
Blue metal–organic framework encapsulated denatured R-phycoerythrin proteins for a white-light-emitting thin film†
Xiaobin
Wang
,
Zhuoyi
Li
,
Wen
Ying
,
Danke
Chen
,
Peipei
Li
,
Zheng
Deng
and
Xinsheng
Peng
 *
*
State Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: pengxinsheng@zju.edu.cn
First published on 19th November 2019
Abstract
Fluorescent proteins (FPs) with good photostability and outstanding photoluminescence features are very promising luminous materials for the fabrication of white-light-emitting diodes (WLEDs). However, the requirement of an aqueous environment and poor thermal stability have strongly restricted their wide applications in lighting devices. In this paper, we present a facile strategy to encapsulate R-phycoerythrin (R-PE) fluorescent proteins into a blue metal organic framework (MOF) HSB-W1 (HSB = hydrogenated Schiff base) thin film through a facile solid-confinement conversion process. As a result, R-PE proteins embedded into HSB-W1 crystals are denatured, but exhibit dual color fluorescence emissions including green (518 nm) and red (600, 647 nm) light, while the original single orange light (578 nm) is significantly suppressed. After careful adjustment of R-PE content, the resulting R-PE@HSB-W1 thin film emits high-quality white light with nearly ideal Commission Internationale de I’Eclairage (CIE) coordinates of (0.33, 0.34), a high color rendering index (CRI) value of approximately 85 and a moderately correlated color temperature (CCT) value of 5740 K. Such a strategy can be widely utilized for other fluorescent molecules and luminescent MOFs to design white-light-emitting materials.
1. Introduction
Over the last few years, research on solid-state white-light-emitting diodes (WLEDs) has become particularly important, since they are regarded as promising lighting sources that can replace traditional incandescent bulbs with low energy efficiency and fluorescent lamps causing serious environmental pollution.1–3 The most straightforward strategy to design WLEDs is the integration of individual blue, green and red LED chips. However, the complicated fabrication process and corresponding low luminous efficiency restrict its wide applications.4,5 An alternative approach commercially accepted relies on combining a single blue- or UV-LED chip with down-conversion phosphors. Although pc-LED is more easily realized without complex control circuitries, it suffers from poor white light performance and chromatic aberration due to the different degradation rates between the chips and phosphors.6–8 Thus, it is urgent to develop single-phase down-conversion materials with white-light emission upon blue- or UV-light excitation.The requirements of suitable down-conversion materials to fabricate WLEDs are (i) a high photoluminescence quantum yield (PLQY), (ii) excellent thermal- and photo-stability, (iii) a wide emission band covering the whole visible spectrum, and (iv) a simple preparation procedure.9 Toward the realization of ideal white-light emitting materials mentioned above, fluorescent proteins (FPs) with good photostability and outstanding photoluminescence features came to our attention.10,11 However, the need for an aqueous environment as well as poor thermal stability strongly restricted the application of FPs in lighting devices.12,13 To circumvent the aforementioned problems, Costa and co-workers developed protein-based coatings by embedding individual blue, green and red FPs into rubber materials. Controlling the thickness of different coatings on UV- or blue-LEDs, the entire light emission of the device could be modulated through a bottom-up energy transfer process.14 Recently, they engineered novel white-emitting fluorescent proteins (WFPs) based on the protein superglue concept, and then prepared single-layered color down-conversion packaging to fabricate FP-based WLEDs. The optimized spectrum corresponds to white light emission with CIE color coordinates of (0.37, 0.38), a CCT of 4300 K and quantum yields up to 26%.15 Instead, Nizamoglu and coworkers suggested another method to integrate dried green- and red-emitting FPs on blue LED chips without using any matrix. The resulting device exhibited cold white light with a CCT of 8400 K and luminous efficiency of 100 lm W−1, which could be employed as a backlight for LCD displays.16,17 These strategies provided new perspectives for the development of FP-based WLEDs, nevertheless, the use of two or three FPs leads to a complex fabrication procedure and relatively low luminous efficiency due to the random spatial distribution of different FPs.15
In order to develop a single FP emitter whose PL results in white light emission, we chose R-phycoerythrin (R-PE), one of the typical fluorescent proteins, carrying two kinds of chromophores including phycoerythrobilin (PEB) and phycourobilin (PUB) as down-conversion materials.18–20 The study found that the absorption peak of R-PE at 498 nm was attributed to PUB, and peaks at 540 as well as 560 nm to PEB.20 In general, R-PE emitted strong fluorescence at 578 nm because of the energy transfer between PEB and PUB,21,22 so that blocking the transfer process might be the right approach to get dual- or three-color emission from R-PE.
Unfortunately, there are many barriers to directly integrate R-PE onto a blue-LED without any matrix. Therefore a suitable substrate, with good thermal stability and a simple preparation process, is significant to our following experiment. In recent years, metal–organic frameworks (MOFs), with microporous structures and exceptional tunability, have been deemed as a potential host for encapsulating functional species.23–25 Owing to the various choices of organic ligands and metal nodes, the luminescence properties of MOFs can be elaborately adjusted and changed.26–29 In 2017, Wu and co-workers designed a novel neutral MOF, HSB-W1 (HSB = hydrogenated Schiff base), which exhibited blue light emission centered at 454 nm upon 365 nm excitation.3 Without regard to the solvent molecules, the accessible void of HSB-W1 is calculated to be 55.4%. Besides, HSB-W1 can be stable up to 230 °C,3 which makes it a promising matrix for R-PE to fabricate WLEDs.
Based on the above considerations, we propose a novel strategy to fabricate WLEDs combining a UV-violet-LED chip with R-PE@HSB-W1 composite films. Following a similar method,30–32 R-PE@HSB-W1 was synthesized by encapsulating R-PE proteins into HSB-W1 crystals through a facile solid-confinement conversion process using zinc hydroxide nano-strands (ZHNs) as a precursor. The nanofibrous highly positively charged ZHNs not only firmly attract and confine R-PE during crystal growth through electrostatic interaction, but also serve as a zinc source to generate HSB-W1 thin films by reacting with 1,2-bis(4′-pyridylmethylamino)ethane (L) and terephthalic acid disodium salt at room temperature. As a result, R-PE proteins encapsulated in HSB-W1 are denatured, but exhibit dual color emission including green (518 nm) and red (600 nm, 647 nm) light, whereas the original single orange (578 nm) emission is dramatically suppressed, probably because of the formation of PEB–Zn and PUB–Zn complexes and the blocking of energy transfer between PEB and PUB derived from the denatured R-PE protein.18,20–22,33–35 The optimized R-PE@HSB-W1 membrane demonstrates a high photoluminescence (PL) quantum yield of 60%, because the chromophore components from denatured R-PE proteins are well isolated in HSB-W1 thin films to restrict the aggregation-caused PL quenching, and the loss in efficiency induced by the energy transfer between PEB and PUB is eliminated.5,8 Furthermore, the composite thin film with remarkable thermal stability exhibits high-quality warm white light with nearly ideal CIE coordinates of (0.33, 0.34), a high CRI of 85 and a moderate CCT value of 5740 K.
2. Experimental details
2.1 Materials
(Zn(NO3)2·6H2O), hydrochloric acid and ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd. Ethanolamine (AE) was purchased from Acros Chemicals. R-phycoerythrin was purchased from BioVision supplied in 100 mM phosphate buffer with a concentration of 12.5 mg mL−1, which emits orange fluorescence (578 nm) upon excitation at 405 nm. Terephthalic acid disodium salt was purchased from Aladdin. 1,2-Bis(4′-pyridylmethylamino)ethane was synthesized by NAFU biology.2.2 Characterization
The morphological features and microstructures of the R-PE@HSB-W1 thin films were observed by field emission scanning electronic microscopy (SEM) (Hitachi S-4800) and transmission electron microscopy (TEM) (Tecnai G2 F20 S-TWIN). The crystalline structure of the membranes was analysed using an X’Pert PRO (SHIMADZU XRD-6000) instrument with Cu Kα radiation in the 2θ range of 8°–42°. The PL spectra of R-PE@HSB-W1 membranes, R-PE aqueous dispersion and ligand L dilute solution were recorded using a spectrophotometer (Edinburgh Instruments FLS920) with a 405 nm laser. The absolute PL quantum yields of R-PE@HSB-W1 membranes, HSB-W1 membranes and R-PE aqueous dispersion were measured using a spectrometer (Hitachi U 4100) equipped with an integrating sphere. The ultraviolet/visible absorption spectra of the R-PE@HSB-W1 membranes and R-PE aqueous dispersion were conducted using a UV/vis spectrometer (Agilent Cary 5000). The circular dichroism spectra were recorded using a JASCO J-815 spectropolarimeter.2.3 Preparation of ZHNs
The synthesis of zinc hydroxide nanostrands (ZHNs) was based on our previous procedure.30–32 The preparation process was as follows: 8 mM Zn(NO3)2 ethanol/water (volume 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solution was quickly mixed with a 1.6 mM AE ethanol/water (volume 2:3) solution, the mixture was stirred for 5 minutes and then kept at room temperature for about 30 minutes.
:3) solution was quickly mixed with a 1.6 mM AE ethanol/water (volume 2:3) solution, the mixture was stirred for 5 minutes and then kept at room temperature for about 30 minutes.
2.4 Preparation of R-PE@HSB-W1
As shown in Scheme 1, R-PE@HSB-W1 membranes were synthesized through a very simple solid-confinement conversion process by encapsulating R-PE proteins into HSB-W1 thin films at room temperature. To obtain R-PE@HSB-W1 membranes named as S2 to S7 with different contents of R-PE, 10 mL prepared ZHNs was mixed with different volumes (0.5 mL, 1 mL, 1.5 mL, 1.8 mL, 2 mL and 2.2 mL) (see details in Table S1, ESI†) of R-PE dilute solution (24.4 μg mL−1) and then stirred for about 2 minutes. During mixing, R-PE proteins were firmly assembled onto the ZHN surface through electrostatic interaction to form R-PE/ZHN composites. R-PE/ZHN thin films were then obtained after filtering these composite nanofibers onto polycarbonate (PC) membranes with a pore size of 200 nm, which were further transferred onto quartz plates by carefully peeling off in ethanol. Eventually, R-PE@HSB-W1 was prepared by immersing R-PE/ZHN thin films into the mixture of 25 μM hydrogenated Schiff base L ethanol/water (with a volume ratio of 1:1) solution and 22 μM terephthalic acid disodium salt N,N-dimethylformamide (DMF)/water (with a volume ratio of 1:2) solution for 24 h at room temperature.
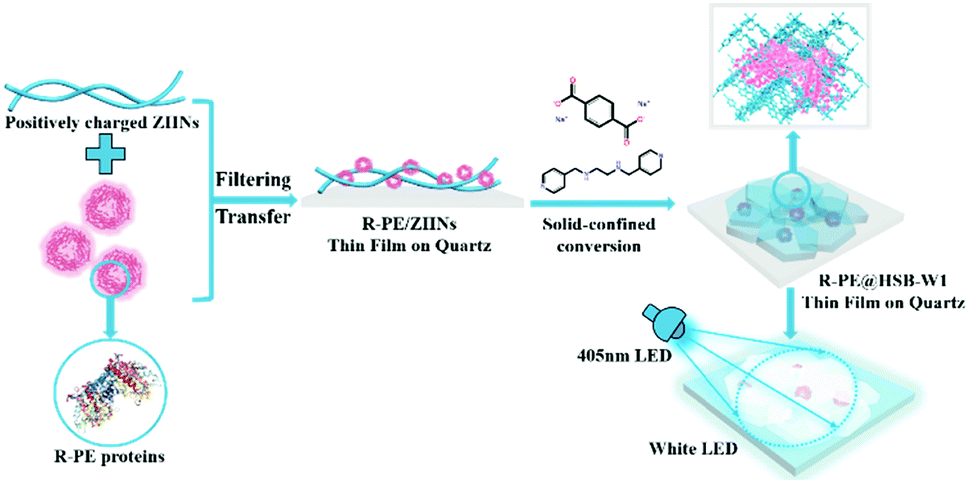 | ||
| Scheme 1 Schematical illustration of the synthesis process of the R-PE@HSB-W1 thin film on quartz for white LEDs. | ||
2.5 Thermal stability, room temperature durability and photo-stability
R-PE@HSB-W1 thin films (S-6) and R-PE aqueous dispersions (24.4 μg mL−1) were treated in air at 80 °C for different times (3 h, 6 h and 12 h). The PL spectra of the resulting R-PE@HSB-W1 thin films and R-PE aqueous dispersions were recorded using a spectrophotometer (Edinburgh Instruments FLS920) with a 405 nm laser. R-PE@HSB-W1 thin films (S-6) were exposed to air under ambient conditions directly (room temperature, humidity from 30 to 70%) for 6 months. R-PE@HSB-W1 thin films (S-6) were continuously illuminated using a 405 nm UV-violet chip operated at 14 mA cm−2 for 6 h, 12 h, 18 h and 24 h. The PL spectra of the resulting R-PE@HSB-W1 thin films were recorded using a spectrophotometer (Edinburgh Instruments FLS-920) with a 405 nm laser.2.6 White light emission demonstration
The optimized R-PE@HSB-W1 thin film (S-6) on a quartz plate prepared from 10 mL ZHNs and a 2.0 mL R-PE aqueous dispersion was placed on a UV-violet-LED chip array (405 nm, purchased from HOYA).3. Results and discussion
To investigate the effect of R-PE concentration on the crystalline structure and PL properties of the resultant films, R-PE@HSB-W1 membranes with different R-PE contents of 0.53, 1.1, 1.6, 1.9, 2.1 and 2.4 wt% were prepared, respectively, named as S-2 to S-7 (Table S1, ESI†). It is clear that all the surfaces of the R-PE@HSB-W1 membranes are smooth, continuous and well-intergrown with layered nanosheets (Fig. 1b and Fig. S1b, c, ESI†). From the cross-section SEM image (Fig. 1c), we can see that the R-PE@HSB-W1 thin film with a thickness of approximately 3 μm present obvious layer structure, which is further confirmed by the cross-section TEM image (Fig. 1d). Obviously, the pure HSB-W1 membrane is porous with randomly standing nanosheets (Fig. 1a and Fig. S1a, ESI†). However, the nanosheets are laid down and orientated well in the case of the R-PE@HSB-W1 composite film, probably because R-PE proteins coated onto ZHNs affect the crystal growth habit of HSB-W1. The cross-section TEM element mapping results (Fig. 1e and f) show the sulfur element from thio-ether bonds of R-PE is uniformly distributed throughout the whole composite film,20 indicating R-PE proteins are evenly embedded into HSB-W1 crystals. The corresponding XRD patterns of R-PE@HSB-W1 membranes (S-3 to S-6) are in alignment with simulated HSB-W1, which demonstrate that the incorporation of R-PE proteins has no effect on the phase of HSB-W1 crystals. | ||
| Fig. 1 Surface SEM images of (a) HSB-W1 (S-1) and (b) R-PE@HSB-W1 (S-6) thin films. (c) Cross-section SEM image of R-PE@HSB-W1 (S-6) thin film. (d) Cross-section TEM image of (b), and the corresponding element mapping images of (e) S and (f) Zn. (g) XRD patterns of simulated HSB-W1 and R-PE@HSB-W1 (S-3 to S-6) thin films. | ||
Similar to the literature, pure HSB-W1 (S-1) emits blue light with CIE coordinates of (0.24, 0.29) centered at 440 nm and 470 nm (Fig. S2, ESI†) upon 405 nm excitation,3 originating from the hydrogenated Schiff base ligand whose emission band is centered at 466 nm. Besides, the PL spectrum of the R-PE/ZHNs membrane was recorded and is shown in Fig. S3a (ESI†), and it is clear that R-PE coated onto zinc hydroxide nanostrands emits strong fluorescence at 468 nm, 518 nm, 600 nm and 647 nm. Fig. 2a demonstrates the photoluminescence (PL) spectra of R-PE@HSB-W1 composite membranes with different R-PE contents. Obviously, the fluorescence emission peaks at 439 nm as well as 471 nm are generated from R-PE proteins and HSB-W1 crystals, whose PL intensities are the superpositions of fluorescence from R-PE and HSB-W1. Other emission peaks at 518 nm and 600 nm, as well as 647 nm, originate from the R-PE proteins. As expected, when the R-PE content increases from 0.53 wt% (S-2) to 2.4 wt% (S-7), the PL intensity of the corresponding composite film rises all along, and HSB-W1 further enhances the fluorescence emission of R-PE molecules through energy transfer. To determine the donor and acceptor units in the R-PE@HSB-W1 composite film, we recorded the UV-vis absorption spectrum of the R-PE/ZHNs membrane and compared it with the PL spectrum of HSB-W1 (Fig. S3b, ESI†). It is obvious that the donor is HSB-W1 and the acceptor is R-PE in the energy transfer process. Table S1 (ESI†) record the CIE coordinates of all the composite membranes (S-2 to S-7), the R-PE content of 2.1 wt% (S-6) with nearly ideal CIE coordinates of (0.33, 0.34) is thus used to fabricate WLEDs.
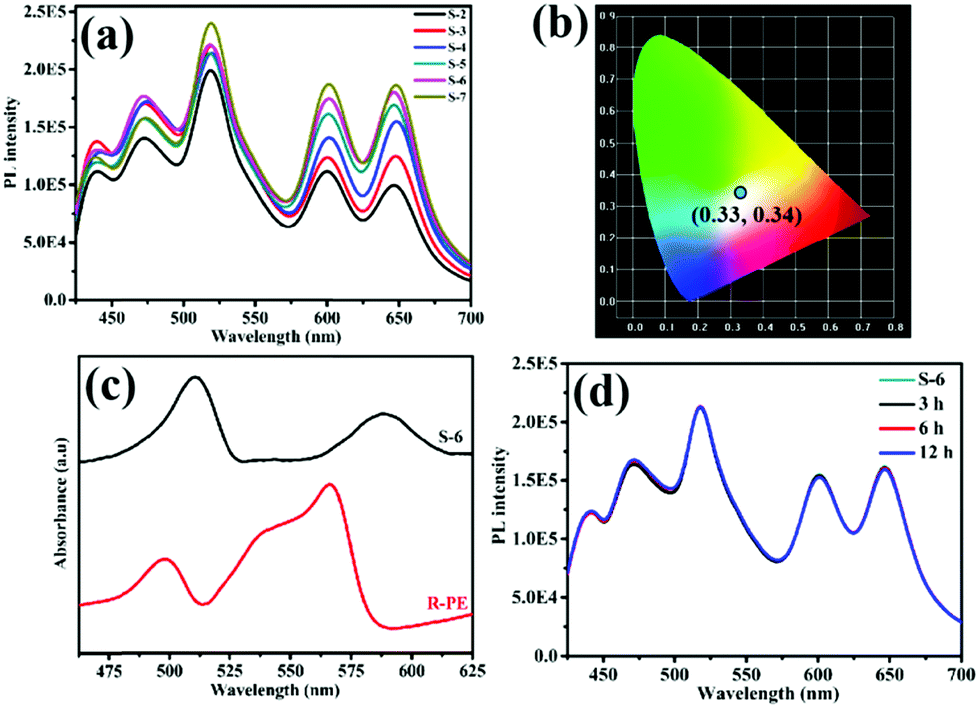 | ||
| Fig. 2 (a) PL spectra of R-PE@HSB-W1 thin films (S-2 to S-7) with different content of R-PE excited at 405 nm. (b) Emission colors in the CIE 1931 chromaticity diagram of the R-PE@HSB-W1 thin film (S-6) excited at 405 nm. (c) The visible range UV-vis absorption spectra of the R-PE solution and the R-PE@HSB-W1 thin film (S-6). (d) The PL stability of the R-PE@HSB-W1 thin film (S-6) treated at 80 °C, excited at 405 nm. | ||
Notably, the R-PE@HSB-W1 composite films emit two new colors of fluorescence including green (518 nm) and red (647 nm) light, whereas the original orange (578 nm) emission of R-PE proteins is dramatically suppressed. The remaining emission peaks at 600 nm is consistent with that of R-PE proteins (Fig. S4a, ESI†). Fig. 2c presents the UV-vis absorption spectra of the R-PE@HSB-W1 composite film (S-6) and R-PE aqueous dispersion. It is obvious that after encapsulation into HSB-W1 crystals, the absorption peaks of R-PE at 495 nm and 565 nm are red shifted to 509 nm and 588 nm, respectively. This is probably because of the interaction between Zn ions from ZHNs and the chromophores of R-PE during the mixing process to form PEB–Zn and PUB–Zn complexes.18,20–22,33–35 The absorption wavelength maximum of PEB red shifted to 588 nm which is quite close to the absorption peak of PEB–Zn salts at 583 nm, and that of PUB red shifted to 509 nm which is the same as that of PUB–Zn salts at 509 nm reported in the literature,33 confirming the formation of PEB–Zn and PUB–Zn. It has been reported that these two complexes reduce the energy of the π* orbital, leading to the red shifts of π → π* and n → π* absorptions.20 In conclusion, the emission peak of R-PE@HSB-W1 at 647 nm is probably generated from PEB chromophore. The local nano-environment also might affect the PL properties,36 resulting in a significant red shift to 647 nm. Generally, due to the energy transfer between PUB and PEB,20–22 R-PE proteins emit single orange fluorescence at 578 nm instead of green light and red light. However, during HSB-W1 crystal growth, the polypeptide structure of R-PE coated onto ZHNs might be pulled apart and denature the R-PE, owing to the electrostatic-assisted assembling along the ZHNs.37 Therefore, the distance between PEB and PUB increases and the energy transfer between PEB and PUB is blocked. R-PE@HSB-W1 thus emits green fluorescence at 518 nm originating from the absorption peak of PUB at 509 nm. Obviously, in this process, the structure of the R-PE proteins is damaged and R-PE is denatured. As shown in Fig. S5 (ESI†), the CD spectrum of pure R-PE solution has negative maxima at 211 nm and 220 nm, which is in accordance with the literature.38 Compared to the R-PE dilute solution, a new negative peak of R-PE@HSB-W1 (S-6) at 234 nm emerges, indicating that the R-PE proteins embedded into the HSB-W1 thin film are denatured. A similar PL spectra of R-PE/ZHNs and R-PE@HSB-W1 (S-6) in the 500 nm to 700 nm range suggested that the denaturation of the R-PE proteins occurred during the mixing of R-PE and ZHNs.
Before using R-PE@HSB-W1 as FP-based WLEDs, the thermal stability, long-term durability and photo-stability of the composite thin film were evaluated. It has been mentioned that HSB-W1 exhibits an excellent thermal stability up to 230 °C,3 and should be a good host for the encapsulation of R-PE based chromophores. However, the PL properties of R-PE, as a kind of fluorescent protein, would be seriously affected at high temperature.38–41 As shown in Fig. S6 (ESI†), after heating at 80 °C for 3 hours, the fluorescence emission of the R-PE aqueous dispersion at 578 nm has been basically degraded. If the heating time goes beyond 12 hours, the fluorescence emission is quenched completely, reaching almost 100% decrement. Fortunately, the PL thermal stability of denatured R-PE encapsulated in HSB-W1 thin film is improved remarkably. Fig. 2d shows the PL spectra of the R-PE@HSB-W1 thin film (S-6) heated at 80 °C for 3, 6 and 12 hours, respectively. Clearly, the PL intensity of the composite film is basically unchanged over different times. The degradation of fluorescence emission at 647 nm is only 2.79% after it is treated at 80 °C for 6 hours, and 5.57% for 12 hours. This means that MOFs provide excellent thermal stability for the chromophores from denatured R-PE. To further clarify this, the PL property of the dilute denatured R-PE solution, containing only α, β and γ monomers of fluorescent proteins,38 after being treated in air at 80 °C for different times was investigated (Fig. S7, ESI†). The denatured R-PE solution emits blue (468 nm, 512 nm) and red (600 nm) light at room temperature, which is very different from the orange fluorescence emission from R-PE proteins at pH 7.0 (578 nm) (Fig. S7, ESI†). After heating for 3 hours, the fluorescence emission of the denatured R-PE solution at 598 nm has degraded basically, and other emission peaks at 467 nm and 504 nm decrease accordingly. It suggests that the fluorescence emission of denatured R-PE proteins is also quenched at high temperatures. Therefore, the excellent PL thermal stability of the MOF-protein complex is derived from MOF confinement. After being encapsulated into HSB-W1 crystals, α, β and γ monomers as well as the chromophores (PEB and PUB) of the denatured R-PE proteins are confined by the MOF framework and not easily influenced by the environment. However, in dilute solution, the corresponding components are flexible, which may be unstable at high temperature. Furthermore, such heat treatment has no effect on the crystal structure of R-PE@HSB-W1. It is obviously comparable to the original R-PE@HSB-W1 thin film, the surface morphological features of the composite films after heating for different times are not changed (Fig. 3).
 | ||
| Fig. 3 Surface SEM images of (a) original R-PE@HSB-W1 thin film (S-6), and membranes after being treated at 80 °C in air for (b) 3 hours, (c) 6 hours and (d) 12 hours. | ||
As illustrated in Fig. S7 (ESI†), the denatured R-PE emits green light at 512 nm and red light at 600 nm. Obviously, the emission peak at 512 nm is derived from PUB, and that at 600 nm is from PEB, so that the origin of the remaining emission peak of R-PE@HSB-W1 at 600 nm is PEB. After the interaction between the Zn ions from ZHNs and the chromophores of R-PE during the mixing process to form PEB–Zn and PUB–Zn complexes, the absorption wavelength maximum of PUB and PEB red shifted, leading to red shifts of the emission peaks of PUB and PEB. Therefore, the origins of the emission peaks at 518 and 647 nm is PUB–Zn and PEB–Zn, which is close to the values reported in the literature.20,33 This further explained the mechanism of the dual-color emission.
Besides, we investigated the long-term durability of the resulting R-PE@HSB-W1 thin film (S-6) by exposing it to air under ambient conditions directly (room temperature, humidity from 30 to 70%) for 6 months. Fig. S8a (ESI†) presents the PL spectra of the R-PE@HSB-W1 thin films (S-6) obtained by the process mentioned above. The PL intensity of fluorescence emission at 600 nm suffers 8.77% loss after 2 months, 16.6% after 4 months and 22.4% after 6 months. The degradation rate is calculated to be about 4% per month. Other fluorescence emissions at 439 nm, 471 nm, 518 nm and 647 nm also degrade after long exposure times. Although the degradation is not significant, the degradation mechanism of PL of R-PE@HSB-W1 (S-6) as the storage time increases was investigated. For comparison, a pre-denatured R-PE@HSB-W1 (S-8) was prepared from 1.5 mL diluted pre-denatured R-PE protein solutions (treated at pH 2) following a similar process (Table S1, ESI†). It is clear that S-8 emits blue (468 nm), green (518 nm) and red (600 nm, 647 nm) light, which is almost identical to R-PE@HSB-W1 (S-2 to S-7) (Fig. S9, ESI†). This further confirmed that the encapsulated R-PE in MOFs is denatured. Afterwards, the S-8 thin film was exposed to air under ambient conditions directly for 1 month (S-9). Fig. S10 (ESI†) indicates that the intensities of the peaks at 518 nm and 647 nm were slightly decreased with the increment of storage time. This is related to the PEB and PUB based chromophores, which might be because of the interaction of species coming from the environment. Interestingly, the fluorescence at 439 nm and 471 nm degrades relatively quicker than other emissions. On one hand, the fluorescence of the chromophores from R-PE degrades as the storage time increases. On the other hand, the PL intensity of the HSB-W1 thin film also declines after being exposed for a long time under atmospheric conditions (Fig. S11, ESI†). Although the degradation occurred slowly, Fig. S8b (ESI†) suggests that the R-PE@HSB-W1 thin film can emit stable white light for a long time.
To study the photo-stability of R-PE@HSB-W1 (S-6), we measured the PL spectra of the composite thin film (S-6) after being continuously illuminated by a 405 nm UV-violet LED chip for 6 hours, 12 hours, 18 hours as well as 24 hours (Fig. S12a, ESI†), and calculated the corresponding CIE coordinates (Fig. S12b, ESI†). It is clear that the PL intensity of the R-PE@HSB-W1 thin film (S-6) after operating for a long time remains basically unchanged, and all the coordinate points are in range of white light. Fig. S13 (ESI†) shows that after being illuminated for 24 hours, the PL intensities of R-PE@HSB-W1 (S-6) at 518 nm, 600 nm and 647 nm remain at 84.8%, 84.9% and 85.5%. This means the R-PE@HSB-W1 thin film has good photo-stability. At last, the absolute PL quantum yields (PLQY) of R-PE@HSB-W1, R-PE aqueous dispersion and HSB-W1 were measured as 60%, 13% and 30%, upon 405 nm excitation within an integrating sphere (Table S2, ESI†). Obviously, the PLQY of R-PE@HSB-W1 is much higher than R-PE as well as HSB-W1. The higher PLQY of the R-PE@HSB-W1 thin film comes from that after embedding R-PE into HSB-W1 crystals, the energy transfer between PEB and PUB is blocked, so that the loss in efficiency of energy transfer process in R-PE is eliminated. Besides, the PEB and PUB related chromophores are uniformly distributed throughout the whole composite film, suppressing the aggregation-caused PL quenching.5,8
R-PE@HSB-W1 thin films with high PLQY, excellent thermal stability and long-term durability are great potential materials for WLED fabrication. Eventually, the optimized composite film (S-6) was prepared from 10 mL ZHNs and 2 mL R-PE aqueous dispersion (24.4 μg mL−1). Fig. 2b records the emission colors in the CIE 1931 chromaticity diagram of this R-PE@HSB-W1 thin film, it is clear that the CIE point of (0.33, 0.34) is in the range of white light, which is very close to ideal white emission (0.33, 0.33). After calculation, the corresponding correlated color temperature (CCT) is 5740 K, and the color rendering index (CRI) is approximately 85. Compared to solar spectrum at 5000 K, the fluorescence emission of R-PE@HSB-W1 is almost consistent with sunlight suitable for human eyes (Fig. S14, ESI†).42,43 To further demonstrate the capability of R-PE@HSB-W1 for practical WLEDs application, the quartz-supported composite thin film (S-6) was directly placed on a UV-violet LED chip array (405 nm). We can see that the resulting WLED device exhibits bright white light when the LED is connected to electrical power (Fig. 4). As illustrated in Fig. S15a (ESI†), upon applying different currents starting from 0 to 18 mA cm−2, the highest EQE value of the UV-violet LED chip is achieved at 14 mA cm−2. Besides, the luminous intensity and luminous efficiency of the S-6 thin film excited by the LED chip increase all along as applied current increases (Fig. S15b and c, ESI†). As the applied current is set to 14 mA cm−2, the luminous efficiency of the resulting WLEDs is 81 lm W−1. Finally, at different applied voltages, there is a slight change in the CIE coordinates of R-PE@HSB-W1 (S-6) (Fig. S15d, ESI†), but all the coordinate points are in the range of white light. Therefore, R-PE@HSB-W1, as a kind of novel single phase white emission material, is very promising for practical lighting applications.
 | ||
| Fig. 4 Photographs of white light-emitting LEDs assembled from a R-PE@HSB-W1 thin film with an UV-violet (405 nm) LED curing chip when the UV-violet LED is turned off and turned on. | ||
4. Conclusion
In summary, we demonstrated a facile solid-confinement conversion process to encapsulate R-PE proteins into HSB-W1 for WLED fabrication. Under optimal conditions, R-PE proteins were evenly distributed and well isolated in the HSB-W1 substrate, resulting in higher PLQY by suppressing the aggregation-caused PL quenching. Interestingly, R-PE proteins embedded into HSB-W1 crystals are denatured and emit two colors of fluorescence including green (518 nm) and red (600 nm, 647 nm) in the visible region instead of the original single orange (578 nm) emission, probably due to the formation of PEB–Zn and PUB–Zn complexes and blocking of energy transfer between PEB and PUB chromophores derived from the denatured R-PE protein. Furthermore, we investigated the thermal stability, long-term durability and photo-stability of these composite films. The results show that R-PE@HSB-W1 can emit high-quality white light at high temperature for a long operation time. Upon 405 nm excitation, the optimized R-PE@HSB-W1 thin film illuminates warm white light with nearly ideal CIE coordinates of (0.33, 0.34), a high CRI of approximately 85 and a moderate CCT of approximately 5740 K, thus suggesting the great potential of single phase down-conversion FP-based materials. With our facile solid-confinement method, in this paper, various fluorescent molecules with high quantum efficiency could be encapsulated into different luminescent MOFs to obtain white-light-emitting composites. This shows the great potential of a large number of novel WLEDs with practical lighting application in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key program of National Natural Science and Foundation (51632008), the National Natural Science Foundation of China (21875212, 21671171), Major R & D plan of Zhejiang Natural Science Foundation (LD18E020001) and the National Key Research and Development Program (2016YF-A0200204).References
- P. Pust, V. Weiler, C. Hecht, A. Tucks, A. S. Wochnik, A. K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS.
- R. Sebastian, L. Frank, S. Gregor, S. Nico, W. Karsten, L. BjoRn and L. Karl, Nature, 2009, 1212, 234–238 Search PubMed.
- Y. Wen, T. Sheng, X. Zhu, C. Zhuo, S. Su, H. Li, S. Hu, Q. L. Zhu and X. Wu, Adv. Mater., 2017, 29, 1700778 CrossRef.
- S. P. Lee, C. H. Huang, T. S. Chan and T. M. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 7260–7267 CrossRef CAS PubMed.
- W. Ying, Y. Y. Mao, X. B. Wang, Y. Guo, H. P. He, Z. Z. Ye, S. T. Lee and X. S. Peng, ChemSusChem, 2017, 10, 1346–1350 CrossRef CAS.
- J. Mckittrick and L. E. Shea-Rohwer, J. Am. Ceram. Soc., 2014, 97, 1327–1352 CrossRef CAS.
- X. G. Hu and X. H. Gao, ACS Nano, 2010, 4, 6080–6086 CrossRef CAS.
- Y. J. Cui, T. Song, J. C. Yu, Y. Yang, Z. Y. Wang and G. D. Qian, Adv. Funct. Mater., 2015, 25, 4796–4802 CrossRef CAS.
- L. Niklaus, S. Tansaz, H. Dakhil, K. T. Weber, M. Pröschel, M. Lang, M. Kostrzewa, P. B. Coto, R. Detsch and U. Sonnewald, Adv. Funct. Mater., 2017, 27, 1601792 CrossRef.
- D. M. Chudakov, M. V. Matz, L. Sergey and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS PubMed.
- K. D. Piatkevich, E. N. Efremenko, V. V. Verkhusha and S. D. Varfolomeev, Russ. Chem. Rev., 2010, 79, 243–258 CrossRef CAS.
- R. D. Costa, V. Fernández-Luna and P. Coto, Angew. Chem., Int. Ed., 2018, 57, 8826–8836 CrossRef PubMed.
- S. R. Meech, Chem. Soc. Rev., 2009, 38, 2922–2934 RSC.
- M. D. Weber, N. Lukas, P. S. Marlene, P. B. Coto, S. Uwe and R. D. Costa, Adv. Mater., 2015, 27, 5493 CrossRef CAS PubMed.
- C. F. Aguiño, M. Lang, V. Fernández-Luna, M. Pröschel, U. Sonnewald, P. B. Coto and R. D. Costa, ACS Omega, 2018, 3, 15829–15836 CrossRef.
- S. Nizamoglu, J. Nat. Appl. Sci., 2016, 20, 490–495 Search PubMed.
- D. A. Press, R. Melikov, D. Conkar, E. N. Firat-Karalar and S. Nizamoglu, Nanotechnology, 2016, 27, 45LT01 CrossRef PubMed.
- J. Dumay, M. Morançais, M. Munier, C. L. Guillard and J. Fleurence, Adv. Bot. Res., 2014, 71, 321–343 Search PubMed.
- K. E. Apt, J. L. Collier and A. R. Grossman, J. Mol. Biol., 1995, 248, 79–96 CrossRef CAS.
- L. J. Cheng, J. S. Ma and L.-C. Chiang, Photochem. Photobiol., 2010, 52, 1071–1076 CrossRef.
- T. Jiang, J. P. Zhang and D. C. Liang, Proteins: Struct., Funct., Bioinf., 1999, 34, 224–231 CrossRef CAS.
- G. C. Wang, B. C. Zhou and C. K. Zeng, Sci. China, Ser. C: Life Sci., 1998, 41, 9–17 CrossRef CAS.
- W. M. Xuan, C. F. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- S. Z. Li and F. W. Huo, Nanoscale, 2015, 7, 7482–7501 RSC.
- H. S. Lu, L. Lu. Bai, W. W. Xiong, P. Z. Li, J. F. Ding, G. D. Zhang, T. Wu, Y. L. Zhao, J. M. Li, Y. H. Yang, B. Y. Geng and Q. C. Zhang, Inorg. Chem., 2014, 53, 8529–8537 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef.
- L. E. Kreno, L. Kirsty, O. K. Farha, A. Mark, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- S. T. Zheng, F. Zuo, T. Wu, I. Burcin, C. Chengtsung, R. A. Nieto, P. Y. Feng and X. H. Bu, Angew. Chem., Int. Ed., 2015, 50, 1849–1852 CrossRef PubMed.
- H. H. Jiang, T. Yoshiro, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS.
- Y. Y. Mao, J. W. Li, W. Cao, Y. L. Ying, P. Hu, Y. Liu, L. W. Sun, H. T. Wang, C. H. Jin and X. S. Peng, Nat. Commun., 2014, 5, 5532 CrossRef PubMed.
- Y. Guo, Y. L. Ying, Y. Y. Mao, X. S. Peng and B. L. Chen, Angew. Chem., Int. Ed., 2016, 55, 15120–15124 CrossRef CAS.
- Z. Y. Li, Y. Guo, X. B. Wang, W. Ying, D. K. Chen, X. Ma, X. Zhao and X. S. Peng, Chem. Commun., 2018, 54, 13865–13868 RSC.
- P. Ocarra, C. Oheocha and D. M. Carrroll, Biochemistry, 1964, 3, 1343–1350 CrossRef CAS.
- W. Cole, C. Gray, D. Nicholson and M. Norman, J. Chem. Soc. C, 1966, 1321–1326 RSC.
- H. Plieninger and K. Stumpf, Chem. Ber., 1970, 103, 2562–2570 CrossRef CAS.
- R. Krishanu, M. H. Chowdhury and J. R. Lakowicz, Anal. Chem., 2008, 80, 6942–6948 CrossRef.
- X. S. Peng, J. Jin, N. Yoshimichi, O. Takahisa and I. Izumi, Nat. Nanotechnol., 2009, 4, 353 CrossRef CAS.
- R. Maccoll and L. E. Eisele, Biophys. Chem., 1996, 61, 161–167 CrossRef CAS.
- A. C. Leney, A. Tschanz and A. J. R. Heck, FEBS J., 2018, 285, 178–187 CrossRef CAS.
- V. Fernández-Luna, D. Sánchez-deAlcázar, J. P. Fernández-Blázquez, A. L. Cortajarena, P. B. Coto and R. D. Costa, Adv. Funct. Mater., 2019, 29, 1904356 CrossRef.
- X. B. Wang, Y. Guo, Z. Y. Li, W. Ying, D. K. Chen, Z. Deng and X. S. Peng, RSC Adv., 2019, 9, 9777–9782 RSC.
- S. P. R. Mallem, K.-S. Im, J.-H. Lee, C. Park and P. Bathalavaram, Opt. Mater., 2019, 95, 109270 CrossRef.
- J. Gotta, T. B. Shalom, S. Aslanoglou, A. Cifuentes-Rius, N. H. Voelcker, R. Elnathan, O. Shoseyov and S. Richter, Adv. Funct. Mater., 2018, 28, 1706967 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc05342e |
| This journal is © The Royal Society of Chemistry 2020 |