
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Tuning aggregative versus non-aggregative lectin binding with glycosylated nanoparticles by the nature of the polymer ligand”†
Panagiotis G.
Georgiou
 a,
Alexander N.
Baker
a,
Sarah-Jane
Richards
a,
Antonio
Laezza
a,
Marc
Walker
b and
Matthew I.
Gibson
*ac
a,
Alexander N.
Baker
a,
Sarah-Jane
Richards
a,
Antonio
Laezza
a,
Marc
Walker
b and
Matthew I.
Gibson
*ac
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK. E-mail: m.i.gibson@warwick.ac.uk
bDepartment of Physics, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK
cWarwick Medical School, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK
First published on 28th November 2019
Abstract
Glycan–lectin interactions drive a diverse range of biological signaling and recognition processes. The display of glycans in multivalent format enables their intrinsically weak binding affinity to lectins to be overcome by the cluster glycoside effect, which results in a non-linear increase in binding affinity. As many lectins have multiple binding sites, upon interaction with glycosylated nanomaterials either aggregation or surface binding without aggregation can occur. Depending on the application area, either one of these responses are desirable (or undesirable) but methods to tune the aggregation state, independently from the overall extent/affinity of binding are currently missing. Herein, we use gold nanoparticles decorated with galactose-terminated polymer ligands, obtained by photo-initiated RAFT polymerization to ensure high end-group fidelity, to show the dramatic impact on agglutination behaviour due to the chemistry of the polymer linker. Poly(N-hydroxyethyl acrylamide) (PHEA)-coated gold nanoparticles, a polymer widely used as a non-ionic stabilizer, showed preference for aggregation with lectins compared to poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA)-coated nanoparticles which retained colloidal stability, across a wide range of polymer lengths and particle core sizes. Using biolayer interferometry, it was observed that both coatings gave rise to similar binding affinity and hence provided conclusive evidence that aggregation rate alone cannot be used to measure affinity between nanoparticle systems with different stabilizing linkers. This is significant, as turbidimetry is widely used to demonstrate glycomaterial activity, although this work shows the most aggregating may not be the most avid, when comparing different polymer backbones/coating. Overall, our findings underline the potential of PHPMA as the coating of choice for applications where aggregation upon lectin binding would be problematic, such as in vivo imaging or drug delivery.
Introduction
Carbohydrates control and direct a myriad of biological processes including: cellular recognition, inflammation, signal transmission and infection of pathogens displayed by them.1,2 However, the intrinsic affinity of a glycan for a lectin target is typically very weak (Kd ∼ mM), which is compensated for on cell surfaces (or for example in glycan arrays3) by the multivalent presentation of multiple copies of the same glycan, which due to the cluster glycoside effect4,5 results in entropy–enthalpy compensation giving a non-linear increase in the observed binding affinity (Kd can be nM or below). Due to this enhancement, there is significant interest in the development of polymeric and nanoparticulate glycosylated materials that benefit from the tuneable display of multiple glycans on scaffolds. For example, sialic acid polymers have been used as decoys for the influenza hemagglutinins enabling nM affinity,6,7 galactosylated polymers as inhibitors of the cholera toxin8,9 and dendrimers to target galectins10 and has been extensively reviewed.11–13Glycomaterials are emerging as tools to modulate complex cellular function: Godula and co-workers remodelled neural progenitor cells with sulphated glycans to control their differentiation,14 mannosylated nanoparticles can potentiate vaccines15 and glycosylated nanomaterials have been used to aid cellular delivery.16–20 For instance, glucose- and galactose-coated iron oxide nanoparticles have been prepared and the influence of the glycan versus poly(ethylene glycol)-coating on the cellular uptake by several cell lines has been studied.17 Glycomaterials have been extensively explored for their application in anti-adhesion therapy to block infectious agents before they can engage the cell.11,21 Kitov et al. used starfish dendrimers to neutralize Shiga-toxin infection,22 and sialic acid polymers can inhibit hemagglutination by influenza.23 Many other glycoconjugates have been explored including galactosylated polymers for their interaction with the cholera toxin,8,9,24,25 fucosylated dendrimers with LecB26 (from Pseudomonas aeruginosa) and mannosylated polymers to target DC-SIGN, DC-SIGN-R and Langerin, which are all found on dendritic cells and form a key part of immune responses.27–29
In addition to binding or delivery, incorporating glycans into materials that can generate a signal output is a promising approach to develop new diagnostics and biosensors.30,31 Gold nanoparticles (AuNPs) in particular offer unique optical properties that have seen them widely employed for the design of biosensors and imaging agents.32–35 Their optical property of localized surface plasmon resonance (LSPR) is determined by the particle size and shape. As the interparticle distance decreases to less than that of the particle diameter, coupling and dipole–dipole interactions between the plasmons of neighboring particles result in a broadening and a shift to longer wavelengths of the surface plasmon absorption band, resulting in the AuNPs appearing blue as opposed to red when not aggregated.
Mirkin et al. exploited DNA-functionalized AuNPs for the colorimetric detection of bacterial DNA based on the ‘red-blue’ color shift upon aggregation of AuNPs.36 The display of glycans on the surface of AuNPs can mimic the cell-surface glycocalyx and hence are ideal probes of glycan recognition processes. In particular, lectins often display multiple binding sites, and hence multivalent glyconanoparticles readily aggregate in the presence of their lectin partner, enabling a simple, label-free read out of binding.37–40 A key challenge, however, in the design and application of colloidal biosensors is retaining colloidal stability in complex media and hence avoiding false positives. Field and co-workers used short PEG linkers to install sialic acids onto AuNPs for the discrimination between avian and human influenza.39 Gibson and co-workers have exploited reversible addition–fragmentation chain transfer (RAFT) polymerization to generate stabilizing poly(N-hydroxyethyl acrylamide) (PHEA) ligands for AuNPs: RAFT installs sulfur containing end-groups which have high affinity for gold surfaces.41–43 By using RAFT agents, with a pentafluorophenyl ester end-group, amino-glycans can be easily attached. The advantage of this controlled radical polymerization44 method is that the length of the linker can be tuned to achieve the delicate balance between stability (favored by longer chain length polymers) and aggregation-response (favored by shorter chain lengths).45–48 This strategy is highly tunable, and has been used to probe the binding of a range of lectins, and also to probe carbohydrate–carbohydrate interactions.49
The above examples make use of the aggregation of AuNPs to give a colorimetric response, but aggregation is not always desirable in other applications such as in vivo, where lectin binding, without macroscopic aggregation would be preferable. Hence there is a need to explore other polymeric coatings, to engineer the glycoparticle interface, and to optimize the solution stability. Biocompatible poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) is an important and frequently employed hydrophilic polymer which has been shown to be a viable alternative to PEG in many nanomedicine applications.50–52 PHPMA demonstrates similar biocompatibility profiles to PEG while also displaying pendent secondary hydroxyl groups that allow, for example, conjugation of targeting moieties and/or drugs via degradable linkages.53,54 PHPMA can be synthesized by a variety of polymerization techniques including conventional free radical polymerization,55 and reversible deactivation radical polymerization (RDRP) techniques such as atom transfer radical polymerization (ATRP),56 and reversible addition–fragmentation chain transfer (RAFT) polymerization of N-(2-hydroxypropyl)methacrylamide (HPMA).57 Some examples of employing HPMA to synthesize glycan-decorated block-copolymers have already been reported.58–60
This work critically compares how methacrylamide, versus acrylamide-based polymer linkers impact the aggregative versus non-aggregative outcomes for glycosylated nanoparticles, with the aim to tune the aggregation independently from the extent of binding. Photo-initiated RAFT polymerization was used to synthesize variable molecular weight galactosamine-functional polymers which were assembled onto gold nanoparticles to give a library of 30 glyconanoparticles. Using the N-acetyl galactosamine binding lectin, soybean agglutinin (SBA), aggregation and binding were investigated using biolayer interferometry and UV-visible analysis. This revealed that PHPMA coatings result in nanoparticles with identical binding affinity to PHEA but avoided all aggregation, showing that subtle changes in the polymer linker can be used to tune the macroscopic response for specific applications. A detailed X-ray photoelectron spectroscopy (XPS) study indicated that intrinsic grafting density differences contribute to this tuneable behaviour, influenced by the chemistry of the linker.
