
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of a supramolecular hydrogel made of phospholipids and oleic acid with a high water content†
Kirian
Talló
 *a,
Manel
Bosch
bc,
Ramon
Pons
a,
Mercedes
Cocera
d and
Olga
López
a
*a,
Manel
Bosch
bc,
Ramon
Pons
a,
Mercedes
Cocera
d and
Olga
López
a
aDepartment of Surfactants and Nanobiotechnology, Institute of Advanced Chemistry of Catalonia (IQAC-CSIC), C/Jordi Girona 18-26, 08034, Barcelona, Spain. E-mail: kirian.tallo@cid.csic.es
bAdvanced Optical Microscopy Unit, Scientific and Technological Centers of the Universitat de Barcelona (CCiTUB), Barcelona, 08028, Spain
cDepartment of Biochemistry and Molecular Biomedicine, Faculty of Biology, Universitat de Barcelona, 08028, Spain
dBicosome S.L., C/Jordi Girona 18-26, 08034, Barcelona, Spain
First published on 18th November 2019
Abstract
A hydrogel formed with phospholipids and fatty acids would be of great interest in the medical field due to the biological relevance that these molecules have in living organisms. However, the tendency of phospholipid mixtures to form vesicular or micellar aggregates at high water content hinders the formation of this type of hydrogel. In this study, a highly hydrated hydrogel (95% water) was formed with hydrogenated phosphatidylcholine and oleic acid. The preparation method involved a freeze-heating cycle of the aqueous lipid mixture, favouring the supramolecular aggregation of these molecules into a microscopic spongy morphology. Confocal fluorescence imaging showed that the microstructure of the hydrogel is made from the aggregation of giant multilamellar vesicles (5–20 μm diameter) while transmission electron microscopy revealed the existence of nanosized unilamellar vesicles (150 nm diameter) coexisting with lipid lamellae. Despite this type of aggregation, X-ray scattering experiments performed on the hydrogel show almost no correlation between lipid membranes. In terms of rheological properties, the material shows a prevalent elastic behaviour and low structural strength, a consequence of non-covalent interactions. With such properties and composition, this structured but easily deformable material might become a useful tool for biomedical applications.
Introduction
Hydrogels can be defined as materials with a high content of water, formed by a three-dimensional network of molecules assembled across an aqueous phase. This type of material acts as a biocompatible alternative to organogels which are composed of an organic solvent instead of water (e.g. lecithin in hydrocarbon solvents).1During the last few decades, polymeric materials have been extensively used to form hydrogels with good mechanical properties.2,3 The covalent crosslinking of monomers grants mechanical strength to the structure of these materials and makes them suitable for devices that require high rigidity and durability.4
Nowadays, other molecules such as saccharides, nucleobases or peptides have been receiving considerable attention because of their ability to form hydrogels that mimic biological systems.5–8 These compounds assemble in water by either physical or chemical interactions to form gels with potential applications in drug delivery, tissue engineering or cell culturing.9–12
Common alternatives to these systems, especially for drug delivery applications, are the ones formed by phospholipids. These molecules can aggregate in water by hydrophobic forces and electrostatic attractions giving rise to structures of different morphologies and phase behaviors.13 It is well known that phospholipids are the basic components of biological membranes and such systems have an excellent biocompatibility.14 However, hydrogels formed by lipids are rarely found in the literature since these molecules are more likely to form vesicular or micellar aggregates at high water content.15 Gelation under these diluted conditions has been achieved with the addition of other molecules such as surfactants and PEGylated lipids or fatty alcohols in high amounts.16,17 These lipid gels present the well-known benefits of liposome preparations, such as the ability to incorporate hydrophobic actives into their lipid membranes, while at the same time they exhibit structural consistency and the high-water environment of a soft supramolecular gel. Nonetheless, it would be of much interest to form a lipid hydrogel free of components such as surfactants, emulsifiers or conjugated polymers by using instead lipids of high biological relevance like fatty acids and phospholipids. A gel system formed exclusively by lipids with high water content would solve formulation challenges and avoid biocompatibility problems.
In a recent work we studied a vesicular system composed of hydrogenated soy phosphatidylcholine (HSPC) and oleic acid (OA) at different pH and molar ratio values. This system exhibited the usual liquid behavior of lipid vesicles in aqueous dispersion.18 In the present work, using similar composition but by means of a different protocol of preparation we report a novel hydrogel system composed only of lipids. This method promotes the supramolecular aggregation of the lipid mixture into a hydrogel system without needing further gelling agents. In this work, several characterization techniques have been used to describe the material microstructure and its rheological behavior.
Experimental
Materials
Hydrogenated soy phosphatidylcholine (HSPC; Phospholipon® 90H) was purchased from Lipoid GmbH (Ludwigshafen, Germany). This product is a mixture of phospholipids that has 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) (ca. 85%) and 1-palmitoyl-2-stearoyl-3-phosphocholine (PSPC) (ca. 15%). 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine Rhodamine B sulfonyl) (ammonium salt) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Oleic acid ≥99% (GC), sodium hydroxide and chloroform were purchased from Merck (Darmstadt, Germany).Preparation of the lipid hydrogel
First, an aqueous dispersion of lipids (0.5% OA and 4.5% HSPC w/w) was formed following a variation of the thin film hydration method as reported previously.18 Roughly, a chloroform solution of HSPC and OA was evaporated to form a thin lipid film that was hydrated with the appropriate volume of deionized water to have a final lipid concentration of 5% w/w. The pH was adjusted to 6.5 with a NaOH solution using an Orion Star A211 Benchtop pH Meter and a ROSS Ultra Semi-Micro pH electrode (both from Thermo Scientific, NA, USA).To form the hydrogel, the lipid dispersion (5% lipids and 95% water w/w) was first stored at −20 °C in a closed vial for one hour to completely freeze the sample. Afterwards, the system was heated up to 70 °C during 10 minutes until the whole dispersion became clear. The system was left cooling at room temperature and stored overnight in the fridge (5 °C). Gelation and absence of water separation was confirmed by turning the vial upside down.
Confocal fluorescence microscopy
To visualize the system in the micrometer range, confocal microscopy was used. The hydrogel was formed following the procedure described above but by adding a lipid fluorescent probe (DPPE-Rhodamine B) into the HSPC/OA chloroform mixture. The final concentration of the lipid probe was 0.02% w/v to ensure enough fluorescent intensity for visualization without compromising the hydrogel composition.Fluorescence images of the systems were acquired using a Zeiss LSM 880 confocal microscope. A 561 nm DPSS laser was used to excite Rhodamine B. The samples were observed using a 10× 0.4 NA dry objective and 25× 0.75 NA and 63× 1.4 NA glycerol immersion objectives. The images were deconvolved using Huygens Essential 3.7.1 (SVI) to compensate for the blur introduced by the spread of light that occurs in optical microscopes. Then, the images were further processed using ImageJ 1.52d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 to compensate for the loss of fluorescence intensity when imaging deep within the samples. With that aim, the brightness and contrast were automatically modified individually in each image of the stack using the B&C tool. Orthogonal views were then generated by interpolation with 1 pixel spacing. Finally, 3D volume rendering of the image stacks was performed using Imaris 7.2.3 (Oxford Instruments).
19 to compensate for the loss of fluorescence intensity when imaging deep within the samples. With that aim, the brightness and contrast were automatically modified individually in each image of the stack using the B&C tool. Orthogonal views were then generated by interpolation with 1 pixel spacing. Finally, 3D volume rendering of the image stacks was performed using Imaris 7.2.3 (Oxford Instruments).
Cryogenic transmission electron microscopy (cryo-TEM)
The samples were imaged following a freeze fracture direct image (FFDI) procedure.20 A hydrogel aliquot of 3 μL was placed onto a glow-discharged Lacey Carbon 300 mesh copper grid (Ted Pella, USA) and was sandwiched between two copper platelets. The specimens were vitrified using a Vitrobot Mark III (FEI Company, Eindhoven, the Netherlands) with its chamber at 25 °C and 100% humidity. Sandwiches were plunge frozen in liquefied ethane and opened under liquid nitrogen. The specimen grids were mounted on a cryotransfer system (Gatan, Pleasanton, USA) and the images were obtained using a microscope Tecnai F20 EM (FEI Company, Eindhoven, the Netherlands). The microscope was operated at 200 kV, under low-dose conditions, and the images were recorded with a 4096 × 4096 pixel CCD Eagle camera (FEI, Eindhoven, the Netherlands). The images presented in this work correspond to a representative fraction of the micrographs saved during the microscopy sessions. The diameter of the vesicles was approximated by examining a total of 6 images from different regions of the sample where multiple vesicles could be seen.X-ray scattering: small (SAXS) and wide angle (WAXS)
X-ray scattering experiments were performed with a SAXS/WAXS S3 MICRO equipped with a linear detector PSD 50M (Hecus GmbH, Austria). The temperature was set to 25 °C and a lipid dispersion was introduced into a glass capillary of 2 mm where gelation took place. A scattering signal was then achieved with an incident X-ray beam from a GENIX-Fox 3D source which produced a Cu Kα line (1.542 Å) directed to the sample and focused on the position sensitive detector (PSD). This setup produces spectra mainly smeared by the detector width. This smear is numerically handled in the model fitting procedure.The intensity of the scattered signal has been plotted as a function of the scattering vector modulus q = (4π/λ)sinθ/2, where θ is the scattering angle and λ the wavelength of the X-ray beam. Experimental SAXS data was analyzed by means of a Gaussian model fitting procedure and a least square Levenberg–Marquardt scheme as previously reported.21,22 For the evaluation of the area per molecule, it was considered that the bilayer was only made of DSPC, which is the main component of the lipid mixture.
Rheological measurements
Oscillatory rheology tests of the hydrogels were performed with an AR-G2 controlled stress rheometer (TA Instruments), equipped with a Peltier temperature control system. A cone-plate geometry of 40 mm diameter, a cone angle of 4° and a gap of 105 μm were used. Measurements were carried out at 25 °C and sample drying was prevented by using a solvent trap. Oscillation amplitude measurements were used to determine the linear viscoelastic region (LVR) of the hydrogel. Then, frequency measurements were performed within the LVR to ensure that the material response in terms of storage (G′) and loss modulus (G′′) was independent of the strain magnitude. Data was analyzed using TRIOS software (TA Instruments, USA).Results and discussion
Insights into the gelation process and relevant parameters
Although the mechanism of gelation is yet to be fully understood, this section aims to provide some information on which parameters are important for the gelation process.As described in the Experimental section, all the results provided in this work are based on a hydrogel composed of HSPC and OA. Nonetheless, we have seen that this type of hydrogel can also be formed with pure phospholipids such as disteroylphosphatidylcholine (DSPC) and other fatty acids like lignoceric acid (data not shown). This means that the formation of the hydrogel does not rely on the specific composition of the Lipoid HSPC extract or the OA but instead is related to the chemical nature of these types of molecules.
One of the most important parameters in terms of achieving gelation and stability is the pH of the lipid dispersion. Even though the pH used in all the experiments was set at 6.5 to have coherence between the results, it is possible to obtain a lipid hydrogel with the pH between 5.5 and 7.5 approximately. Increasing the pH above such a range brings the system into a fluid dispersion due to the high repulsion between the structures caused by a major concentration of oleate ions. On the other hand, lowering the pH below that range promotes aggregation and eventual precipitation of the lipid phase caused by the absence of charge (Fig. S1, ESI†). This phenomenon can also be appreciated when this repulsion between particles is screened by the addition of a salt in the system (e.g. NaCl 1 mg mL−1). Such findings support the idea that the charge of the lipids governs the stability of the hydrogel.
In terms of procedure, we have verified that it is necessary to freeze the sample at −20 °C (or below) in order to end up forming a hydrogel. As reported by other authors, freezing lipid-water dispersion without a cryoprotectant generally leads to membrane deformation caused by dehydration and osmotic stress and eventual rupture of vesicular structures.23,24 We hypothesize that such phenomena might be responsible for the final organization of the lipid structures into a hydrogel. Nonetheless, the mechanics involved during the freezing of lipid solutions are quite complex and vary depending on the composition of the system. In addition, to have a completely firm gel, heating of the sample at 70 °C is necessary to overcome the main phase transition temperature (Tm) of the lipids and restructure the bilayers, completing the gelation process.
Confocal microscopy
Microscopic organization of the hydrogel matrix across the aqueous phase was evaluated with confocal fluorescence microscopy. The signal arising from the lipid phase, marked with DPPE-Rhodamine B, appears in a grey scale while water remains black.The images acquired at low magnification (10×) in Fig. 1 illustrate the microscopic distribution of lipids across water. Orthogonal views in combination with the main image show that the lipid phase expands across all the dimensions in a continuous fashion. This lipid arrangement with differentiated aqueous regions (black areas) gives the hydrogel a 3D sponge-like morphology.
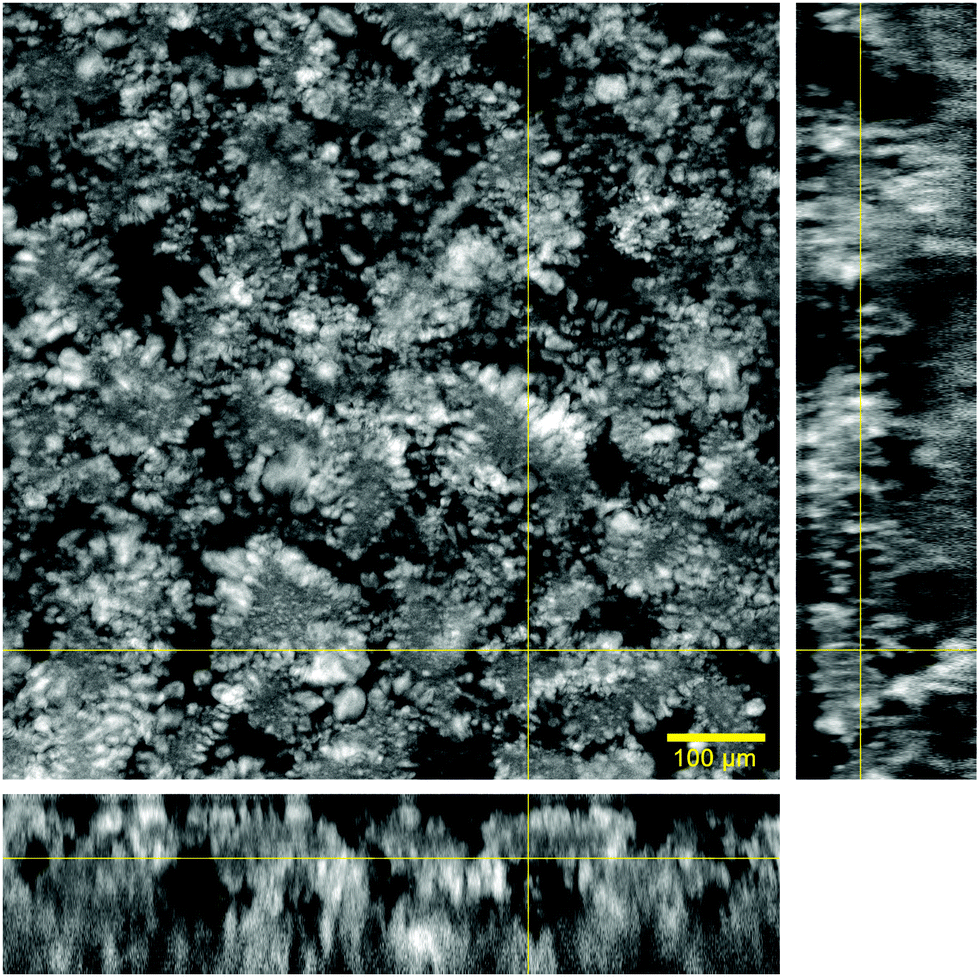 | ||
| Fig. 1 Confocal microscopy image of the hydrogel taken with a 10× objective. This figure comprises an xy slice from a confocal stack of images and the orthogonal projections xz (bottom) and yz (right) obtained at the yellow lines in the xy slice. | ||
Fig. 2 shows in more detail the lipid structures forming the hydrogel matrix and their aqueous surrounding areas. The images in Fig. 2A were acquired with a 25× objective and illustrate several vesicles ranging from 5 to 20 μm in diameter that appear within the aqueous gaps existing between lipid areas. Some of these particles seem to be relatively isolated (white arrows) while there are others that are clustered together and directly attached to the continuous lipid structure (yellow arrows). With further reduction of the background noise and a higher magnification (63×), Fig. 2B illustrates the lamellarity of the system. Giant multilamellar vesicles from 5 to 20 μm in diameter can be clearly identified in several regions of the image (white arrows). The continuous lipid phase that forms the microscopic matrix of the hydrogel seems to be formed by the closely packed structures of multilamellar nature and different morphologies (yellow arrows). The volume rendering and stack animation from these images (Video S1, ESI†) show how lipid structures are widely distributed in the sample.
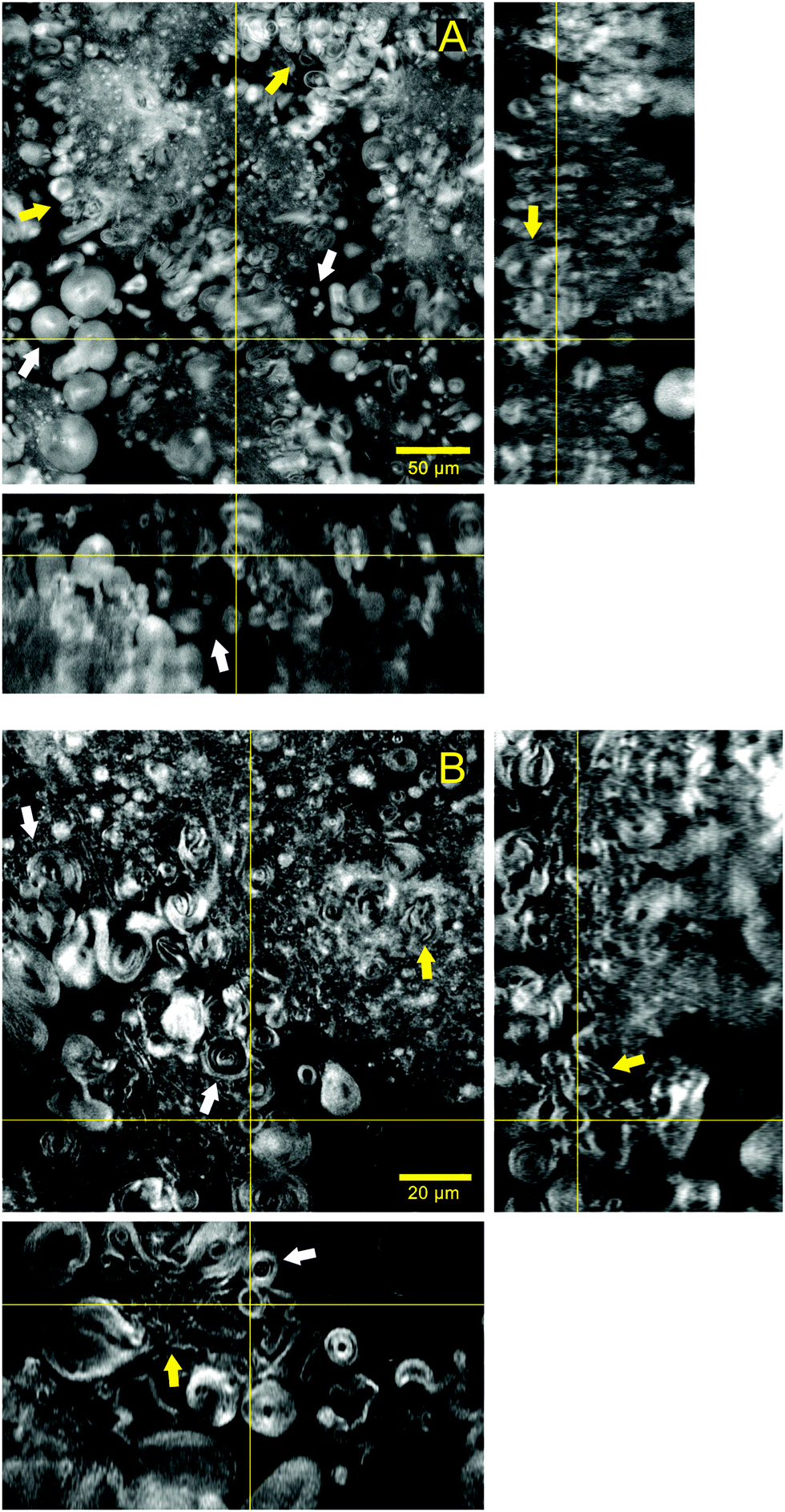 | ||
| Fig. 2 Confocal microscopy images showing in more detail the structures forming the hydrogel lipid matrix. Each panel comprises an xy slice from a confocal stack of images and the orthogonal projections xz (bottom) and yz (right) obtained at the yellow lines in the xy slice. (A) and (B) were taken with 25× and 63× glycerol immersion objectives, respectively. | ||
Increasing the resolution of the images, the regions where lipid structures are clustered together can be better visualized (Video S2, ESI†). This stack animation shows closely how multilamellar vesicles that are merged with lipid lamellae run across the sample volume. Such arrangement allows for a great percentage of water accommodated between lamellae and inside the vesicles.
Comparing this hydrogel with the previously reported ones where surfactants and PEGylated lipids were used,16,17 it is clear how our system not only differs in composition but also in microscopic organization. The reported gels were described as separated bilayered sheets or curved lamellae, and this time the gelation of a lipid hydrogel has been achieved throughout the aggregation of giant multilamellar vesicles co-existing with lipid lamellae. In this regard, such aggregation resembles the one present in colloidal gels where interparticle attraction leads to a mesoscopic three-dimensional network.25
Cryo-TEM
Electron microscopy was used to study the aggregation of the system at the nanoscale. When it comes to cryo-TEM, the high viscosity of hydrogels is generally a problem during the preparation of the specimens for this technique. Without adequate conditions, the vitrified sections after the blotting procedure become too thick or too concentrated to be imaged. For this reason, the FFDI technique was used to obtain thinner sections of the sample into the carbon grids.20 The resulting images from this method have a different appearance than those obtained using a blotting–plunge procedure in a way that the fracture produced across the ice and along lipid lamellae tends to highlight these layered structures.Assuming an even fracture of the ice, Fig. 3 illustrates two different sections of the sample that present darker or brighter areas according to the amount of lipid material present. In this regard, the center of Fig. 3A shows mostly vitrified water (asterisk) while the outer parts of the picture correspond to lipid structures probably stacked in a lamellar arrangement. The dark lines appearing in the left corner of the picture (black arrow) indicate multilamellar sheets oriented perpendicular to the focal plane. The presence of other phases like inverted hexagonal or cubic was dismissed since their distinctive cryo-TEM patterns could not be identified.26,27
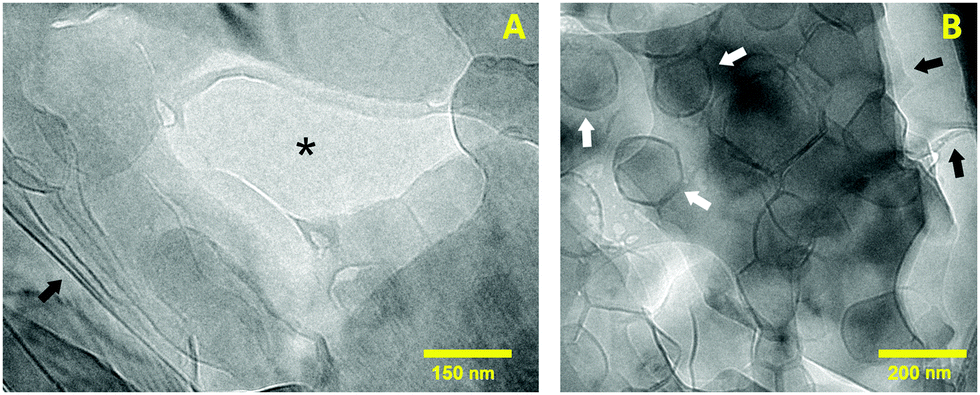 | ||
| Fig. 3 Cryo-TEM micrographs showing the different structures present in the hydrogel at the nanoscale. (A) Highlights several lipid sheets and (B) displays a population of nanosized vesicles. | ||
In Fig. 3B the lamellar organization can be also noticed due to the fracture made across lipid bilayers during the FFDI protocol (black arrows). As stated before, the darker areas of the image are a consequence of a greater amount of lipid sheets overlapped. In addition, this image shows several rounded structures corresponding to nanosized unilamellar vesicles with an average diameter of 150 nm (white arrows). From cryoTEM examination of different specimens of the same sample, it was seen that the particle size ranges from 100 to 250 nm. The irregular shape of these vesicles is caused by the preparation of the samples for CryoTEM at 25 °C. At this temperature the system is clearly below the main phase transition temperature of a mixture of HSPC and OA (Tm ∼ 53 °C).18 As reported by other authors, liposomes below their Tm are generally aggregated in a rigid lamellar gel phase (Lβ) that results in an irregular curvature of the lipid membranes.28–30 In order to form a closed vesicle, the rigidity of the membranes is compensated by the appearance of defects that form this characteristic polyhedral morphology.31
Overall, cryo-TEM shows that the nanostructure of the sample is formed by lipid bilayered sheets and vesicles. This arrangement agrees and complements well with the results from confocal microscopy where multilamellar structures of different shapes constitute the microstructure of the hydrogel. Thus, it is likely that the regions that could not be resolved by confocal microscopy are made of smaller vesicles and lamellar assemblies.
X-ray scattering: SAXS and WAXS
X-ray scattering experiments were used to obtain information related to the organization and phase behavior of lipid aggregates forming the hydrogel matrix.32 The SAXS plot in Fig. 4A (black dots) shows a main band centered at 0.085 Å−1 and two additional lobes at 0.24 and 0.38 Å−1. The shape of this experimental profile is characteristic of a scattering form factor associated with uncorrelated bilayers. The fitting function that best describes this experimental profile (reduced χ2 = 1.70) corresponds to a bilayered system with only a small contribution of correlated lamellae that is seen as a small bump at lower angles (q = 0.022 Å−1i.e. d = 53 nm). The fitting of exclusively uncorrelated bilayers resulted only in slightly worse fit (χ2 = 1.78) with similar parameters for the bilayer structure. Thus, although multilamellar structures were clearly seen by confocal microscopy, the great separation between bilayers does not present a clear correlation peak.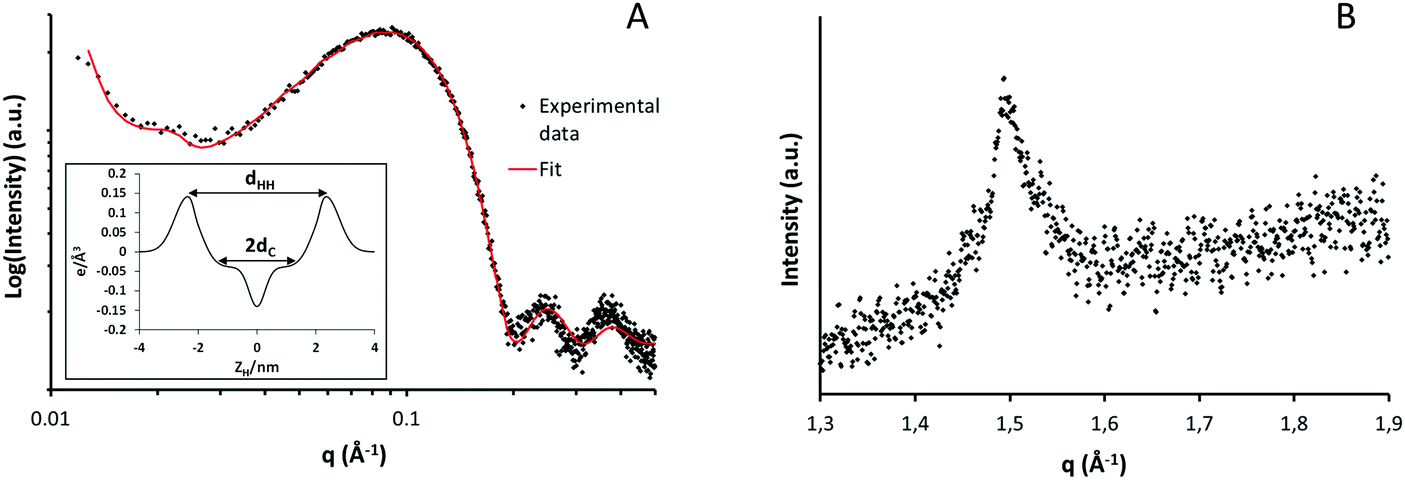 | ||
| Fig. 4 Small (A) and wide (B) angle X-ray scattering intensity profiles of the hydrogel sample at room temperature. | ||
From the least-square fitting of the form factor, a symmetric electron density profile of the lipid bilayer was deduced (inset Fig. 4A). The most relevant parameters obtained from the fitting procedures and the corresponding electronic distributions are shown in Table 1. In X-ray experiments, the best resolved bilayer thickness corresponds to the phosphate headgroup–headgroup separation, dHH.33 In the present case, this value corresponds to 47 Å which falls within the range of the previously reported results for DSPC membrane, which is the main component of the lipid mixture used.34
| Parameters | HSPC/OA hydrogel |
|---|---|
| d HH | 47 Å |
| d C | 14.3 Å |
| σ H | 4.5 Å |
| A | 51 Å2 |
| χ 2 | 1.70 |
Lateral chain packing of the lipid molecules was evaluated using the high angle reflections (WAXS) displayed in Fig. 4B. This plot shows only one band centered at q = 1.49 Å−1 equivalent to a separation of 4.2 Å between hydrocarbon chains. The reflections around 4.1–4.2 Å indicate the presence of a lamellar gel phase organized in a two-dimensional hexagonal lattice.35 It is worth noticing that such crystalline packing is only present below the Tm of the system and disappears above this temperature when the liquid phase is formed.36
Rheological measurements
Most of the time, the mechanical properties of soft materials are directly related to their microscopic structure and molecular organization. Appropriate characterization of the macroscopic behavior of our sample by means of rheological techniques entails a better understanding of the interactions that take place within the hydrogel structure. Considering that most of the gels are viscoelastic materials, the rheological behavior of our sample was evaluated by means of oscillatory measurements.37The plot from the amplitude sweep test in Fig. 5A shows the corresponding variation of elastic modulus (G′) and viscous modulus (G′′) with increasing oscillation strain. As seen from this picture, both dynamic parameters G′ and G′′ are independent of the oscillation amplitude between strains from 0.01 to 0.5%, meaning that the linear viscoelastic region of the sample is present within these values.37,38 Above this region, after enough stress or strain is applied, viscoelastic materials start to flow due to the breakdown of the microstructure. The parameters that define these critical values, known as yield stress and yield strain, give an indication of the structural strength and extent of deformation of the material. These parameters have been estimated from the plot of elastic stress (the product of strain and G′) versus strain as shown in the inset of Fig. 5A.39 The peak value of elastic stress corresponds to a yield stress of 4.8 Pa while the corresponding yield strain value remains around 1.25%. These yield values appear to be considerably low and indicate that our sample presents low structural strength, a characteristic of the weak nature of non-covalent interactions from supramolecular gels.
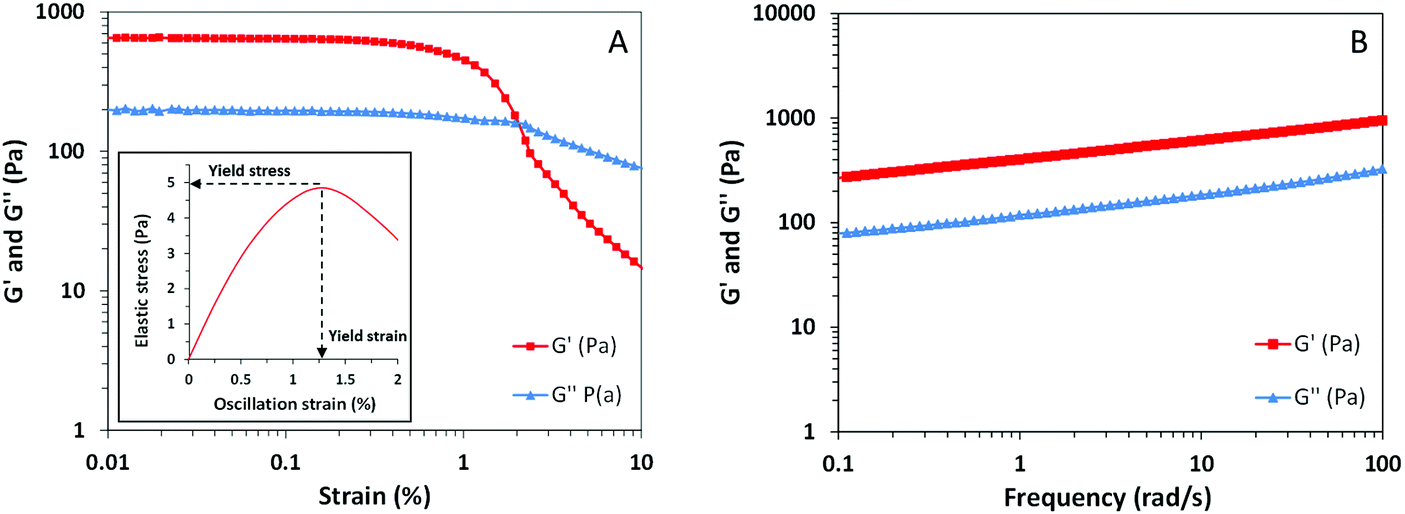 | ||
| Fig. 5 Oscillatory measurements of the HSPC/OA hydrogel sample. Storage modulus and loss modulus are shown as a function of shear stress (A) and frequency (B). Yield values were determined from the plot of elastic stress versus strain (inset in A). | ||
The frequency assay plotted in Fig. 5B illustrates the response of G′ and G′′ of the sample due to the deformations taking place at different timescales. It is known that viscoelastic materials may behave differently depending on whether the stress is applied faster or slower.38 In our results, the values of G′ range from 300 to 1100 Pa and are an indication of the elastic component of the sample. The values from the viscous component (G′′) are lower over the whole frequency range and only range from 80 to 300 Pa. Since the elastic component, related to the ability of the system to store elastic energy, is higher than the viscous one without any crossover point, the material can be considered a structured gel.40 This prevalent elastic behavior appears to be a result of the sponge-like organization of the lamellar sheets and vesicles achieved by means of the gelation protocol. However, the relatively low values of yield stress and G′ indicate that the hydrogel can be easily deformed, presenting low structural strength. Those are the expected properties of a supramolecular hydrogel where the interactions responsible for the assembly of the structure are noncovalent, such as electrostatic forces.11,41 In this regard, a low amount of stress applied to the system is enough to overcome the supramolecular interactions inducing the flow of the material.
Conclusions
In this work, a novel type of hydrogel made with phospholipids and fatty acids has been presented. The method and parameters of preparation used have allowed the gelation of a lipid system with 95% water. Materials with similar rheological behavior and composition were only possible by means of higher lipid concentrations or by adding other molecules like surfactants, emulsifiers or gelling agents.Several techniques used in this work allowed complete characterization of the hydrogel structure at the mesoscale and a description of its rheological behavior. The non-covalent interactions between lipid sheets and vesicles lead to a spongy lipid matrix that endows the material with solid-like behavior. By using phospholipids and fatty acids, molecules of high biological relevance, we believe that such a family of hydrogels will be of great interest in biomedical related fields. The soft nature and low structural strength of the hydrogel matrix makes this material a potential candidate for topical and ocular applications where either lipophilic or hydrophilic drugs could be incorporated into the material.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Jaume Caelles from the SAXS-WAXS service at IQAC for X-ray measurements and Lidia Delgado from CCITUB for cryo-TEM sample preparation. Rheological assays were performed by Susana Vílchez at the Nanostructured Liquid Characterization Unit of CSIC and CIBER-BBN. The authors are also grateful to Lipoid GmbH for kindly providing Phospholipon® 90H and to MINECO for funding this work with the RTC-2016-4957-1 project.Notes and references
- R. Scartazzini and P. L. Luisi, J. Phys. Chem., 1988, 92, 829–833 CrossRef CAS.
- K. Pal, A. K. Banthia and D. K. Majumdar, Des. Monomers Polym., 2009, 12, 197–220 CrossRef CAS.
- B. Ratner and A. Hoffman, Hydrogels Med. Relat. Appl. ACS Symp. Ser., 1976, 31, 1–36 CAS.
- H. J. Kong, E. Wong and D. J. Mooney, Macromolecules, 2003, 36, 4582–4588 CrossRef CAS.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024–2027 RSC.
- N. P. Murphy and K. J. Lampe, J. Mater. Chem. B, 2015, 3, 7867–7880 RSC.
- G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 RSC.
- A. Hong, M. I. Aguilar, M. P. Del Borgo, C. G. Sobey, B. R. S. Broughton and J. S. Forsythe, J. Mater. Chem. B, 2019, 7, 3927–3943 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS.
- X. Li, W. Xue, Y. Liu, D. Fan, C. Zhu and X. Ma, J. Mater. Chem. B, 2015, 3, 4742–4755 RSC.
- P. R. Cullis, M. J. Hope and C. P. S. Tilcock, Chem. Phys. Lipids, 1986, 40, 127–144 CrossRef CAS.
- J. Li, X. Wang, T. Zhang, C. Wang, Z. Huang, X. Luo and Y. Deng, Asian J. Pharm. Sci., 2015, 10, 81–98 CrossRef.
- J. M. Seddon and R. H. Templer, in Structure and Dynamics of Membranes, ed. R. Lipowsky and E. Sackmann, Elsevier B. V., Amsterdam, 1st edn, 1995, vol. 1, pp. 97–160 Search PubMed.
- H. E. Warriner, S. H. J. Idziak, N. L. Slack, P. Davidson and C. R. Safinya, Science, 1996, 271, 969–973 CrossRef CAS PubMed.
- Y. Nakagawa, H. Nakazawa and S. Kato, J. Colloid Interface Sci., 2012, 376, 146–151 CrossRef CAS.
- K. Talló, V. Moner, M. De Cabo, M. Cócera and O. López, Chem. Phys. Lipids, 2018, 213, 96–101 CrossRef.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676 CrossRef CAS.
- L. Belkoura, C. Stubenrauch and R. Strey, Langmuir, 2004, 20, 4391–4399 CrossRef CAS.
- I. Castangia, M. L. Manca, C. Caddeo, A. Maxia, S. Murgia, R. Pons, D. Demurtas, D. Pando, D. Falconieri, J. E. Peris, A. M. Fadda and M. Manconi, Colloids Surf., B, 2015, 132, 185–193 CrossRef CAS.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- J. Wolfe and G. Bryant, Cryobiology, 1999, 39, 103–129 CrossRef CAS.
- T. Kaasgaard, O. G. Mouritsen and K. Jørgensen, Biochim. Biophys. Acta, Biomembr., 2003, 1615, 77–83 CrossRef CAS.
- P. J. Lu, E. Zaccarelli, F. Ciulla, A. B. Schofield, F. Sciortino and D. A. Weitz, Nature, 2008, 453, 499 CrossRef CAS.
- D. P. Siegel, W. J. Green and Y. Talmon, Biophys. J., 1994, 66, 402–414 CrossRef CAS.
- J. Barauskas, A. Misiunas, T. Gunnarsson, F. Tiberg and M. Johnsson, Langmuir, 2006, 22, 6328–6334 CrossRef CAS.
- D. D. Lasic, Liposomes: From Physics to Applications, Elsevier, Amsterdam, 1993 Search PubMed.
- M. Andersson, L. Hammarstroem and K. Edwards, J. Phys. Chem., 1995, 99, 14531–14538 CrossRef CAS.
- M. Johnsson and K. Edwards, Biophys. J., 2003, 85, 3839–3847 CrossRef CAS.
- M. J. Bowick and R. Sknepnek, Soft Matter, 2013, 9, 8088–8095 RSC.
- A. I. I. Tyler, R. Law and J. M. Seddon, in Methods in Membrane Lipids, ed. D. Owen, Humana Press, New York, 2nd edn, 2015, pp. 199–225 Search PubMed.
- N. Kučerka, M. P. Nieh and J. Katsaras, in Advances in Planar Lipid Bilayers and Liposomes, ed. A. Iglic, Elsevier B. V., Amsterdam, 1st edn, 2010, vol. 12, pp. 201–235 Search PubMed.
- D. Marsh, in Handbook of Lipid Bilayers, ed. L. Han, Taylor & Francis Group, Boca Raton, 2nd edn, 2013, pp. 373–437 Search PubMed.
- D. M. Small, J. Lipid Res., 1984, 25, 1490–1500 CAS.
- S. Y. Hou, S. J. Rehfeld and W. Z. Plachy, in Advances in Lipid Research, ed. P. Elias, Elsevier B. V., Amsterdam, 1st edn, 1991, vol. 24, pp. 141–171 Search PubMed.
- H. M. Wyss, in Fluids, Colloids and Soft Materials: An Introduction to Soft Matter Physics, ed. A. Fernandez and A. M. Puertas, John Wiley & Sons, Hoboken, 1st edn, 2016, pp. 149–163 Search PubMed.
- D. T. N. Chen, Q. Wen, P. A. Janmey, J. C. Crocker and A. G. Yodh, Annu. Rev. Condens. Matter Phys., 2010, 1, 301–322 CrossRef CAS.
- H. J. Walls, S. B. Caines, A. M. Sanchez and S. A. Khan, J. Rheol., 2003, 47, 847–868 CrossRef CAS.
- A. Y. Malkin and A. I. Isayev, in Rheology: Concepts, Methods, and Applications, ChemTec Publishing, Toronto, 2nd edn, 2012, pp. 43–126 Search PubMed.
- A. Dawn and H. Kumari, Chem. – Eur. J., 2018, 24, 762–776 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and captions for Videos S1 and S2. Videos S1 and S2 showing volume rendering and stack animations from confocal microscopy images. See DOI: 10.1039/c9tb01599j |
| This journal is © The Royal Society of Chemistry 2020 |