
A zinc selective oxytocin based biosensor†
Evgeniy
Mervinetsky‡
 a,
Israel
Alshanski‡
a,
Kiran Kumar
Tadi
a,
Arezoo
Dianat
b,
Jörg
Buchwald
b,
Rafael
Gutierrez
*b,
Gianaurelio
Cuniberti
bcd,
Mattan
Hurevich
*a and
Shlomo
Yitzchaik
*a
a,
Israel
Alshanski‡
a,
Kiran Kumar
Tadi
a,
Arezoo
Dianat
b,
Jörg
Buchwald
b,
Rafael
Gutierrez
*b,
Gianaurelio
Cuniberti
bcd,
Mattan
Hurevich
*a and
Shlomo
Yitzchaik
*a
aInstitute of Chemistry and The Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: mattan.hurevich@mail.huji.ac.il; shlomo.yitzchaik@mail.huji.ac.il
bInstitute for Materials Science and Max Bergmann Center of Biomaterials, TU Dresden, 01062 Dresden, Germany. E-mail: rafael.gutierrez@tu-dresden.de
cDresden Center for Computational Materials Science, TU Dresden, 01062 Dresden, Germany
dCenter for Advancing Electronics Dresden, TU Dresden, 01062 Dresden, Germany
First published on 20th November 2019
Abstract
Oxytocin is a peptide hormone with high affinity to both Zn2+ and Cu2+ ions compared to other metal ions. This affinity makes oxytocin an attractive recognition layer for monitoring the levels of these essential ions in biofluids. Native oxytocin cannot differentiate between Cu2+ and Zn2+ ions and hence it is not useful for sensing Zn2+ in the presence of Cu2+. We elucidated the effect of the terminal amine group of oxytocin on the affinity toward Cu2+ using theoretical calculations. We designed a new Zn2+ selective oxytocin-based biosensor that utilizes the terminal amine for surface anchoring, also preventing the response to Cu2+. The biosensor shows exceptional selectivity and very high sensitivity to Zn2+ in impedimetric biosensing. This study shows for the first time an oxytocin derived sensor that can be used directly for sensing Zn2+ in the presence of Cu2+.
Introduction
A deficiency or excess in essential trace elements in living organisms has been identified in many diseases.1,2 Zinc is an essential trace element that plays a vital role in many enzymatic and regulatory processes.3 Abnormal low levels of Zn2+ have been associated with diseases such as growth retardation, emotional instability, hyposmia, alopecia and multiple sclerosis.4–7 Monitoring of Zn2+ levels might serve as a valuable medical diagnostic tool. The common techniques for monitoring of Zn2+ ions are flame atomic absorption spectrometry,8 UV-vis spectroscopy,9 and inductively coupled plasma mass spectrometry (ICPMS) in addition to other fluorescence methods.10 However, most of these techniques have limitations due to the complete occupancy of the d-orbitals in zinc ions, which results in the absence of significant magnetic and spectroscopic signals.11Many natural peptides bind metal ions with high selectivity, which makes them attractive receptors for metal ion sensing.12–14 The ability of peptides to bind metal ions depends on their sequence, conformations, and functional groups.15 Peptides can bind metal ions through various functional side chains e.g. the imidazole of histidine,16 or via their backbone atoms e.g. the amide carbonyls.17,18 The complex formation of a peptide with Cu2+ and in some cases with Ni2+ takes place through a unique mechanism in which the protons from the backbone amides are removed via a catalytic binding cascade and replaced by the metal ion under physiological conditions (Fig. 1).19–21 The complex formed by this cascade has a square planar geometry as indicated by studying the crystallographic properties using a penta-glycine peptide model.22
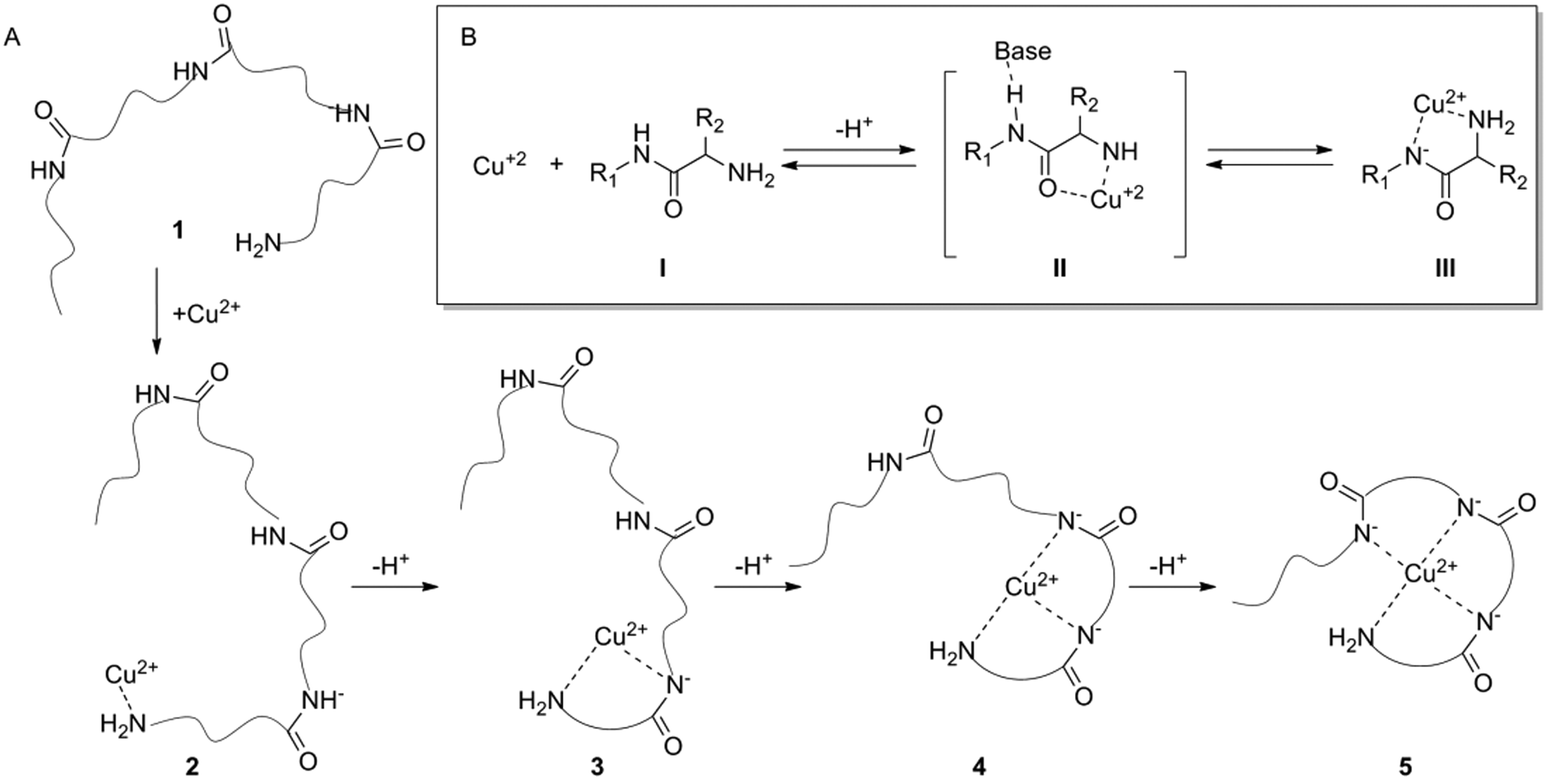 | ||