Experimental section
Materials and methods
Materials and characterization techniques used are given in detail in the ESI.†Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch); 1064 (S–(CS)–S stretch). ESI-MS: m/z [C17H32O2S3 + Na]+ calc. 387.1 g mol−1, exp. 387.61 g mol−1.
O); 1517 (–C6F5); 1064 (S–(CS)–S).
O stretch); 1064 (S–(CS)–S stretch). ESI-MS: m/z [C17H32O2S3 + Na]+ calc. 387.1 g mol−1, exp. 387.61 g mol−1.
O); 1517 (–C6F5); 1064 (S–(CS)–S).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA] ratio of 40. In a typical reaction, N-(2-hydroxypropyl)methacrylamide (HPMA) (0.43 g, 3.01 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropanoic acid pentafluorophenyl ester (PFP-DMP) (0.04 g, 0.075 mmol) were dissolved in 50:50 dioxane:methanol solution (2 mL) in a vial. The resulting solution was degassed by sparging with N2(g) for 15 min and the sealed vial was incubated at 37 °C with magnetic stirring under 460 nm light irradiation for 120 min. After that time, an aliquot of crude polymerization mixture was taken for 1H NMR in methanol-d4 for conversion and Mn,NMR analysis. The reaction was rapidly cooled in liquid nitrogen and precipitated into diethyl ether. The polymer was re-precipitated into diethyl ether from methanol twice to yield a yellow polymer product that was further dried under vacuum. The same procedure was followed for [monomer]:[CTA] ratios of 60, 80, 100 and 120. Conversions were calculated using 1H NMR spectroscopy by comparing the integrations of the HPMA monomer signals (δ 5.73 ppm) with those of the corresponding signals of the polymer (δ 1.31–1.04 ppm, CH3 of PHPMA backbone and CH3 of PHPMA side chain). Mn,NMR was calculated by end-group analysis by comparing the integrations of the –CH3 signals (δ 0.92 ppm) of dodecyl end-group with those of the corresponding signals of the polymer (δ 1.31–1.04 ppm). 1H NMR (400 MHz, CD3OD): δ (ppm) 7.53 (br m, NH of PHPMA side chain), 3.88 (br s, CH of PHPMA side chain), 3.19–3.02 (br m, CH2 of PHPMA sidechain), 2.05–1.79 (br m, CH2 of PHPMA backbone), 1.31–1.04 (br m, CH3 of PHPMA backbone and CH3 of PHPMA side chain), 0.92 (t, 3H, CH2–CH2–CH3 of dodecyl end-group).
:[CTA] ratio of 40. In a typical reaction, N-(2-hydroxypropyl)methacrylamide (HPMA) (0.43 g, 3.01 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropanoic acid pentafluorophenyl ester (PFP-DMP) (0.04 g, 0.075 mmol) were dissolved in 50:50 dioxane:methanol solution (2 mL) in a vial. The resulting solution was degassed by sparging with N2(g) for 15 min and the sealed vial was incubated at 37 °C with magnetic stirring under 460 nm light irradiation for 120 min. After that time, an aliquot of crude polymerization mixture was taken for 1H NMR in methanol-d4 for conversion and Mn,NMR analysis. The reaction was rapidly cooled in liquid nitrogen and precipitated into diethyl ether. The polymer was re-precipitated into diethyl ether from methanol twice to yield a yellow polymer product that was further dried under vacuum. The same procedure was followed for [monomer]:[CTA] ratios of 60, 80, 100 and 120. Conversions were calculated using 1H NMR spectroscopy by comparing the integrations of the HPMA monomer signals (δ 5.73 ppm) with those of the corresponding signals of the polymer (δ 1.31–1.04 ppm, CH3 of PHPMA backbone and CH3 of PHPMA side chain). Mn,NMR was calculated by end-group analysis by comparing the integrations of the –CH3 signals (δ 0.92 ppm) of dodecyl end-group with those of the corresponding signals of the polymer (δ 1.31–1.04 ppm). 1H NMR (400 MHz, CD3OD): δ (ppm) 7.53 (br m, NH of PHPMA side chain), 3.88 (br s, CH of PHPMA side chain), 3.19–3.02 (br m, CH2 of PHPMA sidechain), 2.05–1.79 (br m, CH2 of PHPMA backbone), 1.31–1.04 (br m, CH3 of PHPMA backbone and CH3 of PHPMA side chain), 0.92 (t, 3H, CH2–CH2–CH3 of dodecyl end-group).
FT-IR (neat): ν (cm−1) 3300 (N–H and O–H stretch); 2920 (alkyl C–H stretch); 1775 (C6F5CO stretch); 1630 (amide CO stretch); 1518 (N–H bend); 1443 (alkane); 1200 (C–O stretch); 1080 (C–O stretch); 993 (C–F stretch).
:[CTA] ratio of 100 repeat units. In a typical reaction, N-(2-hydroxyethyl)acrylamide (HEA) (0.868 g, 7.54 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropanoic acid pentafluorophenyl ester (PFP-DMP) (0.04 g, 0.075 mmol) were dissolved in 50:50 dioxane:methanol solution (3.6 mL) in a vial. The resulting solution was degassed by sparging with N2(g) for 15 min and the sealed vial was incubated at 37 °C with magnetic stirring under 460 nm light irradiation for 120 min. After that time, an aliquot of crude polymerization mixture was taken for 1H NMR in methanol-d4 for conversion and Mn,NMR analysis. The reaction was rapidly cooled in liquid nitrogen and precipitated into diethyl ether. The polymer was re-precipitated into diethyl ether from methanol twice to yield a yellow polymer product which was further dried under vacuum. Same procedure was followed for [monomer]:[CTA] ratios of 140, 160, 180 and 200. Conversions were calculated using 1H NMR spectroscopy by comparing the integrations of the HEA monomer signals (δ 5.67 ppm) with those of the corresponding signals of the polymer (δ 2.22–2.04 ppm, CH of PHEA backbone). Mn,NMR was calculated by end-group analysis by comparing the integrations of the –CH3 signals (δ 0.92 ppm) of dodecyl end-group with those of the corresponding signals of the polymer (δ 2.22–2.04 ppm). 1H NMR (400 MHz, CD3OD): δ (ppm) 8.15–8.03 (br m, NH of PHEA side chain), 3.89–3.13 (br m, NH–CH2 and CH2–OH of PHEA side chain), 2.35–2.05 (br m, CH of PHEA backbone), 1.85–1.31 (br m, CH2 of PHEA backbone), 0.92 (t, 3H, CH2–CH3 of dodecyl end-group). FT-IR (neat): ν (cm−1) 3300 (N–H and O–H stretch); 2868 (alkyl C–H stretch); 1772 (C6F5CO stretch); 1638 (amide CO stretch); 1544 (N–H bend); 1438 (alkane); 1216 (C–O stretch); 1060 (C–O stretch); 950 (C–F peak on shoulder of 1060 peak).
O stretch corresponding to the PFP ester.
Results and discussion
To obtain the desired panel of glycosylated AuNPs, photo-initiated reversible addition–fragmentation chain-transfer (photo-RAFT) polymerization was employed. RAFT installs sulfur containing end-groups suitable for conjugation to AuNPs, and also enables installation of a glycan conjugation unit at the opposing end-group.45,62 To enable the role of coating on outputs to be evaluated, two different water soluble, non-ionic, polymers were chosen; N-hydroxyethyl acrylamide (HEA) and N-(2-hydroxypropyl)methacrylamide (HPMA). Rather than traditional thermal RAFT polymerization, photo-RAFT was chosen as it removes the need for heating, it is extremely convenient to undertake in the laboratory and as no external radical source is used, end-group fidelity is maximized.63,64 To the best of our knowledge, this is the first report of HPMA monomer being used to prepare well-defined homopolymers by photo-RAFT.Using 2-(dodecylthiocarbonothioylthio)-2-methylpropanoic acid (PFP-DMP) chain transfer agent (CTA), (see Experimental section for detailed synthetic procedure) as the RAFT agent (suitable for (meth)acrylamide monomers), a range of PHEA and PHPMA telechelic homopolymers with [M]:[CTA] ratios between 25 and 100 were prepared. The polymerization reactions were carried out under 460 nm visible-light irradiation at 37 °C (under N2 atmosphere) in the absence of a photoinitiator or catalyst for 2 h using a mixture of methanol:dioxane (1:1) as the solvent (Scheme 1). Polymerization was stopped at less than 100% conversion to maximise the retention of end-groups. Size exclusion chromatography (SEC) analysis in DMF with 5 mM NH4BF4 revealed narrow monomodal molecular weight distribution peaks of low dispersity for both PHPMA and PHEA homopolymers (Fig. 1).
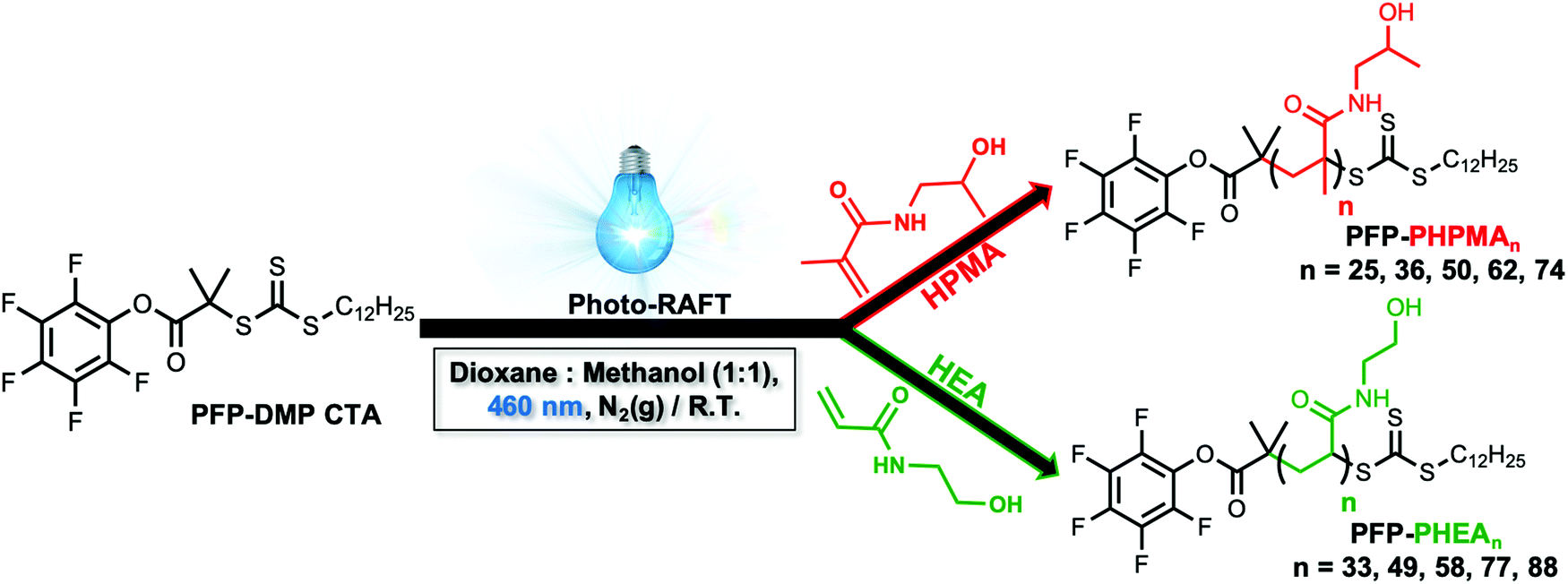 | ||
| Scheme 1 Schematic of the preparation of poly(N-hydroxypropyl methacrylamide) and poly(N-hydroxyethyl acrylamide)-based homopolymers via photo-initiated (460 nm irradiation) reversible addition–fragmentation chain-transfer (photo-RAFT) polymerization at 37 °C. | ||
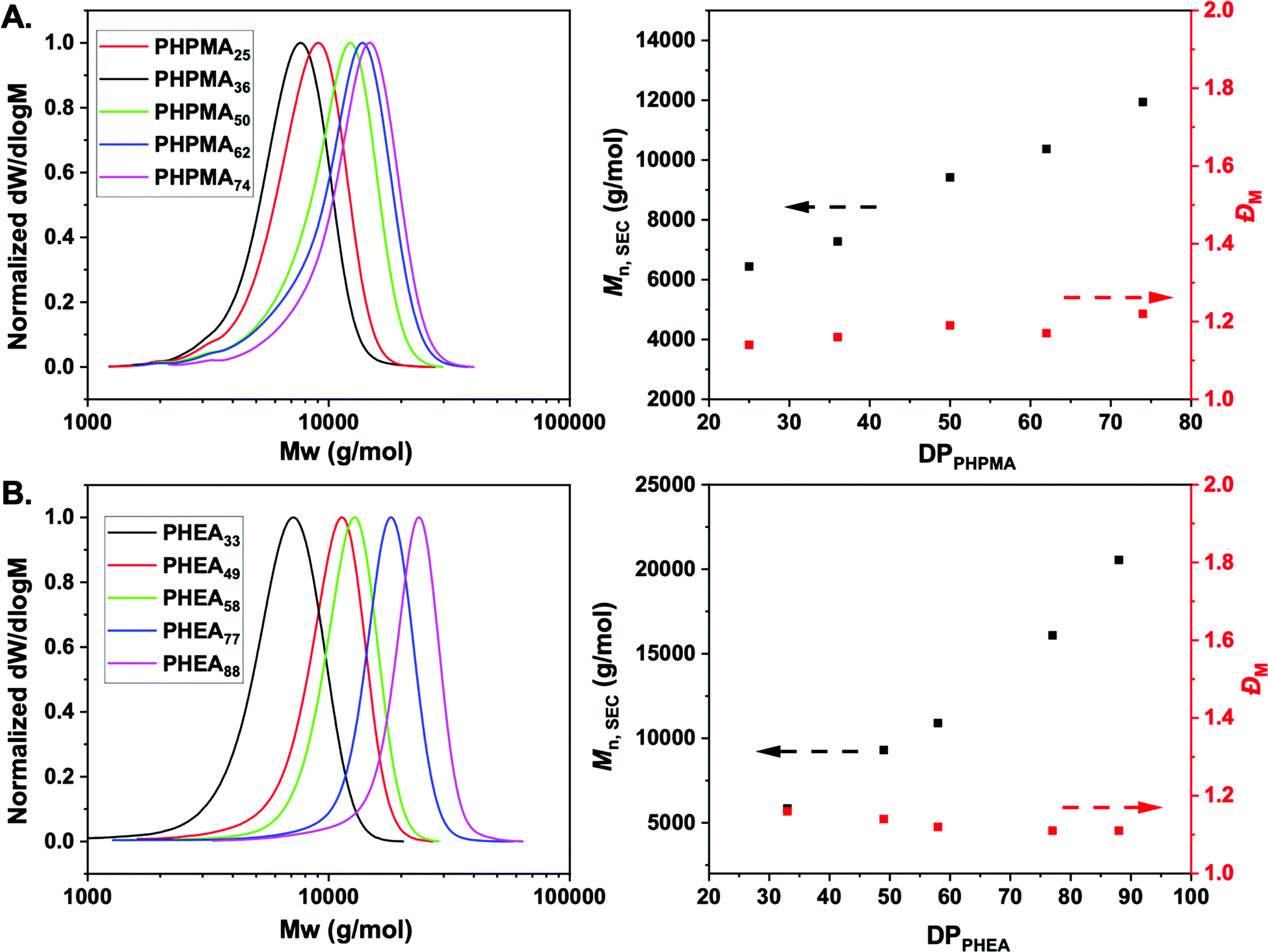 | ||
| Fig. 1 Size exclusion chromatography analysis. Normalized SEC RI molecular weight distributions (left) for (A) PHPMA and (B) PHEA homopolymers. Mn and ĐM as a function of actual degree of polymerization (DP) for the same polymerizations (right). Mn and ĐM values were calculated from PMMA standards using 5 mM NH4BF4 in DMF as the eluent. | ||
1H-NMR spectroscopy in methanol-d4 was used for the determination of the average degree of polymerization (DP) of the final purified homopolymers by comparing the integral ratio of the peak corresponding to –CH3 group of dodecyl end-group at 0.92 ppm to the peak of –CH proton of PHPMA side chain at 3.88 ppm or –CH proton of PHEA backbone at 2.05–2.35 ppm (Fig. S1, ESI†). In all cases, low dispersity values and control of Mn was achieved, indicating a controlled photo-polymerization. Table 1 shows the polymers synthesized and their characterization data. Incorporation of the PFP-group into polymers was confirmed via19F NMR and FT-IR analysis (Fig. S3I and S4, ESI†), showing retention of the group during polymerization. Finally, to examine potential lower critical solution temperature (LCST) behavior for both type of polymers, absorbance points were collected upon heating up to 60 °C in PBS buffer (Fig. S2, ESI†). In both cases, no change in absorbance was detected confirming that the polymers are soluble across a range of temperature and saline conditions, which is essential for ruling out false-positives for binding in the lectin binding assays (below).
| Samplea | [M]:[CTA] |
Conversionb (%) | M n,NMR (g mol−1) | SEC | |
|---|---|---|---|---|---|
| M n,SEC (g mol−1) | Đ M | ||||
| a Sample names are determined according to the number average degree of polymerization (DP) determined by 1H NMR analysis in methanol-d4. b Monomer conversion calculated by comparing the integrations of the monomer with those of the corresponding signals of the polymer. c M n,NMR was calculated by end-group analysis by comparing the integrations of the –CH3 signals (δ 0.92 ppm) of dodecyl end-group with those of the corresponding signals of the polymer. d M n and ĐM values calculated from PMMA standards using 5 mM NH4BF4 in DMF as the eluent. | |||||
| PHPMA25 | 40 | 63 | 4100 | 6400 | 1.14 |
| PHPMA36 | 60 | 60 | 5700 | 7300 | 1.16 |
| PHPMA50 | 80 | 61 | 7700 | 9400 | 1.19 |
| PHPMA62 | 100 | 62 | 9400 | 10400 |
1.17 |
| PHPMA74 | 120 | 62 | 11100 |
11900 |
1.22 |
| PHEA33 | 100 | 33 | 4300 | 5800 | 1.16 |
| PHEA49 | 140 | 35 | 6200 | 9300 | 1.14 |
| PHEA56 | 160 | 35 | 7200 | 10900 |
1.12 |
| PHEA77 | 180 | 42 | 9400 | 16100 |
1.11 |
| PHEA88 | 200 | 43 | 10700 |
20500 |
1.11 |
Glycan installation at the end groups of both PHPMA/PHEA homopolymers was achieved by reaction of the PFP end-group at the α-terminus with D-(+)-galactosamine (GalNH2) (Scheme 2). Successful conjugation of galactosamine was confirmed by 19F-NMR and FT-IR analysis before and after modification (Fig. S3 and S4, ESI†). FT-IR spectroscopy confirmed the disappearance of the characteristic bands of PFP-end group at 950 and 1750 cm−1.
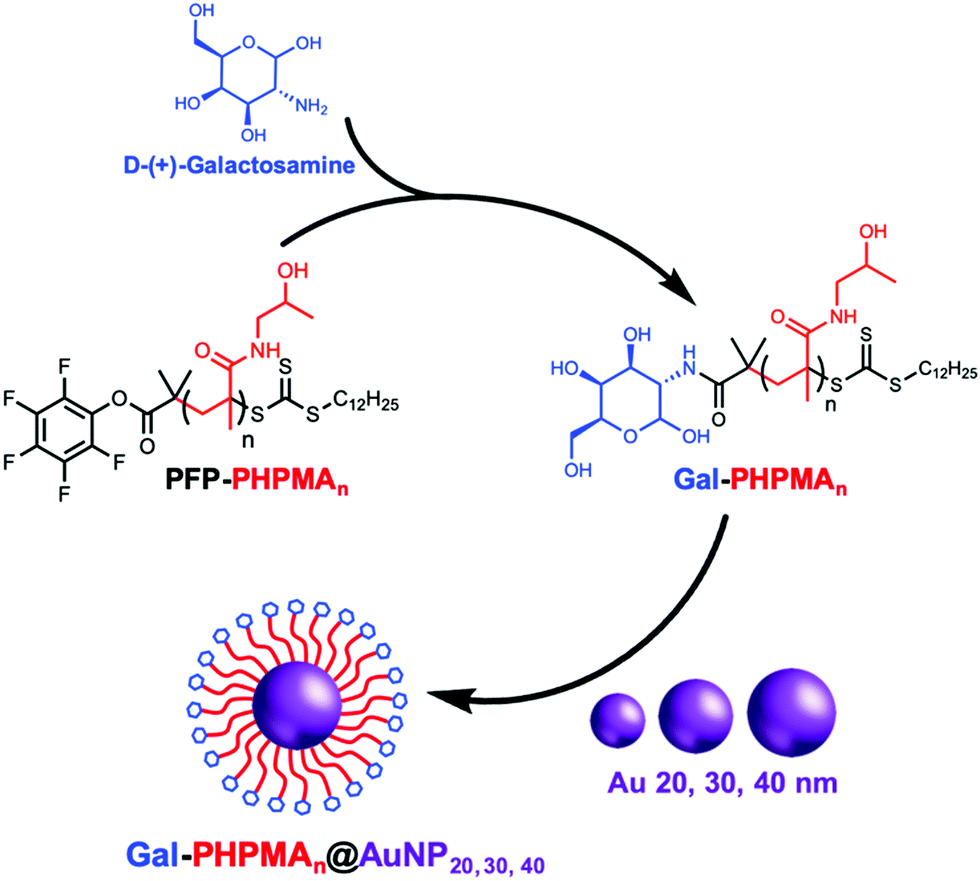 | ||
| Scheme 2 Preparation of galactosamine-poly(hydroxypropyl methacrylamide) (Gal-PHPMAn, n = 25, 36, 50, 62, 74) glycopolymers followed by functionalization onto gold nanoparticles surfaces (AuNP) of 20, 30 and 40 nm diameter. | ||
20, 30 and 40 nm citrate-stabilized gold nanoparticles (AuNP) were synthesized by NaBH4 reduction of HAuCl4 following a seeded growth strategy (see ESI† for detailed synthetic procedure) and subsequently functionalized with thiol-terminated glycopolymers (Gal-PHPMA, Gal-PHEA) by mixing, followed by centrifugation. The resulting polymer-coated nanoparticles were characterized by dry-state transmission electron microscopy (TEM), dynamic light scattering (DLS), zeta-potential and UV-Vis analyses confirming successful addition of the polymer (Table 2 and Table S1, Fig. S5, S6, ESI†). Dynamic light scattering analysis revealed the polymer coated gold particles had increased hydrodynamic diameters compared with the precursor particles, as would be expected (Table 2). In all cases higher hydrodynamic diameter (Dh) and polydispersity (PD) values were measured for PHEA-coated NPs compared to PHPMA-coated NPs. UV-vis analysis for the functionalized AuNPs gave similar results, with ∼2–3 nm SPR-shift for both types of polymer.
| Particle | SPRuncoateda (nm) | SPRpolymercoateda (nm) |
D h (nm) | PDb |
|---|---|---|---|---|
| a SPR maximum recorded by UV-Vis spectroscopy. b D h and PD values determined by DLS (the error represents the standard deviation from 5 repeat measurements). | ||||
| Bare gold 20 nm | 520 | — | 19.7 ± 0.1 | 0.41 ± 0.01 |
| Gal-PHPMA25@AuNP20 | 520 | 523 | 29.7 ± 0.6 | 0.43 ± 0.02 |
| Gal-PHPMA36@AuNP20 | 520 | 523 | 31.2 ± 0.3 | 0.44 ± 0.01 |
| Gal-PHPMA50@AuNP20 | 520 | 524 | 29.0 ± 1.1 | 0.46 ± 0.04 |
| Gal-PHPMA62@AuNP20 | 520 | 523 | 30.2 ± 0.5 | 0.55 ± 0.10 |
| Gal-PHPMA74@AuNP20 | 520 | 524 | 33.6 ± 0.5 | 0.40 ± 0.01 |
| Gal-PHEA33@AuNP20 | 520 | 523 | 26.3 ± 0.5 | 0.55 ± 0.13 |
| Gal-PHEA49@AuNP20 | 520 | 523 | 35.7 ± 0.6 | 0.36 ± 0.01 |
| Gal-PHEA58@AuNP20 | 520 | 523 | 44.2 ± 1.2 | 0.26 ± 0.16 |
| Gal-PHEA77@AuNP20 | 520 | 524 | 52.0 ± 7.1 | 0.37 ± 0.01 |
| Gal-PHEA88@AuNP20 | 520 | 523 | 52.3 ± 1.1 | 0.38 ± 0.01 |
| Bare gold 30 nm | 525 | — | 28.5 ± 0.5 | 0.25 ± 0.01 |
| Gal-PHPMA25@AuNP30 | 525 | 527 | 34.6 ± 1.1 | 0.30 ± 0.03 |
| Gal-PHPMA36@AuNP30 | 525 | 527 | 36.3 ± 0.6 | 0.27 ± 0.01 |
| Gal-PHPMA50@AuNP30 | 525 | 529 | 35.7 ± 0.9 | 0.26 ± 0.01 |
| Gal-PHPMA62@AuNP30 | 525 | 528 | 38.3 ± 1.0 | 0.26 ± 0.01 |
| Gal-PHPMA74@AuNP30 | 525 | 530 | 38.3 ± 0.6 | 0.29 ± 0.01 |
| Gal-PHEA33@AuNP30 | 525 | 528 | 38.6 ± 0.1 | 0.19 ± 0.01 |
| Gal-PHEA49@AuNP30 | 525 | 528 | 43.9 ± 0.5 | 0.18 ± 0.01 |
| Gal-PHEA58@AuNP30 | 525 | 528 | 46.4 ± 0.5 | 0.19 ± 0.01 |
| Gal-PHEA77@AuNP30 | 525 | 527 | 52.2 ± 0.8 | 0.17 ± 0.01 |
| Gal-PHEA88@AuNP30 | 525 | 529 | 57.5 ± 0.8 | 0.19 ± 0.01 |
| Bare gold 40 nm | 530 | — | 40.8 ± 0.5 | 0.26 ± 0.01 |
| Gal-PHPMA25@AuNP40 | 530 | 532 | 38.5 ± 0.7 | 0.41 ± 0.01 |
| Gal-PHPMA36@ AuNP40 | 530 | 532 | 39.4 ± 1.1 | 0.41 ± 0.02 |
| Gal-PHPMA50@ AuNP40 | 530 | 533 | 40.4 ± 1.7 | 0.42 ± 0.02 |
| Gal-PHPMA62@ AuNP40 | 530 | 533 | 43.0 ± 1.3 | 0.38 ± 0.01 |
| Gal-PHPMA74@ AuNP40 | 530 | 532 | 42.8 ± 1.0 | 0.39 ± 0.01 |
| Gal-PHEA33@ AuNP40 | 530 | 531 | 49.4 ± 0.6 | 0.22 ± 0.01 |
| Gal-PHEA49@ AuNP40 | 530 | 531 | 53.2 ± 0.5 | 0.20 ± 0.01 |
| Gal-PHEA58@ AuNP40 | 530 | 531 | 54.8 ± 0.3 | 0.20 ± 0.01 |
| Gal-PHEA77@ AuNP40 | 530 | 531 | 61.5 ± 0.5 | 0.18 ± 0.01 |
| Gal-PHEA88@ AuNP40 | 530 | 531 | 66.1 ± 0.6 | 0.16 ± 0.01 |
To further confirm the presence of glycopolymers on the surface of the AuNPs, XPS analysis was performed for a series of 30 nm Gal-PHPMA/PHEA coated gold nanoparticles (Fig. S7 and S8, ESI†). The presence of N1s amides, that are not present on the naked particles or found commonly in background contaminants, confirmed successful incorporation of the polymers onto the particle surface (Fig. S9C and S10C, ESI†).
A deeper consideration of the particle composition ratios, provided by XPS analysis, highlighted higher grafting densities on the nanoparticle surface in the PHEA systems versus the PHPMA systems. This can be seen in the smaller Au 4f:N1s ratios for PHEA (Table S2, ESI†) compared to the larger PHPMA ratios (Table S3, ESI†). The ratios further indicate that the PHEA glycopolymers are approximately three-times as prevalent on the gold surface than the PHPMA glycopolymers, when comparing similar chain lengths. Whilst these ratios provide only rough estimates of relative grafting densities it does provide evidence for differing surface grafting behaviors between PHEA and PHPMA and shows how simple modification of the polymer ligand can tune the surface and the observed properties (see below).
It is likely that the Gibbs free energy conformations of the dihedral angles in the polymer backbone are influential in determining grafting density. The PHEA backbone can potentially access lower Gibbs free energy conformers compared to PHPMA due to the additional methyl group in the PHPMA backbone increasing steric hinderance. The radius of gyration of PHPMA would therefore be greater than PHEA, leading to less tight packing in PHPMA. In the context of Hill et al.,65 this means that the deflection angle of PHPMA is greater than PHEA so has a lower grafting density. This likely explains the molecular weight effect observed in PHEA. To further understand the complexity in this matter, Barner and co-workers, have recently shown how shorter molecular weight polymers more favourably graft to nanoparticles due to radius of gyration (Rg) effects.66 Previous studies from our group also revealed similar motifs upon comparing water soluble RAFT-derived polymers of poly(vinylpyrrolidone) (PVP) and poly(oligoethyleneglycol methacrylate) (POEGMA). XPS revealed that the sterically bulky POEGMA resulted in far lower grafting densities on gold nanoparticles compared with PVP, suggesting a greater degree of exposed gold surfaces.67
As a next step, saline stability was evaluated by a NaCl titration starting from 0.5 M, Fig. S11 (ESI†). This is essential, as aggregation assays using lectins are evaluated (below) and false positives due to colloidal instability need to be removed. It was observed for PHPMA polymers of DP25 AuNPs of 30 and 40 nm size were unstable and aggregated at high salt (>0.2 M, which is close to physiological (0.137 M)) concentrations (Fig. S13, ESI†), while all Gal-PHEA-coated particles remained stable for all sizes and DPs. This can be understood in terms of the increased ratio of gold to polymer in case of 30 and 40 nm AuNPs and considerably lower grafting densities of PHPMA. While longer polymers (predictably) improved saline stability, it is important to consider the effects of having polymers that are too long; these can prevent/slow the rate of aggregation upon addition of the lectins by steric stabilization.
With this library of nanoparticles varying in the nature of the polymer ligand, their binding to lectins could be assessed. Soybean agglutinin (SBA) was chosen as a model lectin as it has particularly high affinity for N-acetyl galactosamine, which the galactosamine used here gives a mimic of once conjugated as an amide to the polymer end group. Like many members of the legume lectin family SBA possesses a single carbohydrate binding site that requires Mn2+ or Ca2+ for activity.68 However above pH 4.6 (we use pH 7.4 here), SBA is tetrameric allowing for cross-linking and agglutination.69,70 This makes SBA an attractive analyte model lectin for testing glycopolymer-based systems and saccharides for diagnostics of particular interest.71
Initially, the particles were incubated in buffer with SBA from 0–1 mg mL−1. A red-blue color change occurs upon aggregation of gold nanoparticles (Fig. S14 and S15, ESI†) due to coupling of their SPR bands enabling easy read out of aggregation (and indirectly of glycan–lectin binding). Fig. 2 shows the change in absorbance at 700 nm (Abs700) (normalised against the SPR maxima) as a function of SBA for each particle. Rather interestingly, in the case of Gal-PHPMA particles there was no significant increase in Abs700 at any concentration for all the particles, with the exception for Gal-PHPMA74 coated particles which had a (small) shift at the highest SBA concentration. Normally, this would be interpreted as ‘binding not occurring’ and indeed there exist many examples of polymer systems where turbidimetry is used as the output for glycan binding.72–74 However, it is perfectly feasible that binding can occur without cross-linking depending on the architecture of the glycans.75Fig. 2B shows the same experiment but using particles coated with Gal-PHEA. In this case, there was rapid aggregation and clear dose-dependent binding across the entire series. Smaller nanoparticles resulted in less aggregation, as did longer polymer linkers, in line with previous observations.45 This experiment clearly showed that the polymer ligand, as well as the actual glycan, is a key component in the outputs of nanoparticle based sensing systems and needs to be fine-tuned to achieve appropriate outputs.
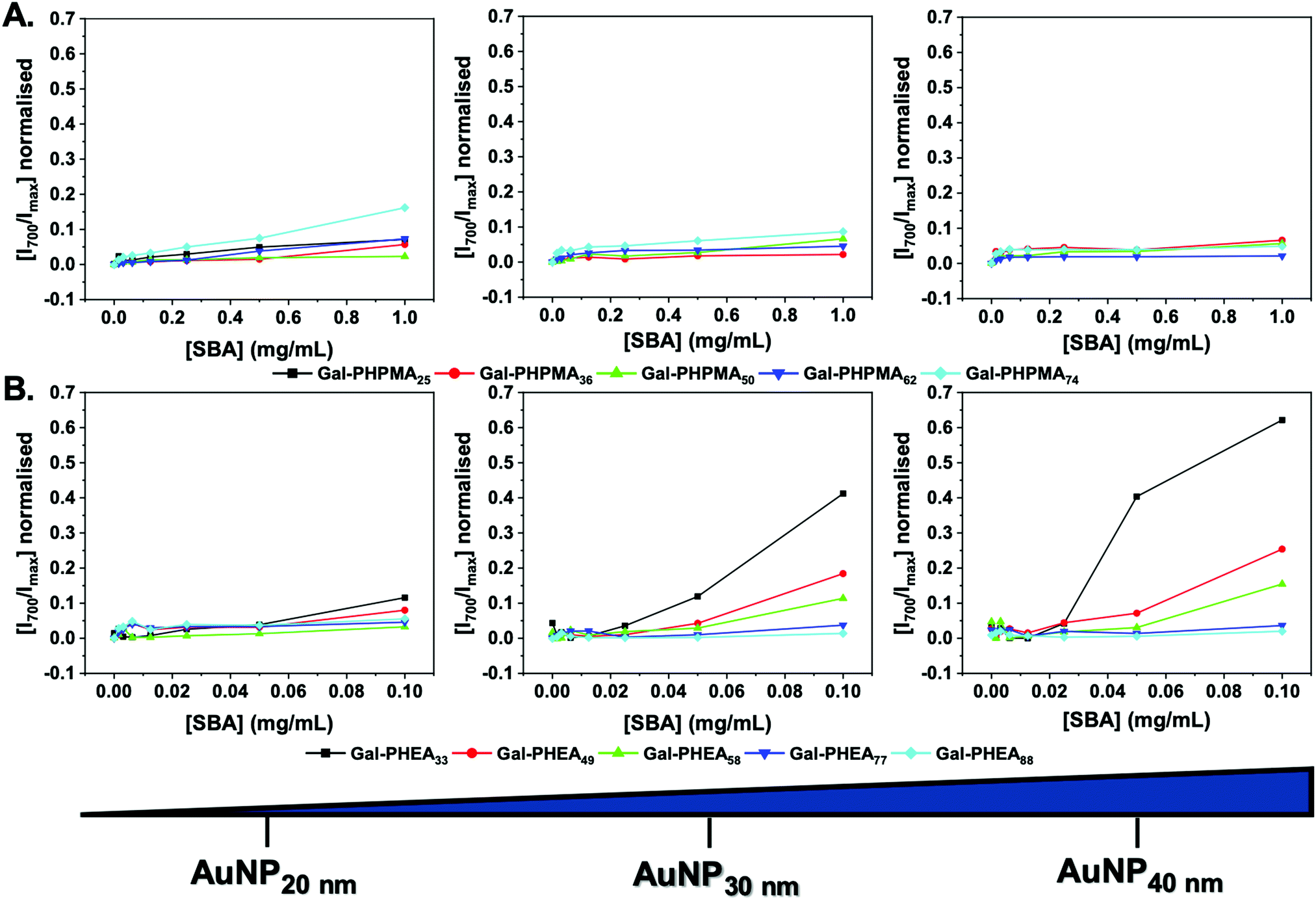 | ||
| Fig. 2 Binding isotherms of nanoparticles with soybean agglutinin, determined by UV-visible spectroscopy. (A) Gal-PHPMA and (B) Gal-PHEA AuNPs of 20, 30 and 40 nm diameter. All samples were incubated for 30 min at 37 °C. Y-Axis is ratio of absorbance at 700 nm, relative to the SPR maximum wavelength of the AuNPs. | ||
To ensure that the above observations (no aggregation of Gal-PHPMA) was not due to inaccessibility of the glycan we used a complementary technique to assess lectin binding. Biolayer interferometry (BLI) was used as a label-free, mass-sensitive method for evaluating biomolecular interactions.76,77 The technology is based on the refraction of white light from two surfaces: a layer of immobilized protein on a biosensor tip, and an internal reference layer. Surface binding to the biosensor tip causes a shift in the interference pattern that can be measured in real-time.78
SBA was modified with biotin-NHS, and immobilized onto streptavidin BLI sensors. Then two different particles were evaluated for dose-dependent binding to the SBA. Gal-PHPMA36@AuNP40 was found not to aggregate in the studies, above, but Gal-PHEA33@AuNP40 did, enabling us to probe if lack of aggregation could rule out binding. Fig. 3 shows the binding data, clearly showing that both particles have affinity towards the SBA with similar dose–response curves being obtained. [Note, kinetic analysis is not possible due to the high valency of these particles.] This experiment proves that the two polymer coatings both present sufficient Gal for strong SBA binding but that the PHPMA coating prevents aggregation from occurring, whereas PHEA encourages it. Taken together this study demonstrates that the chemical nature, as well as the molecular weight, of the polymer linker has a dramatic impact on the outcomes of glyconanoparticle sensing platforms, enabling control over aggregative versus non-aggregative outputs and will help design robust nano-biosensors in the future.
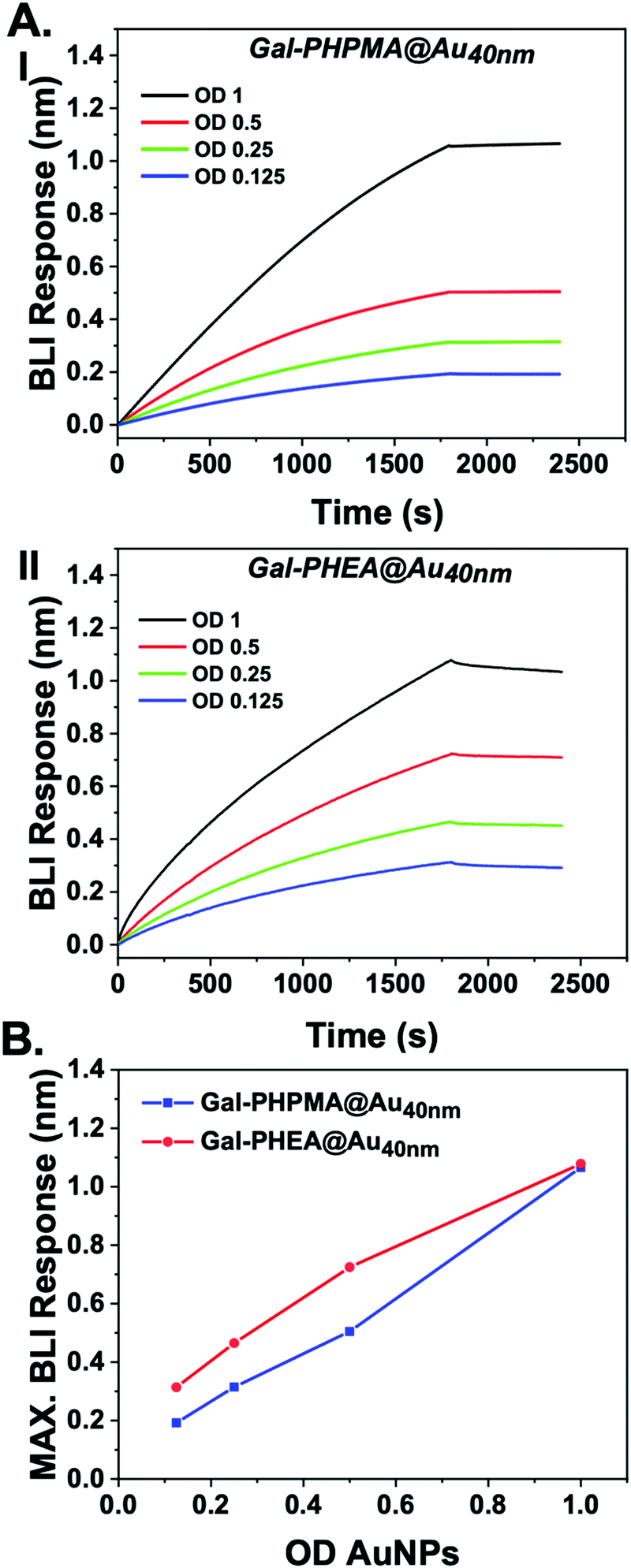 | ||
| Fig. 3 Biolayer interferometry analysis of nanoparticles binding to immobilized SBA. (A) Response of (I) Gal-PHPMA36@AuNP40 and Gal-PHEA33@AuNP40; (B) maximum BLI response vs. AuNP concentration. | ||
Conclusions
Here we demonstrate the crucial impact that the chemical nature, not just the molecular weight, of polymer linkers have in the design of glyconanoparticle sensors. Photo-initiated RAFT polymerization was used to obtain well-defined poly(N-hydroxypropyl methacrylamide), PHPMA and poly(N-hydroxyethyl acrylamide), PHEA bearing pentafluorophenyl ester end groups. Following introduction of a galactosamine unit at the chain ends, the polymers were assembled on 20 to 40 nm gold nanoparticle cores using the terminal thiol group, to give a library of 30 nanoparticles varying in chain length, core size and either a methacrylate or acrylate polymeric backbone. Aggregation assays of these particles using a model lectin showed that, as previously reported, PHEA led to significant aggregation due to lectin-induced cross linking. However, PHPMA nanoparticles showed essentially no response to the lectin, which in traditional aggregation assays would be interpreted as a negative (no binding) result which we show to be incorrect. Using a complementary biolayer interferometry-based assay, it was shown that both types of coatings actually lead to similar lectin binding affinity, even though in the colorimetric aggregation assay there were significant differences. This provides robust evidence that the PHPMA coating approach can prevent aggregation but still present glycans in a manner for lectin binding. XPS showed that the subtle change from acrylamide to methacrylamide changed the grafting density with PHPMA having fewer chains/particle at any given chain length than PHEA, which may contribute to these observations. Overall, the data presented here provides conclusive evidence that the polymer linker itself can play a crucial role in controlling the outputs of glycosylated gold nanoparticles and enable tuning between aggregative and non-aggregative states whilst retaining binding affinity. This simple approach enables selection of the two outputs, depending on the assay or application area (e.g. in vivo versus ex vivo), without requiring significant changes to the design strategy or the glycan conjugation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 814236. MIG holds an ERC starting grant (CRYOMAT 638661). The Royal Society are thanked for funding the cryo-microscopes used in this study. BBSRC/Innovate are thanked for funding the Specialty Glycans project BB/M02878X/1. The BBSRC-funded MIBTP program (BB/M01116X/1) and Iceni Diagnostics ltd are also thanked for a studentship for AB. The Warwick Polymer Research Technology Platform is acknowledged for SEC analysis.Notes and references
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef CAS.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- C. D. Rillahan and J. C. Paulson, Annu. Rev. Biochem., 2011, 80, 797–823 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
- G. B. Sigal, M. Mammen, G. Dahmann and G. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 3789–3800 CrossRef CAS.
- M. L. Huang, M. Cohen, C. J. Fisher, R. T. Schooley, P. Gagneux and K. Godula, Chem. Commun., 2015, 51, 5326–5329 RSC.
- B. D. Polizzotti and K. L. Kiick, Biomacromolecules, 2006, 7, 483–490 CrossRef CAS.
- S.-J. Richards, M. W. Jones, M. Hunaban, D. M. Haddleton and M. I. Gibson, Angew. Chem., Int. Ed., 2012, 51, 7812–7816 CrossRef CAS.
- S. Zhang, R.-O. Moussodia, S. Vértesy, S. André, M. L. Klein, H.-J. Gabius and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5585–5590 CrossRef CAS.
- T. R. Branson and W. B. Turnbull, Chem. Soc. Rev., 2013, 42, 4613–4622 RSC.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- M. L. Huang, R. A. A. Smith, G. W. Trieger and K. Godula, J. Am. Chem. Soc., 2014, 136, 10565–10568 CrossRef CAS.
- J. Conniot, A. Scomparin, C. Peres, E. Yeini, S. Pozzi, A. I. Matos, R. Kleiner, L. I. F. Moura, E. Zupančič, A. S. Viana, H. Doron, P. M. P. Gois, N. Erez, S. Jung, R. Satchi-Fainaro and H. F. Florindo, Nat. Nanotechnol., 2019, 14, 891–901 CrossRef CAS.
- S. Srinivasachari, Y. Liu, G. Zhang, L. Prevette and T. M. Reineke, J. Am. Chem. Soc., 2006, 128, 8176–8184 CrossRef CAS.
- M. Moros, B. Hernáez, E. Garet, J. T. Dias, B. Sáez, V. Grazú, Á. González-Fernández, C. Alonso and J. M. de la Fuente, ACS Nano, 2012, 6, 1565–1577 CrossRef CAS.
- M. Marradi, F. Chiodo, I. García, S. Penadés, G. M. Whitesides, D. Y. Lee, H. Shin, R. J. Pieters, J. M. de la Fuente, S.-H. Nishimura, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, Chem. Soc. Rev., 2013, 42, 4728–4745 RSC.
- N.-C. Reichardt, M. Martín-Lomas and S. Penadés, Chem. Commun., 2016, 52, 13430–13439 RSC.
- C. Van Bruggen, J. K. Hexum, Z. Tan, R. J. Dalal and T. M. Reineke, Acc. Chem. Res., 2019, 52, 1347–1358 CAS.
- S. Bhatia, L. C. Camacho and R. Haag, J. Am. Chem. Soc., 2016, 138, 8654–8666 CrossRef CAS.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS.
- M. Mammen, G. Dahmann and G. M. Whitesides, J. Med. Chem., 1995, 38, 4179–4190 CrossRef CAS.
- M. W. Jones, L. Otten, S.-J. Richards, R. Lowery, D. J. Phillips, D. M. Haddleton and M. I. Gibson, Chem. Sci., 2014, 5, 1611–1616 RSC.
- T. R. Branson, T. E. McAllister, J. Garcia-Hartjes, M. A. Fascione, J. F. Ross, S. L. Warriner, T. Wennekes, H. Zuilhof and W. B. Turnbull, Angew. Chem., Int. Ed., 2014, 53, 8323–8327 CrossRef CAS.
- E. Kolomiets, M. A. Swiderska, R. U. Kadam, E. M. V. Johansson, K. E. Jaeger, T. Darbre and J. L. Reymond, ChemMedChem, 2009, 4, 562–569 CrossRef CAS.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130–15132 CrossRef CAS.
- S. Ordanini, N. Varga, V. Porkolab, M. Thépaut, L. Belvisi, A. Bertaglia, A. Palmioli, A. Berzi, D. Trabattoni, M. Clerici, F. Fieschi and A. Bernardi, Chem. Commun., 2015, 51, 3816–3819 RSC.
- C. M. Jarvis, D. B. Zwick, J. C. Grim, M. M. Alam, L. R. Prost, J. C. Gardiner, S. Park, L. L. Zimdars, N. M. Sherer and L. L. Kiessling, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14862–14867 CrossRef CAS.
- R. Jelinek and S. Kolusheva, Chem. Rev., 2004, 104, 5987–6016 CrossRef CAS.
- S. Cunningham, J. Q. Gerlach, M. Kane and L. Joshi, Analyst, 2010, 135, 2471–2480 RSC.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- Z. Yuan, C.-C. Hu, H.-T. Chang and C. Lu, Analyst, 2016, 141, 1611–1626 RSC.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- D. C. Hone, A. H. Haines and D. A. Russell, Langmuir, 2003, 19, 7141–7144 CrossRef CAS.
- C. L. Schofield, R. A. Field and D. A. Russell, Anal. Chem., 2007, 79, 1356–1361 CrossRef CAS.
- M. J. Marín, A. Rashid, M. Rejzek, S. A. Fairhurst, S. A. Wharton, S. R. Martin, J. W. McCauley, T. Wileman, R. A. Field and D. A. Russell, Org. Biomol. Chem., 2013, 11, 7101 RSC.
- M. Takara, M. Toyoshima, H. Seto, Y. Hoshino and Y. Miura, Polym. Chem., 2014, 5, 931–939 RSC.
- S. O. Pereira, A. Barros-Timmons and T. Trindade, Polymers, 2018, 10, 189 CrossRef.
- M. Beija, J.-D. Marty and M. Destarac, Prog. Polym. Sci., 2011, 36, 845–886 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- S.-J. Richards and M. I. Gibson, ACS Macro Lett., 2014, 3, 1004–1008 CrossRef CAS.
- S.-J. Richards, E. Fullam, G. S. Besra and M. I. Gibson, J. Mater. Chem. B, 2014, 2, 1490–1498 RSC.
- S.-J. Richards, L. Otten and M. I. Gibson, J. Mater. Chem. B, 2016, 4, 3046–3053 RSC.
- L. Otten, D. Vlachou, S.-J. Richards and M. I. Gibson, Analyst, 2016, 141, 4305–4312 RSC.
- S. Won, S. Hindmarsh and M. I. Gibson, ACS Macro Lett., 2018, 7, 178–183 CrossRef CAS.
- B. S. Tucker and B. S. Sumerlin, Polym. Chem., 2014, 5, 1566–1572 RSC.
- M. Talelli, C. J. F. Rijcken, C. F. van Nostrum, G. Storm and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62, 231–239 CrossRef CAS.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2010, 62, 272–282 CrossRef CAS.
- J. Kopeček, Adv. Drug Delivery Rev., 2013, 65, 49–59 CrossRef.
- K. Ulbrich and V. Šubr, Adv. Drug Delivery Rev., 2010, 62, 150–166 CrossRef CAS.
- J. Geng, W. Li, Y. Zhang, N. Thottappillil, J. Clavadetscher, A. Lilienkampf and M. Bradley, Nat. Chem., 2019, 11, 578–586 CrossRef CAS.
- V. Raus and L. Kostka, Polym. Chem., 2019, 10, 564–568 RSC.
- N. Francini, L. Purdie, C. Alexander, G. Mantovani and S. G. Spain, Macromolecules, 2015, 48, 2857–2863 CrossRef CAS.
- P. Bojarová, P. Chytil, B. Mikulová, L. Bumba, R. Konefał, H. Pelantová, J. Krejzová, K. Slámová, L. Petrásková, L. Kotrchová, J. Cvačka, T. Etrych and V. Křen, Polym. Chem., 2017, 8, 2647–2658 RSC.
- D. Roy, B. Ghosn, E.-H. Song, D. M. Ratner and P. S. Stayton, Polym. Chem., 2013, 4, 1153–1160 RSC.
- M. Bartneck, C. T. Schlößer, M. Barz, R. Zentel, C. Trautwein, T. Lammers and F. Tacke, ACS Nano, 2017, 11, 9689–9700 CrossRef CAS PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- S. Won, S.-J. Richards, M. Walker and M. I. Gibson, Nanoscale Horiz., 2017, 2, 106–109 RSC.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–994 CrossRef CAS.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS.
- L. Michalek, K. Mundsinger, C. Barner-Kowollik and L. Barner, Polym. Chem., 2019, 10, 54–59 RSC.
- N. Sze Ieong, C. I. Biggs, M. Walker and M. I. Gibson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1200–1208 CrossRef CAS.
- V. S. R. Rao, K. Lam and P. K. Qasba, J. Biomol. Struct. Dyn., 1998, 15, 853–860 CrossRef CAS.
- M. Huet, Eur. J. Biochem., 1975, 59, 627–632 CrossRef CAS.
- D. K. Mandal, E. Nieves, L. Bhattacharyya, G. A. Orr, J. Roboz, Q.-t. Yu and C. F. Brewer, Eur. J. Biochem., 1994, 221, 547–553 CrossRef CAS.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661 CrossRef CAS.
- M. Kanai, K. H. Mortell and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 9931–9932 CrossRef CAS.
- M. W. Jones, S.-J. Richards, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2013, 4, 717–723 RSC.
- R. Sunasee and R. Narain, Macromol. Biosci., 2013, 13, 9–27 CrossRef CAS.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 15732–15742 CrossRef CAS.
- Y. Ji and R. J. Woods, Glycobiology, Springer, Singapore, 2018, pp. 259–273 Search PubMed.
- L. E. Wilkins, N. Badi, F. Du Prez and M. I. Gibson, ACS Macro Lett., 2018, 7, 1498–1502 CrossRef CAS PubMed.
- H.-M. Schmitt, A. Brecht, J. Piehler and G. Gauglitz, Biosens. Bioelectron., 1997, 12, 809–816 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tb02004g |
| This journal is © The Royal Society of Chemistry 2020 |