
Halogenation on benzo[1,2-b:4,5-b′]difuran polymers for solvent additive-free non-fullerene polymer solar cells with efficiency exceeding 11%†
Enfang
He‡
a,
Yi
Lu‡
a,
Zhi
Zheng
a,
Fengyun
Guo
a,
Shiyong
Gao
 a,
Liancheng
Zhao
a and
Yong
Zhang
*ab
a,
Liancheng
Zhao
a and
Yong
Zhang
*ab
aSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: yongzhang@hit.edu.cn
bSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, China
First published on 21st November 2019
Abstract
In this work, two novel two-dimensional (2D) benzo[1,2-b:4,5-b′]difuran (BDF)-based wide bandgap polymers were designed using a halogenation strategy by incorporating fluorine- and chlorine-substituted conjugated side chains, respectively. With the advantages of low-cost and environment-friendly furan units and halogen atoms, the BDF polymers PFTBDF-FBTA (F10) and PClTBDF-FBTA (F11) possessed lower-lying highest occupied molecular orbital (HOMO) energy levels with a large effect on their optical properties. In addition, the intermolecular interactions induced by F(Cl)⋯H(O) and/or F(Cl)⋯F(Cl) can also promote more preferred polymeric chain stacking behavior. In the application of non-fullerene polymer solar cells (NF-PSCs) with m-ITIC as the electron acceptor in the inverted device structure, the fluorinated F10-based NF-PSC exhibited a power conversion efficiency (PCE) of 10.5% with a higher open circuit voltage (Voc) of 0.908 V, which is much higher than that of the non-halogenated counterpart. In the chlorinated polymer F11:m-ITIC-based device, a further higher Voc of 0.921 V with enhanced current density was achieved, which results in a promising PCE of 11.37%, which is mainly attributed to the lower-lying HOMO and improved intermolecular stacking induced by chlorine. It is also noted that these performances were obtained without using any solvent additive or solvent annealing process, which is attractive to large area printing processing technology. These results demonstrate that halogenation engineering of BDF polymer is a very promising molecular design strategy for BDF polymers to reach the state-of-the-art photovoltaic performance, and these results also show that BDF is an efficient building block to construct highly efficient polymer donors for PSC applications.
Introduction
Polymer solar cells with a p-type conjugated polymer as a donor and an n-type semiconductor as an acceptor have attracted considerable attention from scientific and industrial communities due to their advantages in potentially low-cost, large-area and flexible solar cells through roll-to-roll or printing technologies.1–4 With continuous innovations of material design and device engineering,5–9 especially the rapid advancement of non-fullerene acceptors in the last five years,10–12 the power conversion efficiencies (PCEs) of polymer solar cells (PSCs) have surpassed 16% for single-junction cells and 17% for tandem cells.13,14 The successes of narrow bandgap non-fullerene electron acceptors have motivated scientists to design wide bandgap conjugated polymer donors with the aim of providing complementary absorption to such acceptors to enhance short-circuit current (Jsc), and lower the highest occupied molecular orbital (HOMO) level of the resulting polymer to increase open-circuit voltage (Voc).15,16 To this end, various donor–acceptor type wide conjugated polymers with units such a benzo[1,2-b:4,5-b′]dithiophene (BDT) have been developed because BDT could provide a rigid and planar structure for π–π stacking and high charge carrier mobility, and appropriate energy levels for wide-bandgap polymer donors. Currently, compared with the rapid development and various kinds of efficient non-fullerene acceptors, the efficient polymer donors such as P2F-EHp6 and PBDT-T-2F (PM6)17 mainly rely on BDT-based blocks.4Benzo[1,2-b:4,5-b′]difuran (BDF), which has a similar structure to BDT, has recently attracted wide interest in developing wide bandgap polymer donors. With the incorporation of a smaller furan unit into a polymer skeleton, the BDF-based polymers may form stronger intermolecular interactions and π–π stacking.18 Furthermore, BDF-based polymers will also possess deeper HOMO energy levels compared to their BDT counterparts because of the more electronegative oxygen atom in comparison with the BDT-based polymer, which undoubtedly can generate a higher Voc in PSCs.19 More importantly, furan and its derivatives can be obtained from agricultural plant extracts, which are more environment-friendly and low cost.20 Recent studies have shown that BDF-based polymers can exhibit promising photovoltaic performances in NF-PSCs.21–25 However, in comparison with BDT-based polymers, BDF-based polymers have still undergone insufficient investigations and their photovoltaic performance also has much room to be improved. Hence, it will be highly important and necessary to further fine-tune the BDF-based polymer structure and promote its performances in NF-PSCs.
In polymer donor design for PSCs, halogenation in either donor or acceptor units of the donor–acceptor polymer is an effective molecular design strategy to further downshift the HOMO energy level so as to increase Voc without sacrificing the current density (Jsc), and thus result in a higher photovoltaic efficiency.26,27 Among the halogen atoms, fluorine and chlorine are most widely used in the replacement of aromatic hydrogens due to their strong electronegativity and unreactive activity to aromatic boronic or stannyl monomers in the palladium-catalyzed carbon–carbon cross-coupling reactions, which are the most applied methods in the synthesis of polymers for PSCs. Fluorine has the highest electronegativity of 3.98 and a similar size to hydrogen, whereas chlorine has a weaker electronegativity of 3.16 and a larger atomic radius than that of fluorine. With fluorine substitute in the donor unit, the resulting polymer will possess a lower HOMO energy level and minimized steric effect due to the similar size of fluorine and hydrogen.28,29 However, in the case of a weaker electronegative chlorine substitute, we can find that the HOMO energy level of the target polymer is further lowered than that of the fluorine substituted polymer, which is due to the heavy atom effect and the empty 3d orbitals of the chlorine atom.17,30–32 Beside the significant effect on the HOMO energy levels in these fluorine- and chlorine-substituted polymers, the intermolecular non-covalent interactions such as F(Cl)⋯H(O) or F(Cl)⋯F(Cl) can also affect the molecular stacking of polymer chains as a result of more ordered aggregation in the nanoscale phase-separated bulk-heterojunction film and potentially improve charge carrier mobility, which is beneficial to Jsc.33–36 In addition, the strong intermolecular interactions may also avoid the use of a solvent additive or annealing during processing of the active layer, which will not bring out device complexity and is more preferred for roll-to-roll or printing technology.37–39
Herein, to probe the halogenation effect on BDF polymers and their photovoltaic performance in PSCs, we designed and synthesized two novel two-dimensional polymers, F10 and F11 (see Scheme 1), based on side chain fluorination or chlorination of the BDF donor block, where fluorobenzothiazole (FBTA) was selected as the electron-deficient acceptor unit, which can effectively regulate the LUMO energy level of the resulting polymers.40 The optical properties of F10 and F11 remained similar to that of the non-halogenated one, but significantly downshifted HOMO energy levels in F10 and F11 were achieved. In the NF-PSCs, while selecting m-ITIC as the electron acceptor, the F10:m-ITIC device gave a PCE of 10.5% with a Voc of 0.908 V, a Jsc of 16.89 mA cm−2 and a FF of 69.5%, whereas the F11:m-ITIC device provided a higher Voc of 0.921 V with a Jsc of 17.75 mA cm−2, and a FF of 69.6%, resulting in a promising PCE of 11.37%. These results demonstrate that the BDF unit is a very promising building block and halogenation is an effective design strategy for BDF polymers to construct highly efficient NF-PSCs.
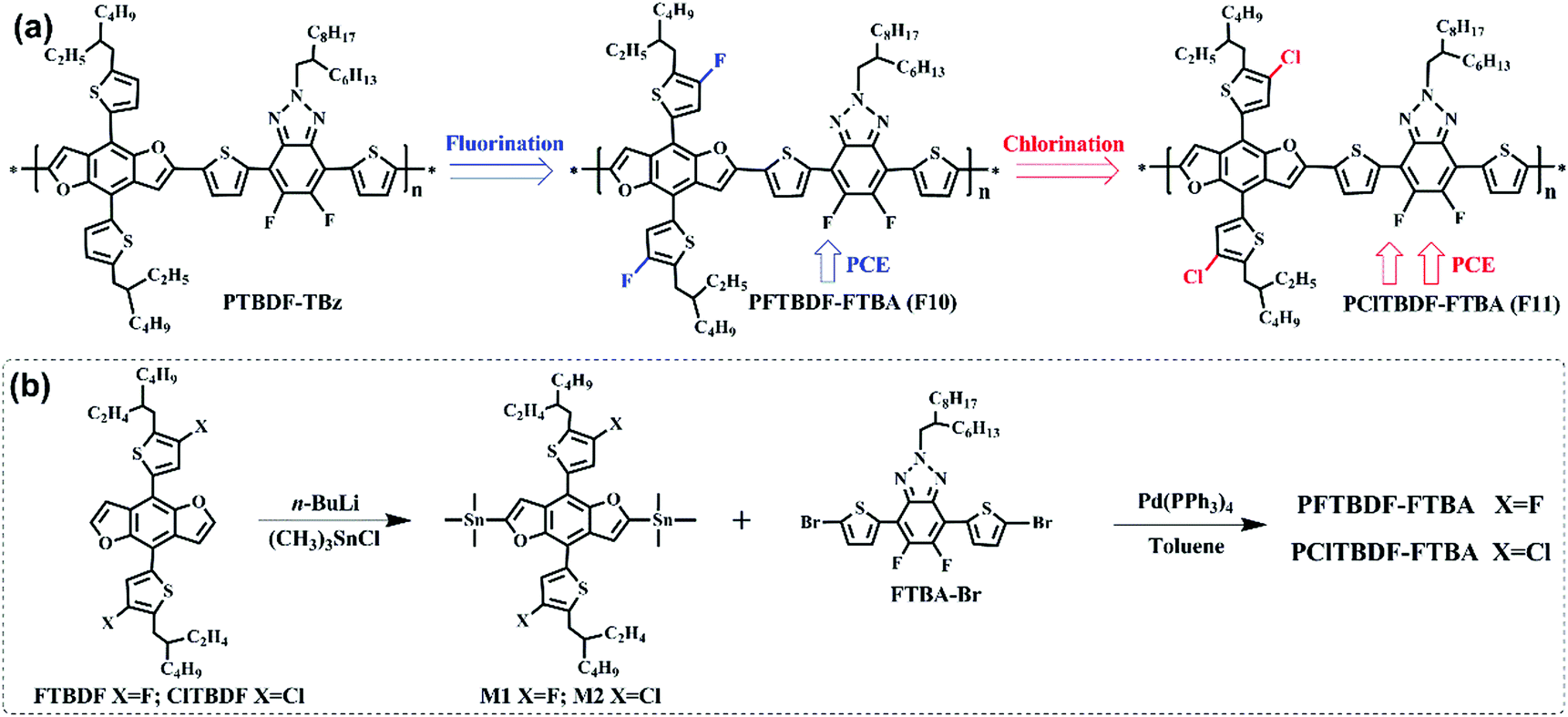 | ||
| Scheme 1 (a) Chemical structures of F10, F11 and PTBDF-TBz; (b) synthetic routes towards polymers F10 and F11. | ||
Results and conclusion
Synthesis
The synthetic route of these two polymers is shown in Scheme 1.41 The procedures for synthesizing 2,6-bis(trimethylstannane)-4,8-bis(5-(2-(2-ethylhexyl)-3-fluorothio phene))benzo[1,2-b:4,5-b′]difuran (M1) and 2,6-bis(trimethylstannane)-4,8-bis(5-(2-(2-ethylhexyl)-3-chlorothiophene))benzo[1,2-b:4,5-b′]difuran (M2) are displayed in the Experimental section. The monomer 4,7-bis(5-bromothiophen-2-yl)-5,6-difluoro-2-(2-hexyldecyl)-4,5-dihydro-2H-benzo-[d][1,2,3]triazole (FBTA-Br) was prepared according to the literature method. The polymers F10 and F11 were synthesized by the copolymerization of M1 or M2 with FBTA-Br via Stille polymerization, respectively. Both polymers have good solubilities in chlorinated solvents, such as chloroform, chlorobenzene, dichlorobenzene, etc. at room temperature. The molecular weights of the polymers were determined by high-temperature gel permeation chromatography at 150 °C using 1,2,4-trichlorobenzene as an eluent. The weight-average molecular weights of F10 and F11 were 41.3 kDa and 38.7 kDa, respectively.Optical properties
The absorption spectra of F10 and F11 in chloroform solutions and film states are displayed in Fig. 1a and b, respectively, and the corresponding data are summarized in Table S1 (ESI†). As shown in Fig. 1a, the absorption spectra of F10 and F11 showed strong 0–0 absorption peaks located at 600 nm with a well-defined 0–1 absorption shoulder, which is ascribed to the strong intramolecular charge transfer (ICT) between BDF and Bz units.42 The molar absorption coefficients of F10 and F11 in solution at 600 nm were measured as 8.7 × 104 M−1 cm−1 and 6.2 × 104 M−1 cm−1 (Fig. S1, ESI†), which is consistent with previous reports. Compared to absorption in solution, the 0–0 absorption peaks of the two polymers in films as shown in Fig. 1b are slightly red-shifted to 604 nm. Moreover, the relative absorption intensity of the 0–0 transition to 0–1 transition in polymers F10 and F11 films is stronger than that of PTBDF-TBz, which may indicate that there are strong intermolecular interactions as induced by F(Cl)⋯H(O) or F(Cl)⋯F(Cl) bonding in comparison with that of non-halogenated PTBDF-TBz.43,44 The absorption edges were found at 644 nm for F10 and 646 nm for F11, and their corresponding optical band-gaps were calculated to be 1.93 eV and 1.92 eV, respectively. The absorption of narrow band-gap electron acceptor m-ITIC is also shown in Fig. 1b, and it can be seen that the absorption of m-ITIC can provide good complementary absorption with F10 and F11 by covering the full solar light of 400–800 nm, which will ensure a potentially high current density in NF-PSCs.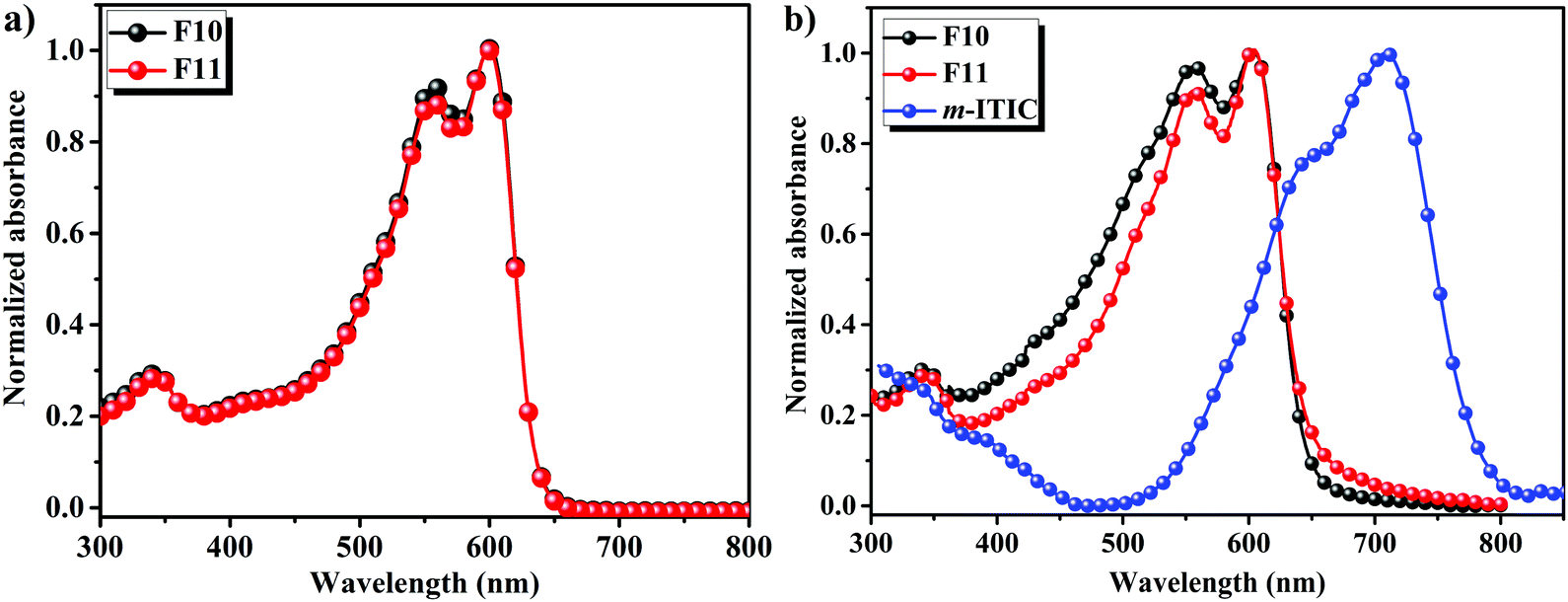 | ||
| Fig. 1 Normalized UV-vis absorption spectra of F10 and F11 (a) in chloroform solutions and (b) film states including m-ITIC in thin film. | ||
Theoretical calculation and electrochemical property
The influence of F and Cl on polymer configuration and frontier molecular orbital distributions was further investigated by theoretical calculations using density functional theory (DFT) at the B3LYP/6-31G(d,p) level with the Gaussian 09 package.45 To simplify the calculation, long alkyl side chains were replaced with methyl groups, which has little effect on the electronic structures of the polymers.46 As seen from Fig. 2a and b, polymers F10 and F11 possessed smaller torsion angles between main chains suggesting that both polymers have better coplanar conjugated backbones. Moreover, F10 and F11 have similar optimized geometries as seen from the side view and top view in Fig. 2a and b and the dihedral angles between side chain and polymer backbone are also very similar though fluorine and chlorine atoms have different atomic radii; the results indicate that the introduction of fluorine and chlorine atoms in the conjugated side chains of polymers F10 and F11 have little influence on the polymeric conformation. As shown in Fig. S2 (ESI†), the HOMO orbital distributions of F10 and F11 are almost delocalized on the BDF unit with minor distributions on the FBTA unit, while the LUMO orbital distributions were delocalized along the polymer main chains. These results are well consistent with well-defined donor–acceptor polymers.47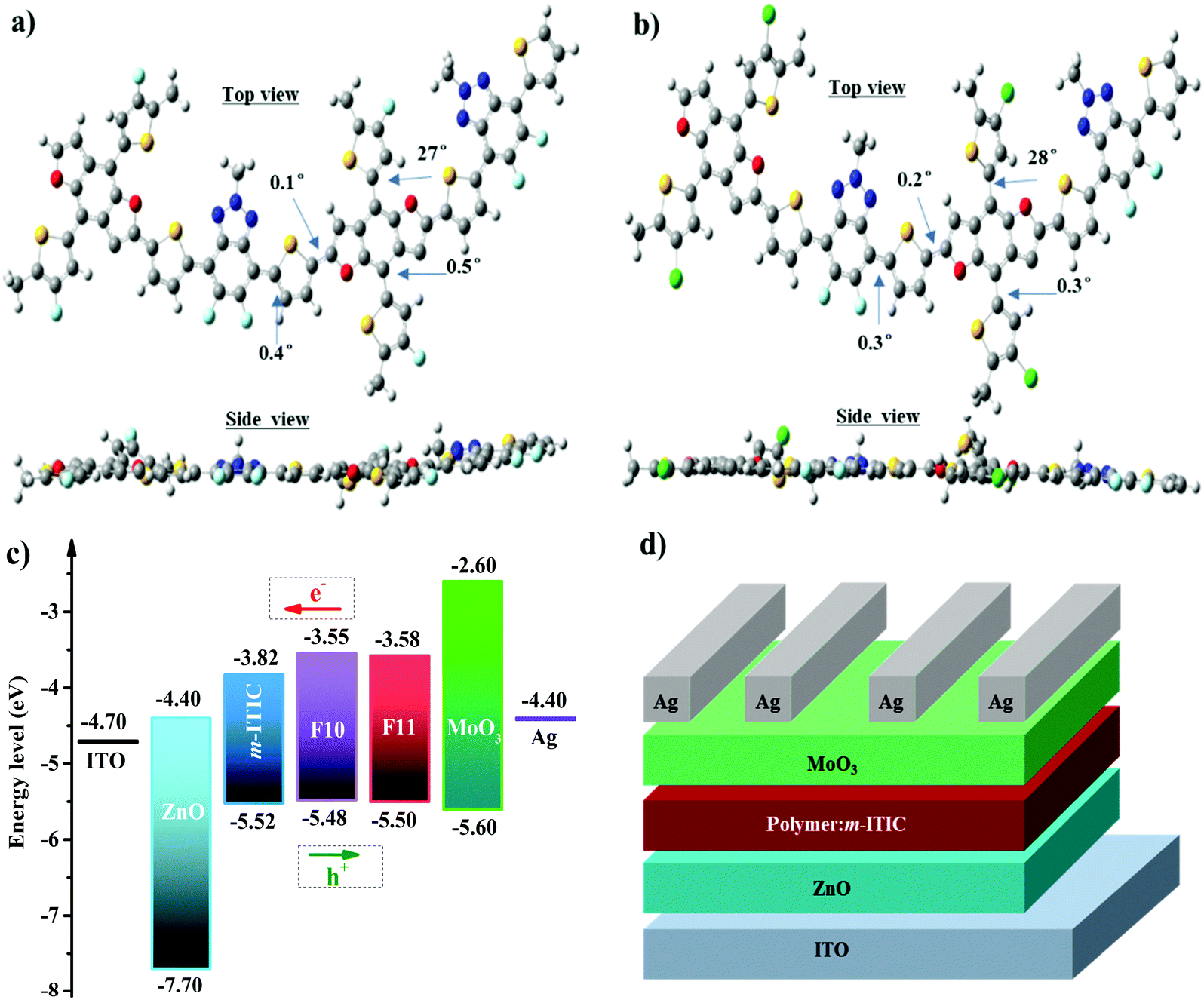 | ||
| Fig. 2 Calculated dihedral angles of F10 (a) and F11 (b) obtained from DFT calculations of the optimized structure (B3LYP/6-31G (d,p)). The S atoms are marked in yellow, O atoms are marked in red, N atoms are shown in blue, F atoms are marked in sky blue, Cl atoms are marked in green, C atoms are shown in gray, and H atoms are marked in white. (c) Energy level diagrams of the donors and acceptor; (d) device structure of NF-PSCs used for F10:m-ITIC and F11:m-ITIC devices. | ||
The energy levels of polymers F10 and F11 were investigated by cyclic voltammetry (CV) according to the equation EHOMO = −e(Vox + 4.80 − Vferro).48 The measured CV curves and corresponding parameters of the two polymers are shown in Fig. S3 and Table S1 (ESI†) and the energy level diagrams of the materials used in PSCs are shown in Fig. 2c. The onset of the oxidation potential (Vox) of F10 and F11 in the film state versus Ag/AgCl is 1.20 V and 1.22 V, respectively, and the onset oxidation potential of FeCp2 (Vferro) is 0.52 V. Thus, the calculated HOMO energy levels are −5.48 eV and −5.50 eV for F10 and F11, respectively, which is markedly downshifted in comparison with that of PTBDF–TBz due to the strong electronegativity of fluorine and chlorine.17,35 For comparison purposes, the HOMO energy levels of J91 and PBT1Cl-Bz,49,50 which are similar BDT counterparts of F10 and F11, respectively, are also listed in Table S1 (ESI†). It is known that the difference of the HOMO energy level of a polymer donor and the LUMO energy level of an electron acceptor is positively proportional to the Voc of PSCs, thus the lower HOMOs in F10 and F11 will be able to harvest a higher Voc. In addition, the LUMO values were calculated from ELUMO = Eopt + EHOMO and were −3.55 eV and −3.58 eV for F10 and F11, respectively. As shown in Fig. 3c, the energy offsets on the LUMOs of F10 (or F11) and m-ITIC are 0.24–0.27 eV, which is enough for electrons, whereas the HOMO offset between F10 (or F11) and m-ITIC is as small as 0.02–0.04 eV, which is unfavorable in fullerene-based PSCs, but recent results have shown that this small driving force is still sufficient for efficient exciton dissociation in NF-PSCs.51
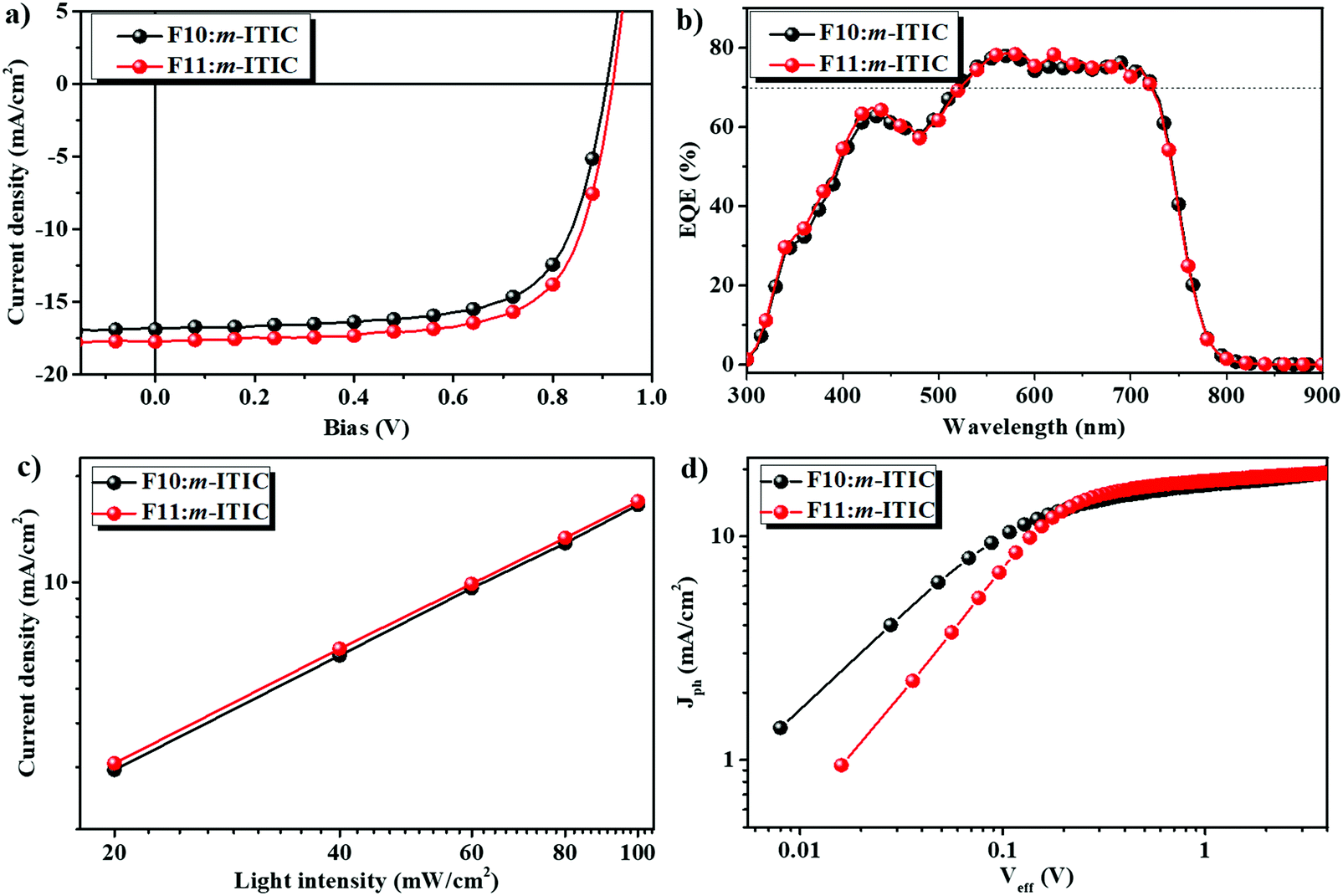 | ||
| Fig. 3 J–V curves (a) and EQE spectra (b) of the devices using F10 and F11 as donor materials under AM 1.5G illumination at 100 mW cm−2; (c) current density-light density curves of F10 and F11; (d) Jph−Veff characteristics of F10- and F11-based devices. | ||
Photovoltaic performance
To investigate the photovoltaic performances of F10 and F11 in NF-PSCs, the devices with the inverted structure of ITO/ZnO/polymer:m-ITIC/MoO3/Ag were fabricated, as shown in Fig. 2d. The optimized devices were obtained at the weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 between F10 (or F11) and m-ITIC after thermal annealing at 130 °C for 2 min. The NF-PSCs without encapsulation were directly tested under AM 1.5 G illumination at 100 mW cm−2. The corresponding parameters are summarized in Table 1. Fig. 3a displays the current density–voltage (J–V) curves of both devices. In the case of fluorinated polymer F10, NF-PSC based on F10:m-ITIC obtained a PCE of 10.50% with a Voc of 0.908 V, a Jsc of 16.89 mA cm−2, and a FF of 69.5%, which were higher than those of non-fluorinated PTBDF-TBz:m-ITIC based devices and were mainly attributed to the simultaneously enhanced Voc and Jsc. Upon chlorination, it is found that the Voc of the F11:m-ITIC device was further increased to 0.921 V as a result of the deeper HOMO energy level, and in combination with the Jsc of 17.75 mA cm−2 and FF of 69.6%, the PCE of F11:m-ITIC was improved to 11.37% due to the enhanced Voc and Jsc. The device performances of the similar BDT counterparts (J91 and PBT1Cl-Bz, Fig. S4, ESI†) of F10 and F11 are also listed in Table 1.49,50 It is noted that J91 has more fluorine substitutes than F10, which should possess a deeper HOMO energy level and higher Voc in NF-PSCs. The results show that the photovoltaic performances of F10:m-ITIC and F11:m-ITIC devices are much higher or comparable with that of J91 and PBT1Cl-Bz-based devices, even though the more efficient electron acceptor IT-4F was applied in the case of PBT1Cl-Bz.50 It is noted that these optimized device performances were achieved without the use of solvent additives or solvent annealing, which will simplify the device fabrication and is more preferred for large area fabrication technologies. The above impressive results indicate that the halogenation strategy in BDF polymers is highly efficient and feasible to further improve the photovoltaic performance of NF-PSCs.
:1.5 between F10 (or F11) and m-ITIC after thermal annealing at 130 °C for 2 min. The NF-PSCs without encapsulation were directly tested under AM 1.5 G illumination at 100 mW cm−2. The corresponding parameters are summarized in Table 1. Fig. 3a displays the current density–voltage (J–V) curves of both devices. In the case of fluorinated polymer F10, NF-PSC based on F10:m-ITIC obtained a PCE of 10.50% with a Voc of 0.908 V, a Jsc of 16.89 mA cm−2, and a FF of 69.5%, which were higher than those of non-fluorinated PTBDF-TBz:m-ITIC based devices and were mainly attributed to the simultaneously enhanced Voc and Jsc. Upon chlorination, it is found that the Voc of the F11:m-ITIC device was further increased to 0.921 V as a result of the deeper HOMO energy level, and in combination with the Jsc of 17.75 mA cm−2 and FF of 69.6%, the PCE of F11:m-ITIC was improved to 11.37% due to the enhanced Voc and Jsc. The device performances of the similar BDT counterparts (J91 and PBT1Cl-Bz, Fig. S4, ESI†) of F10 and F11 are also listed in Table 1.49,50 It is noted that J91 has more fluorine substitutes than F10, which should possess a deeper HOMO energy level and higher Voc in NF-PSCs. The results show that the photovoltaic performances of F10:m-ITIC and F11:m-ITIC devices are much higher or comparable with that of J91 and PBT1Cl-Bz-based devices, even though the more efficient electron acceptor IT-4F was applied in the case of PBT1Cl-Bz.50 It is noted that these optimized device performances were achieved without the use of solvent additives or solvent annealing, which will simplify the device fabrication and is more preferred for large area fabrication technologies. The above impressive results indicate that the halogenation strategy in BDF polymers is highly efficient and feasible to further improve the photovoltaic performance of NF-PSCs.
| Active layer | V oc [V] | J sc [mA cm−2] | FF [%] | PCE [%] | Ref. |
|---|---|---|---|---|---|
| a As-casted film. b Thermal annealing at 150 °C for 2 min. | |||||
| F10:m-ITIC | 0.908 | 16.89 | 69.5 | 10.50 | This work |
| F11:m-ITIC | 0.921 | 17.75 | 69.6 | 11.37 | This work |
| PTBDF-TBz:m-ITIC | 0.85 | 16.63 | 70 | 9.84 | 42 |
| J91:m-ITIC | 0.979 | 14.43 | 42.86 | 6.05a | 49 |
| 0.984 | 18.03 | 65.54 | 11.63b | ||
| PBT1Cl:IT-4F | 0.71 | 16.11 | 63.48 | 7.60 | 50 |
The corresponding external quantum efficiency (EQE) curves of the optimized devices are shown in Fig. 3b. It can be seen that the photo-responses of both devices cover a broad wavelength range of 300–800 nm, which is due to the complementary absorption of F10 (or F11) and m-ITIC, suggesting that both polymer donor and acceptor can absorb solar light efficiently. The EQE peaks for F10:m-ITIC and F11:m-ITIC were 7.96% and 78.59%, respectively, at 570 nm, and the EQE values are more than 70% in the range of 550 nm and 720 nm. The integrated current density values from EQE spectra with an AM 1.5G reference spectrum were 16.732 mA cm−2 and 16.822 mA cm−2 for F11-based and F10-based devices, respectively, which are well consistent with the measured current density from J–V measurements under AM 1.5G illumination.
To understand the charge recombination properties of F10:m-ITIC and F11:m-ITIC devices, we further measured the light intensity (Plight) dependence of Jsc values. In general, the relationship between Jsc and light intensity obeys the relation of Jsc ∝ Plightα, where the exponential factor α indicates the extent of bimolecular recombination: when α as an exponential factor is equal to 1, there is no bimolecular recombination, and when α is smaller than 1, the bimolecular charge recombination happens for the devices.52 As shown in Fig. 3c, both Jsc curves are linear with light intensity, suggesting no significant energy barriers in the device. The α values of F10:m-ITIC and F11:m-ITIC devices are close to 1, indicating that almost all the charges are swept out and collected by the electrode before recombination so as to achieve high Jsc and FF values for both devices.53
The dependence of photocurrent density (Jph) versus the effective voltage (Veff) of the devices was also measured, as shown in Fig. 3d, to study the exciton dissociation of F10- and F11-based devices. The photocurrent density Jph is defined as Jph = JLight − JDark, where JLight and JDark are the photocurrent densities under illumination and in the dark, respectively. The effective voltage Veff is defined as Veff = Voc − Vbias, where Voc is the voltage at which Jph is zero and Vbias is the applied external voltage bias. The exciton dissociation probability of a device can be calculated as Pdiss = Jph/Jsat, where Jsat stands for saturation photocurrent density.54–56 Under short circuit conditions, the Jsat values of F10:m-ITIC- and F11:m-ITIC- based devices were 19.59 and 19.10 mA cm−2, respectively. Thus, the Pdisss of F10:m-ITIC- and F11:m-ITIC- based devices was calculated to be 86.2% and 92.9%, respectively. It is interesting that the chlorinated polymer F11 device showed a more efficient exciton dissociation, which may be attributed to better charge carrier mobility and/or morphology.
To further investigate the charge transport properties of F10:m-ITIC and F11:m-ITIC devices, we measured the charge carrier mobilities by the space charge limit current (SCLC) method. The hole-only device of ITO/PEDOT:PSS/polymer:m-ITIC/MoO3/Ag and electron-only device of ITO/ZnO/polymer:m-ITIC/Ca/Al were fabricated and measured, respectively. The corresponding curves and hole and electron mobilities are summarized in Fig. S4 and Table S2 (ESI†). The hole mobility of the F10:m-ITIC device was found to be 1.27 × 10−4 cm2 V−1 s−1, whereas a higher hole mobility of 1.57 × 10−4 cm2 V−1 s−1 was observed for the F11:m-ITIC device. For the electron mobility, the F11:m-ITIC device also has a higher value of 1.49 × 10−4 cm2 V−1 s−1 than the electron mobility of 1.08 × 10−4 cm2 V−1 s−1 of the F10:m-ITIC device. These results demonstrate that chlorination is more effective in promoting the charge carrier transport property through the plentiful intermolecular non-covalent interactions compared to the fluorination in such polymers.42 The higher hole and electron mobilities in the F11:m-ITIC device as well as its more balanced charge transport than those of the F10 device can therefore support the obtained higher Jsc in F11:m-ITIC solar cells and better exciton dissociation rate.57,58
Surface morphologies
The morphologies of F10:m-ITIC and F11:m-ITIC blend films under optimized device conditions were measured by using atomic force microscopy (AFM) and transmission electron microscopy (TEM). Fig. 4a and b show the AFM images of both blend films. It can be seen that both F10:m-ITIC and F11:m-ITIC films have similar nanoscale aggregations. The root-mean-square (RMS) roughness of the F10:m-ITIC blend film was observed to be 1.69 nm, whereas the F11:m-ITIC blend film has a smaller RMS of 1.27 nm, indicating a better smooth blend film, which is more favorable for exciton separation and thus for a higher Jsc. The TEM images of F10:m-ITIC and F11:m-ITIC blend films are shown in Fig. 4c and d.59 It can be clearly seen that there is no large aggregates observed in both blend films, which indicates that the two blend films are uniform and have good miscibility with nanometer-scale phase-separation between the polymer donor and electron acceptor.22 It can also be found that the F11:m-ITIC film showed a more fibrillary nature than that of F10:m-ITIC, which is similar to the recent report that the fiber-type morphology is more favorable for efficient charge carrier transport and exciton dissociation, and this kind of fibrillary nature in the F11:m-ITIC film should be attributed to the stronger intermolecular non-covalent interactions after introduction of the chlorine atom in polymer F11. | ||
| Fig. 4 AFM and TEM images of F10:m-ITIC (a and c) and F11:m-ITIC (b and d) blend films. | ||
Conclusion
In summary, we have designed and synthesized two novel wide bandgap BDF polymers F10 and F11 with fluorinated and chlorinated conjugated side chains, respectively. The absorption spectra of F10 and F11 show little change compared to that of the non-halogenated counterpart, and the absorption edges of F10 and F11 in film states were at 644 nm and 646 nm, corresponding to the optical bandgaps of 1.93 eV and 1.92 eV, respectively. With the advantages of strong electronegativity of fluorine and chlorine atoms, polymers F10 and F11 exhibited downshifted HOMO energy levels compared to the non-halogenated BDF polymers. The HOMO energy levels of F10 and F11 were at −5.48 eV and −5.50 eV, respectively. The DFT calculations revealed that the polymer backbones of F10 and F11 still have better planarity even with the fluorine and chlorine substitutes. The photovoltaic properties of F10 and F11 in NF-PSCs were investigated under an inverted device structure. The F10:m-ITIC device gave a PCE of 10.5% with a high Voc of 0.908 V, a Jsc of 16.89 mA cm−2 and a FF of 69.5%. The chlorinated F11:m-ITIC device showed a higher Voc of 0.921 V as a result of the lower HOMO energy level after chlorine substitution. The PCE of the F11:m-ITIC device reached 11.37%. It is noted that this performance was achieved without applying a solvent additive and/or solvent annealing process in the processing of the active layer. The charge recombination, charge carrier transport and exciton dissociation and surface morphologies of both devices were also studied to understand the above-mentioned photovoltaic performance. This work demonstrates that the BDF unit is an efficient building block for designing high performance polymer solar cell materials toward low cost commercial NF-PSCs and halogenation engineering of the BDF unit particularly with chlorine is also an outstanding strategy to further promote device performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Natural Science Foundation of China (21644006 and 51403044) and Natural Science Foundation of Heilongjiang Province of China (No. E2018036). Y. Zhang acknowledges the support from the Fundamental Research Funds for the Central Universities (Grant No. HIT. NSRIF.2020001). This work was also supported by the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology, 2019-skllmd-16).References
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- J. S. Wu, S. W. Cheng, Y. J. Cheng and C. S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS.
- B. Fan, X. Du, F. Liu, W. Zhong, L. Ying, R. Xie, X. Tang, K. An, J. Xin, N. Li, W. Ma, C. J. Brabec, F. Huang and Y. Cao, Nat. Energy, 2018, 3, 1051–1058 CrossRef CAS.
- N. A. Ran, S. Roland, J. A. Love, V. Savikhin, C. J. Takacs, Y. T. Fu, H. Li, V. Coropceanu, X. Liu, J. L. Bredas, G. C. Bazan, M. F. Toney, D. Neher and T. Q. Nguyen, Nat. Commun., 2017, 8, 79 CrossRef.
- Y. Yin, Y. Zhang and L. Zhao, Macromol. Rapid Commun., 2018, 39, e1700697 CrossRef.
- M. Liu, Y. Gao, Y. Zhang, Z. Liu and L. Zhao, Polym. Chem., 2017, 8, 4613–4636 RSC.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Yin, Z. Zheng, Y. Lu, D. Chen, M. Liu, F. Guo, S. Gao, L. Zhao and Y. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 29765–29772 CrossRef PubMed.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef.
- B. Liu, X. Chen, Y. Zou, L. Xiao, X. Xu, Y. He, L. Li and Y. Li, Macromolecules, 2012, 45, 6898–6905 CrossRef CAS.
- L. Huo, Y. Huang, B. Fan, X. Guo, Y. Jing, M. Zhang, Y. Li and J. Hou, Chem. Commun., 2012, 48, 3318–3320 RSC.
- A. Gandini, Green Chem., 2011, 13, 1061 RSC.
- Y. Gao, Z. Wang, J. Zhang, H. Zhang, K. Lu, F. Guo, Z. Wei, Y. Yang, L. Zhao and Y. Zhang, Macromolecules, 2018, 51, 2498–2505 CrossRef CAS.
- H. Bin, L. Zhong, Y. Yang, L. Gao, H. Huang, C. Sun, X. Li, L. Xue, Z.-G. Zhang, Z. Zhang and Y. Li, Adv. Energy Mater., 2017, 7, 1700746 CrossRef.
- R. Zhu, Z. Wang, Y. Gao, Z. Zheng, F. Guo, S. Gao, K. Lu, L. Zhao and Y. Zhang, Macromol. Rapid Commun., 2019, 40, 1900227 CrossRef CAS.
- Y. Gao, R. Zhu, Z. Wang, F. Guo, Z. Wei, Y. Yang, L. Zhao and Y. Zhang, ACS Appl. Energy Mater., 2018, 1, 1888–1892 CrossRef CAS.
- Y. Gao, A. Saparbaev, Y. Zhang, R. Yang, F. Guo, Y. Yang and L. Zhao, Dyes Pigm., 2017, 146, 543–550 CrossRef CAS.
- D. Di Nuzzo, G.-J. A. H. Wetzelaer, R. K. M. Bouwer, V. S. Gevaerts, S. C. J. Meskers, J. C. Hummelen, P. W. M. Blom and R. A. J. Janssen, Adv. Energy Mater., 2013, 3, 85–94 CrossRef CAS.
- P. Chao, L. Liu, J. Qu, Q. He, S. Gan, H. Meng, W. Chen and F. He, Dyes Pigm., 2019, 162, 746–754 CrossRef CAS.
- H. Chen, J. Hou, S. Zhang, Y. E. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- Q. Zhang, L. Yan, X. Jiao, Z. Peng, S. Liu, J. Rech, E. Klump, H. Ade, F. So and W. You, Chem. Mater., 2017, 29, 5990–6002 CrossRef CAS.
- Y. Li, J. D. Lin, X. Che, Y. Qu, F. Liu, L. S. Liao and S. R. Forrest, J. Am. Chem. Soc., 2017, 139, 17114–17119 CrossRef CAS.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1800613 CrossRef.
- A. Tang, Q. Zhang, M. Du, G. Li, Y. Geng, J. Zhang, Z. Wei, X. Sun and E. Zhou, Macromolecules, 2019, 52, 6227–6233 CrossRef CAS.
- Q. Fan, W. Su, X. Meng, X. Guo, G. Li, W. Ma, M. Zhang and Y. Li, Sol. RRL, 2017, 1, 1700020 CrossRef.
- Z. Ji, X. Xu, G. Zhang, Y. Li and Q. Peng, Nano Energy, 2017, 40, 214–223 CrossRef CAS.
- P. Chao, Z. Mu, H. Wang, D. Mo, H. Chen, H. Meng, W. Chen and F. He, ACS Appl. Energy Mater., 2018, 1, 2365–2372 CrossRef CAS.
- A. Tang, W. Song, B. Xiao, J. Guo, J. Min, Z. Ge, J. Zhang, Z. Wei and E. Zhou, Chem. Mater., 2019, 31, 3941–3947 CrossRef CAS.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Adv. Energy Mater., 2015, 5, 1500065 CrossRef.
- B. J. Tremolet de Villers, K. A. O’Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876–884 CrossRef CAS.
- H. Zhong, L. Ye, J.-Y. Chen, S. B. Jo, C.-C. Chueh, J. H. Carpenter, H. Ade and A. K. Y. Jen, J. Mater. Chem. A, 2017, 5, 10517–10525 RSC.
- B. Xiao, A. Tang, J. Yang, Z. Wei and E. Zhou, ACS Macro Lett., 2017, 6, 410–414 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS.
- Y. Gao, Z. Wang, J. Zhang, H. Zhang, K. Lu, F. Guo, Y. Yang, L. Zhao, Z. Wei and Y. Zhang, J. Mater. Chem. A, 2018, 6, 4023–4031 RSC.
- J. Wang, M. Xiao, W. Chen, M. Qiu, Z. Du, W. Zhu, S. Wen, N. Wang and R. Yang, Macromolecules, 2014, 47, 7823–7830 CrossRef CAS.
- S. Zhang, Y. Qin, M. A. Uddin, B. Jang, W. Zhao, D. Liu, H. Y. Woo and J. Hou, Macromolecules, 2016, 49, 2993–3000 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- M. Liu, J. Yang, Y. Yin, Y. Zhang, E. Zhou, F. Guo and L. Zhao, J. Mater. Chem. A, 2018, 6, 414–422 RSC.
- T. M. Pappenfus, D. K. Schneiderman, J. Casado, J. T. López Navarrete, M. C. Ruiz Delgado, G. Zotti, B. Vercelli, M. D. Lovander, L. M. Hinkle, J. N. Bohnsack and K. R. Mann, Chem. Mater., 2011, 23, 823–831 CrossRef CAS.
- B. Dandrade, S. Datta, S. Forrest, P. Djurovich, E. Polikarpov and M. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef CAS.
- L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z. Zhang, B. Qiu, X. Li, C. Sun, L. Gao, J. Yao, X. Chen, Y. Yang, M. Xiao and Y. Li, Adv. Mater., 2017, 29, 1703344 CrossRef.
- P. Chao, L. Liu, J. Zhou, J. Qu, D. Mo, H. Meng, Z. Xie, F. He and Y. Ma, ACS Appl. Energy Mater., 2018, 1, 6549–6559 CrossRef.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca, Jr., A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569–4577 CrossRef CAS.
- Q. Fan, W. Su, X. Guo, Y. Wang, J. Chen, C. Ye, M. Zhang and Y. Li, J. Mater. Chem. A, 2017, 5, 9204–9209 RSC.
- L. J. A. Koster, E. C. P. Smits, V. D. Mihailetchi and P. W. M. Blom, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 959–967 CrossRef.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS.
- B. Fan, L. Ying, Z. Wang, B. He, X.-F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243–1251 RSC.
- C. M. Proctor, M. Kuik and T.-Q. Nguyen, Prog. Polym. Sci., 2013, 38, 1941–1960 CrossRef CAS.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganas, M. R. Andersson, R. A. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935–10944 CrossRef CAS PubMed.
- B. Fan, K. Zhang, X.-F. Jiang, L. Ying, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1700746 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc06018a |
| ‡ The authors are equally devoted to the study. |
| This journal is © The Royal Society of Chemistry 2020 |