| Fig. 1 A general mechanism for Cu2+–peptide complex formation. (A) Step-wise formation of four dentate peptide–copper complexes. Copper binding to the terminal amine of peptide 1 forms the peptide:copper intermediate 2. This binding initiates a deprotonation cascade forming eventually the square planar peptide:copper compound 5. (B) Deprotonation mechanism of an amide by copper. The amine binds a copper ion [V] and the copper withdraws the electrons from the carbonyl bond making the amide hydrogen acidic [II]. After three amide protons are removed, copper binds the deprotonated amides to form complex [III]. | ||
Oxytocin (OT) is an important hormone and neurotransmitter that can bind both Cu2+ and Zn2+ with similar affinities, albeit by two different mechanisms.23,24 Zn2+ binds OT through six amide carbonyls in an octahedral complex,18 while copper uses the terminal amine and three deprotonated amides for binding in a square-planar complex.24–27 The high affinity of OT toward the two metal ions suggests that it might serve as an attractive metal ion recognition constituent. However, the similar affinities of OT toward these two ions hamper the attempt to develop a sensor that responds specifically to one in the presence of the other when OT is self-assembled on a gold surface.28
Our previously reported OT sensor, based on a conformational adaptive response,29 was prepared by anchoring the peptide to a glassy carbon surface through the triazole moiety. Although the sensor showed very high sensitivity toward Zn2+ it also showed significant cross-reactivity with Cu2+. We assumed that the reactivity toward Cu2+ is mediated by the triazole ring, which serves as an anchoring moiety, similar to the function of the terminal amine in the native OT. The triazole anchoring event initiates a deprotonation cascade which leads to very strong Cu2+ binding. Metal ion selectivity of the triazole-based OT sensor was gained after using masking agents in the sensing solution, which enabled monitoring of one ion in the presence of the other.30 Alternatively, selective copper binding of the triazole-based OT sensor was achieved at high pH, which presumably assisted the deprotonation steps and decreased the sensitivity toward Zn2+.31 These two studies showed that the triazole-based OT sensor cannot provide agent free zinc selective sensing. We also reported an OT sensor that was based on the direct assembly of the native peptide on a gold surface.28 When native OT was used there was also no selectivity between the two ions because the amine was free to bind Cu2+. Herein, we developed a new OT based sensor that provides sensitivity and selectivity toward Zn2+ ions.
We used molecular modelling to evaluate the contribution of the terminal amine to the Cu2+ binding affinity, and discovered that acetamidation of the terminal amine results in complete insensitivity toward Cu2+ while the binding to Zn2+ is not affected. Based on these studies we designed a new sensing platform in which the OT terminal amine was directly coupled to a lipoic acid (LPA) self-assembled monolayer on a gold surface to form an amide bond. In this case, the possibility of Cu2+ binding to OT was eliminated since there is no anchoring binding cascade initiator. We showed by electrochemical impedance spectroscopy (EIS), which is a sensitive tool to study interactions of a biopolymer with metal ions,32,33 that the Zn2+ binding signal to the OT sensor was preserved because the binding does not rely on the free amine. The new biosensor proved to be highly selective toward Zn2+ and not to other biologically relevant metal ions.
Experimental
Modeling structure and energetics
The ion binding process was addressed for both native and acetylated OT, the former case constitutes a reference point for comparison with OT on surfaces. We used a density functional theory (DFT) approach as implemented in the CP2K software package.34 In particular, we use a DZVP (double zeta for valence electrons plus polarization functions) basis set complemented with a plane-wave basis with an energy cut-off of 350 Ry. Core electrons were taken into account by means of the Goedecker, Teter, and Hutter (GTH) approximation.35 For the exchange–correlation energy, the local density approximation (LDA) was used, with two and eleven explicit valence electrons for Zn2+ and Cu2+ atomic-ions, respectively. Dispersion corrections were included through the standard D2 Grimme parameterization.36 In the case of the isolated molecule, calculations were performed with a supercell with a box size of 40 × 40 × 50 Å. These dimensions ensure avoidance of spurious interactions between neighboring cells when implementing periodic boundary conditions for a finite size system. The ion binding energies EionB were computed as EionB = Ecomplex − EOT+ion. Here, Ecomplex is the energy of the complex OT–ion system, while the second term EOT+ion is the total energy of a simulation, where the ion was placed in the cell at a distance of about 20 Å from the molecule.Au-LPA-OT sensor fabrication
All solutions used in this work were prepared with Milli-Q water (18.3 MΩ cm−1, Millipore Milli-Q system (Bedford, MA)). For pH adjustment we used 50 mM ammonium acetate (pH = 7.0) buffer (AAB) solution (Sigma-Aldrich). Gold surfaces were deep coated in a solution of 1 mM LPA (Sigma-Aldrich) in ethanol for 16 h at 22 °C. The LPA monolayer was coupled with OT (ProSpec-Tany Technogene Ltd, Israel) in acetonitrile (ACN) for 4 h with a (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU) coupling reagent using a standard SPPS protocol.37 Practically, the coupling was done by preparation of 500 μM OT in ACN solution (OT–ACN). 100 μl of this OT–ACN solution was added to a mixture prepared by dissolving 2 mg of COMU reagent in 800 μl ACN with addition of 2 drops triethylamine (Sigma-Aldrich). The Au substrates were dipped in the described solution and placed on a rotatory shaker for 4 h. After the coupling, the substrates were rinsed with ACN and AAB and dried under a dry N2 stream. Following peptide assembly the modified substrates were exposed to Cu(NO3)2 (99.999%, trace metal basis, Sigma-Aldrich) and Zn(NO3)2 (99.999%, trace metal basis, Sigma-Aldrich) AAB solutions in different concentrations.Surface characterization
For surface characterization an Au layer (100 nm) was evaporated on top of a Cr layer (10 nm), which was evaporated on a substrate of a highly-doped n-type Si wafer (〈100〉, R < 0.003 Ω cm−1). An OT layer was adsorbed on this substrate with the same adsorption protocol. Exposure to Cu2+/Zn2+ was done by drop casting of metal ions dissolved in AAB. XPS spectra were recorded using a monochromatic Al Kα X-ray source on a Kratos Axis-HS instrument. XPS analyses were applied to characterize the self-assembled peptide monolayers and the complexation of the peptide with Cu2+/Zn2+ ions. Variable angle spectroscopic ellipsometry (VASE) measurements were carried out with an ellipsometer VB-400 (Woollam Co.) at a Brewster angle of 75°. AFM (Bruker, Innova) was performed in tapping mode in order to monitor the topography homogeneity of the layer as a result of OT adsorption and metal ion chelation.Electrochemical characterization
Electrochemical analyses were conducted with a Bio-Logic SP-300 potentiostat (Bio-Logic Science Instruments, France) utilizing EIS and with the EC-LAB software package. A three-electrode cell was used for the measurements: Ag/AgCl (in 3 M KCl) as the reference electrode (RE), Pt as the counter electrode (CE) and Au as the working electrode (WE). Polycrystalline bulk gold electrodes with a 2 mm diameter were used for electrochemical measurements (CH Instruments). These electrodes were manually-polished using micro-cloth pads (Buehler, Lake Bluff, IL) with a de-agglomerated alumina suspension (Buehler) of decreasing particle size (1.0 and 0.05 μm) and washed with TDW. Then the Au WE was cast by LPA and coupled to OT by the described protocol. The EIS measurements were performed at four stages: the bare gold electrode, after adsorption of the LPA monolayer, after peptide coupling to the monolayer and finally after exposure to metal ions. The frequency range set is from 100 kHz to 0.1 Hz with Ewe = 0.21 V according to a Ag/AgCl (3 M) reference electrode. All EIS scans were done in an EIS solution of 5.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mM K3[Fe(CN)6] and 5.0 mM K4[Fe(CN)6] (RedOx species) with 0.1M of KCl as a supporting electrolyte in 50 mM AAB solution. The results were fitted with the circuit RS[(RCT|W)||Q] where Q is a constant phase element that describes a non-ideal capacitor (Fig. S9, ESI†). For the dose response, exposure of the Au–OT electrode proceeded by dipping of the Au–OT electrode in ion solution in AAB for 10 min, washing with the buffer (by rinsing the electrode with 1 ml AAB using a Pasteur pipette), EIS measurement and further exposure to increasing concentrations of ions in AAB. The presented data are based on statistics of 3 different electrodes. The selectivity analysis is based on comparing the RCT values of EIS after exposure of the Au–LPA–OT electrode to 10–5 M metal ion solution in AAB.
mM K3[Fe(CN)6] and 5.0 mM K4[Fe(CN)6] (RedOx species) with 0.1M of KCl as a supporting electrolyte in 50 mM AAB solution. The results were fitted with the circuit RS[(RCT|W)||Q] where Q is a constant phase element that describes a non-ideal capacitor (Fig. S9, ESI†). For the dose response, exposure of the Au–OT electrode proceeded by dipping of the Au–OT electrode in ion solution in AAB for 10 min, washing with the buffer (by rinsing the electrode with 1 ml AAB using a Pasteur pipette), EIS measurement and further exposure to increasing concentrations of ions in AAB. The presented data are based on statistics of 3 different electrodes. The selectivity analysis is based on comparing the RCT values of EIS after exposure of the Au–LPA–OT electrode to 10–5 M metal ion solution in AAB.
Results and discussion
Modeling of ion binding: structure and energetics
To design a selective OT based biosensor for Zn2+, we aimed to disrupt the mechanism by which OT binds Cu2+ without affecting the Zn2+ binding mechanism. For this purpose we calculated the binding energy of Cu2+ to native OT for four-coordinated binding sites, three deprotonated amides and the terminal amine group. The ion binding energy for this system was determined to be 5.1 eV (Fig. 2). The native OT:Cu2+ binding mechanism (and slightly to nickel) implies that binding takes place only if the free amine (or other amine surrogate entity) of the peptide anchors the ion prior to the deprotonation cascade (Fig. 1). We hypothesized that blocking the terminal amine of OT can interrupt Cu2+ binding. We used an acetylated OT model that doesn't have the free amine moiety. The use of the OT acetylated terminal amine model is aimed at evaluating the binding affinities of the peptide to copper in the case that the ion anchoring to the amine and the subsequent deprotonation cascade do not take place. To perform the DFT calculations of the Cu2+ binding to acetylated OT model, we located a Cu2+ atom close to the acetyl functional group and optimized the geometry assuming that deprotonation does not take place. The calculated binding energy from the optimized structure was determined to be 0.6 eV. This value is much smaller than for the native OT ion binding and suggests that Cu2+ binding to the acetylated OT is rather unlikely. This calculation proves the importance of the amine moiety to the Cu2+ binding mechanism and suggests that OT which does not have a terminal free amine might provide a sensor that is not sensitive to Cu2+.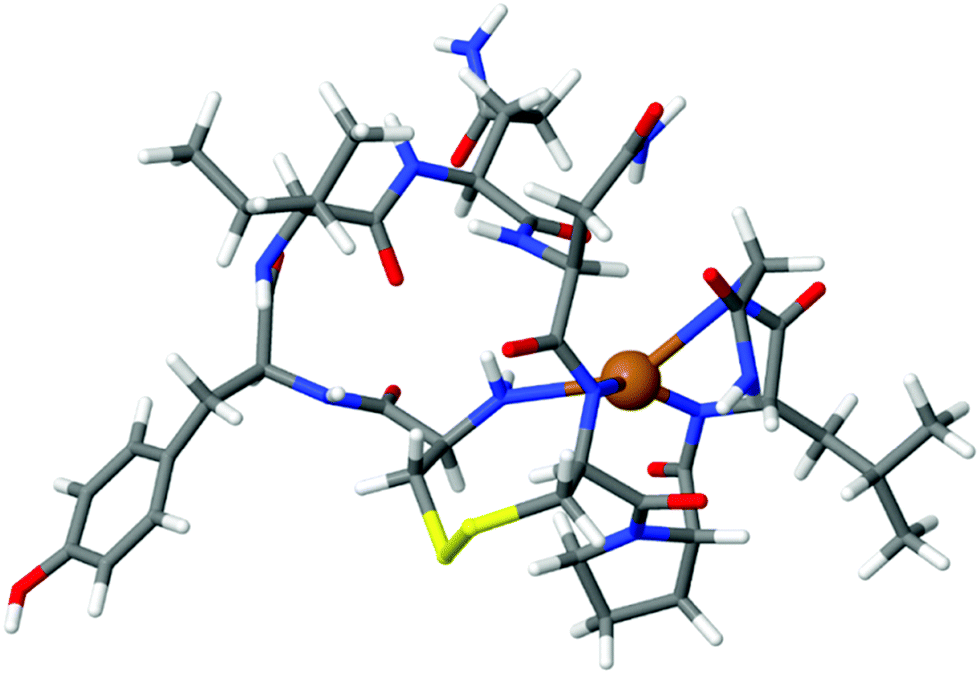 | ||
| Fig. 2 DFT of the OT:Cu2+ complex. The Cu2+ ion is bound to the terminal amine and three deprotonated amides. The binding energy was calculated to be 5.1 eV. | ||
As these DFT results suggest that we can eliminate the Cu2+:OT interaction by acetylation, we wanted to elucidate the effect of OT acetylation also on the Zn2+ binding affinity. The Zn2+:OT binding mechanism suggests that the coordination takes place through several carbonyl oxygens from the peptide backbone. The Zn2+ binding mechanism hence does not rely on the deprotonation cascade and the presence of the free amine is not essential. For the calculation of Zn2+ binding to native OT, three different binding coordination modes were taken into account and the computed binding energies were found to be 2.57 eV, 2.97 eV, and 3.63 eV going from the four-dentate to the six-dentate coordination, respectively (Fig. 3).
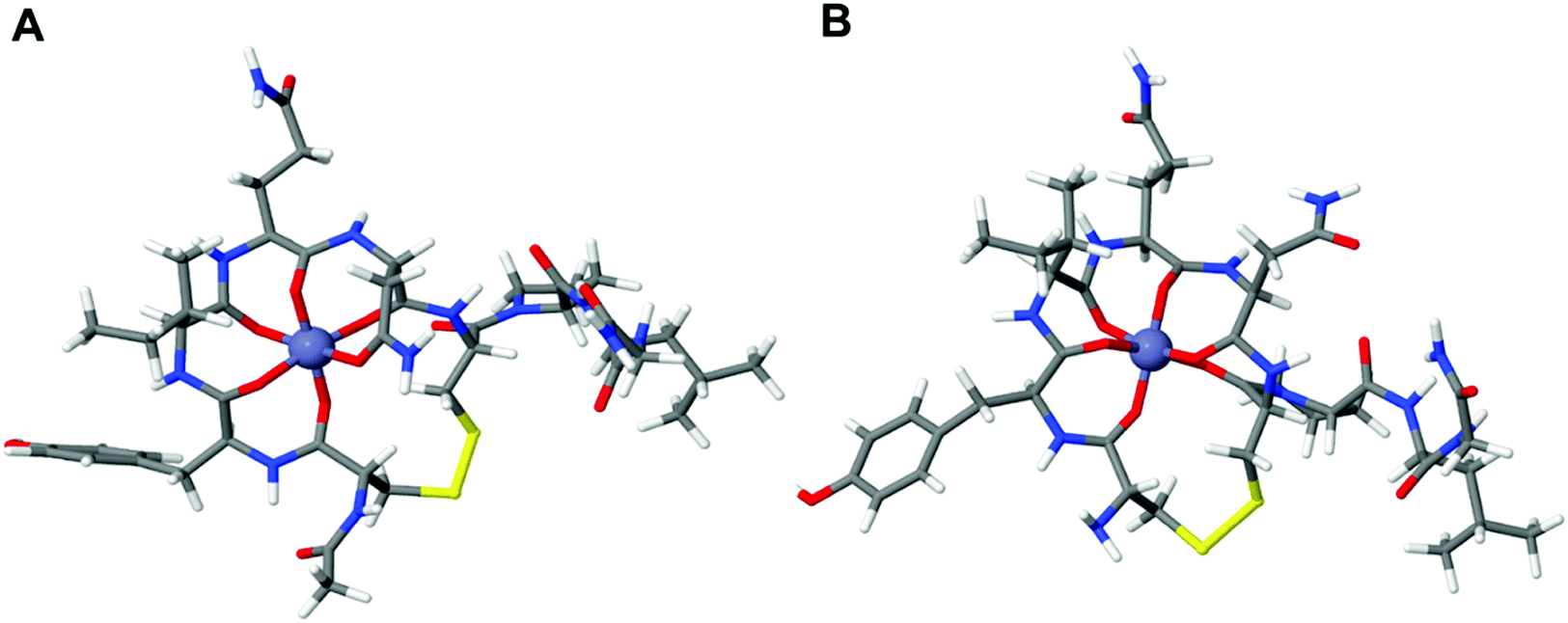 | ||
| Fig. 3 Optimized structure of Zn2+ binding to the (A) acetylated OT and (B) native OT. The difference in the ion binding energy for the two systems is about 0.17 eV. | ||
The calculation shows that the most stable conformation of Zn2+ bound to OT will result in an overall 3.63 eV binding energy (Fig. 3A). In order to evaluate the effect of acetylation on Zn2+ binding, the binding energy of octahedral acetylated OT:Zn2+ coordination was calculated. Our DFT calculations showed that the binding of Zn2+ to acetylated OT results in a 3.46 eV binding energy compared to the 3.63 eV that was calculated for the native OT. Thus, the binding energy of Zn2+ to acetylated OT shows only a slight increase of about 0.17 eV compared to the native OT, which strongly indicates that acetylation is not expected to lead to a dramatic change in the ability of OT binding to Zn2+.
Our DFT calculations imply that acetylated OT might be insensitive toward Cu2+ while its sensitivity to Zn2+ is the same as for native OT. These studies indicate that designing an OT based sensor in which the terminal amine is chemically blocked while the rest of the native functional groups of the peptide are kept intact can potentially provide a selective Zn2+ sensing strategy.
Sensor design on Au surfaces
Based on the DFT calculations, a new OT based sensing platform was designed. The rationale was to anchor the OT to the surface in such a way that the terminal amine will be blocked while the rest of the OT moieties will be intact. We hypothesized that by designing such a sensor, Zn2+ binding can be preserved while Cu2+ binding is eliminated. To attain such a sensor, we sought to anchor the OT to surfaces through alkyl-amidation of the terminal amine. This will achieve two aims: first, the alkyl-amidation will block the Cu2+ binding affinity as was shown for the acetylated OT DFT model. Second, the Zn2+ binding will be maintained since no other functional group of the peptide will be changed and the peptide will be free to adopt the metal binding conformation. The amide on the N-terminal side of OT can be formed by a peptide coupling strategy with a spacer containing carboxylic acid on the surface.37The anchoring of OT to the surface was performed by an amide bond surface formation method that binds the terminal amine of the peptide to a self-assembled monolayer of a carboxyl functionalized surface (SAM, see Fig. 4). For this purpose, a SAM of LPA on a gold electrode was assembled and the carboxylates at the interface were coupled to the OT terminal amide by amidation.38 To confirm that such an Au–LPA–OT electrode can be produced, we prepared OT–LPA gold surfaces and analyzed them by various methods such as XPS. The gold surfaces were modified with LPA to give a SAM on a surface with interfacial carboxylate functionality. The peptide was coupled to the carboxylate modified surface to establish an OT layer. XPS was used to analyze the elemental composition of the layers for each of the assembly steps. The OT–LPA assembly on gold was characterized by a nitrogen related energy of 400.76 eV (Fig. S1 and S4, ESI†), which was not found on the bare Au and Au–LPA samples. Moreover, only for the modified surface was a signal of S 2p (Fig. S2, ESI†) identified, where the peak at 162 eV corresponds to the sulfur atoms bound to the gold surface.39 In addition, carbonyl-related peaks (Fig. S3, ESI†) were found on the Au–LPA–OT surface and not on the bare Au sample. Finally, a Zn 2p related peak was found only on the Au–LPA–OT surface after exposure to Zn2+ solution (Fig. S5, ESI†). These results suggest that this type of fabrication can produce an N-acetylated OT recognition layer for Zn2+ on a gold electrode surface (Au–LPA–OT).
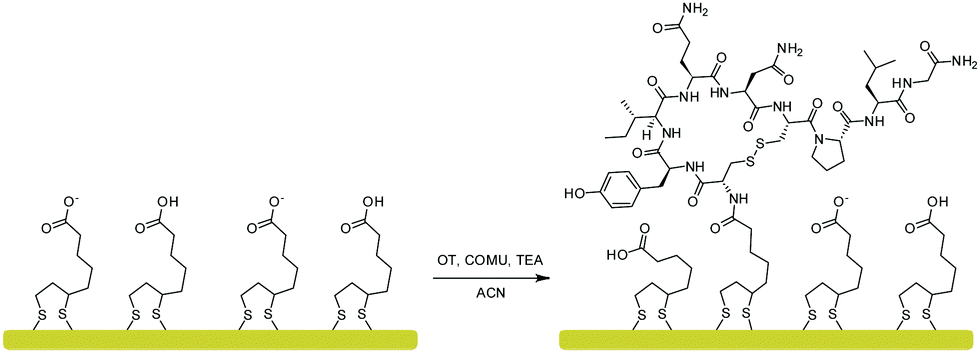 | ||
| Fig. 4 OT coupling to a self-assembled monolayer of LPA on a gold surface via amidation with COMU as a coupling reagent. | ||
The AFM studies prove the formation of a homogenic peptide layer without significant defects or pin-holes (Fig. S6, ESI†). The LPA layer thickness measured by ellipsometry was 9.5 Å, while after OT coupling the layer thickness increased to 18.7 Å. These experimental values are consistent with our calculation of the cross section of the molecules.
Impedimetric evaluation of Au–LPA–OT binding to Zn2+ and Cu2+
To determine the sensitivity of the Au–LPA–OT modified sensor towards Zn2+, the impedimetric response to increasing concentration of Zn2+ was measured. The normalized RCT values (calculated by dividing the measured RCT after ion titration by the RCT measured before titration) were plotted against the concentration of Zn2+ (Fig. 5 and Fig. S7, ESI†). Our results show that the Au–LPA–OT biosensor is sensitive to Zn2+ ions in sub-picomolar to sub-micromolar concentrations. The change in impedance of the Au–LPA–OT sensor in response to increasing concentrations of Cu2+ was also studied (Fig. 5 and Fig. S8, ESI†).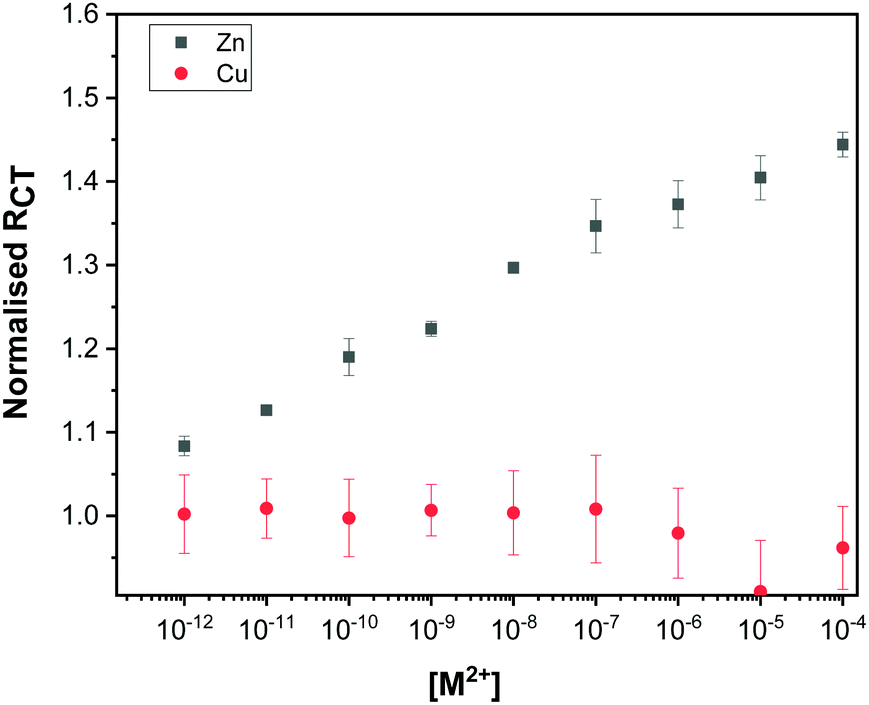 | ||
| Fig. 5 Dose response curve plotted showing the high sensitivity of the sensor towards Zn2+ (black) in comparison to Cu2+ (red). The error bars are derived from measurements of three electrodes. | ||
The differences in the EIS response of the Au–LPA–OT electrode to Zn2+ and Cu2+ show that while the sensor is sensitive toward Zn2+ it is insensitive toward Cu2+.
Selectivity studies
To further evaluate the selectivity of the sensor to Zn2+ and not to other metal ions, we screened the EIS response of the Au–LPA–OT sensor to 10 μM concentrations of various metal ions that are common in biological and environmental fluids (Fig. 6). The sensor clearly shows a highly dominant response to Zn2+ in comparison to other metal ions, since native OT binds Zn2+ in higher affinity compared to other ions.24 The screening analysis proves that we were able to maintain the sensitivity toward Zn2+ without changing the insensitivity toward other metal ions. This analysis demonstrates that we were able to produce an OT based sensor that preserves the unique binding affinity of the peptide toward Zn2+ and eliminates binding capabilities toward Cu2+. This encouraging finding makes Au–LPA–OT an attractive, sensitive and selective Zn2+ sensing platform.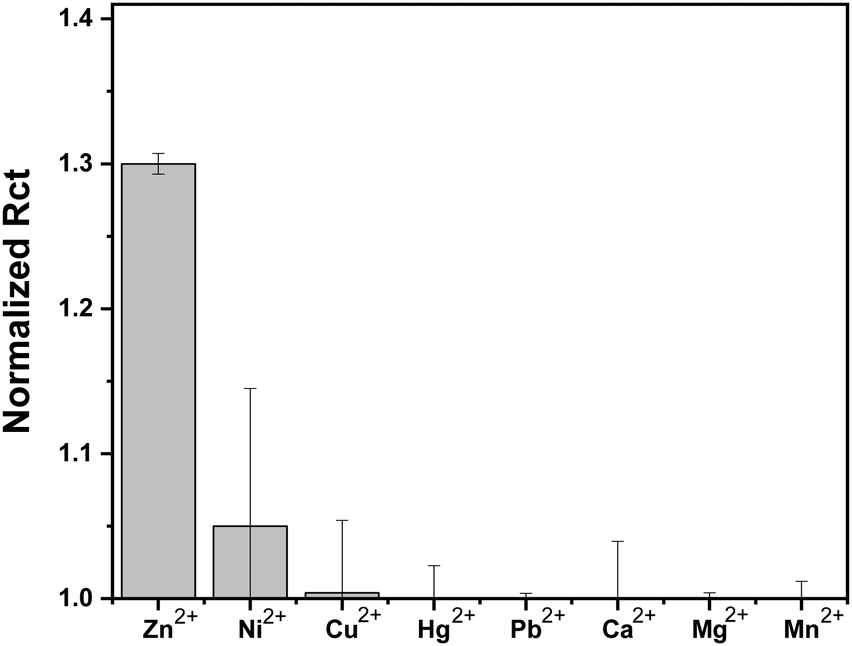 | ||
| Fig. 6 Histogram representing the selectivity of the Au–LPA–OT sensor towards Zn2+ in terms of normalized charge transfer resistance in comparison to various metal ions; [M2+] = 10−5 M, where normalized RCT refers to the ratio of RCT of the sensor exposed to M2+ concentration to RCT of the sensor in buffer. The error bars are derived from measurements of three electrodes. | ||
This work demonstrated that changing molecular features could be used to tune the selectivity of a biological probe. This allows the use of native, highly selective but also very accessible and affordable biological probes without the need to design and synthesize new artificial entities.
Conclusions
The differences between the binding mechanisms of OT to Zn2+ and Cu2+ were elucidated using DFT. We exploited the differences to prepare a selective Zn2+ sensor. Our sensor abolishes the affinity of the OT sensing layer toward Cu2+ while maintaining its Zn2+ binding properties. Modifying the amino-terminal moiety of OT proved to be essential for eliminating Cu2+ binding. We proved that the new sensor does not significantly respond to any metal ion other than Zn2+. This study provided a useful tuning strategy of ion binding by peptides in which the affinity toward Cu2+ can be eliminated by amidation of the free amine that initiates their binding cascade. We assume that such a strategy can be essential for the development of new lines of highly selective peptide-based biosensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the RECORD-IT project. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 664786. We thank Dr Yosef Gofer for XPS analysis and Hiba Elhaj and Rachel Loran for the help in characterization of the biosensor; SY is the Binjamin H. Birstein Chair in Chemistry. This work has also been partly supported by the German Research Foundation (DFG) within the Cluster of Excellence Center for Advancing Electronics Dresden. We acknowledge the Center for Information Services and High Performance Computing (ZIH) at TU Dresden for providing computational resources.Notes and references
- E. L. Que, D. W. Domaille and C. J. Chang, ChemInform, 2008, 39, 1517–1549 CrossRef.
- W. Maret, Adv. Nutr., 2013, 4, 82–91 CrossRef CAS.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- S. Hashmi and S. Al-Salam, Int. J. Clin. Exp. Pathol., 2015, 8, 8786–8796 Search PubMed.
- K. Socha, E. Karpińska, J. Kochanowicz, J. Soroczyńska, M. Jakoniuk, M. Wilkiel, Z. D. Mariak and M. H. Borawska, Nutrition, 2017, 39–40, 76–81 CrossRef CAS.
- C. F. Walker and R. E. Black, Annu. Rev. Nutr., 2004, 24, 255–275 CrossRef CAS.
- H. Yanagisawa, JMAJ, 2004, 47, 359–364 Search PubMed.
- Q. Li, X. Zhao, Q. Lv and G. Liu, Sep. Purif. Technol., 2007, 55, 76–81 CrossRef CAS.
- P. Kaur, S. Kaur, A. Mahajan and K. Singh, Inorg. Chem. Commun., 2008, 11, 626–629 CrossRef CAS.
- Q.-J. Ma, X.-B. Zhang, Y. Zhao, C.-Y. Li, Z.-X. Han, G.-L. Shen and R.-Q. Yu, Spectrochim. Acta, Part A, 2009, 71, 1683–1687 CrossRef.
- M. Abbasi, Z. Ibupoto, M. Hussain, Y. Khan, A. Khan, O. Nur and M. Willander, Sensors, 2012, 12, 15424–15437 CrossRef CAS.
- E. Mervinetsky, I. Alshanski, Y. Hamo, L. M. L. M. Sandonas, A. Dianat, J. Buchwald, R. Gutierrez, G. Cuniberti, M. Hurevich and S. Yitzchaik, Sci. Rep., 2017, 7, 9498 CrossRef.
- W. Yang, D. Jaramillo, J. J. Gooding, D. B. Hibbert, R. Zhang, G. D. Willett and K. J. Fisher, Chem. Commun., 2001, 1982–1983 RSC.
- E. Mervinetsky, I. Alshanski, S. Lenfant, D. Guerin, L. M. Sandonas, A. Dianat, R. Gutierrez, M. Hurevich, S. Yitzchaik and D. Vuillaume, J. Phys. Chem. C, 2019, 123, 9600–9608 CrossRef CAS.
- W. Yang, E. Chow, G. D. Willett, D. B. Hibbert and J. J. Gooding, Analyst, 2003, 128, 712–718 RSC.
- A. Kotynia, Z. Czyznikowska, M. Cebrat, Ł. Jaremko, O. Gładysz, M. Jaremko, A. Marciniak and J. Brasuń, Inorg. Chim. Acta, 2013, 396, 40–48 CrossRef CAS.
- H. Sigel and B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- D. Liu, A. B. Seuthe, O. T. Ehrler, X. Zhang, T. Wyttenbach, J. F. Hsu and M. T. Bowers, J. Am. Chem. Soc., 2005, 127, 2024–2025 CrossRef CAS.
- W. Bal, M. Dyba and H. Kozłowski, Acta Biochim. Pol., 1997, 44, 467–476 CAS.
- D. Peter, K. Varnagy, I. Sovago, S. Istvan, D. Sanna and G. Micera, J. Inorg. Biochem., 1995, 60, 69–78 CrossRef.
- W. Bal, H. Kozlowski, B. Lammek, K. Rolka and L. D. Pettit, J. Inorg. Biochem., 1992, 45, 193–202 CrossRef CAS.
- F. J. Blount, H. C. Freeman, V. R. Holland and W. H. G. Milburn, J. Biol. Chem., 1970, 245, 5177–5185 Search PubMed.
- M. J. Bakermans-Kranenburg and M. H. Van IJzendoorn, Transl. Psychiatry, 2013, 3, 1–14 Search PubMed.
- M. T. Bowers, D. Liu and T. Wyttenbach, J. Am. Chem. Soc., 2008, 130, 1–19 CrossRef.
- L. Joly, R. Antoine, F. Albrieux, R. Ballivian, M. Broyer, F. Chirot, J. Lemoine, P. Dugourd, C. Greco, R. Mitrić and V. Bonačić-Koutecký, J. Phys. Chem. B, 2009, 113, 11293–11300 CrossRef CAS.
- L. Joly, R. Antoine, A. R. Allouche, M. Broyer, J. Lemoine and P. Dugourd, J. Phys. Chem. A, 2009, 113, 6607–6611 CrossRef CAS PubMed.
- H. Wei, X. Luo, Y. Wu, Y. Yao, Z. Guo and L. Zhu, J. Chem. Soc., Dalton Trans., 2000, 4196–4200 RSC.
- E. Mervinetsky, I. Alshanski, J. Buchwald, A. Dianat, I. Lončarić, P. Lazić, Ž. Crljen, R. Gutierrez, G. Cuniberti, M. Hurevich and S. Yitzchaik, Langmuir, 2019, 35(34), 11114–11122 CrossRef CAS.
- S. Yitzchaik, R. Gutierrez, G. Cuniberti and R. Yerushalmi, Langmuir, 2018, 34, 14103–14123 CrossRef CAS PubMed.
- K. K. Tadi, I. Alshanski, E. Mervinetsky, G. Marx, P. Petrou, K. M. Dimitrios, C. Gilon, M. Hurevich and S. Yitzchaik, ACS Omega, 2017, 2, 8770–8778 CrossRef CAS.
- K. K. Tadi, I. Alshanski and M. Hurevich, Surfaces, 2018, 1, 90–95 CrossRef.
- I. Alshanski, J. Blaszkiewicz, E. Mervinetsky, J. Rademann, S. Yitzchaik and M. Hurevich, Chem. – Eur. J., 2019, 25, 12083–12090 CrossRef CAS.
- S. D. Keighley, P. Li, P. Estrela and P. Migliorato, Biosens. Bioelectron., 2008, 23, 1291–1297 CrossRef CAS.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS.
- S. D. Techane, L. J. Gamble and D. G. Castner, J. Phys. Chem. C, 2011, 115, 9432–9441 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS, AFM and EIS. See DOI: 10.1039/c9tb01932d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |