
Solution-processed hybrid hosts: a way to explore high triplet energy with admirable current and power efficiency without outcoupling techniques for phosphorescent OLEDs†
Diksha
Thakur‡
a,
Deepak Kumar
Dubey‡
b,
Rohit Ashok Kumar
Yadav
b,
Mangili
Venkateswarulu
a,
Subrata
Banik
 c,
Jwo-Huei
Jou
*b and
Subrata
Ghosh
*a
c,
Jwo-Huei
Jou
*b and
Subrata
Ghosh
*a
aSBS, IIT Mandi, HP 175005, India. E-mail: subrata@iitmandi.ac.in
bDepartment of Material Science and Engineering, National Tsing Hua University, No. 101, Sec 2, Guang Fu Road, Hsinchu 30013, Taiwan, Republic of China. E-mail: jjou@mx.nthu.edu.tw
cCentral University of Jammu, Jammu 181143, J&K, India
First published on 25th October 2019
Abstract
Host materials with maximum recombination efficiencies and high triplet energies offer the best prospects for high efficacy phosphorescent organic light emitting diodes (PhOLEDs). However, their synthesis is a major challenge due to the constraints associated with existing PhOLEDs. While there exists a flood of vacuum-deposited host materials, the present need is for solution-processed hosts due to their many benefits. Herein, a library of new solution-processed donor–acceptor based highly efficient hosts is developed (molecules DT316, 309, 313, 320, 321) that comprise the electron transporting unit benzimidazole and the hole transporting unit triphenylamine. All the molecules possess good solubility in common organic solvents. Their highly twisted conformations bestow these molecules with high thermal stability and remarkable triplet energies of 2.79–2.87 eV. The use of these new host materials as emissive layers in solution-processed green PhOLEDs with Ir(ppy)3 as the emitter gives excellent efficacy with a maximum power efficiency of 54.4 lm W−1, a current efficiency of 73.1 cd A−1 and a brightness of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880 cd m−2, while with Ir(2-phq)3 for red devices, values of 25.6 lm W−1, 19.3 cd A−1, and 15519 cd m−2, are respectively obtained, without any additional external outcoupling techniques. The effective host-to-guest energy transfer, balanced charge carriers in the recombination zone, and the ability to generate excitons on both the host and the guest account for this marvellous performance.
880 cd m−2, while with Ir(2-phq)3 for red devices, values of 25.6 lm W−1, 19.3 cd A−1, and 15519 cd m−2, are respectively obtained, without any additional external outcoupling techniques. The effective host-to-guest energy transfer, balanced charge carriers in the recombination zone, and the ability to generate excitons on both the host and the guest account for this marvellous performance.
Introduction
Since the first exhibition of electroluminescence (EL) of organic materials by Bernanose et al. in the 1950s, in which high voltage alternating current was applied to an acridine orange thin film, the organic light emitting diode (OLED) industry has shown ever-increasing performances.1,2 The multiple and meritorious advantages of OLEDs, as documented in the literature, have led the display market to incline towards organic materials for solid state lighting.3–19 The first OLED device was invented by Tang et al. in the Eastman Kodak Company and had a device external quantum efficiency (EQE) of 1% and green fluorescence originating from singlet excitons. This proved that organic materials were a realistic option for optoelectronic applications.20 The OLED display and lighting market expects to surge to over multi-billion-dollar value by 2023. However, the low (25%) internal quantum efficiency (IQE) of fluorescent materials and the observation of EL from triplet metal-to-ligand charge-transfer (MLCT) excited states of both Os(II) and Pt(II) complexes persuaded display engineers to devise PhOLEDs. These can bestow 100% IQE by harvesting triplet excitons since the ratio of singlet to triplet excitons is 1:3 upon electrical excitation of organic molecules.21–28 The spin–orbit coupling in organometallic complexes of Ir, Pt, and Os enables the forbidden radiative transition from the triplet excited state to the singlet ground state and hence 100% IQE can be obtained in PhOLEDs.25,29–32 Since the spearheading work of the Forrest, Thompson and Ma groups on PhOLEDs, most studies on high efficiency OLEDs have centered on the development of organic materials and device structures for PhOLEDs.23,24,33 The greatest challenge to high efficiency in PhOLEDs is quenching due to concentration, triplet–triplet annihilation and triplet–polaron quenching at high driving currents.34–38 However, the issue of efficiency roll-off in PhOLEDs can be mitigated to some extent when the emitting layer (EML) of the PhOLEDs is configured with an emitter as a guest which is homogeneously dispersed into a suitable organic host to reduce the possibility of triplet–triplet annihilation.28,39 Although a large number of PhOLEDs have emerged to date, the availability of suitable host materials is still severely limited. The dispersion of a dopant in the host molecule gives a host–guest system with either a Dexter or a Förster energy transfer mechanism, and a well-qualified host material must satisfy a number of constraints.40 To prevent the reverse energy transfer from guest to host and for better transfer of energy from host to guest, the host material must have high singlet and triplet energies.41–44 The energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) must enable the simplistic injection of holes and electrons in the device, and the compound must be thermally and morphologically stable.35,45–50 Thermal evaporation and spin coating are two techniques which are employed in the fabrication of OLED devices.51 Thermal evaporation, an effective technique for realizing high efficacy in small molecule hosts, is restricted in its use due to a multitude of issues such as the requirement for high thermal stability of the organic molecules,52 the low throughput obtained and the comparatively high cost due to the huge wastage of organic materials in the chamber itself. Therefore there is a need for solution-processable OLEDs which could make the resultant devices cost effective and could deliver large area roll-to-roll fabrication.51,53–56 Innumerous polymer host materials, as catalogued in the literature, could be used for large-scale solution-processed PhOLEDs with high film densities and uniform film morphologies but their deep-rooted inadequacies such as their uncertain molecular structures, requirements for harsh purification techniques and lower triplet energies have impelled scientists to search for small molecule host materials that offer certain molecular structures, high purity, and stable thermal properties.57,58 Lin's group achieved an efficacy of 70 lm W−1via a solution process by using 4,4′-bis(carbazol-9-yl)biphenyl (CBP) as the host and bis[5-methyl-7-trifluoromethyl-5H-benzo(c)(1,5)naphthyridin-6-one]iridium(picolinate) ((CF3BNO)2IrPLA) as the green emitter. Jou's group produced an efficacy of 78 lm W−1via spin coating with CBP as the host and bis[5-methyl-8-trifluoromethyl-5H-benzo(c)(1,5)naphthyridin-6-one]iridium(acetylacetonate) ((2-CF3BNO)2Ir(acac)) as the green light emitting guest. However, it is of paramount importance to develop more compounds for the library of host materials.54,56,59,60
Recognizing the present need and the ideal qualities of hosts as intimated above, we developed some benzimidazole based materials which, fortunately, rank among the best reported green and red hosts in terms of efficacy, thermal stability (as evidenced by thermogravimetric analysis (TGA)), morphology, and cost. We utilized a triphenylamine moiety (which is a good hole transporter and has high triplet energy), and a benzimidazole unit (which is a good electron transporting unit), for balanced charge migration to broaden the exciton recombination area and for good solubility in organic solvents which is vital in solution-processed host materials. The steric hindrance among various phenyl rings distorted the structure, as proved by crystallographic analysis of single crystals, which interrupted the π-conjugation and maintained the high triplet energy. After device optimization, the best device performance based on the green PhOLED achieved a maximum power efficiency (PE) of 54.4 lm W−1, a maximum current efficiency (CE) of 73.1 cd A−1 and a maximum luminescence of 21230 cd m−2. The obtained results for PE and CE are better than those of some available commercial hosts (e.g., CBP, TCTA), and are also superior to those of many reported hosts. Similarly, the red device secured values of 25.6 lm W−1, 19.3 cd A−1, and 15519 cd m−2 for maximum PE, CE, and CIE brightness (0.56, 0.44), respectively.
Results and discussion
Optical properties
To gain insight into structure property relationships, the photophysical behaviour of the synthesized molecules (Fig. 1), was scrutinized. The UV-vis absorption behaviour was initially studied in DCM solvent to give an idea of the π-conjugation lengths and charge transfer (CT) characteristics. The UV-vis absorption behaviour of all molecules in the series was the same indicating that substitutional changes at the nitrogen of the imidazole ring did not change the absorption behaviour. Two bands were observed in the range of 270 nm to 395 nm. The high energy band observed with a maximum at 297 nm is due to π to π* transitions. The low energy band with a tail up to 395 nm had a maximum around 341 nm (Fig. 2). The broad nature of the low energy absorption band indicates the presence of CT from donor to acceptor in the synthesized compounds. According to the absorption edges, the optical band gaps (approximate to the S1 level) could be calculated as 3.26 eV, 3.28 eV, 3.25 eV, 3.25 eV, and 3.27 eV for DT316, DT309, DT320, DT321 and DT313, respectively (Table 1), which are higher than that of green and red fluorescent emitters. The triplet energy levels of the compounds DT316, DT309, DT320, DT321 and DT313 calculated by using the B3LYP hybrid functional and 6-311G(d,p) basis set on the Gaussian 03 suite of programs were 2.84 eV, 2.85 eV, 2.86 eV, 2.87 eV and 2.87 eV, respectively. Experimentally, the triplet energy levels calculated from the phosphorescence spectra (Fig. S38, ESI†) at 77 K in THF solvent (10−5 M) were 2.67, 2.69, 2.73, 2.71, and 2.60 eV for compounds DT316, DT309, DT320, DT321 and DT313, respectively, which also qualify these materials as hosts for green and red phosphorescence and EL. The high triplet energy of the host material is a provision for effective confinement of the triplet excitons on the guest and for the consequential prevention of back energy transfer from the dopant to the host material. Further analysis of the excited states of these compounds was done with the help of fluorescence emission spectra. All compounds showed an unstructured emission peak with a maximum at 406 nm (Fig. S2, ESI†) which started from 361 nm and tailed up to 513 nm, and could be assigned to a CT state.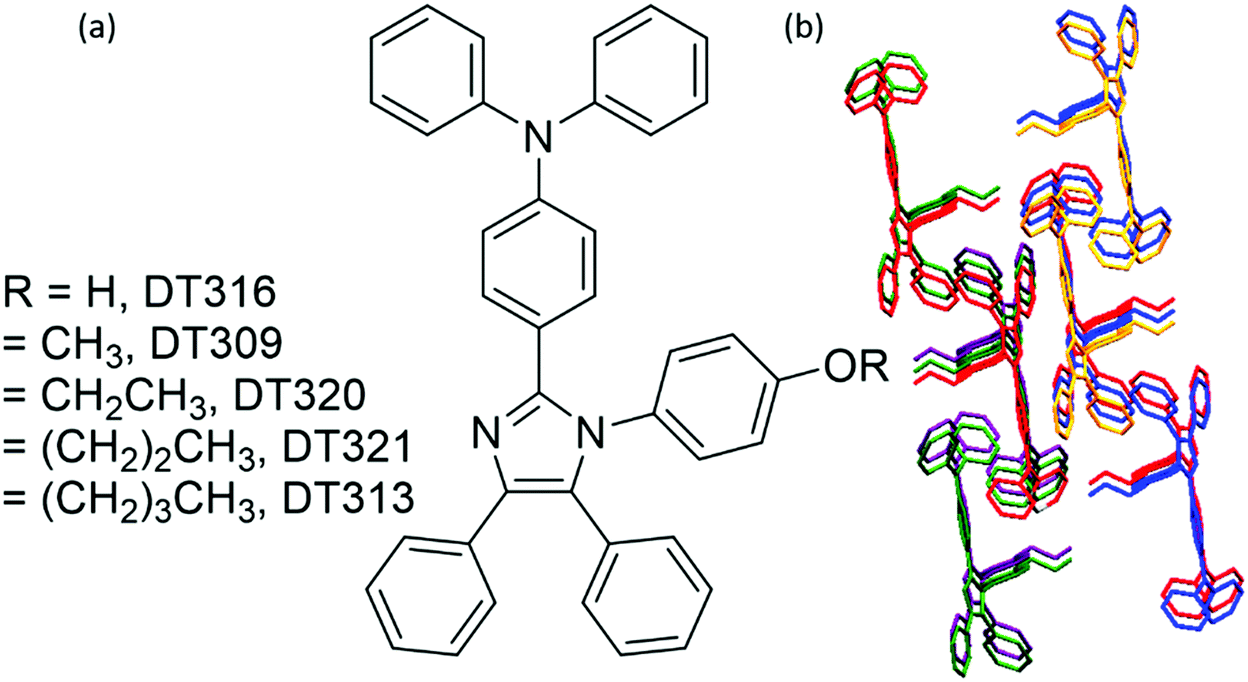 | ||
| Fig. 1 (a) Molecular structures of the designed and synthesized host compounds and (b) the shape packing of DT313. | ||
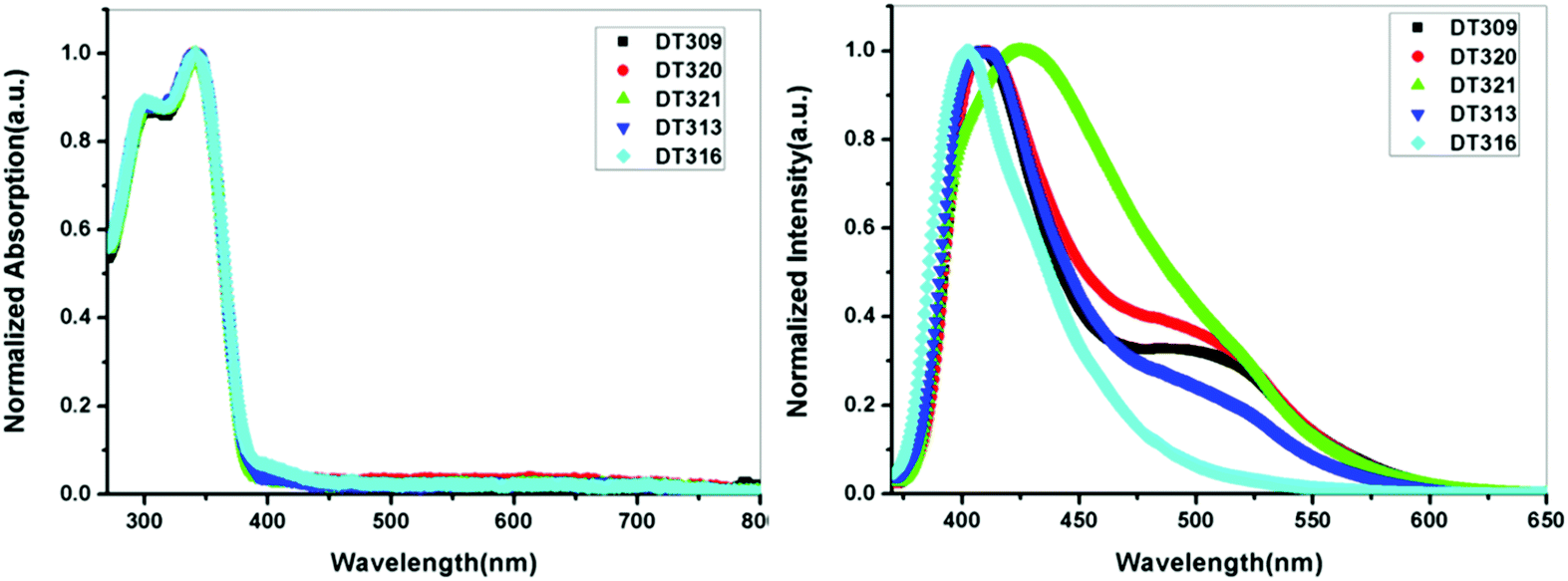 | ||
| Fig. 2 UV-vis absorption (in DCM) and fluorescence emission profiles of thin films of DT309, DT320, DT321, DT313 and DT316. | ||
| a Measured in DCM at 5 × 10−6 M at 298 K. b Optical band gap measured at the onset of the UV-vis absorption spectrum. c HOMO calculated from the oxidation onset of CV, and LUMO calculated from the band gap. d HOMO and LUMO calculated from DFT. e Decomposition temperature estimated from TGA corresponding to 5% weight loss. f Value taken for the second hump in the thin film experiment. | |||||
|---|---|---|---|---|---|
| Compound | DT316 | DT309 | DT320 | DT321 | DT313 |
| Λ abs [nm] | 341 | 341 | 341 | 341 | 341 |
| Λ em(sol)a/(thin film) [nm] | 406/403 | 406/409/497f | 406/412/497f | 406/425 | 406/409/497f |
| Band gapb [eV] | 3.26 | 3.28 | 3.25 | 3.25 | 3.27 |
| HOMO/LUMOc [eV] | −5.21/−1.95 | −5.17/−1.89 | −5.17/−1.92 | −5.15/−1.90 | −5.17/−1.90 |
| HOMO/LUMOd [eV] | −5.03/−1.09 | −5.00/−1.05 | −4.99/−1.04 | −4.99/−1.03 | −4.99/−1.03 |
| T d [°C] | 381.6 | 379.4 | 383.7 | 363.7 | 384.5 |
The thin films are constructed in the layer structure of OLEDs, and therefore, it was obligatory to obtain photoluminescence (PL) spectra in thin films. The synthesized compounds showed differing behaviours in the thin films, unlike in the solution state. The thin film DT309 showed two peaks (Fig. 2), with a high energy emission at 409 nm ranging from 372 nm to 467 nm, and a broader peak at 497 nm starting from 467 nm and tailing up to 612 nm. The presence of extra peaks in the thin film PL spectrum confirms that the packed molecules interact in the solid state. Intermolecular interactions have always led to small red shifts in solid state applications. The solid state emission of DT320 was also observed at 412 nm and 497 nm with the lower energy peak being broadened. It is possible that the extra hump in the low energy region is coming from excimer formation in the solid state.61 Compound DT321 showed a single broadened emission peak from 373 nm to 613 nm with a maximum at 425 nm. Thus the two peaks merged to one broadened peak as the chain length increased from methoxy to propoxy indicating that the chain might be retarding the excimer formation. However, at a chain length of four carbon atoms, i.e. in the butoxy variant, again a small broadened hump appeared at 497 nm and there was a strong peak at 409 nm.
It is possible that in DT313, the chain length leads to a dynamic disorder which may facilitate the excimer formation to some extent. However, this excimer formation was not observed in EL spectra (Fig. S30, ESI†). Instead, the higher energy transfer in the form of holes and electrons from both of the electrodes into the neat film emissive layer under electrical excitation might be responsible for it. DT316 showed only a single peak at 403 nm, blue shifted by 3 nm, which may be assigned to the non-solvent effect.62,63 As DT316 contains an OH moiety, it is possible that the molecules are arranged due to H-bonding such that excimer formation is difficult and the intermolecular interactions lead only to a single emissive peak in the solid state. It is also notable that when the length of the alkyloxy side chain was increased from 1 to 4 carbon atoms, the solid state PL spectrum displayed a red shift compared to that of the solution state. This result can also be explained in terms of long range ordering, packing interactions and solvation effects, which become more dominant with an increase in the length of the alkyl side chain up to 4 carbon atoms, leading to expanded chain structures with long π-conjugation architecture.64–67 To explore the behaviour of emissive states in the synthesized compounds, dipole moment changes in the ground and excited states were monitored by carrying out solvatochromic studies. Low polarity to high polarity solvents (n-hexane to DMSO) were employed for the study. A red shift of 28 nm was observed on going from hexane to DMSO showing that the compounds exhibit very weak CT characteristics (Fig. S4, ESI†). The well-structured emission in hexane comes from a local excited (LE) state since this LE state is stable in a non-polar solvent. From hexane to ether, the well-structured emission merged to almost one broadened band, and in a polar solvent, such as DMSO, the broadened peak maximum shifted to 411 nm from 383 nm. The ground states of all compounds were stable as no solvatochromic behaviour was noticed in the UV-vis absorption of the compounds when shifting from a non-polar solvent to a polar solvent (Fig. S5, ESI†).
Electrochemical and thermal studies
The electrochemical properties of the synthesized compounds were investigated by cyclic voltammetry (CV) in DCM solvent (for DT309, 320, 321 and 313) and in DMF solvent (for DT316). All CV curves are provided in Fig. S6 (ESI†). The oxidation scans of DT309, DT320, DT321, and DT313, in the potential range 0 to 1.7 V, gave two quasi-reversible peaks in the ranges 0.79 V to 0.81 V and 1.10 V to 1.15 V. However, DT316 gave irreversible peaks in the potential range 0 to 1.7 V at 1.05 V with an onset at 0.87 V. The oxidation onset potential for DT316 is high compared to those for the rest of the materials. The high value of the oxidation onset potential for DT316 may be due to the presence of hydrogen bonding involving the OH group attached to the para position of the benzene ring of the N1 atom of the imidazole core.From the onset potentials of the oxidation peaks of these compounds the ionization potentials were measured to be −5.21 eV, −5.17 eV, −5.17 eV, −5.15 eV, and −5.17 eV, for DT316, DT309, DT320, DT321 and DT313, respectively. The band gaps (Eg) obtained from the onset absorption of UV light were used to provide electron affinity (IP) values of −1.95 eV, −1.89 eV, −1.92 eV, −1.90 eV, and −1.90 eV for DT316, DT309, DT320, DT321, and DT313, respectively. The lifetime of an OLED device depends on the thermal stability of a material. To investigate the thermal stability of the compounds, TGA experiments were carried out. The TGA graphs are represented in Fig. S7 (ESI†). The extrapolated onset temperatures were considered to be the decomposition temperatures of the materials. DT316 showed a low level of degradation at 51.4 °C due to the solvent, with a decomposition temperature of 381.6 °C corresponding to a 5% weight loss. DT309 showed an extrapolated onset decomposition temperature of 371.7 °C, however, the decomposition temperature corresponding to 5% weight loss was 379.4 °C. Similarly DT321 and DT313 showed extrapolated onset decomposition temperatures of 355.7 °C and 372.84 °C, respectively. The decomposition temperatures corresponding to 5% weight loss were 363.7 °C and 378.2 °C for DT321 and DT313, respectively. DT320 showed a two-step degradation with the first onset arising at 201.50 °C and 5% weight loss at 383.70 °C. Thus it was concluded that all of the synthesized materials are suitable for inclusion in the library of OLED materials in terms of their thermal stabilities. The robust thermal characteristics of the compounds may facilitate excellent film integrity under electrical excitation because the organic materials are less vulnerable to heat and form stable amorphous films with increased glass transition temperatures (Tg). In contrast, materials with low Tg values crystallize easily, which affects the film homogeneity, and crystal boundaries raise the electrical resistance to the carriers. Therefore, high Tg materials result in better film integrity and lead to better device performances. The Tg values for DT316, DT309, DT320, DT321, and DT313 are 280 °C, 215 °C, 219 °C, 240 °C and 232 °C, respectively (Fig. S8, ESI†). These high Tg values imply that these materials are potential candidates for use in OLED device fabrication.
The theoretical level of understanding
Spectral properties and molecular orbital analyses can be better understood with the help of density functional theory (DFT) calculations. Thus theoretical calculations using the B3LYP hybrid functional and 6-311G(d,p) basis set on the Gaussian 03 suite of programs were carried out to analyse the HOMO–LUMO energy levels and electron distributions over the synthesized compounds.68 There was no specific trend observed in the theoretical HOMO–LUMO levels of the compounds. The HOMO energy levels of DT316, DT309, DT320, DT321 and DT313, are −5.03 eV, −5.00 eV, −4.99 eV, −4.99 eV and −4.99 eV, while the LUMO levels are −1.09 eV, −1.05 eV, −1.04 eV, −1.03 eV and −1.03 eV, respectively. The optimized structures and HOMO–LUMO distributions for DT309 and DT320 are given in Fig. 3 (for other compounds, see Fig. S36 and S37, ESI†). The theoretical HOMO energy levels approximately match the experimental energy levels, but there is a great mismatch between the theoretical LUMO and experimental LUMO energy levels which was expected because the theoretical calculations were carried out for a single gas molecule while the experimental results were extracted from concentrated solutions of the compounds. UV-vis absorption properties for all of the optimized compounds were calculated at the TD-B3LYP/6-311G(d,p) level of DFT. The UV-vis absorptions (Fig. S31–S35, ESI†) of all compounds were in close agreement with the experimental absorption spectra. The optimized structures were also utilized to obtain higher triplet state energies in order to obtain the theoretical energy gaps between the first excited singlet states and first excited triplet states (ΔEST).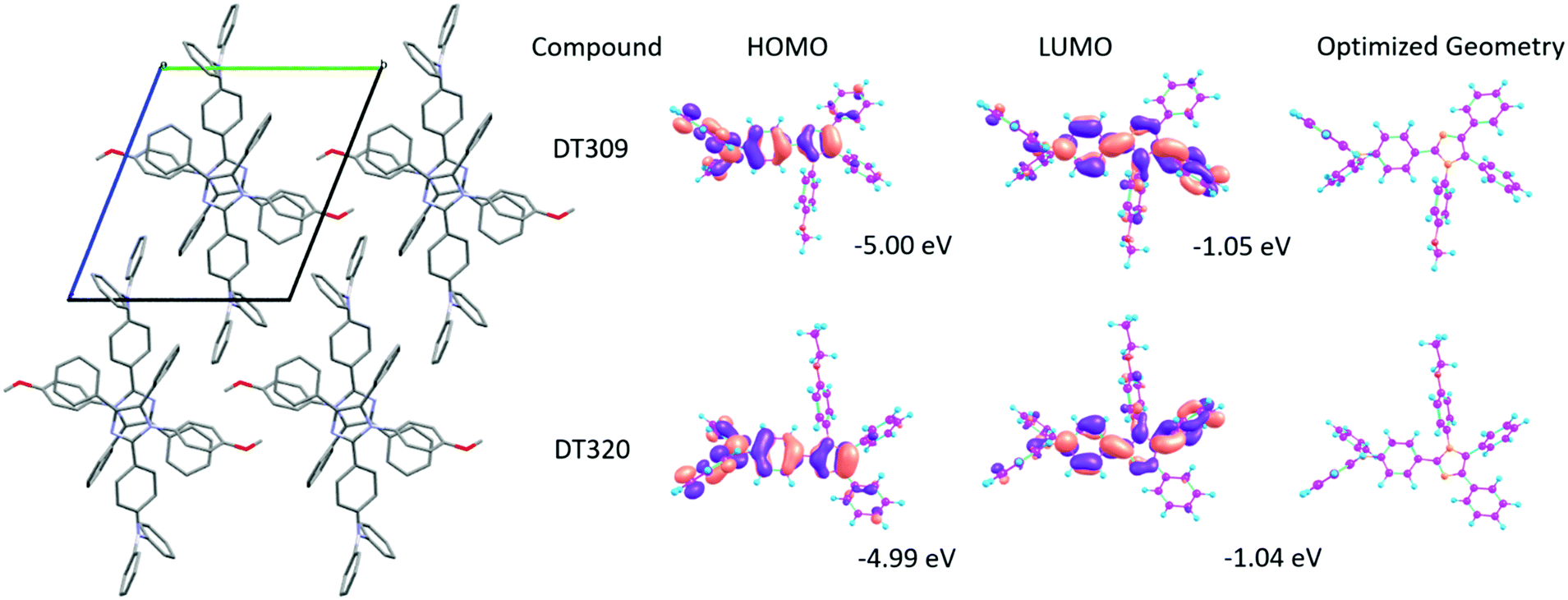 | ||
| Fig. 3 Crystal packing diagram for DT309, and optimized geometries of DT309 and DT320. | ||
The molecular level understanding
To establish the relationship between fluorescence emission and structure at the molecular level, it was desirable to grow crystals of the studied compounds. A thorough study was done using crystals of the four compounds grown in DCM:MeOH solution by the slow evaporation method. Crystal refinement data are tabulated in Tables S1–S4 (ESI†). The ORTEP diagrams of DT309, DT320, DT321, and DT313 are provided in Fig. 4. The crystal structure data for all four compounds helped to prove that no significant π–π stacking exists between neighbouring molecules. This is evidenced by the fact that the alkoxy-substituted phenyl ring is tilted to the imidazole ring plane at a large angle (80.63° in DT309 (Fig. S11, ESI†), 79.89° in DT320 (Fig. S15, ESI†), 89.97° in DT321 (Fig. S19, ESI†), and 83.82° in DT313 (Fig. S23, ESI†)). These various tilted phenyl rings barred neighbouring molecules from stacking with each other and hence aided the solid state emission. Instead, various CH–π interactions exist between the neighbouring molecules. The miscellaneous distances of the short range contacts in DT309 are 2.837, 2.882, 2.596, 2.866, and 2.855 Å (Fig. S10, ESI†) and in DT320 they are 2.877, 2.807, 2.624, 2.888, and 3.341 Å (Fig. S13, ESI†). Similarly, in DT321, the short range contacts are 2.351, 2.773, and 2.649 Å (Fig. S17, ESI†) and in DT313 they are 2.814, 2.620, and 2.874 Å (Fig. S21, ESI†). All these interactions are beneficial for maintaining molecular stiffness and solid state emission. Fig. 3 and Fig. S14, S18, S22 (ESI†) show the crystal packing diagrams of DT309, DT320, DT321, and DT313, respectively. One common feature in the packing of all of the molecules is that the adjacent layers are packed in opposite directions: triphenylamine is packed in the first layer on the head side and the imidazole ring is, again, seated on the head side in second layer. The propensity towards the formation of excimer decreases from DT309 to DT321 and inclination towards the one broadened peak instead of two increases from DT309 to DT321 (i.e. one carbon to three carbon chain length), which can be explained by the crystal data. The distance between the imidazole rings of the two closely spaced molecules in the case of the methoxy substituted DT309 is 4.165 Å (Fig. S9, ESI†). In DT320, with ethoxy substitution, the two imidazole rings sit as two stairs placed on one another (Fig. S12, ESI†), hence, the distance between the imidazole rings of the two molecules increases to 8.752 Å which hinders the formation of the excimer leading to an almost broadened peak in the thin film PL spectrum. In the case of DT321, the two neighbouring molecules are not packed like stairs, and the distance between the two imidazole rings is larger (4.297 Å, Fig. S16, ESI†) than that of DT309 which again, probably, restricts the excimer formation resulting in the disappearance of the second hump. In contrast, in the case of the butoxy substituted DT313, instead of a single broad peak we observed the appearance of the second hump again. The probable reason for this is the dynamic disorder in DT313 as indicated in the ORTEP diagram (Fig. 4) which facilitates excimer formation resulting in the double peak spectrum.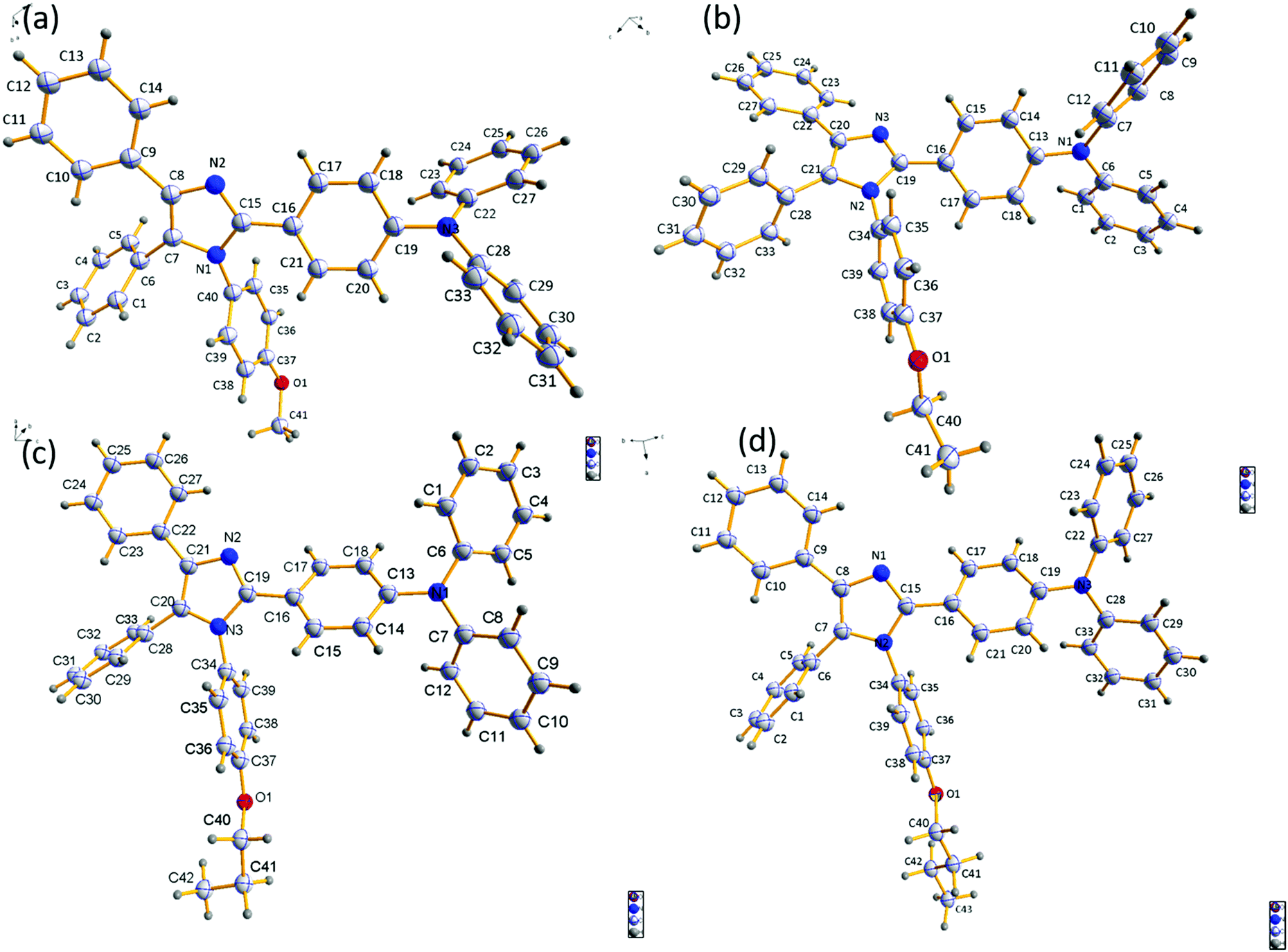 | ||
| Fig. 4 ORTEP diagrams of compounds DT309, DT320, DT321 and DT313. | ||
Carrier-transporting properties
In order to understand the bipolar charge-transporting characteristics of the synthesized host materials, single carrier devices were fabricated with the following device structures.Hole only device (HOD): ITO(125 nm)/PEDOT:PSS(30 nm)/TAPC(35 nm)/HOST(20 nm)/TAPC(35 nm)/LiF(1.0 nm)/Al(150 nm); electron only device (EOD): ITO(125 nm)/TPBi(30 nm)/HOST(20 nm)/TPBi(35 nm)/LiF(1.0 nm)/Al(150 nm). Fig. 5 shows the current density–voltage properties of the single carrier devices. It is interesting to note that the hole and electron current densities both displayed an increasing and comparable trend with the increase of voltage, which implies that the synthesized host compounds showed bipolar carrier-transporting characteristics. Both the hole and the electron current densities were higher in the molecules DT309, DT320, DT321, and DT313 than in compound DT316. This can be explained by the shallower HOMO levels of the molecules DT309, DT320, DT321, and DT313 compared to that of DT316, which significantly reduce the hole trap between PEDOT:PSS and the host, and noticeably enhance the hole blocking ability at the interface of the host and electron transport layer (TPBi). Moreover, the optimized chain length of the alkyl unit may contribute to the higher and balanced carrier charge mobility of these molecules. However, several previous studies have shown that appropriate chain length modifications of alkyl units that are associated with conjugated frameworks and polymer semiconductors can promote intermolecular/interchain packing order and molecular orbital overlapping, and hence enhance the carrier charge mobility and solubility of the molecules.69–73 Furthermore, it is worth mentioning that the electron current densities of all of the compounds were found to be slightly higher than the hole current densities at the same voltages.
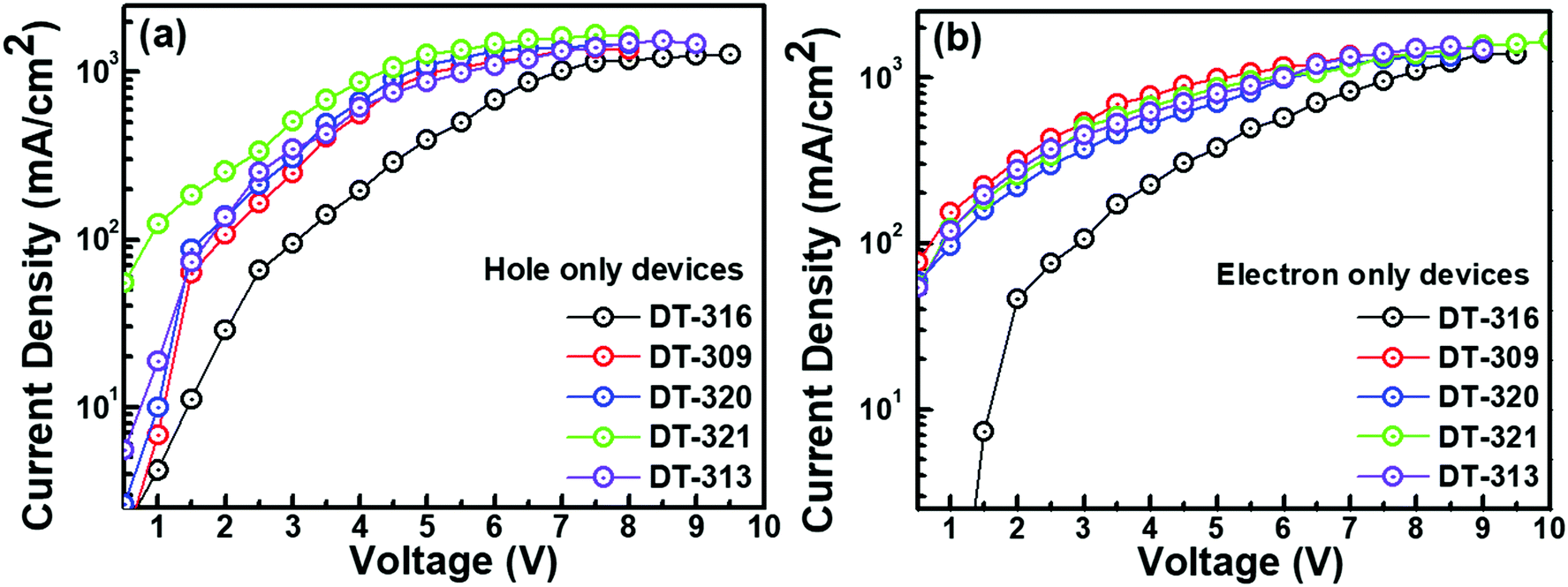 | ||
| Fig. 5 Current density–voltage plots of hole (a) and electron (b) single carrier devices of the molecules DT316, DT309, DT320, DT321, and DT313. The hole and electron densities of the molecules are comparable to each other, which confirms the bipolarity of the synthesized compounds. | ||
Electroluminescence properties of devices
We were encouraged by the following characteristics of the designed and synthesized organic molecules: triplet energies that were higher than that of phosphor to prevent reverse energy transfer from guest to host; suitable molecular orbital energy levels of the HOMOs and LUMOs for effectively collecting the holes and electrons from adjacent hole and electron transport layers, respectively; and good thermal and morphological stabilities. Therefore, all of the compounds were further evaluated as hosts in solution-processed green and red phosphorescent OLEDs using the following device configuration: ITO/PEDOT:PSS(30 nm)/EML(20 nm)/TPBi(35 nm)/LiF(1 nm)/Al(100 nm). The recognized phosphorescent green and red emitters, Ir(ppy)3 and Ir(2-phq)3, respectively, were selected as the emissive materials because of their suitable triplet energies, band gaps, and HOMO–LUMO levels when compared with those of the synthesized host molecules.74 To confirm our results, control devices were also fabricated with CBP and TCTA as the host materials with the same device structure. In all of the fabricated OLED devices, PEDOT:PSS and LiF assisted as the hole injection layer and electron injection layer, respectively. TPBi was used as the electron transporting layer and the hole blocking layer because of its deep HOMO level. In OLED device fabrication schemes, we adopted a solution-process over a dry-process because of its cost-effectiveness, suitability for continuous roll-to-roll manufacturing of large areas, low-material consumption rate, and simple device structure features.The energy level diagrams of all of the devices are depicted in Fig. S29 (ESI†). All of the performances of the fabricated green OLED devices are displayed in Fig. 6, and their collected key EL data are summarized in Table 2. For the 17.5 wt% green emitter Ir(ppy)3, the OLED device with the DT321 host demonstrated a maximum current efficiency (ηCE,max) of 73.1 cd A−1, a maximum power efficiency (ηPE,max) of 54.4 lm W−1, a maximum external quantum efficiency (ηEQE,max) of 10.9% and a maximum luminance (Lmax) of 21230 cd m−2, while the D320 host gave values of ηCE,max = 68.7 cd A−1, ηPE,max = 53.9 lm W−1, ηEQE,max = 10.3% and Lmax = 25740 cd m−2. For the same doping concentration, the OLED device based on host DT316 displayed ηCE,max = 42.8 cd A−1 (ηPE,max = 33.3 lm W−1, ηEQE,max = 6.7% and Lmax = 14400 cd m−2), host DT309 gave ηCE,max = 63.3 cd A−1 (ηPE,max = 50.0 lm W−1, ηEQE,max = 12.1% and Lmax = 24250 cd m−2) and host DT313 gave ηCE,max = 42.0 cd A−1 (ηPE,max = 37.4 lm W−1, ηEQE,max = 7.9% and Lmax = 18510 cd m−2). The devices with DT316, DT309 and DT313 had low turn-on voltages (Von, to deliver a brightness of 1 cd m−2) of 3.7, 3.6 and 3.2 V, respectively. The apparent higher brightness, lower turn-on voltage and better efficiency of the OLED device incorporating the DT321 host might be attributed to the fact that the hole mobility and electron mobility of DT321 are better balanced and higher than those of its counterparts.
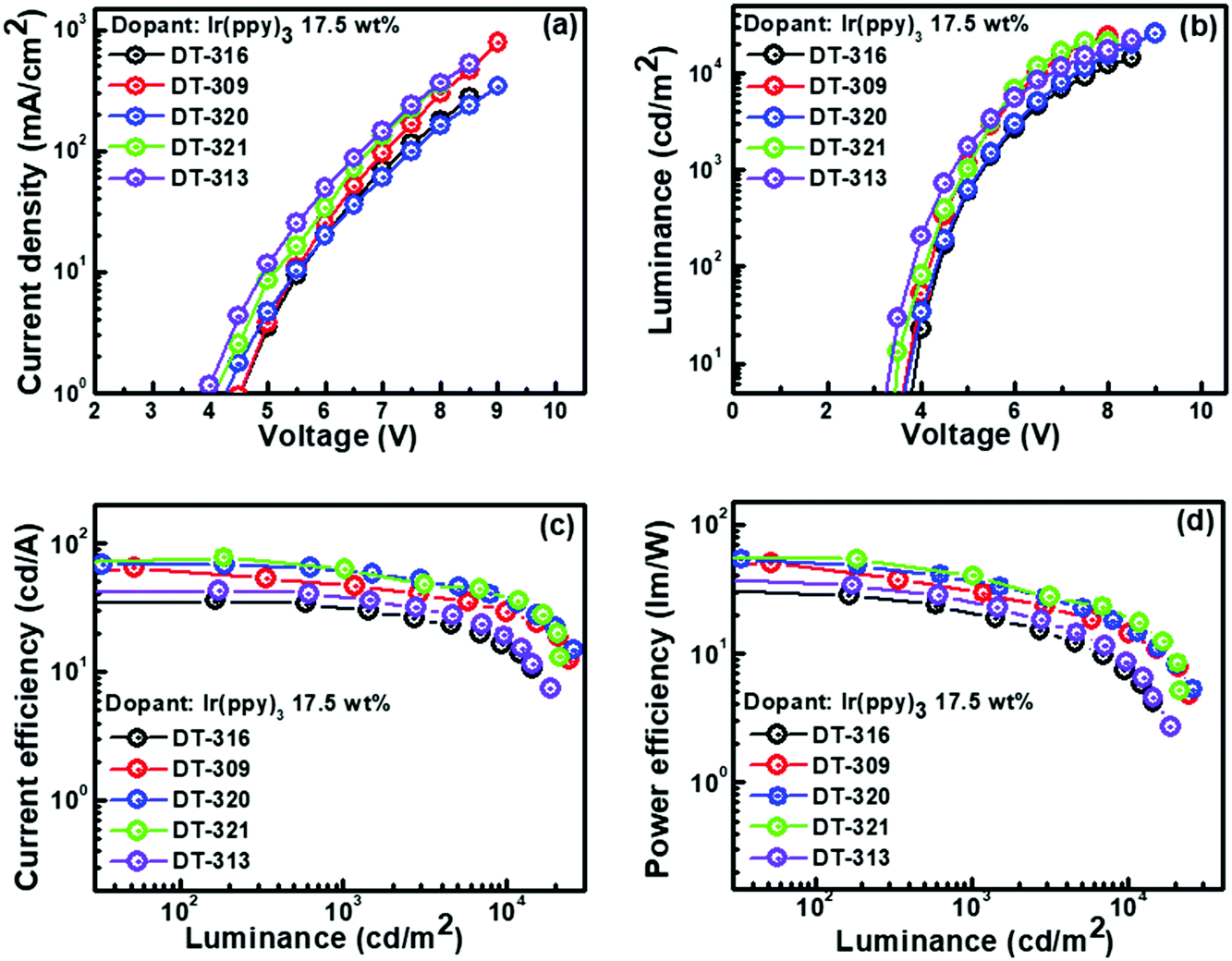 | ||
| Fig. 6 Device performances of solution-processed phosphorescent green OLED devices based on the newly synthesized host materials DT316, DT309, DT320, DT321, and DT313: (a) current density vs. voltage, (b) luminance vs. voltage, (c) current efficiency vs. luminance, and (d) power efficiency vs. luminance. | ||
| Host | Dopant (wt%) | OV (Von) | PE100/CE100/EQE100 (lm W−1/cd A−1/%) | PE1000/CE1000/EQE1000 (lm W−1/cd A−1/%) | PEmax/CEmaxa/EQEmaxa (lm W−1/cd A−1/%) | CIExy coordinates | Max. lum. (cd m−2) |
|---|---|---|---|---|---|---|---|
| a CEmax and EQEmax at the PEmax. | |||||||
| DT-316 | 7.5 | 4.5 | 15.6/19.4/5.2 | 9.0/14.5/3.9 | 19.0/20.3/5.1 | (0.31, 0.62) | 13280 |
| 12.5 | 4.0 | 22.2/30.1/5.3 | 15.4/26.5/4.7 | 23.5/30.0/5.0 | (0.31, 0.62) | 14130 |
|
| 17.5 | 3.7 | 30.9/41.8/6.5 | 20.4/33.9/5.3 | 33.3/42.8/6.7 | (0.31, 0.62) | 14400 |
|
| 22.5 | 3.5 | 20.6/23.9/3.3 | 15.9/23.2/3.4 | 21.1/23.6/3.3 | (0.31, 0.62) | 14030 |
|
| DT-309 | 7.5 | 4.3 | 19.1/30.5/8.1 | 11.3/22.0/5.8 | 26.0/37.2/10.2 | (0.31, 0.62) | 17020 |
| 12.5 | 4.0 | 27.8/40.6/10.8 | 18.1/32.0/8.5 | 34.1/43.5/12.0 | (0.31, 0.62) | 21290 |
|
| 17.5 | 3.6 | 48.1/62.2/11.9 | 31.1/48.1/8.6 | 50.0/63.3/12.1 | (0.31, 0.62) | 24250 |
|
| 22.5 | 3.4 | 28.4/34.4/9.6 | 20.5/29.7/8.3 | 31.3/35.3/9.9 | (0.31, 0.62) | 22010 |
|
| DT-320 | 7.5 | 4.5 | 17.1/27.7/7.4 | 20.4/20.4/5.1 | 23.5/33.8/9.3 | (0.31, 0.62) | 19950 |
| 12.5 | 4.0 | 25.5/37.0/10.1 | 17.7/31.8/8.6 | 28.4/36.8/9.9 | (0.32, 0.62) | 20530 |
|
| 17.5 | 3.6 | 51.4/68.7/10.4 | 37.4/61.8/9.3 | 53.9/68.7/10.3 | (0.31, 0.61) | 25740 |
|
| 22.5 | 3.5 | 27.5/33.6/9.4 | 20.2/29.4/8.2 | 29.7/33.5/9.4 | (0.31, 0.63) | 23610 |
|
| DT-321 | 7.5 | 4.5 | 22.9/36/10.2 | 16.8/30.3/9.0 | 23.1/36.8/10.1 | (0.29, 0.64) | 21060 |
| 12.5 | 4.1 | 32.3/46.7/10.4 | 25.1/41/9.1 | 33.3/46.8/10.7 | (0.30, 0.64) | 21880 |
|
| 17.5 | 4.0 | 54.3/73/10.6 | 40.3/63.8/10.5 | 54.4/73.1/10.9 | (0.30, 0.64) | 21230 |
|
| 22.5 | 3.6 | 30.4/41.9/13.5 | 22.9/34.1/12.5 | 35.8/49.0/11.8 | (0.29, 0.64) | 21340 |
|
| DT-313 | 7.5 | 3.7 | 20.7/28.6/5.9 | 12.0/21.3/4.4 | 25.0/28.2/5.4 | (0.31, 0.62) | 11540 |
| 12.5 | 3.5 | 23.8/30.5/6.4 | 15.7/25.3/6.1 | 25.3/28.3/7.2 | (0.31, 0.62) | 16460 |
|
| 17.5 | 3.2 | 35.7/42.5/7.5 | 25.8/38.6/6.8 | 37.4/42.0/7.9 | (0.31, 0.62) | 18510 |
|
| 22.5 | 3.1 | 20.6/23.9/3.3 | 15.9/23.2/3.4 | 21.1/23.9/3.4 | (0.31, 0.62) | 14030 |
|
| CBP | 12.5 | 3.1 | 41.5/46.6/12.9 | 32.9/42.8/11.5 | 41.8/46.7/12.8 | (0.32, 0.62) | 16710 |
| TCTA | 12.5 | 3.2 | 31.1/39.8/11.5 | 29.1/43.7/14.8 | 36.4/41.1/11.9 | (0.32, 0.62) | 29890 |
Inspired by the excellent EL performance of the green phosphorescent OLEDs, red phosphorescent OLEDs were fabricated using a suitable Ir-based red emitter Ir(2-phq)3, with an optimized doping concentration of 10.0 wt%. The performances of the fabricated OLED devices are shown in Fig. 7, and the data are collected in Table 3. For example, the best performing OLED device containing the DT321 host exhibited a ηPE of 21.0 lm W−1, ηCE of 19.9 cd A−1 and ηEQE of 7.5% at 100 cd m−2. At 1000 cd m−2, this device exhibited a ηPE of 13.7 lm W−1, ηCE of 16.2 cd A−1, and ηEQE of 9.0%. The device also exhibited a low turn-on voltage of 3.6 V and maximum luminance of 15519 cd m−2, which are extremely good for solution-processed devices. Furthermore, ηmax,CE was 17.8 cd A−1 when the DT313 host was employed and this increased to 18.7, 23.1, and 23.8 cd A−1 for the hosts DT320, DT309 and DT316, respectively. The resulting ηmax,EQE was 6.8% when the DT313 host was used, and increased to 8.5, 8.7 and 8.8% for the hosts DT320, DT309 and DT316, respectively. All these results are based on the same doping concentration, i.e., 10 wt%.
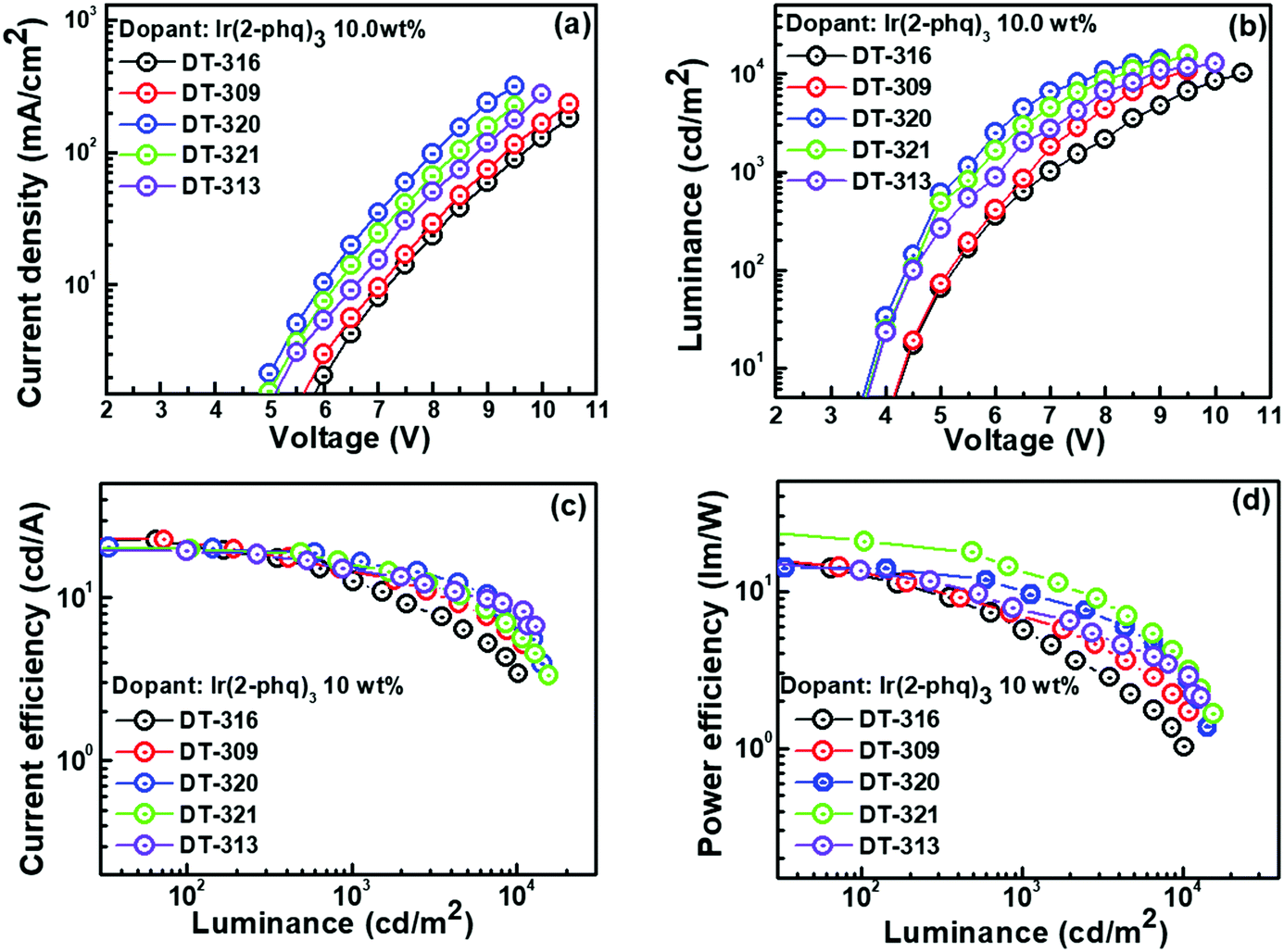 | ||
| Fig. 7 The effects of the newly synthesized host materials on (a) current density vs. voltage, (b) luminance vs. voltage, (c) current efficiency vs. luminance, and (d) power efficiency vs. luminance plots of the solution-processed phosphorescent red OLED devices based on newly synthesized host materials. | ||
| Host | OV (Von) | PE100/CE100/EQE100 (lm W−1/cd A−1/%) | PE1000/CE1000/EQE1000 (lm W−1/cd A−1/%) | PEmax/CEmaxa/EQEmaxa (lm W−1/cd A−1/%) | CIExy coordinates | Max. lum. (cd m−2) |
|---|---|---|---|---|---|---|
| a CEmax and EQEmax at the PEmax. b Ref. J.-H. Jou et al., Org. Electron., 2015, 24, 254–262. | ||||||
| DT-316 | 4.2 | 13.1/21.4/8.3 | 5.8/12.8/5.0 | 18.0/23.8/8.8 | (0.56, 0.44) | 10191 |
| DT-309 | 4.1 | 13.6/22/8.5 | 7.0/14.7/6.8 | 17.8/23.1/8.7 | (0.56, 0.44) | 10922 |
| DT-320 | 3.6 | 14.8/20.1/7.6 | 10.1/17.2/9.6 | 16.7/18.7/8.5 | (0.56, 0.44) | 14290 |
| DT-321 | 3.6 | 21.0/19.9/7.5 | 13.7/16.2/9.0 | 25.6/19.3/8.3 | (0.56, 0.44) | 15519 |
| DT-313 | 3.7 | 13.5/19.3/7.3 | 7.7/14.9/6.2 | 15.7/17.8/6.8 | (0.56, 0.44) | 13030 |
| CBPb | — | 13.5/18.1/8.1 | 8.5/16.4/7.3 | —/—/— | (0.57, 0.42) | 13040 |
The doping concentration of emitters greatly affects the performance of OLEDs,75 as shown in Fig. S24–S28 (ESI†) and Table 2. For example, when using the best performing host DT321 for the green OLED device, ηmax,PE ranged from 23.1 lm W−1 to 54.4 lm W−1 (an increment of 135%), as the doping concentration was increased from 7.5 wt% to 17.5 wt%. Interestingly, when the concentration was increased to 22.5 wt%, ηmax,PE dropped to 35.8 lm W−1, a decrement of 34% when compared to the OLED device fabricated with a 17.5 wt% doping concentration. The doping concentration of the emitter also profoundly influenced the maximum luminance and turn-on voltage of the OLED devices.
For example, the green OLED device based on the DT320 host exhibited a maximum luminance of 19950 cd m−2 and a turn-on voltage of 4.5 V at 7.5 wt%, which were correspondingly increased and decreased to 25740 cd m−2 and 3.6 V, as the doping concentration was changed to 17.5 wt%. Interestingly, as the doping concentration was further enhanced, the maximum luminance of OLED device showed a decline, which may be attributed to triplet–triplet annihilation as well as concentration-induced quenching resulting from the self-aggregation of the emitter at high concentrations.55,74,75
Furthermore, we measured the PL quantum yields (PLQYs) of neat and doped films of the host materials following the procedures described by Dai et al. in both air and a nitrogen (N2) environment.76 Neat films of the molecules DT316, DT321 and DT313 respectively showed PLQYs of 64.23%, 80.62% and 76.45% in air, and 71.55%, 84.31% and 78.65% in the N2 environment.
The PLQYs of 17.5 wt% Ir(ppy)3-doped host films were 86.03% (DT316), 99.47% (DT321) and 90.07% (DT313) in N2. The PLQYs of 10 wt% Ir(2-phq)3-doped DT316, DT321 and DT313 host films were 89.05%, 91.80% and 91.61%, respectively, in the same environment. The measurements of PLQYs of the synthesized molecules confirm our aforementioned results.
All the synthesized materials behave as excellent matrixes in the designed device structure; the obtained results are either better or comparable to the conventionally used host materials such as CBP and TCTA, as shown in Table 2. This may be attributed to up to four efficiency-enhancement factors resulting from either the molecular design of the hosts or the designed device structures or a combination of both. First, as demonstrated in Fig. S29 (ESI†), the device structure significantly favours the injection of holes to the synthesized hosts because they exhibit hole injection barriers of 0.24 to 0.31 eV, which are 0.39 to 0.46 eV lower than that of the emitter Ir(ppy)3 (0.70 eV). Meanwhile, the same device structure favours the injection of electrons to the emitter because there exists a −0.60 eV electron trap, while there is a barrier of 0.75 to 0.83 eV for the electrons to enter into the hosts. Similarly, for the red phosphorescent device, the resultant device structure favours the injection of holes to both the host and Ir(2-phq)3 because of their very similar energy barriers (0.20 eV for the guest and 0.24 to 0.31 eV for the host), whereas the electrons predominantly enter into the emitter due to its −0.10 eV electron trap. Thus excitons are generated on both the host and the guest, giving the high device efficiency. Second, all the designed host materials possess very low LUMO levels, which significantly confine the electrons into the desired emissive zone and prevent an overflow of the electrons into the undesirable hole injection layer.77 Third, Fig. 8(a) and (c) show the proposed energy transfer routes between the hosts and guests. The higher first singlet energy state (S1) of the host favours significant Förster energy transfer between S1 of the host and S1 of the guest, which is further transferred to the first triplet energy state (T1) of the guest through an intersystem crossing process. Moreover, T1 of the host transfers energy to T1 of the guest via a Dexter energy transfer process because of the higher doping concentration. It is also noteworthy that the triplet energy of all of the synthesized hosts is sufficiently higher than those of both of the employed emitters Ir(ppy)3 and Ir(2-phq)3, to effectively enable the host-to-guest energy transfer, hence the higher device performances. Fourth, Fig. 8(b) and (d) show the EL spectra of the reported five different molecular hosts doped with 17.5 wt% Ir(ppy)3 or 10.0 wt% Ir(2-phq)3. The EL maximum of the green OLEDs is centered at 512 nm, while for the red OLEDs, it is at 568 nm. Notably, no emission relating to the host in the deep-blue region can be observed, indicating an efficient host-to-guest energy transfer and also confirming the aforementioned hypothesis. All these factors reveal that these five small molecules may serve as suitable hosts for highly-efficient solution-processed OLED devices.
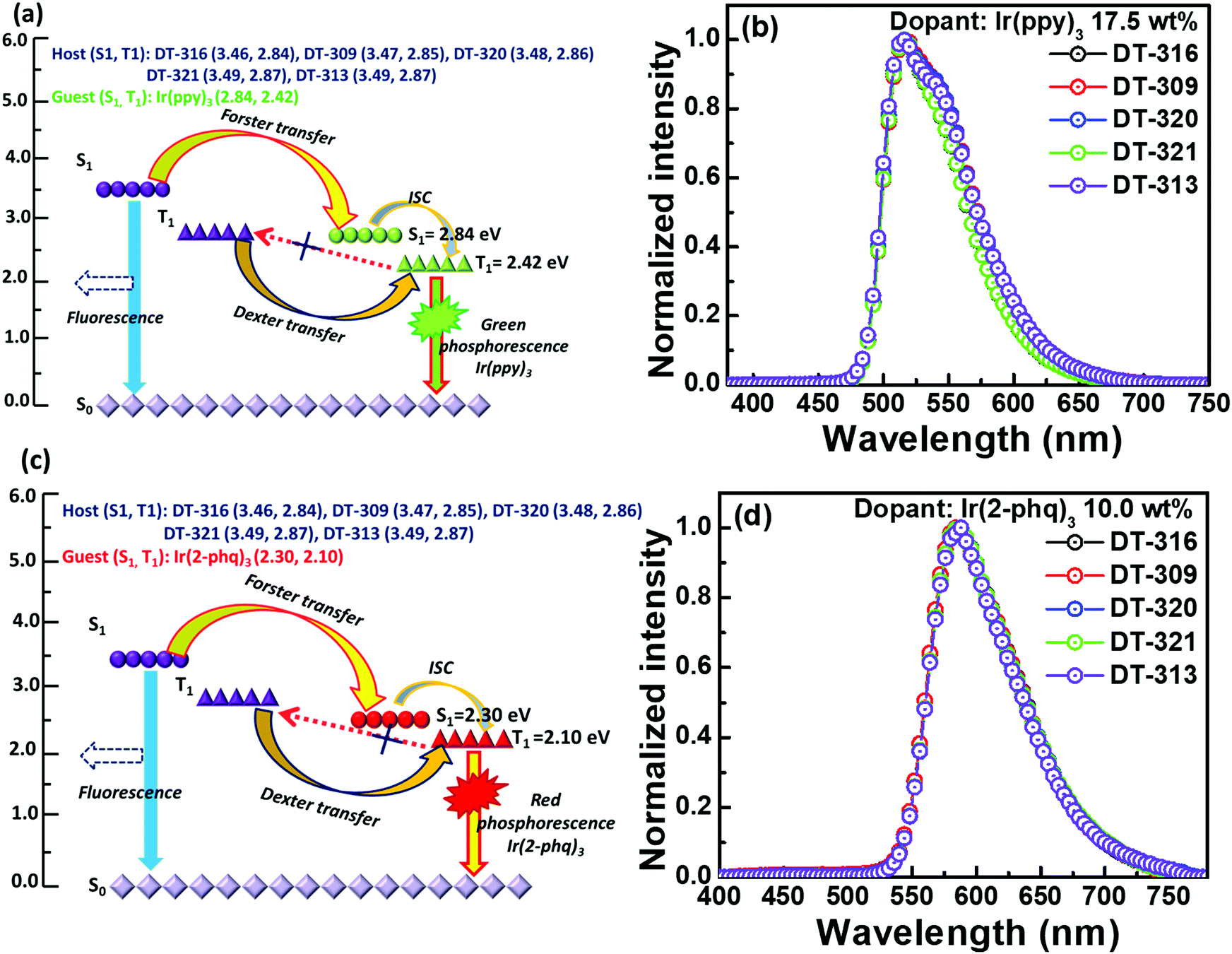 | ||
| Fig. 8 A schematic diagram of the energy-level alignment of the singlet excited states (S1), triplet excited states (T1) and ground states (S0), as well as the energy transfer and light-emission processes in the newly synthesized host materials DT316, DT309, DT320, DT321, and DT313 with (a) phosphorescent green Ir(ppy)3 and (c) red Ir(2-phq)3 emitters; (b and d) show the recorded EL spectra for the optimized green and red OLED devices, respectively. | ||
Conclusions
In summary, a small library of bipolar benzimidazole-triphenylamine integrated hybrid host materials was developed. They had a desirable twisted conformation which furnished the solution-processed hosts with high triplet energies, elevated currents and power efficiencies and high thermal stabilities for green and red phosphorescent OLEDs. The DT-321 host based device displayed the highest efficacy with the structure ITO/PEDOT:PSS(30 nm)/EML(20 nm)/TPBi(35 nm)/LiF(1 nm)/Al(100 nm) exhibited a maximum power efficiency of 54.4 lm W−1, a current efficiency of 73.1 cd A−1 and a maximum brightness of 21880 cd m−2 for the green device (emitter, Ir(ppy)3), while the red device (emitter Ir(2-phq)3) provided 25.6 lm W−1, 19.3 cd A−1, and 15519 cd m−2, respectively, without any external outcoupling techniques. The high PLQYs of the host compounds with the emitters is a result of the propeller type structure of the materials, as evidenced by the crystal structures. The proposed Dexter and Förster mechanisms for energy transfer which led to effective host-to-guest energy transfer, balanced charge carriers in the recombination zone with bipolarity of the developed host material, and the ability to generate excitons on both host and guest are key factors for high efficiency device. It is noteworthy that the designed hosts, which emit from their weak CT states, display better performances than commercial hosts such as CBP. Thus the designed and synthesized host materials are helpful in enlarging the library of high performing green and red PhOLEDs.
Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
Financial support from UAY (Grant no. IITMandi_005) is thankfully acknowledged. The research facility of AMRC is acknowledged.References
- K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218–4233 CrossRef CAS PubMed.
- A. Bernanose, Br. J. Appl. Phys., 1955, 6, S54 CrossRef.
- M. Gross, D. C. Müller, H.-G. Nothofer, U. Scherf, D. Neher, C. Bräuchle and K. Meerholz, Nature, 2000, 405, 661 CrossRef CAS.
- L. Hung and C. Chen, Mater. Sci. Eng., R, 2002, 39, 143–222 CrossRef.
- J. Cui, Q. Huang, J. C. Veinot, H. Yan, Q. Wang, G. R. Hutchison, A. G. Richter, G. Evmenenko, P. Dutta and T. J. Marks, Langmuir, 2002, 18, 9958–9970 CrossRef CAS.
- S. Tokito, T. Iijima, Y. Suzuri, H. Kita, T. Tsuzuki and F. Sato, Appl. Phys. Lett., 2003, 83, 569–571 CrossRef CAS.
- M. T. Lee, C. H. Liao, C. H. Tsai and C. H. Chen, Adv. Mater., 2005, 17, 2493–2497 CrossRef CAS.
- C. Tang, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- H.-N. Peng, Y. Yu, X.-H. Liu, J. Zheng, H. Cheng and X. Hu, J. Chem. Res., 2018, 42, 20–23 CrossRef CAS.
- A. Maheshwaran, V. G. Sree, H. Y. Park, H. Kim, S. H. Han, J. Y. Lee and S. H. Jin, Adv. Funct. Mater., 2018, 28, 1802945 CrossRef.
- F. Wang, D. Liu, J. Li and M. Ma, Adv. Funct. Mater., 2018, 28, 1803193 CrossRef.
- J. Liang, C. Li, X. Zhuang, K. Ye, Y. Liu and Y. Wang, Adv. Funct. Mater., 2018, 28, 1707002 CrossRef.
- Y. K. Wang, Q. Sun, S. F. Wu, Y. Yuan, Q. Li, Z. Q. Jiang, M. K. Fung and L. S. Liao, Adv. Funct. Mater., 2016, 26, 7929–7936 CrossRef CAS.
- D. Zhang, X. Song, M. Cai, H. Kaji and L. Duan, Adv. Mater., 2018, 30, 1705406 CrossRef.
- T. Huang, W. Jiang and L. Duan, J. Mater. Chem. C, 2018, 6, 5577–5596 RSC.
- S. Grigalevicius, D. Tavgeniene, G. Krucaite, R. Griniene, W.-C. Li, D. Luo and C.-H. Chang, Dyes Pigm., 2018, 152, 100–104 CrossRef CAS.
- C. Wu, B. Wang, Y. Wang, J. Hu, J. Jiang, D. Ma and Q. Wang, J. Mater. Chem. C, 2019, 7, 558–566 RSC.
- X. Cai, R. Liu, H. Shi, C. Li and H. Zhu, Dyes Pigm., 2017, 143, 196–202 CrossRef CAS.
- T. Yang, H. Xu, K. Wang, P. Tao, F. Wang, B. Zhao, H. Wang and B. Xu, Dyes Pigm., 2018, 153, 67–73 CrossRef CAS.
- P. Xiao, J. Huang, Y. Yu, J. Yuan, D. Luo, B. Liu and D. Liang, Appl. Sci., 2018, 8, 1449 CrossRef.
- A. Brown, K. Pichler, N. Greenham, D. Bradley, R. H. Friend and A. Holmes, Chem. Phys. Lett., 1993, 210, 61–66 CrossRef CAS.
- M. Baldo, D. O’brien, M. Thompson and S. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422 CrossRef CAS.
- Y. Ma, H. Zhang, J. Shen and C. Che, Synth. Met., 1998, 94, 245–248 CrossRef CAS.
- M. A. Baldo, D. O'brien, Y. You, A. Shoustikov, S. Sibley, M. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- M. Baldo, S. Lamansky, P. Burrows, M. Thompson and S. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908 CrossRef CAS PubMed.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- S. Grigalevicius, D. Tavgeniene, G. Krucaite, R. Griniene, Y.-P. Wang, S.-R. Tsai and C.-H. Chang, Dyes Pigm., 2018, 159, 173–178 CrossRef CAS.
- L. J. Sicard, H. C. Li, Q. Wang, X. Y. Liu, O. Jeannin, J. Rault-Berthelot, L. S. Liao, Z. Q. Jiang and C. Poriel, Angew. Chem., Int. Ed., 2019, 58, 3848–3853 CrossRef CAS.
- L. Ding, J.-N. Wang, X.-Y. Liu, H. Chen, F. Igbari and L.-S. Liao, Org. Electron., 2019, 71, 258–265 CrossRef CAS.
- J. Tagare and S. Vaidyanathan, J. Mater. Chem. C, 2018, 6, 10138–10173 RSC.
- M. Godumala, S. Choi, S. Y. Park, M. J. Cho, H. J. Kim, D. H. Ahn, J. S. Moon, J. H. Kwon and D. H. Choi, Chem. Mater., 2018, 30, 5005–5012 CrossRef CAS.
- S. Jhulki, A. Ghosh, T. J. Chow and J. N. Moorthy, RSC Adv., 2015, 5, 101169–101176 RSC.
- S. Reineke, K. Walzer and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125328 CrossRef.
- D. Song, S. Zhao, Y. Luo and H. Aziz, Appl. Phys. Lett., 2010, 97, 268 Search PubMed.
- C. Xiang, X. Fu, W. Wei, R. Liu, Y. Zhang, V. Balema, B. Nelson and F. So, Adv. Funct. Mater., 2016, 26, 1463–1469 CrossRef CAS.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS.
- D.-Y. Zhou, H. Zamani Siboni, Q. Wang, L.-S. Liao and H. Aziz, J. Phys. Chem. C, 2014, 118, 24006–24012 CrossRef CAS.
- W.-Y. Hung, L.-C. Chi, W.-J. Chen, E. Mondal, S.-H. Chou, K.-T. Wong and Y. Chi, J. Mater. Chem., 2011, 21, 19249–19256 RSC.
- E. Mondal, W. Y. Hung, Y. H. Chen, M. H. Cheng and K. T. Wong, Chem. – Eur. J., 2013, 19, 10563–10572 CrossRef CAS.
- E. Mondal, W. Y. Hung, H. C. Dai and K. T. Wong, Adv. Funct. Mater., 2013, 23, 3096–3105 CrossRef CAS.
- S.-H. Cheng, W.-Y. Hung, M.-H. Cheng, H.-F. Chen, A. Chaskar, G.-H. Lee, S.-H. Chou and K.-T. Wong, J. Mater. Chem. C, 2014, 2, 8554–8563 RSC.
- S.-W. Li, C.-H. Yu, C.-L. Ko, T. Chatterjee, W.-Y. Hung and K.-T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 12930–12936 CrossRef CAS.
- P.-I. Shih, C.-H. Chien, C.-Y. Chuang, C.-F. Shu, C.-H. Yang, J.-H. Chen and Y. Chi, J. Mater. Chem., 2007, 17, 1692–1698 RSC.
- F. M. Hsu, C. H. Chien, C. F. Shu, C. H. Lai, C. C. Hsieh, K. W. Wang and P. T. Chou, Adv. Funct. Mater., 2009, 19, 2834–2843 CrossRef CAS.
- Y. Tao, S. Gong, Q. Wang, C. Zhong, C. Yang, J. Qin and D. Ma, Phys. Chem. Chem. Phys., 2010, 12, 2438–2442 RSC.
- I. Neogi, S. Jhulki, A. Ghosh, T. J. Chow and J. N. Moorthy, ACS Appl. Mater. Interfaces, 2015, 7, 3298–3305 CrossRef CAS PubMed.
- S. Jhulki, S. Seth, A. Ghosh, T. J. Chow and J. N. Moorthy, ACS Appl. Mater. Interfaces, 2016, 8, 1527–1535 CrossRef CAS.
- J.-H. Jou, S. Kumar, D. Tavgeniene, C.-C. An, P.-H. Fang, E. Zaleckas, J. V. Grazulevicius and S. Grigalevicius, J. Mater. Chem. C, 2014, 2, 8707–8714 RSC.
- T. W. Lee, T. Noh, H. W. Shin, O. Kwon, J. J. Park, B. K. Choi, M. S. Kim, D. W. Shin and Y. R. Kim, Adv. Funct. Mater., 2009, 19, 1625–1630 CrossRef CAS.
- J. Chen, C. Shi, Q. Fu, F. Zhao, Y. Hu, Y. Feng and D. Ma, J. Mater. Chem., 2012, 22, 5164–5170 RSC.
- J.-H. Jou, M.-F. Hsu, W.-B. Wang, C.-L. Chin, Y.-C. Chung, C.-T. Chen, J.-J. Shyue, S.-M. Shen, M.-H. Wu and W.-C. Chang, Chem. Mater., 2009, 21, 2565–2567 CrossRef CAS.
- J.-H. Jou, C.-J. Li, S.-M. Shen, S.-H. Peng, Y.-L. Chen, Y.-C. Jou, J. H. Hong, C.-L. Chin, J.-J. Shyue and S.-P. Chen, J. Mater. Chem. C, 2013, 1, 4201–4208 RSC.
- J. H. Jou, Y. M. Yang, S. Z. Chen, J. R. Tseng, S. H. Peng, C. Y. Hsieh, Y. X. Lin, C. L. Chin, J. J. Shyue and S. S. Sun, Adv. Opt. Mater., 2013, 1, 657–667 CrossRef.
- J. H. Burroughes, D. D. Bradley, A. Brown, R. Marks, K. Mackay, R. H. Friend, P. Burns and A. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- G. Gustafsson, Y. Cao, G. Treacy, F. Klavetter, N. Colaneri and A. Heeger, Nature, 1992, 357, 477 CrossRef CAS.
- J.-H. Jou, S. Sahoo, S. Kumar, H.-H. Yu, P.-H. Fang, M. Singh, G. Krucaite, D. Volyniuk, J. V. Grazulevicius and S. Grigalevicius, J. Mater. Chem. C, 2015, 3, 12297–12307 RSC.
- J.-H. Jou, S.-M. Shen, S.-H. Chen, M.-H. Wu, W.-B. Wang, H.-C. Wang, C.-R. Lin, Y.-C. Chou, P.-H. Wu and J.-J. Shyue, Appl. Phys. Lett., 2010, 96, 72 CrossRef.
- S. Kumar, P. Singh, R. Srivastava, R. R. Koner, A. Pramanik, J. Mathew, S. Sinha, M. Rawat, R. Anand and S. Ghosh, J. Mater. Chem. C, 2014, 2, 6637–6647 RSC.
- Z. Zhao, B. He, H. Nie, B. Chen, P. Lu, A. Qin and B. Z. Tang, Chem. Commun., 2014, 50, 1131–1133 RSC.
- R. Rao, C.-W. Liao, W.-L. Su and S.-S. Sun, J. Mater. Chem. C, 2013, 1, 5491–5501 RSC.
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904–912 CrossRef CAS.
- R. D. McCullough, S. Tristram-Nagle, S. P. Williams, R. D. Lowe and M. Jayaraman, J. Am. Chem. Soc., 1993, 115, 4910–4911 CrossRef CAS.
- C.-K. Shin and H. Lee, Synth. Met., 2004, 140, 177–181 CrossRef CAS.
- B. Liu, J. Lin, M. Yu, B. Li, L. Xie, C. Ou, F. Liu, T. Li, D. Lu and W. Huang, J. Phys. Chem. C, 2017, 121, 19087–19096 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, J. Montgomery Jr, T. Vreven, K. Kudin and J. Burant, Gaussian 03, Revision D.01, Gaussian Inc., Wallingford CT, 2013 Search PubMed.
- C. Wang, Y. Qin, Y. Sun, Y.-S. Guan, W. Xu and D. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 15978–15987 CrossRef CAS PubMed.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- J. S. Lee, S. K. Son, S. Song, H. Kim, D. R. Lee, K. Kim, M. J. Ko, D. H. Choi, B. Kim and J. H. Cho, Chem. Mater., 2012, 24, 1316–1323 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner and W. Zhang, Nat. Mater., 2006, 5, 328 CrossRef CAS PubMed.
- Z. Liu, G. Zhang and D. Zhang, Acc. Chem. Res., 2018, 51, 1422–1432 CrossRef CAS PubMed.
- J.-H. Jou, S. Sahoo, D. K. Dubey, R. A. K. Yadav, S. S. Swayamprabha and S. D. Chavhan, J. Mater. Chem. C, 2018, 6, 11492–11518 RSC.
- J.-H. Jou, S.-C. Fu, C.-C. An, J.-J. Shyue, C.-L. Chin and Z.-K. He, J. Mater. Chem. C, 2017, 5, 5478–5486 RSC.
- S. W. Dai, B. W. Hsu, C. Y. Chen, C. A. Lee, H. Y. Liu, H. F. Wang, Y. C. Huang, T. L. Wu, A. Manikandan and R. M. Ho, Adv. Mater., 2018, 30, 1705532 CrossRef PubMed.
- J.-H. Jou, T.-H. Li, S. Kumar, C.-C. An, A. Agrawal, S.-Z. Chen, P.-H. Fang, G. Krucaite, S. Grigalevicius and J. Grazulevicius, Org. Electron., 2015, 24, 254–262 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1897654–1897657. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9tc04484a |
| ‡ Both have equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |