
Enhanced bactericidal activity of brucite through partial copper substitution†
Batiste
Clavier
a,
Téo
Baptiste
a,
Florian
Massuyeau
b,
Alain
Jouanneaux
a,
Amandine
Guiet
a,
Fabien
Boucher
c,
Vincent
Fernandez
b,
Christine
Roques
d and
Gwenaël
Corbel
 *a
*a
aInstitut des Molécules et Matériaux du Mans (IMMM), UMR-6283 CNRS, Le Mans Université, Avenue Olivier Messiaen, 72085 Le Mans Cedex 9, France. E-mail: gwenael.corbel@univ-lemans.fr; Fax: +33 (0)2 43 83 35 06; Tel: +33 (0)2 43 83 26 48
bInstitut des Matériaux Jean Rouxel (IMN), UMR-6502 CNRS, Université de Nantes, 2 rue de la Houssinière, BP32229, 44322 Nantes cedex 3, France
cInstitut Universitaire de Technologie du Mans, Le Mans Université, Avenue Olivier Messiaen, 72085 Le Mans Cedex 9, France
dLaboratoire de Génie Chimique, UMR-5503 CNRS, Faculté de Pharmacie, Université Paul Sabatier - Toulouse III, 35, chemin des maraîchers, 31 062 Toulouse Cedex 4, France
First published on 13th November 2019
Abstract
Brucite Mg(OH)2 belongs to a family of two-dimensional compounds with a CdI2-type structure built up from layers of edge-sharing octahedra delineating 2D galleries. In the current study, nanometer-sized platelets of copper substituted Mg(OH)2 were prepared by co-precipitation at room temperature in mixed alkaline (NaOH/Na2CO3) medium. Very weak substitution of a few hydroxyl ions by carbonate groups was highlighted at first by infrared spectroscopy and then quantified by thermogravimetric (TG) and mass spectrometric (MS) evolved gas analyses. The presence in a very low amount of water molecules in the galleries induces disorder in the stacking of layers of edge-sharing octahedra along the c-axis. The dehydration of the hydroxides taking place below 225 °C preserves the brucite-type structure of the samples while suppressing the stacking disorder. Copper substitution greatly enhances the bactericidal activity of nanometer-sized platelets of brucite against two bacteria frequently involved in healthcare-associated-infections. 10 mol% of cupric ions in Mg(OH)2 (a copper loading of 0.102 mg mL−1 in the suspension) were sufficient to induce, after 3 h in contact, 100% and 99.3% reductions in viability of Gram-negative E. coli and Gram-positive S. aureus, respectively (reductions as low as 23% and 48% are reported for the parent compound Mg(OH)2 in the same conditions). A good compromise between fast bactericidal kinetics and a high reduction in viability is reached by the 15 mol% copper-substituted Mg(OH)2 hydroxide. Its use gives the opportunity to five-fold reduce the copper loading of the bactericidal agent while being at least equally or even more efficient compared to the conventional CuO (a Cu loading of 0.799 mg mL−1 and 0.154 mg mL−1 in the suspension of CuO and 15 mol% copper substituted Mg(OH)2 particles, respectively).
1. Introduction
Bacterial pathogens causing healthcare-associated-infections (HAIs) pose ongoing and increasing challenges to hospitals, both in the clinical treatment of patients and in the prevention of their person-to-person transmission. In 2012, a report of the French Institute for Public Health Surveillance1 pointed out that three pathogens are responsible for more than half of the number of HAIs in France: the Gram-positive Staphylococcus aureus (S. aureus, 15.9%) and two Gram-negative bacteria Escherichia coli (E. coli, 26%) and Pseudomonas aeruginosa (P. aeruginosa, 8.4%). By designing bactericidal compounds having no or very low ecotoxicity, the impact of engineered materials on the environment and the risk of resistance acquisition to antimicrobial agents will be reduced by limiting their uses. Certain oxides such as periclase MgO2–4 and tenorite CuO5–7 exhibit bactericidal activity towards these pathogenic bacteria. Due to their long-term geological availabilities, these oxides could therefore be substituted, with advantages, for metallic silver, frequently used as a bactericidal agent. But CuO has ecotoxic effects while MgO has not.8 However, hydration of MgO by water vapour9,10 or in aqueous solution11,12 occurs leading with time to the growth onto the grain surface of a layer of brucite Mg(OH)2. Then, the hydration of the grain core occurs thanks to a diffusion process from the outer surface. The hydration rate depends to a great extent on the particle size of MgO and the temperature. For nanometer-sized particles of MgO with a specific surface area of ≈81 m2 g−1 (corresponding to an average ∅ ≈ 20 nm), Smithson and Bakshi11 showed that complete conversion into Mg(OH)2 occurred after ≈100 min of immersion in water at 38 °C. Makhluf et al.4 have evaluated the activity of MgO particles towards E. coli in the culture medium at 37 °C. These authors showed that the activity is only significant when the size of MgO particles is lower than 10 nm,4 which in turn drastically increases its sensitivity to moisture. Sawai et al.13 considered that the alkaline effect caused by the hydration of MgO is one of the primary factors in its antibacterial activity against yeasts (Saccharomyces cerevisiae and Candida albicans) and filamentous fungi (Aspergillus niger and Rhizopus stolonifer). Dong et al.14 have reported that nanometer-sized platelets of Mg(OH)2 (a thickness of ≈10 nm and a face diameter of ≈200–300 nm) also exhibit bactericidal activity towards E. coli at 37 °C. A complete eradication of E. coli is reached after 24 h of bacteria incubation with Mg(OH)2 particles. Since Mg(OH)2 is active and rapidly formed when nanometer-sized particles of oxide were immersed in the culture medium, the bactericidal activity of MgO particles mainly originates from the hydroxide formed at the surface. Akhavan et al.15 evaluated the antibacterial activity of nano-structured CuO layers grown on the surface of copper foil by immersion in a diluted sodium hydroxide solution (0 to 3 × 10−2 mol L−1) maintained at 60 °C for 20 h. High antibacterial activity against E. coli (with a reduction in viability around 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 or 99.999% after 90 min) was observed when a NaOH concentration of 3 × 10−2 mol L−1 was used. These authors ascribed this very high activity to the presence of Cu(OH)2 detected in a large amount at the foil surface by XPS analysis. The limit for using Mg(OH)2 as a bactericidal agent is that its performance depends on the shape and aggregation of particles with a reduction in E. coli viability not exceeding 90% in the best case after 4 h.16 The partial substitution of magnesium ions with cupric ones could greatly enhance the bactericidal activity of Mg(OH)2 particles, whether in terms of kinetics or the reduction level in cell viability.
log10 or 99.999% after 90 min) was observed when a NaOH concentration of 3 × 10−2 mol L−1 was used. These authors ascribed this very high activity to the presence of Cu(OH)2 detected in a large amount at the foil surface by XPS analysis. The limit for using Mg(OH)2 as a bactericidal agent is that its performance depends on the shape and aggregation of particles with a reduction in E. coli viability not exceeding 90% in the best case after 4 h.16 The partial substitution of magnesium ions with cupric ones could greatly enhance the bactericidal activity of Mg(OH)2 particles, whether in terms of kinetics or the reduction level in cell viability.
While apparently simple, such an approach has not been yet reported in the literature because the synthesis of Mg1−xCux(OH)2 particles is challenging. The synthetic route to prepare Mg(OH)2 nanoparticles consists in precipitating them by dropwise addition of an alkaline solution (ammonia or sodium hydroxide) to a solution of magnesium salt (chloride, sulfate or nitrate) maintained at constant temperature, typically in the range RT to 60 °C.17 Unfortunately, Cudennec et al.18 observed that precipitation carried out at room temperature from a solution of copper salt with sodium hydroxide solution transiently gives rise to weakly crystallized particles of copper hydroxide which quickly transform into CuO within a few hours of ageing. For these authors, progressive dissolution of Cu(OH)2 particles occurs with time in hydroxide ion rich-medium, leading to the formation of the soluble square planar [Cu(OH)4]2− complex. This anion is considered as the elementary building unit which enables the precipitation of copper oxide by condensation/dehydration.19
In the present study, nanometer-sized particles of several copper-substituted magnesium hydroxides Mg1−xCux(OH)2 (0 ≤ x ≤ 0.15) are successfully prepared at room temperature by adding Na2CO3 to the precipitating sodium hydroxide solution. Only the combination of temperature-controlled X-ray diffraction, infrared spectroscopy, optical absorption spectroscopy, thermogravimetric (TG) and mass spectrometric (MS) evolved gas analyses and X-ray photoelectron spectroscopy enables one to determine the chemical composition of the as-prepared samples (Mg1−xCux(OH)2−2y(CO3)y·zH2O). For each composition, evaluation of its bactericidal activity is carried out against two bacteria frequently involved in healthcare-associated-infections: the Gram-positive S. aureus and the Gram-negative E. coli. Because the morphology, size, specific surface area and dissolution of particles are parameters which can possibly affect their bactericidal activity, these features are determined by transmission electron microscopy, X-ray diffraction, N2 adsorption and atomic emission spectroscopy, respectively. A bactericidal mechanism is finally proposed.
2. Results and discussion
2.1 Sample identification
Within the synthesis conditions described in the ESI† (Section 1.2), the XRPD patterns of raw powders show a single Mg(OH)2 brucite type phase up to a copper content of x = 0.15 (Fig. 1a). For x = 0.2, a trace of Cu(OH)2 is detected (one peak at around 24° in 2θ), thus indicating that the limit of the solid solution “Mg1−xCux(OH)2” is exceeded. The structure of brucite, Mg(OH)220 (Fig. 2), is built up from layers of edge-sharing octahedra occupied by magnesium cations (6-fold coordinated to hydroxyl groups). The hydroxyl groups OH−, which are directed along the threefold axes of the structure, point out of both sides of each layer. The stacking along the c direction of these layers delineates infinite bi-dimensional galleries (Fig. 2).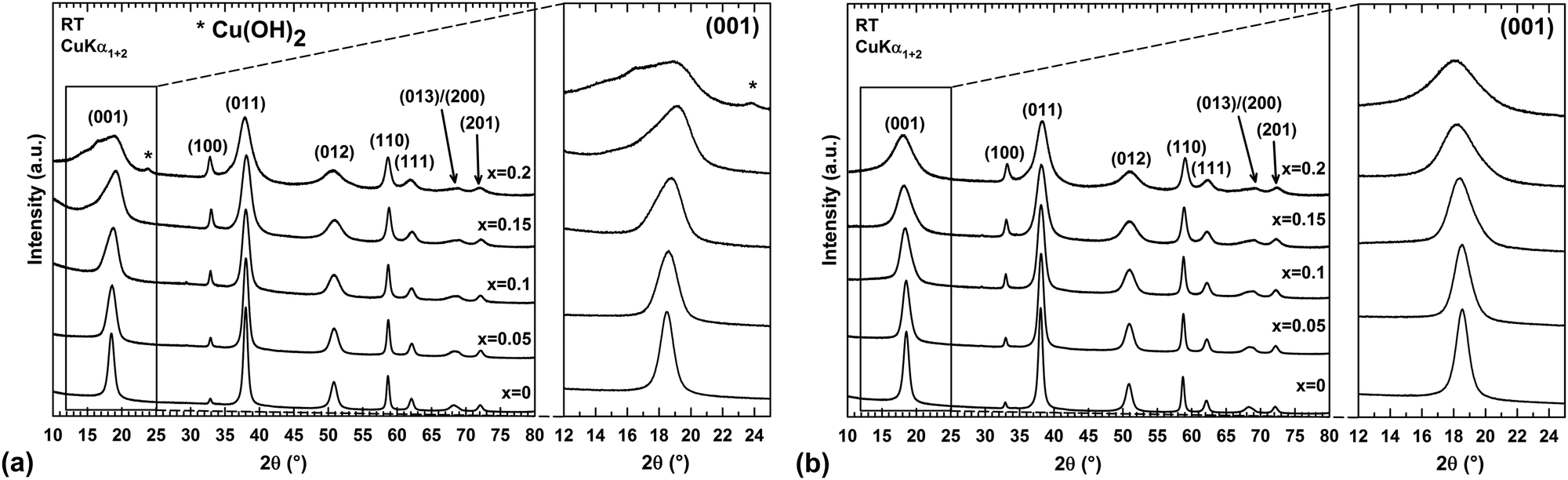 | ||
| Fig. 1 Room-temperature (RT) XRPD patterns of as-prepared (a) and dehydrated (b) “Mg1−xCux(OH)2” powder samples for different copper contents x. Insets: Effect of the sample dehydration on the profile shape of the (001) diffraction peak. | ||
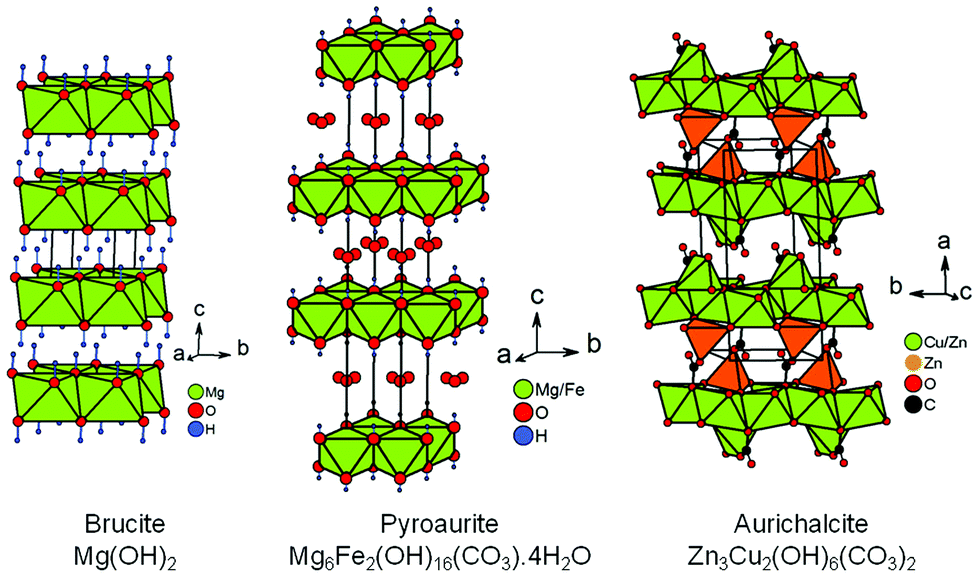 | ||
| Fig. 2 Perspective view of the crystal structures of brucite, pyroaurite and aurichalcite. The location of the hydrogen atoms in the structure of aurichalcite is not reported in ref. 21. | ||
As shown in Fig. 1a, the narrow and symmetric (001) peak of Mg(OH)2 (x = 0) phase transforms into a broad and asymmetric peak upon substitution, thus revealing disorder in the stacking of the layers along the c direction. A close structural inspection of two layered hydroxides, Mg6Fe2(OH)16(CO3)·4H2O22,23 and Zn3Cu2(OH)6(CO3)2,21 has helped us to find an explanation for this disorder.
The rhombohedral structure of pyroaurite Mg6Fe2(OH)16(CO3)·4H2O22,23 (Fig. 2) is built up from layers of edge-sharing octahedra as in brucite, except that a quarter of these octahedra are occupied by Fe3+ ions. To balance the positive charge in the brucite-type layers [Mg6Fe2(OH)16]2+ generated by this partial aliovalent substitution, carbonate anions are hosted in the galleries delineated by these layers stacked along the c direction. The orientation of triangular CO32− anions is parallel to the brucite-type layers. Water molecules also find a place in these galleries. A second form with hexagonal symmetry, named sjögrenite,23,24 is reported in the literature for the Mg6Fe2(OH)16(CO3)·4H2O hydroxycarbonate. Both structures only differ in the stacking sequences of brucite-type layers and oxygen atoms (from carbonate ions and water molecules) located in the interlayer space along the c direction. The sequences are called three-layer rhombohedral (3R) and two-layer hexagonal (2H) with reference to the pyroaurite and sjögrenite polytypes, respectively.25 Whatever the polytype, Mg6Fe2(OH)16(CO3)·4H2O belongs to the large family of layered double hydroxides (LDH)26 with the general formula [M1−xIIMxIII(OH)2][An−]x/n·mH2O, where MII is a divalent metal cation which can be partially substituted by a MIII trivalent cation, and An− is a charge compensating anion such as NO3− or CO32− for instance. Disorder in the stacking frequently occurs in LDH.27 Stacking faults can originate from a difference in water content from one interlayer gallery to the others. Since the amount of intercalated anions An− in the gallery is linked to the content of trivalent cations, stacking faults can help the structure to locally adapt to a non homogeneous cationic distribution between brucite-type layers.
To some extent, aurichalcite Zn3Cu2(OH)6(CO3)221 is structurally related to brucite (Fig. 2). The monoclinic unit cell of aurichalcite contains four crystallographically independent metal sites (M1–M4). The M3 site is only occupied by zinc and both divalent cations are equally distributed on the three remaining metal sites. The M1 and M2 sites are both 6-fold coordinated to oxygen atoms in octahedral coordination (green octahedra in Fig. 2), while the M3 and M4 sites are in tetrahedral and 5-fold (3+2) distorted coordination (orange polyhedra in Fig. 2), respectively. In aurichalcite, the octahedra share edges to form infinite layers parallel to the (b, c) basal plane. However, these layers of edge-sharing octahedra are not as dense as in brucite. When compared to a brucite-type layer, one-fifth of the octahedra are empty in layers of aurichalcite. The remaining M3 and M4 sites are both located outside these cationic deficient layers in such way that the two opposite triangular faces of each empty octahedron correspond to a triangular face of the coordination polyhedra of M3 and M4 sites. In aurichalcite (Fig. 2), all carbonate ions are bidentate and share one oxygen atom with three octahedra of a deficient brucite-type layer. Half of the carbonate ions share a second oxygen atom with [M3O4] tetrahedra in order to form pillars linking two adjacent layers of edge-sharing octahedra. The remaining carbonate ions share a second oxygen with [M4O5] polyhedra lying on the layer to which they are already linked.
It is worth noting that the precipitation of mixed hydroxydes occurred when a solution of magnesium and copper nitrates is added dropwise to a mixture of NaOH and Na2CO3 solutions. From the above structural considerations, one cannot rule out that carbonate or nitrate anions as well as water molecules might be incorporated into the galleries of the brucite-type hydroxide “Mg1−xCux(OH)2” we obtained. To detect the presence of foreign anions and water, infrared spectra were collected at room temperature on raw powders. Fig. 3a and b show the superimposition of the different spectra as a function of the copper content x.
 | ||
| Fig. 3 (a) and (b) Selected portions of infrared transmission spectra collected at room temperature on “Mg1−xCux(OH)2” raw powder samples showing the bands arising from the stretching vibrations ν of hydroxyl OH− ions, the bending vibration δ of water molecules and stretching vibrations ν of either carbonate or nitrate ions. (c) Variation with the copper content x of the ratio between integrated band areas of carbonate/nitrate and hydroxyl ions. | ||
In each IR spectrum displayed in Fig. 3a, the intense vibration band observed at around 3700 cm−1 is attributed to the stretching vibration ν of the hydroxyl group.28 The frequency of the OH stretching vibration in the x = 0 sample is in very good agreement with the value of 3697 cm−1 determined by Martens and Freud on Mg(OH)2 powder prepared by vapour phase hydroxylation of MgO.29 In Fig. 3b, the weak absorption band at around 1650 cm−1 would correspond to the bending vibration δ of water molecules28 either present in a low amount in the interlayer gallery or physisorbed at the grain surface. This point will be clarified later. Two additional bands are also detected at lower frequencies in each IR spectrum shown in Fig. 3b. These bands could be ascribed to stretching vibrations ν of either carbonate or nitrate groups.28 Along the series, the ratio between the integrated band areas of carbonate/nitrate and hydroxyl ions first slightly increases in the substitution range 0 ≤ x ≤ 0.1 before nearly doubling between x = 0.1 and 0.15. It clearly shows that the amount of foreign anions in the sample increases with the copper content x without being directly proportional to it.
The nature of the foreign anions was determined by analyzing the gas released when the sample decomposes upon heating. For this purpose, a thermogravimetric analyser coupled to a mass spectrometer was used. In parallel, the thermal decomposition has been followed by temperature-controlled XRPD to provide information about the transformation process. Thermograms, mass spectra and thermal evolutions of diffraction patterns for three selected samples x = 0, 0.1 and 0.2 are all displayed in Fig. 4. The thermograms and mass spectra recorded on the two remaining samples of the series (x = 0.05 and 0.15) are similar to those of the x = 0 and 0.1 samples. These data are thereby not displayed in Fig. 4. In Fig. 4, two distinct weight losses are clearly observed in the thermograms of the x = 0 and x = 0.1 samples. A first small weight loss takes place in the temperature range 100–225 °C, which ends up with a plateau. This weight loss is only associated with release of water molecules since a bump is observed at around 125 °C in the m/z = 18 spectrum. Temperature-controlled diffraction reveals that the brucite-type structure is preserved in this temperature range (Fig. 4). Because a bending vibration δ of water molecules has been detected by IR spectroscopy for all samples, this first weight loss can only be associated with a dehydration process. In addition to the bump at around 125 °C ascribed to the dehydration of the brucite-type hydroxide, a second bump is clearly observed at around 175 °C in the m/z = 18 spectrum of the x = 0.2 sample. As shown in the room temperature diffraction patterns displayed in Fig. 1a and 4, a secondary phase Cu(OH)2 is present in a very low amount with a major brucite-type hydroxide for the as-prepared x = 0.2 sample. Cu(OH)2 is a metastable phase which decomposes into copper oxide CuO at 145–160 °C,30 which is a very low decomposition temperature for a hydroxide-type compound. In Fig. 4, the only diffraction peak of Cu(OH)2 at around 24° in 2θ disappears at 175 °C, which corresponds to the temperature at which the second bump is detected in the m/z = 18 spectrum. No trace of copper oxide is however detected in the diffraction patterns collected on the x = 0.2 sample in the temperature range 175–225 °C within which the dehydrated brucite-type hydroxide still exists. In their study, Günter et al.30 have observed that the diffraction lines of CuO, resulting from the decomposition of Cu(OH)2, are broad, which means that the coherent diffraction domains are small in size. Starting with a very low amount of Cu(OH)2 present in the x = 0.2 sample, it is not so surprising that no diffraction lines of CuO are detected in the background of the XRPD pattern collected in the temperature range 175–225 °C because of the small amount and the small grain size of the copper oxide produced. Finally, the absence of a plateau in the thermogram of the x = 0.2 sample originates from the successive weight losses caused by the dehydration of the brucite-type hydroxide and the decomposition of Cu(OH)2. In the RT–225 °C range, the successive dehydration of the brucite-type hydroxide and the decomposition of Cu(OH)2 do not allow one to determine the water content z of the x = 0.2 sample. In Table 1, the water content z determined from the first weight loss of the thermograms increases with the copper content x but remains relatively small even for highly copper substituted magnesium hydroxides (z ≈ 0.11 for a copper content x = 0.15).
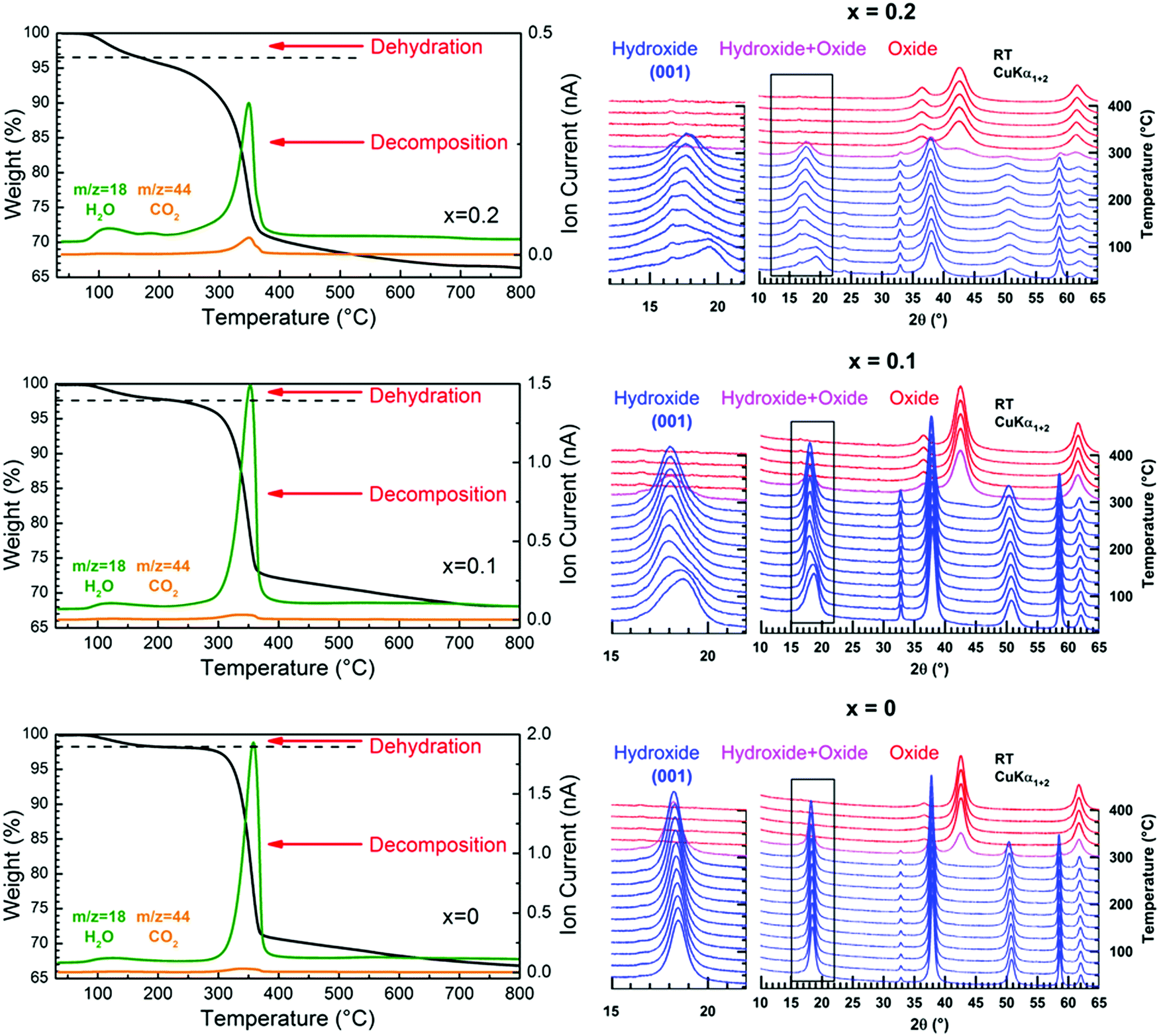 | ||
| Fig. 4 (Left) TGA thermograms and mass spectra of released water and carbon dioxide collected upon heating in argon on “Mg(OH)2”, “Mg0.9Cu0.1(OH)2” and “Mg0.8Cu0.2(OH)2” raw powder samples. (Right) Thermal evolution of the XRPD patterns of the same samples when annealed in air from RT to 400 °C. | ||
| Copper content x | Dehydration | Decomposition | ||
|---|---|---|---|---|
| Weight loss % | z(H2O) | Weight loss % | y(CO3) | |
| 0 | 1.84 | 0.062 | 31.33 | 0.034 |
| 0.05 | 1.91 | 0.066 | 30.57 | 0.043 |
| 0.1 | 2.36 | 0.085 | 29.56 | 0.045 |
| 0.15 | 3.51 | 0.114 | 30.12 | 0.129 |
In layered double hydroxides (LDH), water molecules are both physisorbed at the grain surface and located within the interlayer galleries.31,32 The former are easily removed by drying the raw powder at around 60 °C, while subsequent heating above 250 °C is necessary for releasing the latter.32 Only inter-layered water molecules are counted in the unit formula of LDH with a metal/H2O ratio that does not exceed 0.5, as for instance in pyroaurite Mg6Fe2(OH)16(CO3)·4H2O.22,23 For the “Mg1−xCux(OH)2” samples, there are concerns about whether the dehydration taking place in the temperature range 100–225 °C corresponds to release of water molecules located within the interlayer galleries of the brucite-type structure or adsorbed at the particle surface or both together. This question is of vital importance since the temperature at which the dehydration occurs in “Mg1−xCux(OH)2” is lower than the temperature characteristic of the release of inter-layered water molecules in LDH. As mentioned in the ESI† (Section 1.5), raw “Mg1−xCux(OH)2” powders were dried in static air at 60 °C prior to any analysis, which normally allows the desorption of water molecules physisorbed at the particle surface. When in situ studying the dehydration of the hydroxides by temperature-controlled diffraction, the asymmetric broadening of the (001) diffraction peak progressively disappears as the temperature increases in the range RT–225 °C in Fig. 4. Because the layers of edge-sharing octahedra are stacked along the c axis of the Mg(OH)2 structure, this change in the peak profile is a sign that the dehydration affects this stacking, a process that any desorption of water molecules physisorbed at the particle surface cannot obviously cause. Thereby, the observed dehydration should mainly correspond to the loss of water molecules located in a low amount within the interlayer galleries. Now, why does the dehydration affect the stacking of layers? With such a small content of water in the dried samples (Table 1), certain galleries of the mixed hydroxides are certainly free of water molecules, while others are partially filled. Note that the height of the galleries in fully dehydrated Mg(OH)2 is around 2.689 Å,20 which is more than 2 times smaller than the height of the galleries (5.688 Å) in pyroaurite Mg6Fe2(OH)16(CO3)·4H2O,22,23 which contain a huge amount of water molecules. To accommodate these water molecules and the variation in water content from one gallery to the others, layers of edge-sharing octahedra cannot be stacked in an orderly manner as in an idealized brucite structure (i.e. an increase in the height of galleries containing water molecules along the c axis). Disorder in the stacking then exists involving the asymmetric broadening of the (001) peak towards low 2θ scattering angles observed in the XRPD patterns (Fig. 1a). In this figure, the broadening becomes significant for a copper content higher than x = 0.1 when the water content exceeds ≈8 mol% (Table 1). Note that the position of the (001) peak for a given composition has slightly shifted towards low scattering 2θ angles after dehydration. It means that the interlayer galleries weakly expand in height along the c axis when removing water molecules in Mg1−xCux(OH)2−2y(CO3)y·zH2O hydroxides. With the bactericidal properties of materials being not related to their thermal dehydration, the origin of the lattice expansion when dehydrating samples is only discussed in the ESI† (Section 2).
Above 250–300 °C, the sample starts decomposing by simultaneously releasing water and a few CO2 molecules as shown at m/z = 18 and m/z = 44 in the mass spectra displayed in Fig. 4. No traces of NO2 molecules resulting from thermal decomposition of nitrates anions are detected by mass spectrometry (not shown in Fig. 4). Thereby, carbonate anions are present in the interlayer galleries within the structure of the mixed hydroxides obtained. Because of the isovalent character of the magnesium ion substitution by divalent copper, the layers of edge-sharing octahedra in mixed “Mg1−xCux(OH)2” hydroxides are neutral in charge, contrary to what is observed in pyroaurite and sjögrenite polytypes of Mg6Fe2(OH)16(CO3)·4H2O. The presence of carbonate anions in “Mg1−xCux(OH)2” then implies that they share oxygen atoms between layers, as observed in the structure of aurichalcite Zn3Cu2(OH)6(CO3)221 for instance (Fig. 2). The consequence of that is a partial substitution of a few hydroxyl ions by carbonate groups in the formulation of mixed hydroxides as follows: Mg1−xCux(OH)2−2y(CO3)y·zH2O. Note that the metal/H2O ratio in pyroaurite Mg6Fe2(OH)16(CO3)·4H2O (= 0.5) is more than 4 times larger than that found in Mg0.85Cu0.15(OH)1.742(CO3)0.129·0.114H2O for a similar metal/CO32− ratio (≈1/8). Finally, the XRPD pattern collected above 300 °C (Fig. 4) shows that the only decomposition product is a single mixed periclase-type oxide Mg1−xCuxO even for the composition x = 0.2, which originally contains a trace of copper hydroxide Cu(OH)2. From the above thermal and temperature-controlled diffraction analyses, the dehydration and decomposition reactions are the following:
| Mg1−xCux(OH)2−2y(CO3)y·zH2O → Dehydration → Mg1−xCux(OH)2−2y(CO3)y + zH2O(g) |
| Mg1−xCux(OH)2−2y(CO3)y → Decomposition → Mg1−xCuxO + yCO2(g) + (1 − y) H2O(g) |
From the structural correlation with aurichalcite Zn3Cu2(OH)6(CO3)2,21 a small fraction of divalent cations must be 4-fold or 5-fold coordinated to oxygen atoms and outside the brucite-type layers in order to take into account the partial substitution of a few hydroxyl ions by carbonate groups in Mg1−xCux(OH)2−2y(CO3)y·zH2O compounds. The small content y of carbonate ions calculated from the second weight loss of the thermograms (Fig. 4 and Table 1) means that this fraction of Mg2+/Cu2+ cations in 4-fold or 5-fold coordination should be also small. The coordination environment of a transition metal ion such as Cu2+ in the crystal lattice directly influences the energy levels of its d-orbitals through crystal field splitting (i.e. octahedral splitting, tetrahedral splitting). The position in wavelength of the absorption band in the UV-visible-NIR spectrum is directly related to the energy required for d–d electronic transitions to occur. Absorption spectroscopy can be then used to probe the coordination of copper ions in Mg1−xCux(OH)2−2y(CO3)y·zH2O compounds. The visible-NIR absorption spectra of all compositions of the series are displayed in Fig. 5a. For the white x = 0 sample (i.e. Mg(OH)2), only two weak absorptions occurred in the NIR region of the spectrum with maxima at around 7150 and 7250 cm−1. Martens and Freund29 showed that these bands correspond to the first fundamental overtones of the stretching vibrations ν of hydroxyl OH− ions observed for Mg(OH)2 in the range 3500–4000 cm−1. When cupric ions are tetrahedrally coordinated to oxygen atoms in copper doped ZnO33 or in CuAl2O4 spinel,34 a broad absorption is measured in the wavenumber range 5550 cm−1 (1800 nm) to 8330 cm−1 (1200 nm). Apart from the overtones of νOH vibrations, no additional absorption band is detected in this NIR region of the spectra displayed in Fig. 5a, thus clearly indicating that no cupric ions are 4-fold or 5-fold coordinated to oxygen atoms in Mg1−xCux(OH)2−2y(CO3)y·zH2O compounds. However, a broad absorption band appears at higher wavenumbers in the spectra of the x > 0 samples with a maximum at around 13900 cm−1 (≈720 nm). Its intensity grows as the copper content x increases in the series (hyperchromic effect). It means that copper containing samples absorb light in the red region of the visible absorption spectrum, which is in agreement with the complementary blue color of the powder (Fig. 5b). In the hexaaquacuprate complex [Cu(H2O)6]2+, the transition of one electron from the Eg orbital to the T2g orbital of divalent copper (3d9) in an octahedral crystal field gives rise to a single absorption at about 12500 cm−1 (≈800 nm).35 In the spectrochemical series, the strength of the hydroxo ligand is slightly weaker than the strength of the aqua ligand. By replacing six aqua ligands with six hydroxo ligands, the absorption is expected to occur at a wavelength lower than 800 nm. Thereby, the absorption band at around 13900 cm−1 (≈720 nm) corresponds to the 2Eg(D) → 2T2g(D) electronic transition of divalent copper 6-fold coordinated by hydroxyl groups. The optical absorption spectrum of malachite Cu2(CO3)(OH)236 shows two broad bands in the UV-visible-NIR range, an intense band centered at around 12346 cm−1 (810 nm) with a weak band at lower wavenumber 8696 cm−1 (1150 nm). In the structure of malachite,37 divalent copper resides in an octahedron with two copper–oxygen distances longer than the four remaining ones. This tetragonal distortion [4+2], frequently encountered for an octahedron occupied by Cu2+ ions, is ascribed to a Jahn–Teller effect. In the spectra displayed in Fig. 5a, no additional absorption band or shoulder is present in the range of wavenumbers 7500–10000 cm−1. Furthermore, the band at around 13900 cm−1 has an almost symmetric shape. Both observations suggest that the Jahn–Teller distortion of the octahedra occupied by Cu2+ ions in Mg1−xCux(OH)2−2y(CO3)y·zH2O compounds, if it exists, is weak in magnitude. The above analyses of diffuse reflectance spectra clearly demonstrate that all cupric ions are located within layers of edge-sharing octahedra of the structure, as are most of the magnesium ions. Thereby, one has to admit that the divalent ions located outside the layers, in a small amount and in 4-fold or 5-fold coordination, are Mg2+.
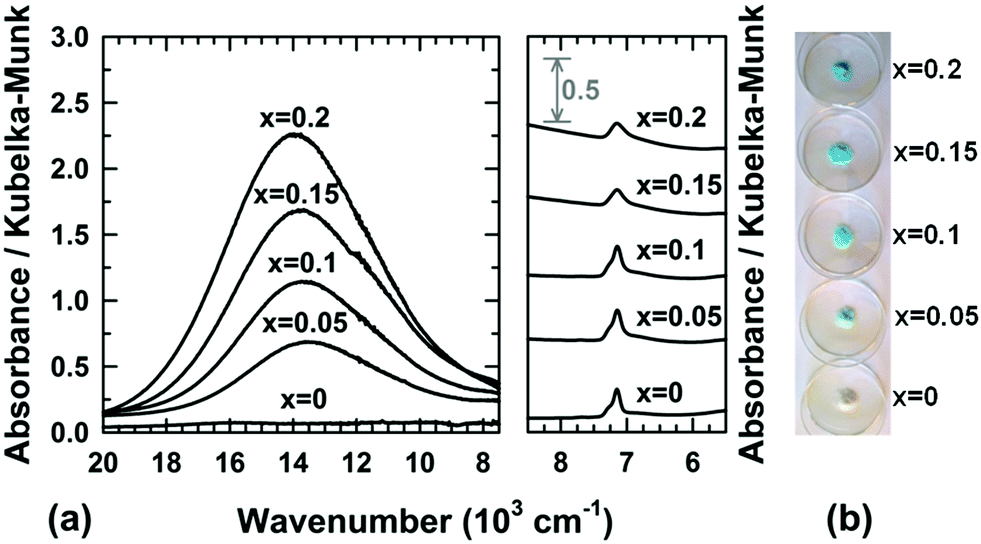 | ||
| Fig. 5 (a) UV-visible-NIR spectra collected at room temperature and (b) pictures of raw powder Mg1−xCux(OH)2−2y(CO3)y·zH2O samples. | ||
The morphology and crystallinity of hydroxide particles were determined by transmission electron microscopy (TEM) and selected area electron diffraction (SAED), respectively. TEM micrographs reveal that particles of magnesium hydroxide (x = 0) have smooth edges and a platelet-like shape. The particles have a face diameter ranging from ≈10 to 50 nm with a thickness lower than ≈10 nm. For copper containing samples, the platelets tend to aggregate in a face-to-face fashion and rest on the edge when deposited on the TEM grid, making difficult their observations, as shown in Fig. 6b and Fig. 6c. Platelets tend to form larger and larger shapeless aggregates as their copper content x increases (Fig. 6d). All SAED patterns of copper substituted hydroxide appear similar and show four distinct rings. These four rings are well indexed to the crystal planes (001), (011), (012) and (110) of the brucite-type hydroxide, confirming the high crystallinity of the as-prepared particles. For each single phase hydroxide (x = 0–0.15), the copper to magnesium atomic ratio, determined by EDX analysis, is in very good agreement with the nominal ratio (Fig. 6f). Both the absorption spectrum collected on the colorless supernatant (Fig. S2 in the ESI†) and the EDX analyses of the platelets clearly demonstrate that divalent copper and magnesium ions present in the mixed nitrate solution completely precipitate after its addition to 150 mL of the mixed NaOH/Na2CO3 solution.
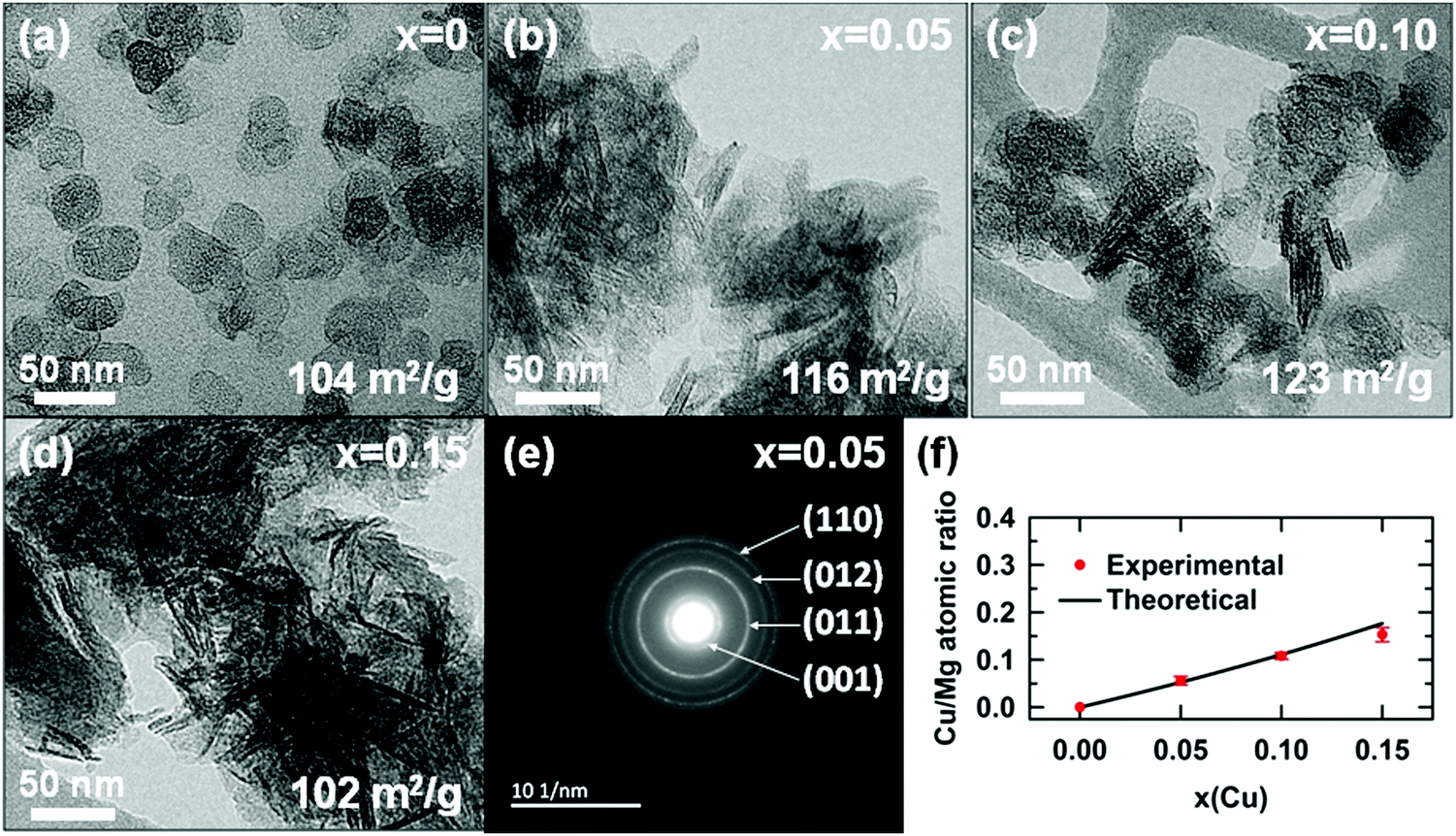 | ||
| Fig. 6 (a–d) TEM micrographs of raw powder Mg1−xCux(OH)2−2y(CO3)y·zH2O samples. (e) SAED pattern of the x = 0.05 sample. Specific surface areas determined from N2 adsorption measurements are given. (f) Evolution of the Cu/Mg atomic ratio determined by EDX analysis as a function of the nominal copper content x. | ||
2.2 Effect of copper substitution on the brucite Mg(OH)2 structure
The disorder in the stacking of layers of edge-sharing octahedra along the c-axis for hydrated and highly copper substituted hydroxides induces a strong asymmetric broadening of the (001) diffraction peak (Fig. 1a). This disorder makes impossible the refinement of the unit cell parameters through diffraction pattern fitting by the Le Bail method.38 Temperature-controlled XRPD and thermal analyses have revealed that this broadening disappears and the brucite-type structure is preserved when the samples are dehydrated in the temperature range RT–225 °C (Fig. 4). With the aim of determining the unit cell parameters from the diffraction data, sample dehydration was undertaken under static air conditions. In order to avoid any decomposition of the brucite-type hydroxide, the temperature at which the dehydration has been carried out was chosen 25 °C below the end of the plateau at 225 °C observed in the thermograms displayed in Fig. 4. Thus, all powder samples were dehydrated in air for 4 h at 200 °C. XRPD patterns were then collected at room temperature on the dehydrated powders and are shown in Fig. 1b.In Fig. 1b, an anisotropic line broadening is highlighted for the dehydrated powders with a significantly narrower line breath for the (100) and (110) reflections than for the (001), (011) and (012) ones. This anisotropic broadening is inherent to the platelet-like shape of the particles. From the TEM analyses and the larger breath of the (00l) line, one can conclude that the crystallographic c axis is perpendicular to the platelet faces and the platelet thickness 〈H〉 is smaller than the long diagonals 〈D〉 of hexagonal platelet faces (Fig. 7a). Moreover all the Bragg peaks have a symmetric Voigt-type profile, which makes possible a microstructure analysis from XRPD data using the integral breadth method.39 Attempts to satisfactorily fit the diffraction patterns by the Le Bail method38 were then undertaken selecting the “platelet vector size” model of the Fullprof program40 with a [00l] direction normal to the hexagonal platelet faces. In this model, a single hexagonal platelet can be circumscribed into a cylinder. The long diagonals 〈D〉 of the hexagonal platelet faces correspond to the diameter of this cylinder, while the platelet thickness 〈H〉 is the height of this cylinder. In the microstructural analysis, the peak broadening is assumed to only originate from the nanometer size of the particles (the “size broadening” effect) rather than lattice distortion (the “microstrain broadening” effect). The two parameters of the Lorentzian component of the TCH-Z profile shape function were only refined to calculate the average integral-breadth apparent size 〈Sβ〉 for each (hkl) reflection. As shown in Fig. 7a, the patterns can be satisfactorily fitted with this model. The average diameter 〈D〉 and thickness 〈H〉 of the platelet-like particles were determined from the apparent sizes 〈Sβ〉 along the [hh0] and [00l] directions, respectively. The variations of the unit cell parameters and 〈D〉 and 〈H〉 for the dehydrated Mg1−xCux(OH)2−2y(CO3)y compounds are shown in Fig. 7b as a function of the copper content x.
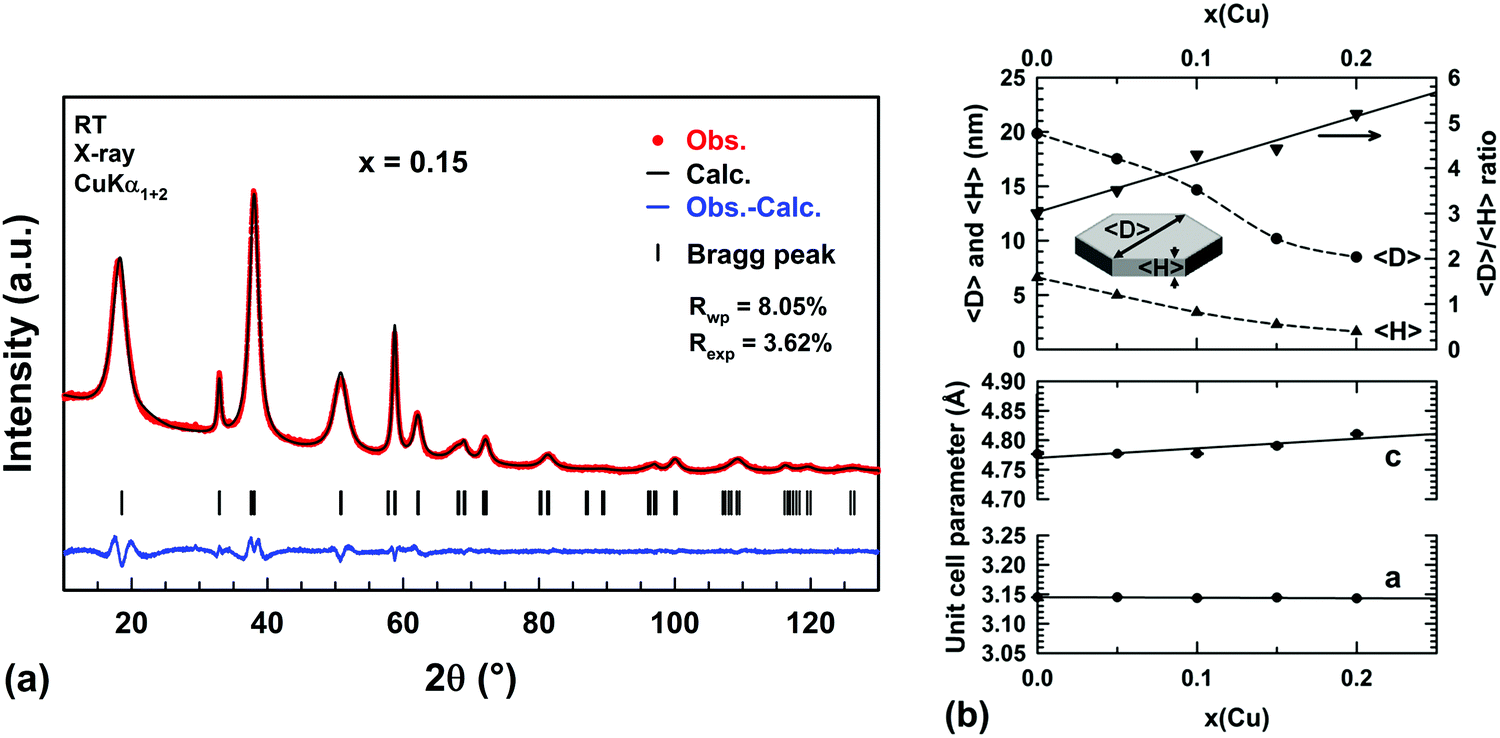 | ||
Fig. 7 (a) Comparison of the observed (red dots) X-ray powder diffraction pattern of the dehydrated Mg0.85Cu0.15(OH)2−2y(CO3)y (x = 0.15) sample with the pattern calculated (black line) through fitting by the Le Bail method and assuming a hexagonal platelet-like shape of nanometer-sized particles. Vertical markers give the Bragg peak positions of the trigonal space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 (no 164). (b) Variations of the average diagonal 〈D〉 and thickness 〈H〉 of hexagonal platelets as well as the unit cell parameters a and c with the copper content x in dehydrated Mg1−xCux(OH)2−2y(CO3)y raw powder samples. m1 (no 164). (b) Variations of the average diagonal 〈D〉 and thickness 〈H〉 of hexagonal platelets as well as the unit cell parameters a and c with the copper content x in dehydrated Mg1−xCux(OH)2−2y(CO3)y raw powder samples. | ||
Very small linear expansions of both cell parameters a and c are expected since divalent copper is slightly larger than the magnesium ion in 6-fold coordination (ionic radii 0.73 Å versus 0.72 Å41). In Fig. 7b, only the cell parameter c exhibits such an increase, the cell parameter a remaining constant in the whole compositional range. It is worth noting that in aurichalcite Zn3Cu2(OH)6(CO3)2,21 [M3O4] tetrahedra and carbonate ions form pillars linking two adjacent layers of octahedra. In aurichalcite, the average height of the galleries is ≈5.1 Å, which is almost two times the height of the galleries in dehydrated brucite Mg(OH)2 (2.689 Å20). In Fig. 2, the layers of octahedra in aurichalcite are not as flat as in brucite but are slightly corrugated where three octahedra share one oxygen atom with the carbonate ion of one pillar. Within a layer, this corrugation shortens and lengthens the distances between cations residing in the octahedra. In the dehydrated Mg1−xCux(OH)2−2y(CO3)y compounds, the presence of bridging carbonate ions must induce a similar deformation of the Mg(OH)2-type structure (i.e. expansion in height of the interlayer galleries and corrugation of layers). The deformation is expected to be small in magnitude due to the low amount of carbonate ions present in copper substituted magnesium hydroxides. In our opinion, the elongation of the unit cell along the c axis with x (Δc ≈ +0.034 Å over the whole compositional range) reflects the combined effects of copper substitution and the expansion in height of the interlayer galleries caused by the increasing amount of bridging carbonate ions. The absence of variation of the a parameter suggests that the shortening and lengthening of the cation–cation distances in the corrugated layer are fully balanced. One plausible reason for this behavior lies in the flexibility of divalent copper coordination polyhedra.
In Fig. 7b, a homothetic size reduction of the platelet-like particles is noted as the copper content x increases along the series. Indeed, both the average diameter 〈D〉 and thickness 〈H〉 dropped by half for samples ranging from x = 0 to x = 0.2. The particle shape is therefore significantly affected by the substitution, as shown by the increase of the 〈D〉/〈H〉 ratio with the copper content x in Fig. 7b. For the x = 0.1 and 0.15 samples, the average thicknesses of platelet-like particles deduced from their XRPD patterns are 〈H〉 ≈34 Å and ≈23 Å, respectively. These values are in good agreement with the average thicknesses of the black edges in the TEM images ≈40 Å and ≈28 Å for the x = 0.1 (Fig. 6c) and x = 0.15 (Fig. 6d) samples, respectively. It confirms that the size of coherent diffraction domains corresponds to the size of the platelets. As the 〈D〉/〈H〉 ratio increases, the platelets tend to rest on the edge when deposited on the TEM grid.
2.3 Bactericidal activity
The bactericidal activity of the as-prepared hydroxides was determined in sterile distilled water rather than in growth medium, as frequently done in numerous studies (MgO,4 Mg(OH)214 or ZnO42 nanoparticles in lysogeny broth (LB)). The reason is that any direct interaction between particles and peptides/proteins present in nutrient media must be avoided since it has been suspected to weaken and even to inhibit the adherence of the particles to the bacteria cells, thus decreasing their bactericidal activity.4 This direct interaction of particles with the bacteria cells is of vital importance in the present study because Dong et al.14 and Pan et al.16 have demonstrated that Mg(OH)2 particles can only kill E. coli by adhering to its cell membrane. Another point to be considered is the temperature and the relative humidity (RH) at which the antibacterial activity is determined. Michels et al.43 have clearly demonstrated that silver ion-containing materials, as a conventional antibacterial agent, exhibit no activity towards methicillin resistant S. aureus (MRSA) at T ≈ 22 °C and RH ≈ 50% within the first 360 min of exposure. A measurable efficacy (reduction in bacteria viability >6log10) was however observed by these authors at a higher humidity level (RH > 90%), higher temperature (35 °C) and longer contact time (24 h), the conditions recommended in Japanese Industrial Standard methodology JIS Z 2801 for antibacterial surfaces. At T ≈ 22 °C and RH ≈ 50%, Michels et al.43 have found that copper alloys exhibit a reduction in bacteria viability greater than 7log10 within 75 min of exposure. When tested under the JISZ 2801 test conditions, this high antibacterial activity of copper alloys was preserved. From the above considerations and for application purposes, the efficacy of any bactericidal surface must, in our opinion, be evaluated at a temperature typical of indoor environments such as hospitals (≈20 °C), directly in sterile distilled water (to avoid variations in RH during the test) and for a short contact time.
In the present study and for comparison purposes, the concentration of 1 mg mL−1 of particles within the water suspension of bacteria was identical to the one chosen by Dong et al.14 and Makhluf et al.4 to determine the antibacterial activity of nanometer-sized particles of Mg(OH)2 and MgO, respectively. For each mixed hydroxide tested, the sterile water suspension of particles in contact with a bacterial strain was maintained at 20 °C for 15, 30, 60 and 180 min. After each contact time t, a counting of the number Nt of CFU was performed to estimate the reduction in cell viability. For each mixed hydroxide tested, the number Nt of CFU is displayed in Table 2 at the different contact times for both E. coli and S. aureus. For three contact times 15, 60 and 180 min, the percentage reduction in viability of both E. coli and S. aureus is plotted as a function of the copper content x in Fig. 8.
| Copper content x/composition | Particle size D/H (nm) | Contact time (min) | E. coli | S. aureus | ||||
|---|---|---|---|---|---|---|---|---|
| Log10N0 | Log10(N0/Nt) | Reduction (%) | Log10N0 | Log10(N0/Nt) | Reduction (%) | |||
| 15 | 0 | 0 | 6.26 ± 0.01 | 0.04 ± 0.02 | 9.6 ± 4.5 | |||
| 0 | 10–50/10 | 30 | 6.31 ± 0.03 | 0 | 0 | 0.13 ± 0.06 | 25.2 ± 9.7 | |
| 60 | 0 | 0 | 0.20 ± 0.05 | 36.9 ± 6.8 | ||||
| 180 | 0.11 ± 0.02 | 23.2 ± 2.8 | 0.29 ± 0.07 | 48.4 ± 7.9 | ||||
| 15 | 6.31 ± 0.03 | 0.24 ± 0.04 | 42.6 ± 5.8 | 6.26 ± 0.01 | 0.34 ± 0.08 | 53.6 ± 9.0 | ||
| 0.10 | 10–50/10 | 30 | 0.37 ± 0.01 | 57.0 ± 0.8 | 0.69 ± 0.05 | 79.3 ± 2.5 | ||
| 60 | 1.19 ± 0.09 | 93.4 ± 1.3 | 1.17 ± 0.09 | 93.2 ± 1.3 | ||||
| 180 | 6.31 | 100 | 2.14 ± 0.01 | 99.3 ± 0.1 | ||||
| 15 | 6.31 ± 0.03 | 0.49 ± 0.15 | 66.5 ± 11.4 | 6.26 ± 0.01 | 0.65 ± 0.09 | 77.2 ± 4.8 | ||
| 0.15 | 10–50/10 | 30 | 0.86 ± 0.05 | 86.2 ± 1.5 | 0.91 ± 0.19 | 87.1 ± 5.4 | ||
| 60 | 2.98 ± 0.10 | 99.9 ± 0.1 | 1.23 ± 0.10 | 94.0 ± 1.4 | ||||
| 180 | 6.31 | 100 | 2.01 ± 0.16 | 99.0 ± 0.4 | ||||
| 15 | 6.31 ± 0.03 | 0.67 ± 0.12 | 78.5 ± 5.8 | 6.26 ± 0.01 | 0.73 ± 0.03 | 81.5 ± 1.3 | ||
| 0.20 | (biphasic) | 30 | 0.87 ± 0.06 | 86.3 ± 2.0 | 0.80 ± 0.08 | 84.2 ± 2.7 | ||
| 60 | 3.02 ± 0.10 | 99.9 ± 0.1 | 1.24 ± 0.24 | 93.8 ± 3.3 | ||||
| 180 | 6.31 | 100 | 1.91 ± 0.03 | 98.8 ± 0.1 | ||||
| 15 | 6.52 ± 0.02 | 2.30 ± 0.11 | 99.5 ± 0.1 | 6.18 ± 0.12 | 0.07 ± 0.06 | 13.8 ± 11.9 | ||
| CuO | 50/50 | 30 | 2.43 ± 0.03 | 99.6 ± 0.1 | 0.08 ± 0.16 | 11,3 ± 32.6 | ||
| 60 | 2.91 ± 0.01 | 99.9 ± 0.1 | 0.07 ± 0.12 | 12.7 ± 23.6 | ||||
| 180 | 4.70 ± 0.01 | 100 | 2.01 ± 0.49 | 98.7 ± 1.2 | ||||
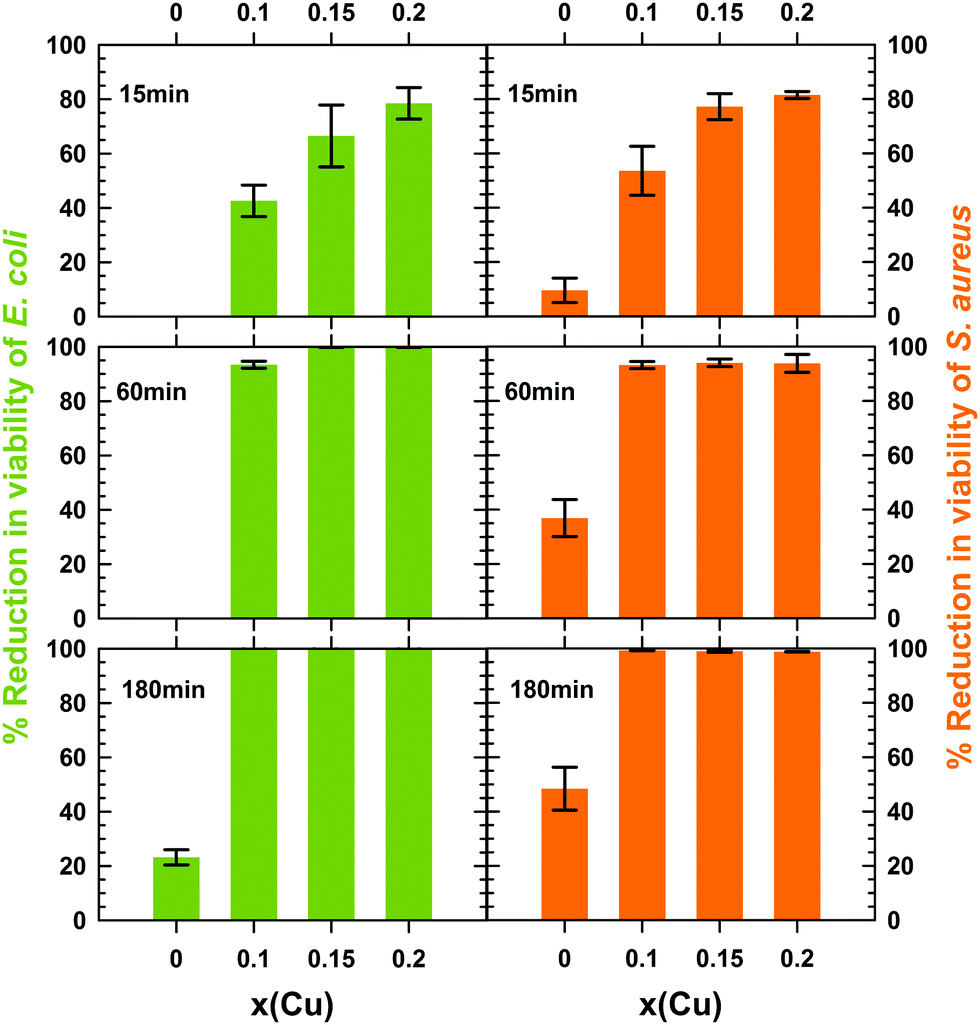 | ||
| Fig. 8 Time dependence of the reduction in viability of E. coli and S. aureus in contact with nanometer-sized Mg1−xCux(OH)2−2y(CO3)y·zH2O particles with different copper content x (% average value ± standard deviation over 2 assays). | ||
All copper containing hydroxides exhibit a reduction in viability of E. coli greater than 40% within the first 15 min, the higher the copper content the larger the amount of killed bacteria (Table 2 and Fig. 8). At the same time, the pure magnesium hydroxide remains completely inactive. After 1 h, 6.6% of E. coli bacteria stay alive in contact with the x = 0.1 mixed hydroxide, while this percentage drastically drops to less than 0.1% (a 3log10 reduction) for the x = 0.15 and 0.2 samples (Table 2 and Fig. 8). After 3 h, the culture medium is completely free of E. coli and a reduction higher than 6.3log10 was achieved whatever the copper content x of the mixed hydroxides (Table 2). In contrast, the reduction in viability of E. coli does not exceed 23% after 3 h in contact with pure magnesium hydroxide nanometer-sized particles (Table 2 and Fig. 8). Such low bactericidal activity towards E. coli has been reported by Pan et al.16 on hexagonal Mg(OH)2 nanoplatelets (∅ ≈ 700 nm) forming small micrometer-sized aggregates (≈ 1.2 μm) and very recently by Halbus et al.44
To the best of our knowledge, the antibacterial activity of nanometer-sized brucite Mg(OH)2 particles towards Gram-positive S. aureus has been only reported in the literature by Vatsha et al.45 The activity of polyester fabrics coated by micrometer-sized Mg(OH)2 particles (average size > 2.5 μm) was also evaluated by Wang et al.46 In this latter study, the reduction in cell viability was 92 to 97% (1log10 to 1.58log10) after 24 h in contact with micrometer-sized Mg(OH)2 particles at 37 °C. Our results displayed in Table 2 and Fig. 8 show that the bactericidal activity of nanometer-sized brucite Mg(OH)2 particles is slightly higher towards Gram-positive S. aureus than towards Gram-negative E. coli bacteria. However, half of S. aureus bacteria still stay alive after 3 h in contact with the particles (reduction in cell viability ≈48%). This low activity indicates that the bactericidal kinetics of brucite Mg(OH)2 remains very low despite the use of nanometer-sized particles. A longer contact time would be therefore required to inactivate the whole S. aureus culture, as in the study of Wang et al.46 In Fig. 8, copper substitution enhances the bactericidal activity of Mg(OH)2 in a similar trend to that observed for Gram-negative E. coli: the higher the copper content x the faster the bactericidal kinetics. A huge reduction in the counts of viable S. aureus bacteria is observed after 3 h when at least 10 mol% (x = 0.1) of magnesium in brucite Mg(OH)2 is substituted by cupric ions (<1% of viable cells for x = 0.1 and ≈48% for Mg(OH)2). When comparing the activities of copper-substituted Mg(OH)2 nanoparticles towards the two types of bacteria tested in the present study, one can note that the reduction in viability after 3 h in contact is however only around 2log10 for S. aureus bacteria while it was higher than 6.3log10 for E. coli ones.
Makhluf et al.4 have evaluated the activity of MgO particles with different sizes (8, 11, 15, 18 and 23 nm) towards E. coli and S. aureus in the culture medium at 37 °C after 1 h or 4 h in contact. The activity is only significant (reduction in cell viability >95%) when the size of the particles is around 8 nm and the contact time is at least 4 h. For nanometer-sized particles of MgO with a specific surface area of ≈81 m2 g−1 (corresponding to an average ∅ ≈ 20 nm), Smithson and Bakshi11 showed that complete conversion into Mg(OH)2 occurred after only ≈100 min of immersion in water at 38 °C. Any size reduction of MgO particles below ≈20 nm inevitably speeds up the hydration kinetics, the time required for the full conversion into Mg(OH)2 being shorter than 100 min in that way. One has to therefore consider the bactericidal activities of “MgO” nanoparticles reported by Makhluf et al.4 after 4 h in contact as those of Mg(OH)2 nanoparticles. Note that the size of Mg(OH)2 platelets we have prepared by precipitation (Fig. 7b 〈D〉 ≈ 20 nm and 〈H〉 ≈ 7 nm) is roughly equal to the largest particles in Makhluf's study (23 ± 2 nm), thus allowing a direct comparison of their activities. In Makhluf's study, the reduction in cell viability is slightly higher for S. aureus than for E. coli at the same time of treatment and it remains below 50% for both bacteria after 4 h of treatment. These results are very similar to those displayed in the present study for Mg(OH)2. Nevertheless, our study shows that copper substitution can enhance these activities, in particular towards E. coli, thus greatly exceeding the 3log10 reduction reported by Makhluf et al.4 for 8 nm-sized particles of hydrated MgO.
The bactericidal performance of nanometer-sized particles of the mixed hydroxides Mg1−xCux(OH)2−2y(CO3)y·zH2O (0 ≤ x ≤ 0.2) was then compared with that of the conventional inorganic antibacterial agent CuO. For this purpose, the bactericidal activity of CuO particles must be determined in conditions similar to those used for the mixed hydroxides (1 mg mL−1) and ideally with particles of comparable size. 50 nm-sized CuO particles have been therefore synthesized by thermally decomposing nanometer-sized Cu(OH)2 particles. The copper hydroxide was prepared beforehand by the same synthetic route used for the mixed hydroxides. The evaluation of the bactericidal activity of those CuO particles has been then performed and the reduction in viability of both E. coli and S. aureus is displayed in Table 2 as well. At first, the reduction in viability of E. coli does not exceed 4.7log after 3 h in contact with those 50 nm-sized particles of CuO, which is lower than the 6.3log reductions determined with all copper-substituted Mg(OH)2 particles at t = 3 h (i.e. the x = 0.1, 0.15 and 0.2 samples in Table 2). Secondly, the reduction in viability of S. aureus remains below ≈14% within the first hour in contact with 50 nm-sized CuO particles. This reduction drastically rises to ≈99% (2log) after 3 h in contact, thus overtaking the bactericidal performance of all copper-substituted Mg(OH)2 (i.e. the x = 0.1, 0.15 and 0.2 samples in Table 2). As stated above, all copper-substituted Mg(OH)2 hydroxides, whatever their copper content, kill more than half of the S. aureus inoculum within the first 15 min in contact with a reduction in viability continuing to gradually rise with the time. By discarding the biphasic x = 0.2 sample, a good compromise between fast bactericidal kinetics and a high reduction in viability is reached by the 15 mol% copper-substituted Mg(OH)2 hydroxide (Table 2). Its use gives the opportunity to five-fold reduce the copper loading of the bactericidal agent while being at least equally or even more efficient compared to the conventional CuO (a Cu loading of 0.799 mg mL−1 and 0.154 mg mL−1 in the suspension of CuO and 15 mol% copper substituted Mg(OH)2 particles, respectively). This is a significant step towards the design of more environmentally friendly bactericidal agents than the ecotoxic copper oxide.
2.4 Bactericidal mechanism
Makhluf et al. and Applerot et al. have determined the bactericidal mechanism of nanometer-sized MgO4 and CuO5 particles, respectively. For both oxides, the mechanism would result from the following successive actions: (i) generation of exogenic Reactive Oxygen Species (ROS) by the particles attached to the bacterial cell membrane, (ii) an increase in the permeability of the cell envelope due to the damage induced by exogenic ROS, (iii) penetration of individual particles into the cell and (iv) cell lysis with loss of vital intracellular content (proteins, DNA and RNA) damaged by endogenic ROS generation. In ref. 47, it was postulated that the ROS production by metal oxide nanoparticles originates from the electron-donating nature of the surface defect sites (superoxide anion ˙O2− radicals in nanometer-sized MgO4 and CuO5). Pan et al.16 showed that large micrometer-sized aggregates of Mg(OH)2 nanoparticles prepared by precipitation from MgCl2 salt (average ∅ ≈ 5.5 μm and a reduction in E. coli viability around 90% after 24 h) have higher bactericidal activity than poorly agglomerated nanoplatelets resulting from the hydration of MgO particles (aggregates of ∅ ≈ 1.2 μm and a reduction in viability around 50% after 24 h). These authors relate the high activity of large micrometer-sized aggregates of particles to their better adsorption onto the cell envelopes of E. coli, highlighted by imaging the bacteria exposed to particles with an electron microscope. After exposition to Mg(OH)2 slurries, observations made by fluorescence microscopy on stained bacteria revealed that lethality of E. coli was due to cell lysis. Both observations made by Pan et al.16 are similar to those of Applerot et al.5 for CuO particles. It gives clear evidence that Mg(OH)2 and CuO particles exhibit the same bactericidal mechanism. In the Applerot et al. study,5 the reductions in viability of E. coli and S. aureus after 3 h in contact with ≈7 nm-sized CuO particles were 99.9% and 97%, respectively. These reductions are similar to those reported by Makhluf et al.4 after 4 h in contact with ≈8 nm-sized hydrated MgO particles (i.e. Mg(OH)2). In addition, the bactericidal kinetics of CuO nanoparticles is as fast as that of Mg(OH)2 ones. It would mean that the ability of CuO and Mg(OH)2 nanoparticles to be adsorbed onto the cell envelope of bacteria and to produce ROS is comparable when they are of equivalent size. Cu+2O also exhibits antibacterial activity towards E. coli and S. aureus.48 This activity originates from the ability of the oxide to strongly adsorb and damage fumarase A enzymes found in the cell membrane of bacteria. A significant decrease of enzyme catalytic activity, vital for the bacteria, is then observed, which is ascribed to a displacement of iron from the catalytic [4Fe-4S] clusters by cuprous ions (thiophilic character).49 This mechanism was later confirmed by Meghana et al.50Although the bactericidal mechanism of the Mg1−xCux(OH)2−2y(CO3)y·zH2O samples should be similar to that of the brucite Mg(OH)2, one might wonder why the copper substitution greatly enhances the bactericidal properties, whether in terms of the kinetics or of the activity level: the higher the copper content the faster the kinetics and the higher the activity level.
Nano-structuring is known to promote the formation of surface defects (an incomplete coordination sphere of cations at the grain surface), which could also lead locally to a slight reduction of cupric ions to cuprous ones. A thorough examination of the chemical state of the surface was performed on all Mg1−xCux(OH)2−2y(CO3)y·zH2O samples by X-ray photoelectron spectroscopy (XPS) in order to determine the oxidation state(s) of the copper ions. The results are presented in the ESI† (section 3). From this XPS study, the presence of cuprous ions detected at the grain surface would result from partial photo-reduction of cupric ions when the samples are exposed to X-rays in the secondary vacuum condition. Because of the absence of cuprous ions at the grain surface of as-prepared powders, the enhancement of the bactericidal activity of Mg(OH)2 through copper substitution cannot be ascribed to a decrease of the enzyme catalytic activity.
Sawai et al.13 considered that the alkaline effect caused by the hydration of MgO is one of the contributing factors to its biocidal activity against eukaryotic cells like yeasts (Saccharomyces cerevisiae and Candida albicans) and filamentous fungi (Aspergillus niger and Rhizopus stolonifer). Mendonca et al.51 showed that the reduction in viability was around 8log10 after only 15 min in contact with a sterile NaHCO3–NaOH buffer at pH = 11, while it was only 0.5log10 at pH = 10. This latter study demonstrates the deleterious effect of one pH unit increase on the cell viability. The significant enhancement of the bactericidal activities through copper substitution in brucite Mg(OH)2 could originate from a copper dependent increase in pH of the water suspension of nanoparticles. To clarify this point, the pH was measured for water suspensions of x = 0 and x = 0.15 nanoparticles at the same specified time and concentration (1 mg mL−1) used in the antibacterial tests. The values are gathered together in Table 3. For both samples, a strong rise in pH occurs in the first 15 min after immersing the nanoparticles in water. After 180 min of immersion, the pH values are around 11 and 10.4 for the x = 0 and x = 0.15 samples, respectively. This is in good agreement with the value of 10 and 10.6 measured on broth media containing Mg(OH)252 and MgO4 nanoparticles, respectively. At this basic pH, the reduction in cell viability determined by Makhluf et al.4 was however zero for E. coli and less than 6% for S. aureus after 4 h in contact with hydrated MgO nanoparticles. Thereby, the high antibacterial activity of copper-substituted Mg(OH)2 does not originate from a stronger alkaline effect because copper substitution tends to slightly lower the pH of the water suspension of particles (Table 3).
| Immersion time (min) | x = 0 | x = 0.15 | |||
|---|---|---|---|---|---|
| pH | Mg (mg L−1) | pH | Mg (mg L−1) | Cu (mg L−1) | |
| 0 | 5.7 | — | 5.7 | — | — |
| 15 | 9.54 | — | 8.18 | — | — |
| 30 | 9.85 | — | 8.35 | — | — |
| 60 | 9.62 | — | 8.70 | — | — |
| 180 | 10.96 | 7.35 | 10.38 | 7.46 | 0.44 |
| 1440 | 10.51 | 9.86 | 9.85 | 10.52 | 0.10 |
It is well known that free hydrated cupric ions can catalyze ROS generation from aqueous H2O2 through Fenton-like redox cycling between Cu2+ and Cu+ ions.53,54 Any release of cupric ions due to the partial dissolution of particles in the culture medium could catalyze exogenic ROS production from H2O2, a by-product of the normal metabolism of oxygen. The higher the release, the higher the bactericidal activity. This however presupposes that the dissolution rate of Mg1−xCux(OH)2−2y(CO3)y·zH2O particles would rise as the copper content x increases. Atomic emission spectroscopy has been therefore used for measuring the amount of magnesium and copper elements possibly released from particles of the Mg(OH)2−2y(CO3)y·zH2O (x = 0) and Mg0.85Cu0.15(OH)2−2y(CO3)y·zH2O (x = 0.15) hydroxides when they are immersed in deionized water at the tested concentration of 1 mg mL−1. The measurement was performed on the supernatant after 3 h and 24 h of immersion. Table 3 displays the results of these analyses. For both the x = 0 and x = 0.15 samples, the magnesium concentration in the supernatant almost reaches the same value of ≈7.4 mg L−1 and ≈10 mg L−1 after 3 h and 24 h of immersion, respectively. The solubility of Mg(OH)2 in pure water calculated from its solubility product (Ksp = 5.61 × 10−12 in ref. 55) cannot theoretically exceed ≈1.12 × 10−4 mol L−1 or ≈6.53 mg L−1. Our measurements show that this solubility limit is attained for an immersion time of 3 h (Table 3). A slight acidification of water occurs with time (Table 3), which could explain why the magnesium concentration is slightly higher than the theoretical solubility limit for 24 h. When Mg0.85Cu0.15(OH)2−2y(CO3)y·zH2O particles start dissolving in the supernatant, it is expected that the amount of released copper ions is proportional to that of released magnesium ions in compliance with the Cu/Mg ratio of the composition. If it is respected, the copper concentration in the supernatant calculated from the magnesium concentration should be equal to ≈3.44 mg L−1 and ≈4.85 mg L−1 after 3 h and 24 h of immersion, respectively. Surprisingly, the copper concentration in the supernatant is only around 0.5 mg L−1 after 3 h and even drops to 0.1 mg L−1 after 24 h. These low copper concentrations in the supernatant are certainly related to the solubility product of Cu(OH)2 in pure water (Ksp = 2.2 × 10−20 in ref. 56), which is much lower than that of Mg(OH)2. With magnesium ions being released from the particles in a larger amount that copper ions, it would mean that surface enrichment of copper of the Mg1−xCux(OH)2−2y(CO3)y·zH2O particles or precipitation of Cu(OH)2 from the solvated copper ions occurs with the immersion time. In our opinion, the second hypothesis remains the most likely because the copper concentration of the supernatant decreases with the immersion time (Table 3). The presence of Cu(OH)2 in the powder recovered by filtration of the water suspension of Mg0.85Cu0.15(OH)2−2y(CO3)y·zH2O particles was not however detected by XRPD certainly because of its small amount. It is worth noting that the magnesium concentration in the supernatant is very close for both Mg(OH)2−2y(CO3)y·zH2O (x = 0) and Mg0.85Cu0.15(OH)2−2y(CO3)y·zH2O (x = 0.15) hydroxides despite their different magnesium contents. Copper substitution would therefore increase the dissolution rate of the nanoparticles in water and at the same time would favor the precipitation of Cu(OH)2 particles: the higher the copper content x the larger the amount of precipitated Cu(OH)2 particles. Akhavan et al.15 have demonstrated that copper hydroxide exhibits bactericidal activity. In that way, the precipitation of Cu(OH)2 particles could be one factor contributing to the enhancement of the bactericidal activity of brucite Mg(OH)2. But, the low amount of Cu(OH)2 particles in the water suspension makes too small its contribution to the activity. The majority of ROS are therefore produced from copper-substituted Mg(OH)2 particles rather than from the very low amount of free hydrated cupric ions or Cu(OH)2 particles.
In Fig. 7b, a homothetic size reduction of the platelet-like particles is noted as the copper content x increases along the Mg1−xCux(OH)2−2y(CO3)y·zH2O series. Despite that, all raw powders have similar specific surface area whatever their copper content x (Fig. 6a–d). As the TEM images showed (Fig. 6), this feature originates from the tendency of platelets to form larger and larger shapeless aggregates as the platelet shape changes with the copper content x (an increase of the 〈D〉/〈H〉 ratio with the copper content x in Fig. 7b). The agglomeration thus annihilates the beneficial effect of the size reduction on the specific surface area. Pan et al.16 showed that the bactericidal activity of Mg(OH)2 nanoparticles can be greatly enhanced when they are mass adsorbed onto cell envelopes of E. coli bacteria. In this latter study, poorly agglomerated and large nanoplatelets (∅particle ≈ 700 nm, ∅aggregate ≈ 5.5 μm) exhibit lower bactericidal activity than large and shapeless micrometer-sized aggregates of small nanoflakes (∅particle ≈ 10 nm, ∅aggregate ≈ 5.5 μm). In the present study, copper substitution enhances the antibacterial activity of Mg(OH)2 nanoparticles by promoting the formation of shapeless aggregates with better mass adsorption onto cell envelopes of bacteria. It has been shown that the carotenoid pigments, giving the golden color to the S. aureus bacteria envelope, have antioxidant properties.57 These pigments can help these bacteria to detoxify ROS produced by copper-substituted Mg(OH)2 nanoparticles adsorbed onto their envelopes, thus decreasing their susceptibility to antimicrobial treatment, especially for contact times as long as 3 h (Table 2). Deprived of this antioxidant molecule, no Gram-negative E. coli survive to 3 h of treatment with copper-substituted Mg(OH)2 nanoparticles (Table 2).
3. Conclusion and perspective
Improvement of the surface driven properties of a material originates from the combination of its appropriate chemical modification with the nano-structuring of particles. Copper substitution was here applied with the aim to increase the bactericidal performance of brucite-type magnesium hydroxide nanoparticles. The partial substitution of magnesium ions in Mg(OH)2 by Cu2+, which cannot exceed ≈15 mol%, promotes the reduction in size and the aggregation of nanometer-sized platelets. Because the platelets are prepared at room temperature by a precipitation route in mixed alkaline (NaOH/Na2CO3) medium, a very few hydroxyl ions are substituted by carbonate groups, which form pillars between two consecutive layers of edge-sharing octahedra in the brucite-type structure of the mixed hydroxides. This controlled incorporation of cupric ions in Mg(OH)2 nanoparticles significantly enhances their bactericidal performance against two bacteria frequently involved in healthcare-associated-infections. 10 mol% of cupric ions in Mg(OH)2 (a copper loading of 0.102 mg mL−1 in the suspension) were sufficient to induce after 3 h in contact 100% and 99.3% reductions in viability of Gram-negative E. coli and Gram-positive S. aureus, respectively (reductions as low as 23% and 48% are reported for the parent compound Mg(OH)2 in the same conditions). This higher antibacterial activity of copper-substituted Mg(OH)2 nanometer-sized platelets is likely due to their better mass adsorption onto cell envelopes of bacteria. A good compromise between fast bactericidal kinetics and high reduction in viability is reached by the 15 mol% copper-substituted Mg(OH)2 hydroxide. Its use gives the opportunity to five-fold reduce the copper loading of the bactericidal agent while being at least equally or even more efficient compared to the conventional CuO (a Cu loading of 0.799 mg mL−1 and 0.154 mg mL−1 in the suspension of CuO and 15 mol% copper substituted Mg(OH)2 particles, respectively). This is a significant step towards the design of more environmentally friendly bactericidal agents than the ecotoxic copper oxide. Due to the long-term geological availabilities of magnesium and copper together with the simplicity and the low-cost of their preparation, these copper-substituted magnesium hydroxides have great application potential as bactericidal materials for reducing the risk of healthcare-associated-infections (HAIs). However, their human toxicity must be assessed prior to any practical use. Such a study will be the object of a subsequent investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French Ministry of Higher Education, Research and Innovation and Le Mans University are acknowledged for providing a doctoral grant to BC.References
- IVS, (Institut de Veille Sanitaire)-Réseau d’alerte, d’investigation et de surveillance des infections nosocomiales (Raisin). Enquête nationale de prévalence des infections nosocomiales et des traitements anti-infectieux en établissements de santé, France, mai-juin 2012 (http://www.invs.sante.fr/enp) 2013, 1–186.
- J. Sawai, H. Kojima, H. Igarashi, A. Hashimoto, S. Shoji, T. Sawaki, A. Hakoda, E. Kawada, T. Kokugan and M. Shimizu, World J. Microbiol. Biotechnol., 2000, 16, 187–194 CrossRef CAS.
- L. Huang, D.-Q. Li, Y.-J. Lin, M. Wei, D. G. Evans and X. Duan, J. Inorg. Biochem., 2005, 99, 986–993 CrossRef CAS.
- S. Makhluf, R. Dror, Y. Nitzan, Y. Abramovich, R. Jelinek and A. Gedanken, Adv. Funct. Mater., 2005, 15, 1708–1715 CrossRef CAS.
- G. Applerot, J. Lellouche, A. Lipovsky, Y. Nitzan, R. Lubart, A. Gedanken and E. Banin, Small, 2012, 8, 3326–3337 CrossRef CAS.
- A. Azam, A. S. Ahmed, M. Oves, M. S. Khan and A. Memic, Int. J. Nanomed., 2012, 7, 3527–3535 CrossRef CAS.
- A. F. Halbus, T. S. Horozov and V. N. Paunov, ACS Appl. Mater. Interfaces, 2019, 11, 12232–12243 CrossRef CAS.
- V. Aruoja, S. Pokhrel, M. Sihtmae, M. Mortimer, L. Madler and A. Kahru, Environ. Sci.: Nano, 2015, 2, 630–644 RSC.
- R. I. Razouk and R. S. Mikhail, J. Phys. Chem., 1958, 62, 920–925 CrossRef CAS.
- G. K. Layden and G. W. Brindley, J. Am. Ceram. Soc., 1963, 46, 518–522 CrossRef CAS.
- G. L. Smithson and N. N. Bakhshi, Can. J. Chem. Eng., 1969, 47, 508–513 CrossRef.
- J. Blaha, Ceram.-Silik., 1995, 39, 45–51 CAS.
- J. Sawai and T. Yoshikawa, J. Appl. Microbiol., 2004, 96, 803–809 CrossRef CAS.
- C. Dong, D. Song, J. Cairney, O. L. Maddan, G. He and Y. Deng, Mater. Res. Bull., 2011, 46, 576–582 CrossRef CAS.
- O. Akhavan, R. Azimirad, S. Safa and E. Hasani, J. Mater. Chem., 2011, 21, 9634–9640 RSC.
- X. Pan, Y. Wang, Z. Chen, D. Pan, Y. Cheng, Z. Liu, Z. Lin and X. Guan, ACS Appl. Mater. Interfaces, 2013, 5, 1137–1142 CrossRef CAS.
- C. Henrist, J. P. Mathieu, C. Vogels, A. Rulmont and R. Cloots, J. Cryst. Grow., 2003, 249, 321–330 CrossRef CAS.
- Y. Cudennec, A. Lecerf and Y. Gerault, Eur. J. Solid State Inorg. Chem., 1995, 32, 1013–1022 CAS.
- Y. Cudennec, A. Riou, Y. Gérault and A. Lecerf, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 661–666 CrossRef CAS.
- L. Desgranges, G. Calvarin and G. Chevrier, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 82–86 CrossRef.
- M. M. Harding, B. M. Kariuki, R. Cernik and G. Cressey, Acta Crystallogr., Sect. B: Struct. Sci., 1994, 50, 673–676 CrossRef.
- R. Allmann, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 972–977 CrossRef CAS.
- L. Ingram and H. F. W. Taylor, Mineral. Mag., 1967, 36, 465–479 CrossRef CAS.
- R. Allmann and H.-H. Lohse, Neues Jahrb. Mineral., Monatsh., 1966, 6, 161 Search PubMed.
- R. Allmann, Chimia, 1970, 24, 99 CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- V. A. Drits and A. S. Bookin, in Layered Double Hydroxides: Present and Future, ed. V. Rives, Nova Science, New York, 2001 Search PubMed.
- F. A. Miller and C. H. Wilkins, Anal. Chem., 1952, 24, 1253–1294 CrossRef CAS.
- R. Martens and F. Freund, Phys. Status Solidi A, 1976, 37, 97–104 CrossRef CAS.
- J. R. Gunter and H. R. Oswald, J. Appl. Crystallogr., 1970, 3, 21–26 CrossRef.
- A. De Roy and J. P. Besse, Solid State Ionics, 1989, 35, 35–43 CrossRef.
- S. K. Yun and T. J. Pinnavaia, Chem. Mater., 1995, 7, 348–354 CrossRef CAS.
- R. Pappalardo, J. Mol. Spectrosc., 1961, 6, 554–571 CrossRef CAS.
- J. J. Freeman and R. M. Friedman, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 758–761 RSC.
- D. N. Sathyanarayana, Electronic Absorption Spectroscopy and Related Techniques, Universities Press, 2001.
- M. Behrens and F. Girgsdies, Z. Anorg. Allg. Chem., 2010, 636, 919–927 CrossRef CAS.
- P. Süsse, Acta Crystallogr., 1967, 22, 146–151 CrossRef.
- A. Le Bail, H. Duroy and J. L. Fourquet, Mater. Res. Bull., 1988, 23, 447–452 CrossRef CAS.
- J. I. Langford, International Conference “Accuracy in Powder Diffraction II”, May 26-29, 1992 at NIST, Gaithersburg, MD, USA, 1992, 110–126.
- J. Rodriguez Carvajal, Phys. B, 1993, 192, 55–69 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- V. Lakshmi Prasanna and R. Vijayaraghavan, Langmuir, 2015, 31, 9155–9162 CrossRef CAS.
- H. T. Michels, J. O. Noyce and C. W. Keevil, Lett. Appl. Microbiol., 2009, 49, 191–195 CrossRef CAS.
- A. F. Halbus, T. S. Horozov and V. N. Paunov, Biomimetics, 2019, 4, 41 CrossRef.
- B. Vatsha, P. Tetyana, P. Shumbula, J. Ngila, L. Sikhwivhilu and R. Moutloali, J. Biomater. Nanobiotechnol., 2013, 4, 365–373 CrossRef.
- Y. Wang, L. Sha, J. Zhao, Q. Li, Y. Zhu and N. Wang, Appl. Surf. Sci., 2017, 400, 413–419 CrossRef CAS.
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grätzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1999, 99, 77–174 CrossRef CAS.
- K. Sunada, M. Minoshima and K. Hashimoto, J. Hazard. Mater., 2012, 235–236, 265–270 CrossRef CAS.
- L. Macomber and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8344–8349 CrossRef CAS.
- S. Meghana, P. Kabra, S. Chakraborty and N. Padmavathy, RSC Adv., 2015, 5, 12293–12299 RSC.
- A. F. Mendonca, T. L. Amoroso and S. J. Knabel, Appl. Environ. Microbiol., 1994, 60, 4009–4014 CAS.
- C. Dong, J. Cairney, Q. Sun, O. Maddan, G. He and Y. Deng, J. Nanopart. Res., 2010, 12, 2101–2109 CrossRef CAS.
- A. N. Pham, G. Xing, C. J. Miller and T. D. Waite, J. Catal., 2013, 301, 54–64 CrossRef CAS.
- T. T. M. Nguyen, H.-J. Park, J. Y. Kim, H.-E. Kim, H. Lee, J. Yoon and C. Lee, Environ. Sci. Technol., 2013, 47, 13661–13667 CrossRef CAS.
- D. R. Lide, ed. CRC Handbook of Chemistry and Physics 2008-2009: A Ready-Reference Book of Chemical and Physical Data, CRC Press, Boca Raton, Florida, USA, 2008 Search PubMed.
- Handbook of Inorganic Chemicals, ed. P. Patnaik, McGraw-Hill, USA, 2003 Search PubMed.
- G. Y. Liu, A. Essex, J. T. Buchanan, V. Datta, H. M. Hoffman, J. F. Bastian, J. Fierer and V. Nizet, J. Exp. Med., 2005, 202, 209–215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Experimental. (2) Origin of the lattice expansion when dehydrating samples. (3) Chemical state of surface particles probed by X-ray Photoelectron Spectroscopy (XPS). See DOI: 10.1039/c9tb01927h |
| This journal is © The Royal Society of Chemistry 2020 |