
Synthesis of CsPbBr3 perovskite nanocrystals with acoustically actuated millisecond mixing†
Chun Kiu
Ng‡
 a,
Hao
Deng‡
b,
Hanchen
Li
a,
Wenping
Yin
a,
Tuncay
Alan
*b and
Jacek J.
Jasieniak
*a
a,
Hao
Deng‡
b,
Hanchen
Li
a,
Wenping
Yin
a,
Tuncay
Alan
*b and
Jacek J.
Jasieniak
*a
aARC Centre of Excellence in Exciton Science, Department of Materials Science and Engineering, Faculty of Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: Jacek.Jasieniak@monash.edu
bDepartment of Mechanical and Aerospace Engineering, Laboratory for Microsystems, Faculty of Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: Tuncay.Alan@monash.edu
First published on 8th December 2020
Abstract
The rapid nucleation and growth dynamics of metal halide perovskite nanocrystals (PNCs) present major challenges in controlling their ensemble evolution and scaling using typical batch syntheses. Continuous flow approaches are an alternative scalable and high-throughput process that can address these limitations by enabling rapid mixing and more precise control of reaction profiles. In this work, we use an acoustically actuated mixer within a continuous microfluidic (MF) platform to study the effect of precursor mixing rate on the synthesis of CsPbBr3 PNCs using the ligand assisted reprecipitation (LARP) approach. It is found that near complete mixing can be achieved at <23 ms timescales under highly efficient acoustic mixing. This enables superior control of the nanocrystal formation stages compared to both non-actuated MF and the standard batch LARP syntheses, which can take more than 2 s to achieve complete mixing with magnetic stirring. Through the study of the temporal evolution of the optical properties in the MF system, it is further found that nanoplatelet formation dominates during early reaction times <200 ms, which subsequently transform into nanocuboids. This complex colloidal evolution of PNCs further validates the need to achieve mixing at the millisecond timescale to afford structurally controlled PNCs with minimal polydispersity.
Introduction
Colloidal all-inorganic cesium lead halide perovskites (CsPbX3, X = Cl−, Br−, I−) have attracted great interest since they were first reported in 2015.1 These materials possess attractive optoelectrical properties arising from their flexible composition, including tunable optical bandgaps across the entire visible spectrum,1,2 narrow photoluminescence (PL) emission full-width half maximum (FWHM),3 near-unity PL quantum yields4 and low-cost solution processability.5 These properties are being harnessed in various applications, such as lasing,6,7 light emitting diodes,8,9 photodetectors,10 and photovoltaic cells with great success.11,12 To date, most of these applications have focused on colloidal perovskite nanocube (NCu) derivatives. However, the optical properties of perovskite nanocrystals (PNCs) also strongly depend on their size and morphology,1 with quantum confinement yielding large hypsochromic shifts of the optical properties. Hence, effective control of their size and uniformity is crucial to take advantage of their full potential.The established hot-injection (HI) synthesis of PNCs requires vacuum, inert conditions, high temperatures and rapid thermal quenching. All of these limit their ability to be upscaled.13,14 While these can be partially addressed in non-injection synthesis techniques, they nonetheless remain fundamentally limited by the slow heat-up rates leading to more inhomogeneous dispersions.15 The ligand assisted reprecipitation (LARP) approach,16 is an attractive alternative for PNC synthesis since it is compatible with ambient conditions and can be more readily upscaled.17,18 LARP utilizes simple solvent control to induce a supersaturated state for the nucleation of CsPbX3. For inorganic PNCs,4 dimethylformamide (DMF) is used to solubilize cesium halide (CsX) and lead halide (PbX2) salts. In addition, oleic acid (OA) and oleylamine (OLA) organic ligands are also included to form the precursor solution. This is then injected into a vigorously stirred toluene (TOL) anti-solvent for the rapid nucleation of PNCs. The oleylammonium–oleate (OLA+–OA−) ligand pair or oleylammonium bromide (OLABr) provide surface passivation and stabilizes the PNCs in a colloidal state.19,20 Notably, HI and similar heat-up syntheses have traditionally produced higher quality PNCs than from LARP, as they are performed under more favorable reaction conditions (high temperature, non-polar solvents and Pb-rich conditions). Synthetic advances in LARP, such as the use of HI,21 are rapidly closing the difference in crystal quality between these reaction approaches. This makes LARP a highly promising approach going forward.
Solution-based synthesis of PNCs through LARP is considered to follow the classical LaMer theory for nucleation and growth of nanoparticles.22 Accordingly, the formation of these colloids occurs in three stages (Fig. 1a): (I) the build-up of monomers/reactants; (II) a spontaneous burst of nuclei once a certain degree of supersaturation is exceeded; and (III) growth of these nuclei through monomer addition via Ostwald ripening or oriented attachment (Fig. 1b and c).23,24 During the process, effective and rapid mixing of the injected reagents is crucial to control the supersaturation condition, which in turn affects the nucleation of the nanoparticles.4 However, in its conventional form, LARP is a batch process on a milliliter scale that relies on external magnetic stirring. As shown in Video S1 (ESI†), the mixing of such large volumes is not instantaneous. The reactant concentrations fluctuate for seconds until the injected solution is fully homogenized. This local variation in the precursor concentration invariably results in (i) a polydisperse nanoparticle distribution and (ii) unwanted by-products, both of which compromise the effectiveness of the synthetic process.4
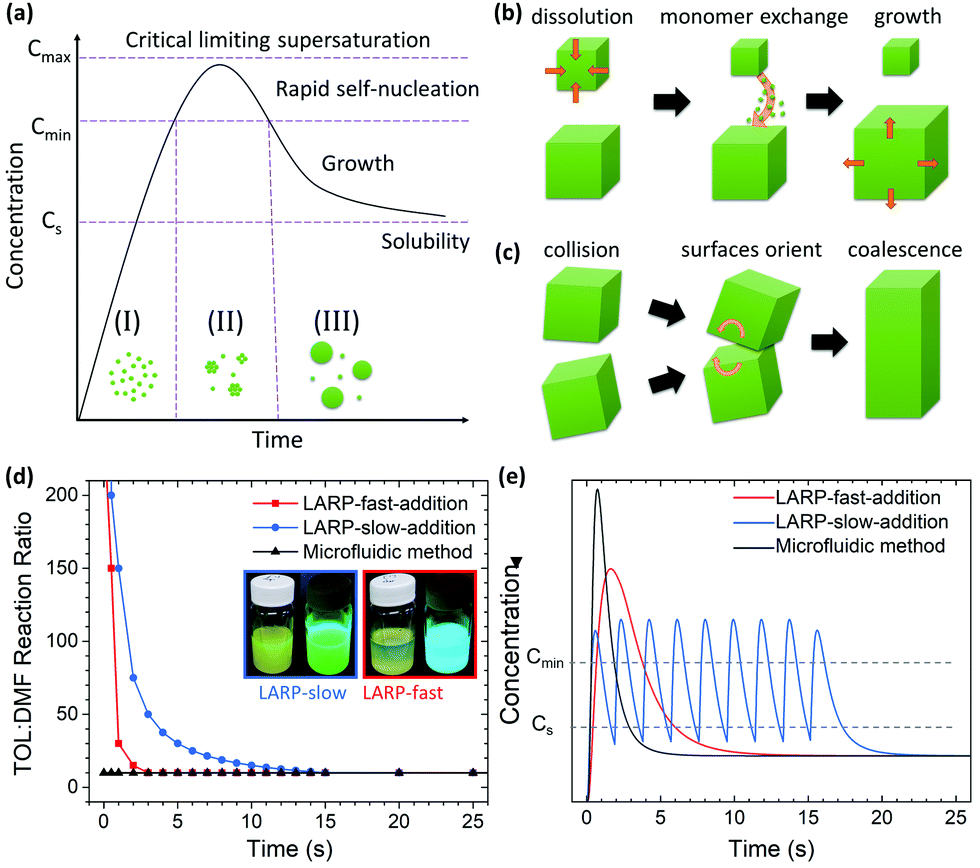 | ||
| Fig. 1 (a) The classical LaMer nucleation model of nanoparticles comprises of three phases: (I) monomer generation; (II) rapid self-nucleation at a critical monomer supersaturation concentration; and (III) nanocrystal growth via diffusion. Schematic of nanocrystal growth from (b) Ostwald ripening and (c) oriented attachment. (d) The precursor-to-toluene ratio for the different synthetic approaches, with the two insets showing the resultant LARP solutions using a fast/slow addition of the precursor under white and UV-light (λ = 365 nm). (e) A theoretical depiction of the expected differences in monomer concentration over time for the corresponding synthetic methods in Fig. 1d, where the CsPbBr3 solubility (Cs) and minimum concentration for nucleation (Cmin) are shown. | ||
In the case of the salt metathesis reaction of forming CsPbBr3 PNCs (eqn (1)), a commonly observed by-product is the zero-dimensional Cs4PbBr6 material.25 There are several driving forces that contribute to this: (i) a Cs+ rich environment will favor Cs4PbBr6 formation (eqn (2));26 (ii) a rich DMF environment preferably dissolves CsPbBr3 crystals over Cs4PbBr6 (eqn (3));27 and (iii) an amine rich environment can leech out PbBr2 and transform CsPbBr3 to Cs4PbBr6 (eqn (4)).28,29 Our own investigations see that both DMF and amine ligands are important factors in the generation of Cs4PbBr6 (Fig. S1 and S2, see ESI,† for more details). What is needed to suppress Cs4PbBr6 formation and obtain pure, monodisperse CsPbBr3 PNCs is a modified LARP process that can offer instantaneous mixing for rapid homogenization and precise control of the reaction parameters at high throughputs.
| CsBr + PbBr2 →CsPbBr3 | (1) |
| 4CsBr + PbBr2 →Cs4PbBr6 | (2) |
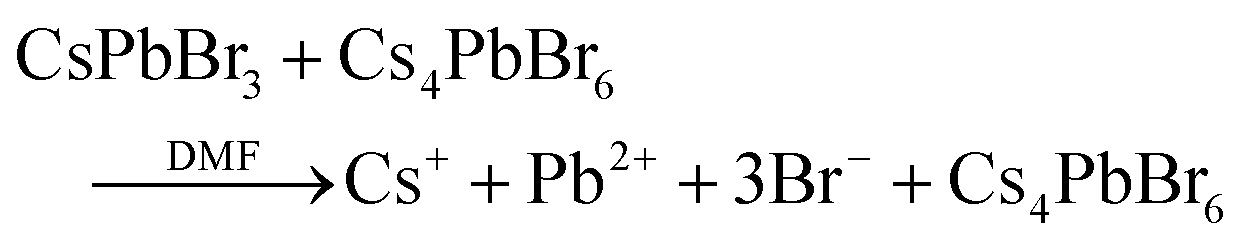 | (3) |
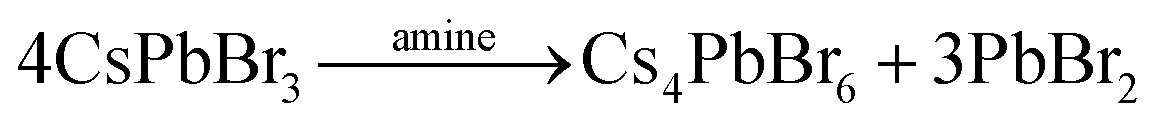 | (4) |
More recently, microfluidic (MF) techniques have been able to mix reagents in small droplets with a modified HI method.30–34 These droplet based systems offer precise control and fast mapping of parameters, such as the molar ratio of chemicals, reaction temperature, and reaction time.30,35 This high degree of control is advantageous for the study of nanomaterials in a laboratory environment.35,36 However, the droplet based methods employing pico-to-nano liter volume samples cannot easily accommodate high throughput operations even with considerable upscaling. This is a significant drawback for most typical MF approaches. Commonly, the high precision and control they offer are often accompanied by flowrates in the order of 10 μL min−1. Ideally, continuous-flow devices which can tolerate high flow rates and are compatible with different solvents and multistep reactions are needed for the production of complex structures.37 Prior microfluidic synthesis of organic nanoparticles suggests that enhanced mixing helps reduce particle size and polydispersity.38–40 To test if this holds true for PNCs, the synthesized CsPbBr3 PNCs from a continuous MF method with an active micromixer is characterized and compared to conventional batch LARP synthesized PNCs.
Here, we demonstrate for the first time that an acoustically actuated MF mixer can synthesize highly uniform CsPbBr3 PNCs at high throughputs and with negligible by-products. This provides a foundation for a scalable continuous synthetic method for PNCs and an alternative pathway to batch synthetic methods. Central to this new approach is a micro-mechanical oscillator that vibrates at MHz frequencies to rapidly homogenize two solutions in under 23 ms at 1.1 mL min−1 flow rate. This MF platform can synthesize highly crystalline CsPbBr3 cubic/orthorhombic phased PNCs that are superior to their conventional LARP synthesized analogues and contain significantly less Cs4PbBr6 by-products. The technique allows for the investigation of various reaction parameters such as the precursor concentrations, flow rates, the degree of mixing, and provides an insight into the early time evolution of the colloidal perovskite crystals.
Results & discussion
Theoretical basis from LaMer nucleation theory
Before examining PNC syntheses in the MF platform, the influence of different mixing conditions on the nucleation and growth processes in batch LARP is first studied as a baseline. For this, standard LARP using both fast and slow injection of the precursor solution into a stirred toluene anti-solvent are considered. According to LaMer theory, fast injection of the precursor solution should result in the most homogeneous nucleation event to yield the highest quality CsPbBr3 PNCs. However, this is rarely the case, as the highly localized injection results in significant amounts of Cs4PbBr6 dispersed alongside CsPbBr3 NCu.26 This is a result of the poor rate of mixing (i.e. time to homogenize) from the magnetic stirring of the injected precursor solution, thus creating favorable conditions for Cs4PbBr6 to form viaeqn (2) and (4). Under a slower precursor addition rate, the rate of mixing is markedly improved due to the staggered addition of smaller precursor solution volumes. This is reflected in Fig. 1d where the LARP slow addition reaches the final TOL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF ratio of 10:1 more slowly than the fast addition. The rapid homogenization of each successive volume of precursor solution throughout the anti-solvent results in more favorable conditions for eqn (1) to proceed, thus resulting in a predominantly CsPbBr3 NCu dispersion. The differences in monomer concentration between both precursor addition rates is visually depicted in Fig. 1e where the slow addition has multiple smaller supersaturation events leading to nucleation.
:DMF ratio of 10:1 more slowly than the fast addition. The rapid homogenization of each successive volume of precursor solution throughout the anti-solvent results in more favorable conditions for eqn (1) to proceed, thus resulting in a predominantly CsPbBr3 NCu dispersion. The differences in monomer concentration between both precursor addition rates is visually depicted in Fig. 1e where the slow addition has multiple smaller supersaturation events leading to nucleation.
For the MF platform and its synthesis of CsPbBr3 PNCs, the inclusion of the micromixer is expected to result in the near instantaneous mixing of the precursor with the anti-solvent. Theoretically, this would enhance the degree of supersaturation and, therefore, the rate of homogeneous nucleation compared to traditional LARP. These changes compared to conventional LARP are reflected in Fig. 1d and e, where the TOL:DMF immediately reaches the final 10:1 ratio and a higher degree of supersaturation is observed, respectively. As a result of these synthetic enhancements, adopting an MF platform with micromixing should yield PNCs with a smaller average crystal size, a reduction to the final ensemble polydispersity and a reduction in unwanted by-products compared to batch processed LARP PNCs.
Microfluidic mixer – effect of actuation parameters on mixing efficiency
Conventional mixers based uniquely on polydimethylsiloxane (PDMS) are prone to suffer from severe swelling when exposed to non-polar solvents, such as TOL, and are therefore incompatible with conventional LARP synthesis. As such, the mixer employed in this study consists of a silicon oscillator which capitalizes on the robust nature of silicon against both polar and non-polar solvents. The acoustically actuated, silicon micromechanical oscillator is sandwiched between two polymeric MF channels which guide the reagents in and out of the system (Fig. 2a).41 When a 1.06 MHz-frequency AC signal is applied to a piezoelectric transducer, attached onto the system, the silicon oscillator starts vibrating. This vibration generates a strong acoustic streaming field in the fluid medium, which provides highly efficient mixing, thus enabling very rapid homogenization of the reagents.42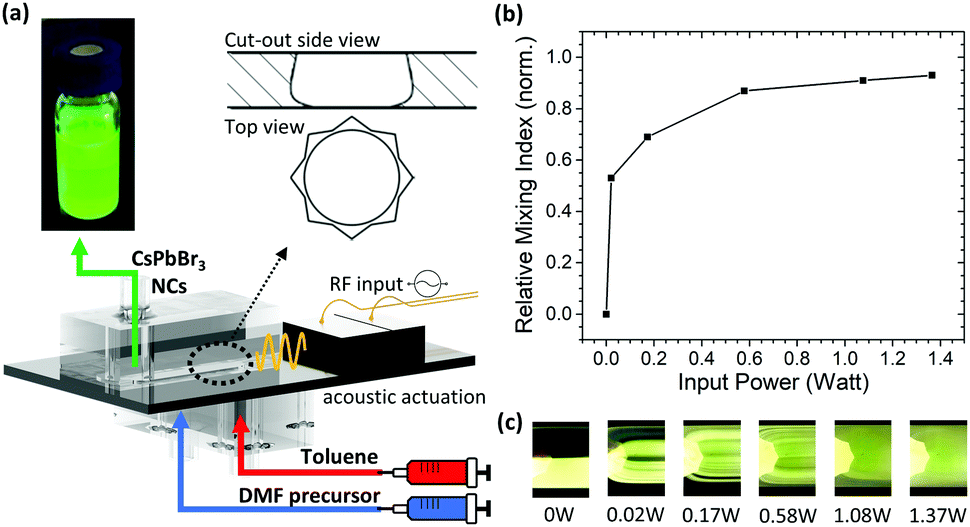 | ||
| Fig. 2 (a) A schematic of the star-shaped MF mixer and its components, with a sketch showing the side and top views of the through-hole and the silicon oscillator, and the collected CsPbBr3 PNCs under ambient and UV-light (λ = 365 nm). (b) The relationship between the RMIN and input power for a water and dye mixture, (c) with corresponding photographs under λ = 450–490 nm illumination immediately after passing the through-hole. | ||
As a first step in the study, the effect of the actuation parameters on the mixing efficiency was characterized using deionized (DI) water and a fluorescent dye as input solutions, introduced at a 1.1 mL min−1 flow rate. As the input power to the piezoelectric transducer (Pin) was gradually varied, the flow profiles were imaged and the relative mixing index (RMIN) was calculated for each step (Fig. 2b and c). RMIN is a non-dimensional number indicating the mixing efficiency; RMIN = 0 and RMIN = 1 correspond to no mixing and complete mixing, respectively (see ESI,† for more details). When Pin = 0 W (i.e. passive operation), the water and dye streams flow in parallel, with a distinct interface (RMIN = 0) as shown in Fig. 2c. Increasing Pin perturbs the laminar flow profile and enhances mixing. For every increasing increment of Pin, RMIN increases until near full homogenization (RMIN = 0.91, i.e. 91% efficiency) is achieved at Pin = 1.37 W. This shows that the mixing efficiency can be effectively tuned and the mixing time reduced to <23 ms by solely controlling Pin. This can be calculated from the known flow rate and estimated fluid path length through the oscillator through-hole (Fig. S3, see ESI,† for details). Moreover, this is achieved independently of other parameters and at relatively high flow rates. The throughput here is approximately 3 times higher than that offered by conventional microfluidic techniques used for PNC synthesis.30 Furthermore, this level of control is not achievable by batch processes, for which the homogenization time is approximately 100 times slower (Fig. S4, Video S2, ESI†).
Perovskite nanocrystal synthesis – batch method
As a baseline, LARP synthesis of CsPbBr3 PNCs under both slow and fast precursor addition rates have been conducted. Under slow precursor addition rates, CsPbBr3 NCu are the preferred products, as indicated by the dominant absorption onset at ∼510 nm (Fig. 3a).4 However, there is also appreciable Cs4PbBr6 formation, which is evident by their characteristic absorption peak at ∼314 nm.26 Meanwhile, the fast precursor addition rates see significantly more Cs4PbBr6 impurities in the samples, with the NCu being additionally more polydisperse based on the broadened absorption onset. These trends are consistent for syntheses conducted at precursor concentrations of 0.040 M and 0.010 M, with the latter being the optimal concentration for the MF system (vide infra). Notably, the polydispersity of these PNCs is not well reflected in their PL spectra due to energy transfer mechanisms from the smaller nanoplatelets (NPL) to larger NCu.43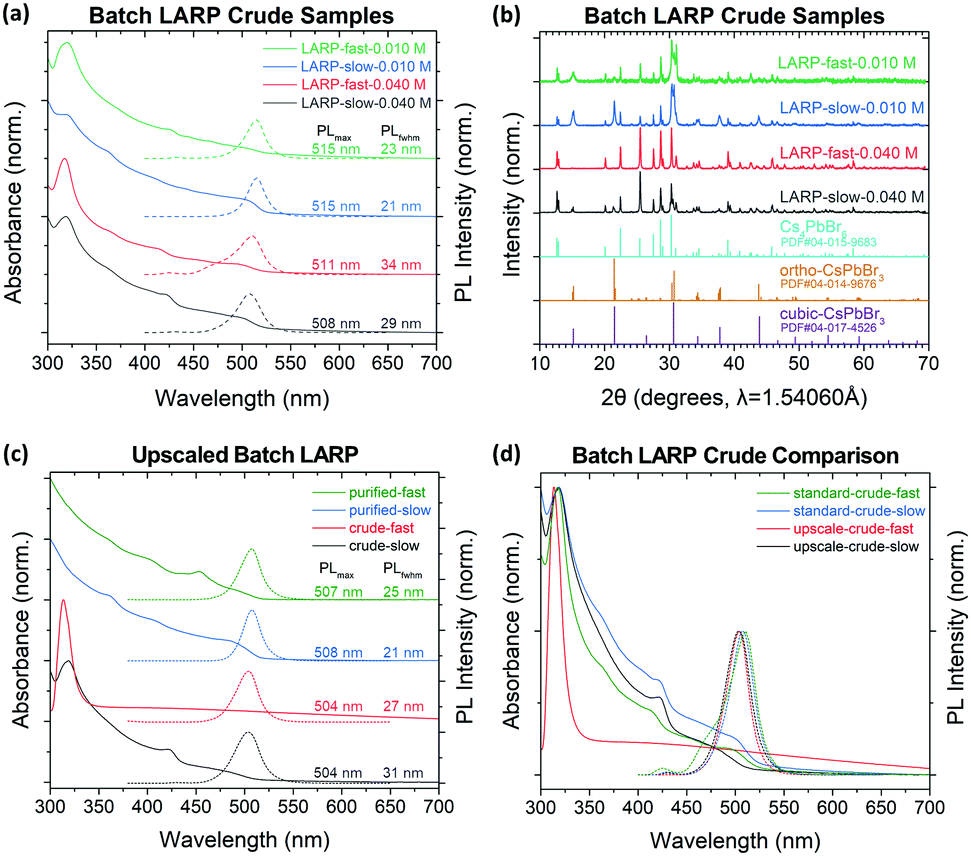 | ||
| Fig. 3 (a) UV-Vis (solid line) and PL (dotted line) spectra of CsPbBr3 crude solutions synthesized through LARP at different precursor concentrations and addition rates. (b) XRD spectra of the crude LARP solution's crystal products and reference spectra of their products. (c) UV-Vis and PL spectra of PNC dispersions from mild 5× upscaling of batch LARP synthesis with 0.040 M precursor solution; and (d) a comparison between standard and upscaled batch CsPbBr3 LARP crude reaction volumes. | ||
To better estimate the product ratios between CsPbBr3 and Cs4PbBr6 from the batch LARP reactions in Fig. 3a, the synthesized PNCs were fully precipitated and analysed using powder XRD (Fig. 3b). The broad peak at 2θ = 15.2° suggests cubic (α) CsPbBr3 PNCs (PDF#00-054-0752) with unit cell dimensions of a = b = c = 5.83 Å and a Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Given that the majority of precipitated CsPbBr3 crystals are too large to be colloidally stable, the orthorhombic (γ) CsPbBr3 (PDF#01-085-6500) is the most likely phase, with unit cell dimensions of a = 8.250 Å, b = 11.753 Å, c = 8.203 Å, and a Pnma space group. Since the two phases differ only by a slight octahedral tilt of the [PbBr6]4− octahedra, the identification of their phase is often difficult due to their similar XRD patterns and that the two phases can co-exist within a single particle.44,45 The remaining peaks clearly correspond to rhombohedral Cs4PbBr6 (PDF#04-015-9683), with unit cell dimensions of a = b = 13.722 Å, c = 17.315 Å with a Rc space group. This phase is most easily identifiable through its twin peaks at 2θ = 12.6 and 12.9°. For both 0.040 M and 0.010 M precursor concentrations, a stronger CsPbBr3 peak is observed for the slow addition samples, with the fast addition sample at 0.040 M showing predominantly Cs4PbBr6.
m space group. Given that the majority of precipitated CsPbBr3 crystals are too large to be colloidally stable, the orthorhombic (γ) CsPbBr3 (PDF#01-085-6500) is the most likely phase, with unit cell dimensions of a = 8.250 Å, b = 11.753 Å, c = 8.203 Å, and a Pnma space group. Since the two phases differ only by a slight octahedral tilt of the [PbBr6]4− octahedra, the identification of their phase is often difficult due to their similar XRD patterns and that the two phases can co-exist within a single particle.44,45 The remaining peaks clearly correspond to rhombohedral Cs4PbBr6 (PDF#04-015-9683), with unit cell dimensions of a = b = 13.722 Å, c = 17.315 Å with a Rc space group. This phase is most easily identifiable through its twin peaks at 2θ = 12.6 and 12.9°. For both 0.040 M and 0.010 M precursor concentrations, a stronger CsPbBr3 peak is observed for the slow addition samples, with the fast addition sample at 0.040 M showing predominantly Cs4PbBr6.
These experiments clearly indicate that mixing rates strongly affect the PNC dispersions in LARP, which emerges as a major challenge when upscaling reactions. Even a mild five-fold upscaling (i.e. 5 mL of 0.040 M precursor added to 50 mL of TOL) for both fast and slow addition rates reveals these difficulties (Fig. 3c and d). This is most obvious for the upscaled crude solution with a fast precursor addition rate, for which there are no distinct absorption peaks from CsPbBr3 and instead the spectrum is dominated by the Cs4PbBr6 peak at ∼314 nm. Given this preferred reaction mode, the final reaction yield for CsPbBr3 is reduced by ∼87% compared to a standard LARP synthesis performed at the normal reaction scale (i.e. 1 mL of 0.040 M precursor added to 10 mL of TOL). Meanwhile, under a slow precursor addition rate, the upscaled crude solution's absorption spectra does not significantly differ from its normal scale counterpart in Fig. 3a. After purification, the upscaled PNCs show a characteristic CsPbBr3 NCu absorption curve, albeit with the final product yield being 35% lower compared to normal reaction volume. The loss in product yield is attributed to excessive CsPbBr3 growth times, which results in more CsPbBr3 crystals coarsening beyond colloidally stable size regimes.
Perovskite nanocrystal synthesis – microfluidic mixer
The highly efficient mixing capabilities of the MF mixer provides a distinct opportunity in overcoming the mixing challenges associated with batch LARP. To ascertain its limits, the LARP reaction has been adapted to the MF platform and optimized for CsPbBr3 NCu synthesis by controlling the precursor concentrations, flow rates, and actuation parameters.:10 ratio as in batch LARP.
The crude solution absorption and PL spectra of the MF synthesized particles under active (1.37 W) and passive (0.0 W) modes of operation are shown in Fig. 4a and b, respectively. XRD analysis shows that active mixing can drastically reduce the presence of Cs4PbBr6 within its crude solution compared to passive or batch LARP prepared samples (Fig. S6, ESI†). At 0.040 M precursor concentrations, dominant CsPbBr3 NPL formation is observed under active and passive mixing. Compared to its batch LARP equivalent under identical reaction conditions, i.e. the 0.040 M batch LARP sample under fast addition in Fig. 3, the absorption peak from Cs4PbBr6 is significantly weaker than its CsPbBr3 absorption peaks. At lower precursor concentrations, the passive mixed samples exhibit NPL and/or Cs4PbBr6 as the dominant products or impurities within the NCu dispersion. Meanwhile, a progressive improvement in the size and shape homogeneity is observed for the active mixed samples, with a precursor concentration of 0.010 M showing a pure CsPbBr3 NCu dispersion.
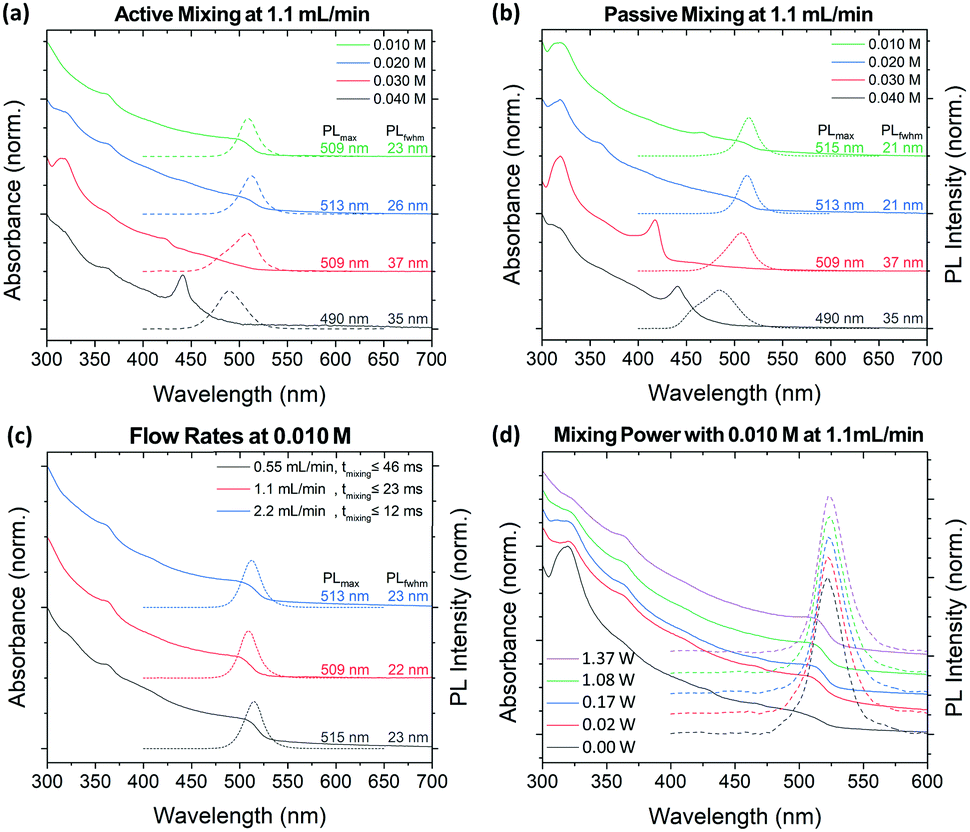 | ||
| Fig. 4 UV-Vis (solid line) and PL (dotted line) spectra of CsPbBr3 crude samples synthesized with the MF platform examining the dependence of: the precursor concentration under (a) active 1.37 W mixing and (b) passive 0 W mixing; (c) the various flow rates operating with a precursor concentration of 0.010 M; and (d) power dependence from 0 to 1.37 W at a precursor concentration of 0.010 M. | ||
These synthetic trends are well correlated to the extent of precipitation of the OLA+–OA− salts across this investigated concentration regime. At concentrations of 0.030 M and above, highly polydispersed colloidal PNCs are observed, which is commensurate with the presence of insolubilized ligand salt pairs. Meanwhile, the fully solubilized salts in the 0.010 M precursor solutions at room temperature, evidently enable the most homogeneous CsPbBr3 NCu to be achieved. Furthermore, the existence of Cs4PbBr6 and the inhomogeneity of the NCu in the passive mixed sample, clearly demonstrates the importance of active mixing in achieving rapid homogenization to achieve high quality PNCs.
Perovskite nanocrystal synthesis – batch method
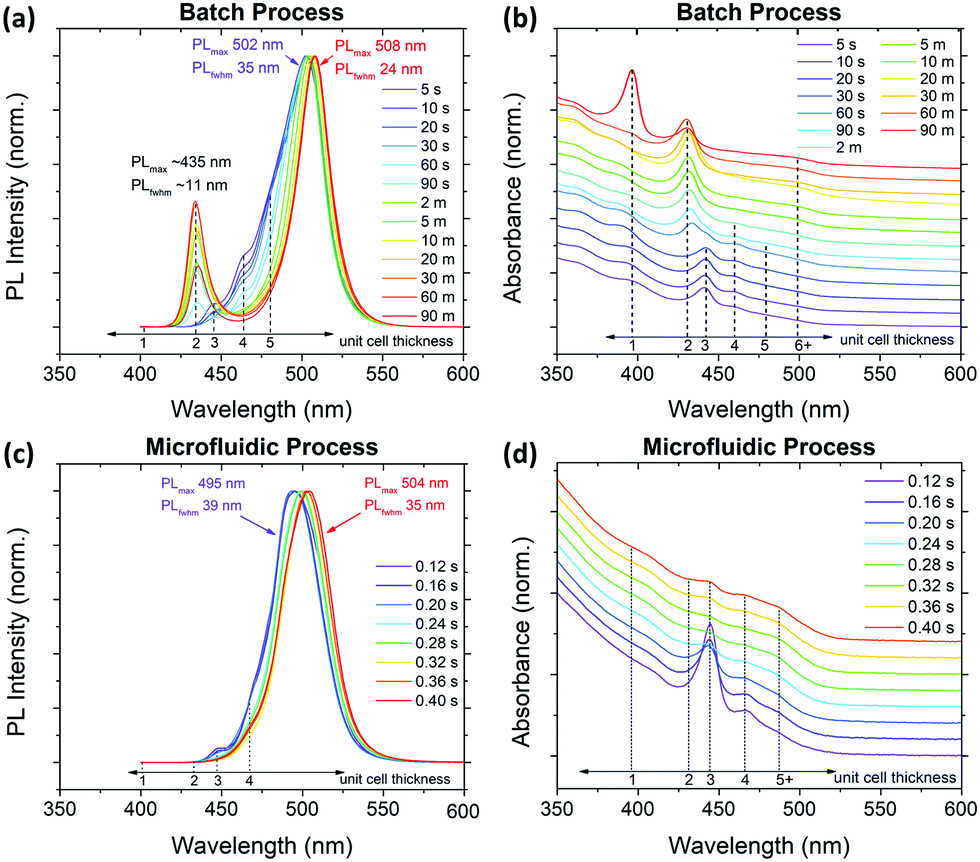 | ||
| Fig. 5 (a) Time dependent PL and (b) absorption spectra of a batch LARP fast 0.040 M precursor injection reaction, and (c) time dependent PL and (d) absorption spectra of a MF platform examining <0.5 s growth times under active (1.37 W) mixing with a 0.020 M precursor solution. | ||
Comparison between purified MF and batch LARP PNCs
For a direct comparison between the synthesized PNCs, precursor solutions of 0.010 M have been used for both slow/fast injection batch and passive/active MF LARP methodologies. The synthesized CsPbBr3 PNCs from these four synthetic approaches were identically purified, without further fractionation to bias the crystal size distributions, and analyzed using both XRD and transmission electron microscopy (TEM) to determine their phase and crystal size distributions, respectively.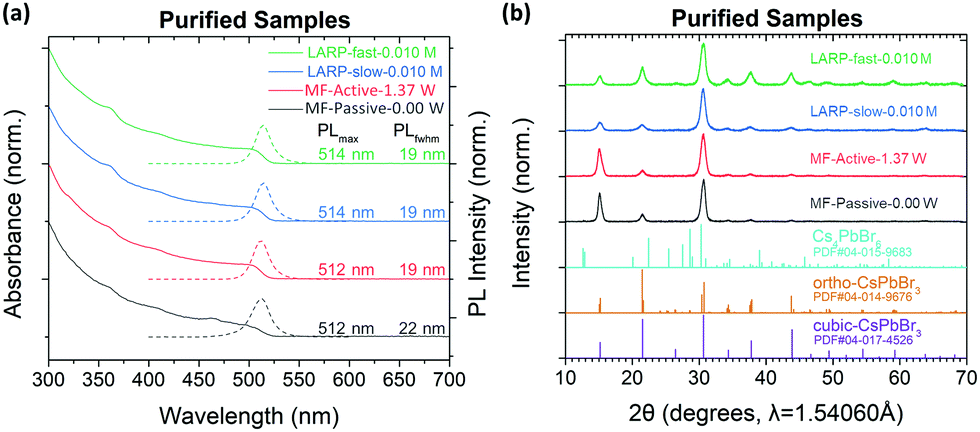 | ||
| Fig. 6 (a) UV-Vis (solid line) and PL (dotted line) spectra of purified CsPbBr3 samples synthesized from the batch LARP and MF methods, all using a precursor concentration of 0.010 M; and (b) their respective XRD patterns and references. | ||
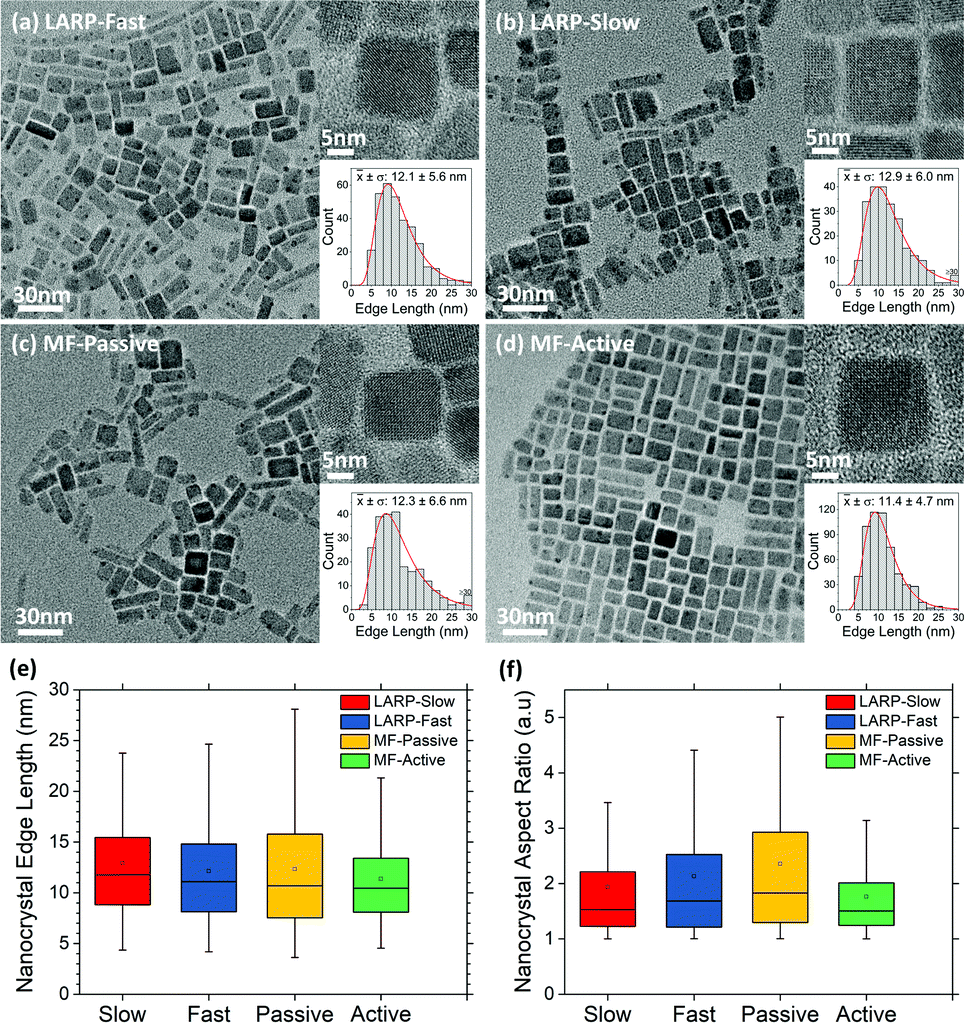 | ||
| Fig. 7 TEM images with insets of high-resolution TEM and crystal edge length histograms of purified CsPbBr3 samples synthesized from (a) LARP-fast-addition, (b) LARP-slow-addition, (c) MF passive 0 W mixing and (d) MF active 1.37 W mixing. Histograms of TEM imaged PNCs populations and their corresponding nanocrystal (e) edge lengths and (f) aspect ratios. | ||
The TEM results show that the two batch LARP samples have similar size distributions, with the fast addition variant forming slightly smaller crystals, but with a higher aspect ratio. In contrast, vastly different structural characteristics are observed for the passive and actively mixed MF samples. The passively mixed PNCs are the most polydisperse, exhibiting the worst distributions for both edge lengths and aspect ratios across all four TEM samples. Meanwhile, the actively mixed MF sample sees significant improvements over its batch LARP counterparts, exhibiting smaller average crystal sizes, narrower size distributions and more uniform aspect ratios that are closer to 1. This is attributed to the micromixer's ability to rapidly and uniformly mix the precursor reagents with the anti-solvent to produce higher quality nanocrystal dispersions, as is expected from LaMer theory.
Conclusions
The rapid nucleation and growth dynamics of PNCs requires a transition away from simple lab-based batch synthetic approaches to those that inherently feature more controllable mixing in order to achieve major advances in their structural and compositional control. The work presented herein demonstrates the advantages of such a transition by incorporating an active mixing element into a microfluidic platform for the synthesis of PNCs based on the LARP reaction. The application of an external force for mixing, advantageously enables the mixing efficiency to be independently controlled by manipulating the driving power of the piezoelectric transducer. Homogeneous mixing has been achieved under nearly ambient temperatures within 23 ms compared to more than 2 s for a batch process. It is shown that under such homogeneous and fast mixing times, optimized precursor concentrations and flow rates enable the formation of more monodisperse CsPbBr3 nanocrystals without the presence of Cs4PbBr6 by-products. This demonstrates that the underlying microfluidics platform with actuated mixing provides a pathway for the continuous generation of homogeneous and phase pure CsPbBr3 PNCs. We envisage that this novel approach towards the synthesis of these PNCs can have far reaching consequences for all other perovskite and alternative composition colloids that inherently exhibit fast nucleation and growth dynamics.Experimental
Materials
Cesium bromide (CsBr, 99.999%), lead bromide (PbBr2, 99.999%), oleylamine (OLA, 70% technical grade), oleic acid (OA, 90% technical grade), dimethyl formamide (DMF, 99.8% anhydrous), dimethyl sulfoxide (DMSO, 99.5%) are sourced from Sigma-Aldrich. Toluene (TOL, ≥99.9%), ethyl acetate (EA, ≥99.5%), ethanol (EtOH, ≥99.9%) are sourced from Merck. All reagents are used without further purification; the exceptions are oleylamine and oleic acid, which is degassed at 120 °C for 30 min before being stored for later use.Characterisation equipment
Ultraviolet-Visible (UV-Vis) absorption spectroscopy of colloidally dispersed PNCs in toluene are analyzed with a PerkinElmer Lambda 950 UV-Vis-NIR spectrometer. Photoluminescence (PL) spectroscopy of colloidally dispersed PNCs in toluene is analyzed with a Horiba Fluoromax-4 Spectrofluorometer. Additional time-dependent PL measurements are recorded with the commercial spectrometer from the company StellarNet in the US, with CCD camera (Silver-Nova-TEC-X2), under 390 nm excitation (SL1-LED). A Beckman Coulter Allegra TM X-22R centrifuge machine is used for purification steps. X-ray Diffraction (XRD) spectra are recorded using a Bruker D8 Advance X-ray diffractometer with a Lynxye XE 1D detector using a Cu source (Kα = 1.54060 Å) radiation at 40 kV, 40 mA. XRD films are prepared by drop-casting the purified nanocrystal solutions dispersed in toluene and gently dried under flowing gas at 50 °C. Scanning electron microscopy (SEM) images are obtained on an FEI Magellan 400 Field Emission Scanning Electron Microscope equipped with a Schottky-type field emission gun and in-lens secondary electron detector. Transmission electron microscopy (TEM) images are obtained on a FEI Tecnai G2 T20 Transmission Electron Microscope equipped with a LaB6 electron source operated at 200 kV. Images are acquired by either a Gatan Orius SC200D CCD camera or a Gatan Orius SC600 CCD camera. Optical microscope images are obtained on a Nikon ECLIPSE-CI optical microscope at 5× magnification, illumination by an Lh-M100Cb-1 fluorescence mercury lamp powered by Nikon C-SHG1 100 W Super High-Pressure Mercury Lamp Power Supply. Images are acquired by Dino-lite Digital Microscope AM4023CT camera. The piezoelectric transducer (PZ26, Ferroperm Piezoceramics, Meggitt Pty) is powered by Stanford Research Systems DS345 signal generator and T&C Power Conversion, Inc. AG 1006 amplifier. Kd Scientific KDS-101-CE Syringe pumps are used for delivering reagents to the MF device. The input power is measured by Rohde & Schwarz NRP 18S-25 Power gauge. The temperature of the microfluidic system during synthesis was measured with a FLIR i7 thermal imager.Microfluidic platform
The MF device displayed in Fig. 2a consists of three layers. The bottom layer is made from polydimethylsiloxane (PDMS), has a 5 mm × 2 mm × 100 μm MF channel, patterned using a standard soft lithography process. Holes have been punched at both ends of the channel act as inlets for the reagents being mixed. The top layer is also a PDMS MF channel, containing an outlet for the mixed solution. A microfabricated Si structure forms the intermediate layer, the mixer, sandwiched between the two channels. The mixer consists of a variable thickness plate with through-hole at its centre. It is fabricated via a modified deep reactive ion etching (DRIE) process.36 The through-hole is aligned with the closed end of the top channel, and the midpoint of the bottom channel and the three layers are bonded together through a standard plasma bonding process. A piezoelectric transducer (PZ26, Ferroperm Piezoceramics, Meggitt Pty) is attached to the silicon chip using epoxy glue (Araldite 5 Minute Epoxy Adhesive, Selleys). The piezoelectric transducer is connected to a signal generator (Stanford Research Systems DS345) outputting a 1.06 MHz sine function signal, and an amplifier (T&C Power Conversion, Inc. AG 1006) providing 0.1% gain. Upon receiving the signal, the piezoelectric transducer will vibrate and resonate the oscillator on the chip, thereby generates a strong acoustic streaming field, which will efficiently homogenize the fluids flowing through the hole within milliseconds. The optimal operating frequency of the device is obtained experimentally, by performing a frequency sweep and the frequency providing the best mixing performance is chosen to be used through all other experiments.Synthetic procedures
:DMF ratio of 10:1 for 2 min under ambient conditions to precipitate CsPbBr3 PNCs. Ethyl acetate is then added to a 2:1 ratio of crude:EA to assist in precipitation. This mixture is then centrifuged at 8000 rpm for 5 min. The supernatant is discarded, and the pellet dispersed with toluene to be centrifuged again at 3000 rpm for 2 min. This supernatant is taken for further measurements and stored under ambient conditions in the dark.
:10. After being mixed by the acoustic micromixer, the product solution will flow through another section of PTFE tubing that connects the outlet port with the collection valve. To avoid cross-contamination between samples, the channel is being flushed with DMSO (to remove any Cs4PbBr6), ethanol, and toluene between each experiment. In addition, samples are collected after 30 s of operation to allow for the removal of any air bubbles and flow stabilization. Purification of the crude PNCs were similarly followed as above for consistency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Australian Research Council funded Center of Excellence in Exciton Science (Grant No. CE170100026). This research used equipment funded by Australian Research Council grant LE130100072. The authors acknowledge use of facilities within the Monash X-ray Platform (MXP), the Monash Centre for Electron Microscopy (MCEM). This work was performed in part at the Melbourne Centre for Nanofabrication (MCN) in the Victorian Node of the Australian National Fabrication Facility (ANFF).References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- Q. A. Akkerman, V. D’Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS.
- X. Li, K. Zhang, J. Li, J. Chen, Y. Wu, K. Liu, J. Song and H. Zeng, Adv. Mater. Interfaces, 2018, 5, 1800010 CrossRef.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. V. Gustafsson, M. T. Trinh, S. Jin and X. Y. Zhu, Nat. Mater., 2015, 14, 636–642 CrossRef CAS.
- L. J. Chen, J. H. Dai, J. De Lin, T. S. Mo, H. P. Lin, H. C. Yeh, Y. C. Chuang, S. A. Jiang and C. R. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 33307–33315 CrossRef CAS.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS.
- Z. Shi, S. Li, Y. Li, H. Ji, X. Li, D. Wu, T. Xu, Y. Chen, Y. Tian, Y. Zhang, C. Shan and G. Du, ACS Nano, 2018, 12, 1462–1472 CrossRef CAS.
- P. Ramasamy, D.-H. Lim, B. Kim, S.-H. Lee, M.-S. Lee and J.-S. Lee, Chem. Commun., 2016, 52, 2067–2070 RSC.
- Q. A. Akkerman, M. Gandini, F. Di Stasio, P. Rastogi, F. Palazon, G. Bertoni, J. M. Ball, M. Prato, A. Petrozza and L. Manna, Nat. Energy, 2017, 2, 16194 CrossRef CAS.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, eaao4204 CrossRef.
- C. K. Ng, C. Wang and J. J. Jasieniak, Langmuir, 2019, 35, 11609–11628 CrossRef CAS.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS.
- J. van Embden, A. S. R. Chesman and J. J. Jasieniak, Chem. Mater., 2015, 27, 2246–2285 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS.
- S. Wei, Y. Yang, X. Kang, L. Wang, L. Huang and D. Pan, Chem. Commun., 2016, 52, 7265–7268 RSC.
- C. K. Ng, W. Yin, H. Li and J. J. Jasieniak, Nanoscale, 2020, 12, 4859–4867 RSC.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. ten Brinck, P. Aellen, A. Süess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C.-J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS.
- H. Huang, J. Raith, S. V. Kershaw, S. Kalytchuk, O. Tomanec, L. Jing, A. S. Susha, R. Zboril and A. L. Rogach, Nat. Commun., 2017, 8, 996 CrossRef.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS.
- H. Yang, Y. Zhang, J. Pan, J. Yin, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2017, 29, 8978–8982 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS.
- W. Wang, D. Wang, F. Fang, S. Wang, G. Xu and T. Zhang, Cryst. Growth Des., 2018, 18, 6133–6141 CrossRef CAS.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S. T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. Kazes, Chem. Mater., 2018, 30, 84–93 CrossRef CAS.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. Demello and M. V. Kovalenko, Nano Lett., 2016, 16, 1869–1877 CrossRef CAS.
- R. M. MacEiczyk, K. Dümbgen, I. Lignos, L. Protesescu, M. V. Kovalenko and A. J. Demello, Chem. Mater., 2017, 29, 8433–8439 CrossRef CAS.
- I. Lignos, L. Protesescu, D. Börte Emiroglu, R. Maceiczyk, S. Schneider, M. V. Kovalenko and A. J. Demello, Nano Lett., 2018, 18, 1246–1252 CrossRef CAS.
- L. Bezinge, R. M. Maceiczyk, I. Lignos, M. V. Kovalenko and A. J. Demello, ACS Appl. Mater. Interfaces, 2018, 10, 18869–18878 CrossRef CAS.
- I. Lignos, V. Morad, Y. Shynkarenko, C. Bernasconi, R. M. Maceiczyk, L. Protesescu, F. Bertolotti, S. Kumar, S. T. Ochsenbein, N. Masciocchi, A. Guagliardi, C. J. Shih, M. I. Bodnarchuk, A. J. Demello and M. V. Kovalenko, ACS Nano, 2018, 12, 5504–5517 CrossRef CAS.
- R. W. Epps, K. C. Felton, C. W. Coley and M. Abolhasani, Lab Chip, 2017, 17, 4040–4047 RSC.
- Y. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698–711 CrossRef CAS.
- T. W. Phillips, I. G. Lignos, R. M. Maceiczyk, A. J. Demello and J. C. Demello, Lab Chip, 2014, 14, 3172–3180 RSC.
- N. H. A. Le, H. Van Phan, J. Yu, H. K. Chan, A. Neild and T. Alan, Int. J. Nanomed., 2018, 13, 1353–1359 CrossRef CAS.
- P. Huang, S. Zhao, H. Bachman, N. Nama, Z. Li, C. Chen, S. Yang, M. Wu, S. P. Zhang and T. J. Huang, Adv. Sci., 2019, 6, 1900913 CrossRef CAS.
- H. Bachman, C. Chen, J. Rufo, S. Zhao, S. Yang, Z. Tian, N. Nama, P. H. Huang and T. J. Huang, Lab Chip, 2020, 20, 1238–1248 RSC.
- N. H. An, Le, H. Deng, C. Devendran, N. Akhtar, X. Ma, C. Pouton, H. K. Chan, A. Neild and T. Alan, Lab Chip, 2020, 20, 582–591 RSC.
- H. Van Phan, M. B. Coşkun, M. Şeşen, G. Pandraud, A. Neild and T. Alan, Lab Chip, 2015, 15, 4206–4216 RSC.
- C. De Weerd, L. Gomez, H. Zhang, W. J. Buma, G. Nedelcu, M. V. Kovalenko and T. Gregorkiewicz, J. Phys. Chem. C, 2016, 120, 13310–13315 CrossRef CAS.
- Y. Yu, D. Zhang, C. Kisielowski, L. Dou, N. Kornienko, Y. Bekenstein, A. B. Wong, A. P. Alivisatos and P. Yang, Nano Lett., 2016, 16, 7530–7535 CrossRef CAS.
- M. C. Brennan, M. Kuno and S. Rouvimov, Inorg. Chem., 2019, 58, 1555–1560 CrossRef CAS.
- M. Koolyk, D. Amgar, S. Aharon and L. Etgar, Nanoscale, 2016, 8, 6403–6409 RSC.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. Zheng, Z. Yang and E. H. Sargent, Adv. Mater., 2017, 29, 1605945 CrossRef.
- C. De Weerd, J. Lin, L. Gomez, Y. Fujiwara, K. Suenaga and T. Gregorkiewicz, J. Phys. Chem. C, 2017, 121, 19490–19496 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Further detailed information on synthesis, calculations, photographs, and characterizations (UV-Vis, PL, TR-Abs, TR-PL, AFM). See DOI: 10.1039/d0tc04519e |
| ‡ C.K. Ng and H. Deng contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |