chassagne, C. Fontana, C. Guerreiro, C. Gauthier, A. Phalipon, G. Widmalm and L. A. Mulard, Eur. J. Org. Chem., 2013, 4085–4106 CrossRef CAS
- E. Eckhardt and T. Ziegler, Carbohydr. Res., 1994, 264, 253–269 CrossRef CAS PubMed
- T. Ziegler, E. Eckhardt, J. Strayle and H. Herzog, Carbohydr. Res., 1994, 253, 167–183 CrossRef CAS PubMed
- H. Kubo, K. Ishii, H. Koshino, K. Toubetto, K. Naruchi and R. Yamasaki, Eur. J. Org. Chem., 2004, 1202–1213 CrossRef CAS
- A. G. Pearson, M. J. Kiefel, V. Ferro and M. Von Itzstein, Carbohydr. Res., 2005, 340, 2077–2085 CrossRef CAS PubMed
- B. Yu and H. Tao, J. Org. Chem., 2002, 67, 9099–9102 CrossRef CAS PubMed
- B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS
- H. B. Pfister and L. A. Mulard, Org. Lett., 2014, 16, 4892–4895 CrossRef CAS PubMed
- K. Wright, C. Guerreiro, I. Laurent, F. Baleux and L. A. Mulard, Org. Biomol. Chem., 2004, 2, 1518–1527 RSC
- C. Gauthier, P. Chassagne, F. X. Theillet, C. Guerreiro, F. Thouron, F. Nato, M. Delepierre, P. J. Sansonetti, A. Phalipon and L. A. Mulard, Org. Biomol. Chem., 2014, 12, 4218–4232 RSC
- Z. Hu, M. Benkoulouche, L. A. Barel, G. Le Heiget, A. Ben Imeddourene, Y. Le Guen, N. Monties, C. Guerreiro, M. Remaud-Siméon, C. Moulis, I. André and L. A. Mulard, J. Org. Chem., 2021, 86, 2058–2075 CrossRef CAS PubMed
- F. Bélot, D. Rabuka, M. Fukuda and O. Hindsgaul, Tetrahedron Lett., 2002, 43, 7743–7747 CrossRef
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed
- P. Ghosh, N. M. Dahms and S. Kornfeld, Nat. Rev. Mol. Cell Biol., 2003, 4, 202–213 CrossRef CAS PubMed
- J. Klumperman, A. Hille, T. Veenendaal, V. Oorschot, W. Stoorvogel, K. Von Figura and H. J. Geuze, J. Cell Biol., 1993, 121, 997–1010 CrossRef CAS PubMed
- G. Griffiths, B. Hoflack, K. Simons, I. Mellman and S. Kornfeld, Cell, 1988, 52, 329–341 CrossRef CAS PubMed
- F. Huang, A. Nesterov, R. E. Carter and A. Sorkin, Traffic, 2001, 2, 345–357 CrossRef CAS PubMed
- L. F. Leloir, Science, 1971, 172, 1299–1303 CrossRef CAS PubMed
- J. E. Heidlas, K. W. Williams and G. M. Whitesides, Acc. Chem. Res., 1992, 25, 307–314 CrossRef CAS
- H. Kondo, S. Aoki, Y. Ichikawa, R. L. Halcomb, H. Ritzen and C. H. Wong, J. Org. Chem., 1994, 59, 864–877 CrossRef CAS
- D. L. Boger and T. Honda, J. Am. Chem. Soc., 1994, 116, 5647–5656 CrossRef CAS
- H. I. Duynstee, E. R. Wijsman, G. A. Van Der Marel and J. H. Van Boom, Synlett, 1996, 313–314 CrossRef CAS
- T. Inazu, H. Hosokawa and Y. Satoh, Chem. Lett., 1985, 297–300 CrossRef CAS
- S. Hashimoto, T. Honda and S. Ikegami, J. Chem. Soc., Chem. Commun., 1989, 685–687 RSC
- M. M. Sim, H. Kondo and C. H. Wong, J. Am. Chem. Soc., 1993, 115, 2260–2267 CrossRef CAS
- G. Böhm and H. Waldmann, Liebigs Ann. Chem., 1996, 613–619 Search PubMed
- O. J. Plante, R. B. Andrade and P. H. Seeberger, Org. Lett., 1999, 1, 211–214 CrossRef CAS PubMed
- M. W. Weishaupt, H. S. Hahm, A. Geissner and P. H. Seeberger, Chem. Commun., 2017, 53, 3591–3594 RSC
- P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 19–28 RSC
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424–7427 CrossRef CAS PubMed
- A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798–6799 CrossRef CAS PubMed
- R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed
- Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051–15053 CrossRef CAS PubMed
- S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS PubMed
- A. G. Wenzel and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 12964–12965 CrossRef CAS PubMed
- M. S. Taylor and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 10558–10559 CrossRef CAS PubMed
- S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198–7199 CrossRef CAS PubMed
- M. Kotke and P. R. Schreiner, Tetrahedron, 2006, 62, 434–439 CrossRef CAS
- M. Kotke and P. Schreiner, Synthesis, 2007, 779–790 CAS
- Y. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 10089–10092 CrossRef CAS PubMed
- E. I. Balmond, D. M. Coe, M. C. Galan and E. M. McGarrigle, Angew. Chem., Int. Ed., 2012, 51, 9152–9155 CrossRef CAS PubMed
- K. A. D’Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058–11066 CrossRef PubMed
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed
- A. Nakagawa, M. Tanaka, S. Hanamura, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2015, 127, 11085–11089 CrossRef
- M. Tanaka, A. Nakagawa, N. Nishi, K. Iijima, R. Sawa, D. Takahashi and K. Toshima, J. Am. Chem. Soc., 2018, 140, 3644–3651 CrossRef CAS PubMed
- M. Tanaka, J. Nashida, D. Takahashi and K. Toshima, Org. Lett., 2016, 18, 2288–2291 CrossRef CAS PubMed
- N. Nishi, J. Nashida, E. Kaji, D. Takahashi and K. Toshima, Chem. Commun., 2017, 53, 3018–3021 RSC
- N. Nishi, K. Sueoka, K. Iijima, R. Sawa, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2018, 57, 13858–13862 CrossRef CAS PubMed
- M. Tanaka, D. Takahashi and K. Toshima, Org. Lett., 2016, 18, 5030–5033 CrossRef CAS PubMed
- S. M. Levi, Q. Li, A. R. Rötheli and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 35–39 CrossRef CAS PubMed
- G. A. Bradshaw, A. C. Colgan, N. P. Allen, I. Pongener, M. B. Boland, Y. Ortin and E. M. McGarrigle, Chem. Sci., 2019, 10, 508–514 RSC
- S. Medina, M. J. Harper, E. I. Balmond, S. Miranda, G. E. M. Crisenza, D. M. Coe, E. M. McGarrigle and M. C. Galan, Org. Lett., 2016, 18, 4222–4225 CrossRef CAS PubMed
- C. Palo-Nieto, A. Sau, R. Williams and M. C. Galan, J. Org. Chem., 2017, 82, 407–414 CrossRef CAS PubMed
- C. Palo-Nieto, A. Sau and M. C. Galan, J. Am. Chem. Soc., 2017, 139, 14041–14044 CrossRef CAS PubMed
- T. Kimura, M. Sekine, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2013, 52, 12131–12134 CrossRef CAS PubMed
- S. Tomita, M. Tanaka, M. Inoue, K. Inaba, D. Takahashi and K. Toshima, J. Org. Chem., 2020, 85, 16254–16262 CrossRef CAS PubMed
- G. Báti, J.-X. He, K. B. Pal and X.-W. Liu, Chem. Soc. Rev., 2019, 48, 4006–4018 RSC
- P. J. Garegg, J.-L. Maloisel and S. Oscarson, Synthesis, 1995, 409–414 CrossRef CAS
- A. Maggi and R. Madsen, Eur. J. Org. Chem., 2013, 2683–2691 CrossRef CAS
- T. Ogawa, K. Katano and M. Matsui, Carbohydr. Res., 1978, 64, C3–C9 CrossRef CAS
- C. Cruzado, M. Bernabe and M. Martin-Lomas, Carbohydr. Res., 1990, 203, 296–301 CrossRef CAS
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed
- Y. Ye, S. Kim, R. P. King, M. Baik and S. L. Buchwald, Angew. Chem., Int. Ed., 2023, 62, e202300109 CrossRef CAS PubMed
-
R. F. Heck, Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1982, pp. 345–390 Search PubMed
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS
- R. F. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS
- D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050–17051 CrossRef CAS PubMed
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed
- F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS
- E. I. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2017 CrossRef CAS PubMed
- A. O. King, N. Okukado and E. I. Negishi, J. Chem. Soc., Chem. Commun., 1977, 683–684 RSC
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed
- B. D. Sherry, R. N. Loy and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4510–4511 CrossRef CAS PubMed
- K. N. Baryal, S. Adhikari and J. Zhu, J. Org. Chem., 2013, 78, 12469–12476 CrossRef CAS PubMed
- O. Boutureira, M. Isabel Matheu, Y. Díaz and S. Castillón, RSC Adv., 2014, 4, 19794–19799 RSC
- A. Sau, R. Williams, C. Palo-Nieto, A. Franconetti, S. Medina and M. C. Galan, Angew. Chem., Int. Ed., 2017, 56, 3640–3644 CrossRef CAS PubMed
- R. S. Babu, M. Zhou and G. A. O’Doherty, J. Am. Chem. Soc., 2004, 126, 3428–3429 CrossRef CAS PubMed
- R. S. Babu and G. A. O’Doherty, J. Am. Chem. Soc., 2003, 125, 12406–12407 CrossRef CAS PubMed
- E. T. Sletten, Y. J. Tu, H. B. Schlegel and H. M. Nguyen, ACS Catal., 2019, 9, 2110–2123 CrossRef CAS PubMed
- E. T. Sletten, S. K. Ramadugu and H. M. Nguyen, Carbohydr. Res., 2016, 435, 195–207 CrossRef CAS PubMed
- E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288–14302 CrossRef CAS PubMed
- E. A. Mensah and H. M. Nguyen, J. Am. Chem. Soc., 2009, 131, 8778–8780 CrossRef CAS PubMed
- M. Zhou and G. A. O’Doherty, Org. Lett., 2006, 8, 4339–4342 CrossRef CAS PubMed
- R. Balamurugan and S. R. Koppolu, Tetrahedron, 2009, 65, 8139–8142 CrossRef CAS
- S. A. Thadke, B. Mishra, M. Islam, S. Pasari, S. Manmode, B. V. Rao, M. Neralkar, G. P. Shinde, G. Walke and S. Hotha, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed
- Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem. – Eur. J., 2010, 16, 1871–1882 CrossRef CAS PubMed
- S. Hotha and S. Kashyap, J. Am. Chem. Soc., 2006, 128, 9620–9621 CrossRef CAS PubMed
- Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS
- G. Sureshkumar and S. Hotha, Tetrahedron Lett., 2007, 48, 6564–6568 CrossRef CAS
- D. Zhu, M. Geng and B. Yu, Angew. Chem., Int. Ed., 2022, 61, e202203239 CrossRef CAS PubMed
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Nat. Commun., 2020, 11, 1–7 CrossRef PubMed
- P. Grice, S. V. Ley, J. Pietruszka, H. M. I. Osborn, H. W. M. Priepke and S. L. Warriner, Chem. – Eur. J., 2006, 3, 431–440 CrossRef
- A. Kozioł, A. Lendzion-Paluch and A. Manikowski, Org. Process Res. Dev., 2013, 17, 869–875 CrossRef
- D. Dhara, A. Dhara, P. V. Murphy and L. A. Mulard, Carbohydr. Res., 2022, 521, 108644 CrossRef CAS PubMed
- C. Crawford and S. Oscarson, Eur. J. Org. Chem., 2020, 3332–3337 CrossRef CAS
- C. J. Crawford, Y. Qiao, Y. Liu, D. Huang, W. Yan, P. H. Seeberger, S. Oscarson and S. Chen, Org. Process Res. Dev., 2021, 25, 1573–1578 CrossRef CAS PubMed
- C. Reichardt, Angew. Chem., Int. Ed. Engl., 1965, 4, 29–40 CrossRef
- C. Reichardt, Org. Process Res. Dev., 2007, 11, 105–113 CrossRef CAS
- T. Tyrikos-Ergas, E. T. Sletten, J. Y. Huang, P. H. Seeberger and M. Delbianco, Chem. Sci., 2022, 13, 2115–2120 RSC
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Nat. Commun., 2020, 11, 1–7 Search PubMed
- C. Hu, S. Wu, F. He, D. Cai, Z. Xu, W. Ma, Y. Liu, B. Wei, T. Li and K. Ding, Angew. Chem., Int. Ed., 2022, 61, e202202554 CrossRef CAS PubMed
- D. B. Werz, B. Castagner and P. H. Seeberger, J. Am. Chem. Soc., 2007, 129, 2770–2771 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |