
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical dealloying as a tool to tune the porosity, composition and catalytic activity of nanomaterials†
Christian
Rurainsky
a,
Alba Garzón
Manjón
b,
Felix
Hiege
a,
Yen-Ting
Chen
c,
Christina
Scheu
*b and
Kristina
Tschulik
 *a
*a
aFaculty of Chemistry and Biochemistry, Chair of Analytical Chemistry II, Ruhr University Bochum, Bochum, Germany. E-mail: kristina.tschulik@rub.de
bMax-Planck-Institut für Eisenforschung GmbH, Max-Planck-Straße 1, 40237 Düsseldorf, Germany. E-mail: scheu@mpie.de
cCenter for Solvation Science (ZEMOS), Ruhr University Bochum, Bochum, Germany
First published on 18th August 2020
Abstract
Electrochemical dealloying as a post-treatment can greatly improve the catalytic activity of nanoparticles. To date, selecting suitable conditions to reach desired porosity, composition and catalytic activity is based on trial-and-error-attempts, due to insufficient understanding of the electrochemically induced morphological and compositional changes of the nanoparticles. These changes are elucidated here by combining electrochemistry with identical location electron microscopy analyses and linking them to the electrocatalytic properties of the obtained nanocatalysts. Using AgAu alloy nanoparticles and the hydrogen evolution reaction as a model system, the influence of cyclic voltammetry parameters on the catalytic activity upon electrochemical dealloying is investigated. Increasing the number of cycles initially results in a decreased Ag content and a sharp improvement in activity. Additional dealloying increases the nanoparticle porosity, while marginally altering their composition, due to surface motion of atoms. Since this is accompanied by particle aggregation, a decrease in catalytic activity results upon extensive cycling. This transition between porosity formation and particle aggregation marks the optimum for nanocatalyst post-production. The gained insights may aid speeding up the development of new materials by electrochemical dealloying as an easy-to-control post-processing route to tune the properties of existing nanoparticles, instead of having to alter usually delicate synthesis routes as a whole.
Introduction
The partial dissolution of an element, in most cases a metal, out of a composite or alloy has usually been considered undesirable, for instance in the context of corrosion. This view has changed in the early 2000s when researchers started to purposely remove Ag from AgAu bulk alloys to create nanoporous gold (NPG) and studied the associated evolution of nanoporosity.1 NPG and therefore the precursor alloys, mostly AgAu, have been investigated for use as a catalyst, for instance in alcohol coupling2,3 and oxidation.4 Methanol oxidation in fuel-cell applications5 and proton reduction for hydrogen production6 are the most prominent examples for this. Progressing this approach from bulk alloys to nanoparticles has the potential to further increase the specific activity through both improved mass transport and decreased material amounts, consequently reducing the cost of these catalysts. Thus, we present here a systematic in-depth study into the behavior of AgAu nanoparticles to elucidate this potential and identify underlying material property–activity relations.The intentional selective dissolution of components from an alloy is called dealloying and has been deployed for various bi- and multi-metallic systems.7–11 The general mechanistic understanding of the evolution of porosity and the morphological changes during dealloying are comparably well understood for bulk systems and micrometer scale thin films. There is a well-established model for the evolution of pores12 and the control over ligament sizes and their coarsening has been widely researched.13–16 Following the discovery that dealloyed CuPt nanoparticles showed excellent catalytic properties towards the oxygen reduction reaction (ORR),17 dealloying of nanoparticles has attracted more interest in the last decade. Since then, chemical and electrochemical dealloying of nanoparticles has been done for a variety of different alloys and for different catalytic reactions.18–22 For some reactions like the hydrogen evolution reaction (HER)23 and ORR24 a structure–performance relationship could even be established for dealloyed materials. Simulations have shown that in order to get percolation dealloying, which is needed to produce porosity, the alloy should at least contain 60 at% of the less noble metal.25 Experimentally it was found that for AgAu about 55 at% Ag is required in the alloy, which is in good agreement with the theoretical model. In addition to composition, for nanoparticles the size is an important factor for the formation of porosity. Nanoparticles that are too small do not show porosity after dealloying. They only get rougher and enriched in the more noble metal component on the surface, because forming pores are too quickly closed by atoms of the more noble metal diffusing on the surface at small length scale.18 The dealloying behavior is thus governed by the relationship of dissolution speed and surface diffusion of the more noble metal component. The size requirement for porosity formation differs for different systems; for NiPt it was found to be around 15 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and for AgAu around 25 nm26 in diameter, respectively.
8 and for AgAu around 25 nm26 in diameter, respectively.
The advantage of electrochemical over chemical dealloying is the ability to steer the process more precisely. Besides the electrolyte composition and time the dealloying potential can also be controlled during potentiostatic dealloying.21 In addition, utilizing techniques like cyclic voltammetry (CV), the applied potential and the speed at which it is altered during dealloying can be adjusted. This grants more control over the partial dissolution and surface reorganization of the nanoparticles, and hence over the composition and porosity of the formed material.26
An in-depth study of the dealloying behavior of AgAu nanoparticles during potentiostatic dealloying has been reported by Erlebacher, Sieradzki et al.26 They observed pore formation under these constant potential conditions and suggested similar behavior for dealloying by cyclic voltammetry at scan rates below 0.1 V s−1. In contrast to that, the authors predicted the formation of hollow core–shell nanoparticles with an Au enriched outer shell for higher scan rates.
Due to this potentially greater control over the produced particle morphology based on the experimental parameters, in this work we focus on cyclic voltammetry to dealloy AgAu nanoparticles. Varying cycling numbers are used to determine the effects of extended dealloying on nanoparticles and find out whether this is beneficial to gain certain morphologies and for catalytic applications. We use the HER as an industrially important benchmark reaction to link the evolving catalyst activity to the morphology, composition and possible porosity of dealloyed nanoparticles. Based on these results, we provide guidelines on how to select dealloying parameters to obtain high porosities and/or high catalytic activity.
Experimental section
The synthesis of Ag, Au and AgAu nanoparticles was carried out according to Grasmik et al.27 based on a modified version of the method by Turkevich et al.28 The monometallic nanoparticles were made as a comparison to make sure the observed effects are only present in the alloy nanoparticles.Silver–gold nanoparticles
50 mL of ultra-pure water (Thermo Scientific Barnstead GenPure xCAD Plus, 0.055 μS cm−1 at 25 °C) was heated in a 100 mL round bottom flask until boiling. 100 μL of 10 mM HAuCl4 (99.99% metal basis, Alfa Aesar) and 400 μL of 10 mM AgNO3 (99.9995% metal basis, Alfa Aesar) solution were quickly added, followed by 500 μL of a 60 mM trisodium citrate (AnalaR NORMAPUR, VWR Chemicals) solution, while vigorously stirring the solution. The mixture was boiled under reflux for 1 h and then quickly cooled down to room temperature with an ice bath, while adding another 500 μL of the 60 mM trisodium citrate solution. Afterwards, the mixture was centrifuged (Centrifuge 5810 R, Eppendorf) for 20 min at 20000 rcf and the supernatant liquid was removed until less than 200 μL was left and then refilled with ultra-pure water to 2 mL.
Silver nanoparticles
50 mL of ultra-pure water was heated in a 100 mL round bottom flask until boiling. 500 μL of 10 mM AgNO3 solution followed by 500 μL of a 60 mM tri-sodium citrate solution were quickly added under vigorous stirring. The mixture was boiled under reflux for 1 h and no color change was observed. Afterwards, another 500 μL of the 60 mM tri-sodium-citrate solution followed by 2 mg D-(+)-glucose (AnalaR NORMAPUR, VWR Chemicals) were added to the mixture to initiate nucleation. The mixture was boiled and stirred for another 1 h. Afterwards it was cooled down to room temperature using an ice bath. The mixture was then centrifuged for 20 min at 20000 rcf and the supernatant solution was removed until about 200 μL was left; the suspension was refilled to 2 mL using ultra-pure water.
Gold nanoparticles
50 mL of ultra-pure water was heated in a 100 mL round bottom flask until boiling. 500 μL of 10 mM HAuCl4 solution and 500 μL of 60 mM trisodium citrate solution were quickly added under vigorous stirring. The mixture was boiled under reflux for 1 h and afterwards quickly cooled down to room temperature using an ice bath. During the cooling another 500 μL of the 60 mM trisodium citrate solution was added. Afterwards the mixture was centrifuged for 20 min at 20000 rcf and the supernatant liquid was removed until about 200 μL was left. The remaining particle suspension was then refilled to 2 mL with ultra-pure water.
Electrochemical setup
For all electrochemical measurements, a glassy carbon electrode (GCE) (d = 4 mm) was used as a working electrode (WE). The electrode was polished to a mirror finish with 1 μm, 0.3 μm and 0.05 μm Al2O3 particle suspensions (LECO Instruments GmbH), before measurements. After polishing, the electrode was cleaned by ultrasonication (Elmasonic S 100H) in ultra-pure water for 3 min. Next, 4 μL of the nanoparticle suspension were dropcast onto the electrode and dried in an Ar stream. A graphite rod (d = 6 mm) was used as the counter electrode (CE) and either a Ag/AgCl (3 M KCl) (E = 207 mV29vs. standard hydrogen electrode (SHE), SI Analytics GmbH) or a mercury–mercurous sulfate electrode (MSE, E = 658 mV29vs. SHE, SI Analytics GmbH) for chloride free electrolytes was used as a reference electrode (RE). Measurements were run at 25 °C using an AutoLab PGStat 12 potentiostat.
Electrochemical nanoparticle characterization was performed following our previously developed protocol.27 Cyclic voltammetry (CV) measurements were performed in 0.1 M HCl, which was diluted from 37% HCl (AnalaR NORMAPUR, Reag. Ph. Eur.) using ultra-pure water, between −0.2 V and 1.25 V vs. Ag/AgCl (3 M KCl) at a scan rate of 0.05 V s−1.
To determine the electrochemically accessible surface area of nanoparticles before and after dealloying, under potential deposition (UPD) of Pb2+ on the nanoparticles was done in de-oxygenated 0.1 M NaOH (VWR chemicals, Reag. Ph. Eur.) solution with 1 mM Pb(NO3)2 (Riedel-de-Haen). CVs were recorded between −0.44 V and −1.16 V vs. MSE at 0.1 V s−1 for the UPD.
Electrochemical dealloying of the nanoparticles was done in 1.0 M HClO4, which was diluted from 70% HClO4 (AnalaR NORMAPUR) with ultra-pure water. The used potential window was 0.00 V to 1.25 V vs. MSE at a scan rate of 1.0 V s−1. All measurements were scanned in positive directions initially and were stopped after the potential reversal in the final scan at 0.7 V (instead of 0.0 V) to avoid re-deposition of silver ions leached before. Dealloying was achieved using half a cycle, 100, 500, 1000, 2000 and 4000 cycles, respectively.
The hydrogen evolution reaction (HER) was investigated using linear sweep voltammetry (LSV) between −0.6 V and −1.1 V vs. MSE in 0.5 M H2SO4 (made from 95–97%, Fisher Scientific, pH = 0.47) at a scan rate of 0.002 V s−1. The potential was later calculated using the Nernst equation to get the potential against the reversible hydrogen electrode (RHE). All measurements were performed under an Ar atmosphere and the solution was deoxygenated prior to the measurements. Before the catalytic measurements, the catalyst-modified electrode was conditioned by cycling between −0.6 and −1 V at 0.1 V s−1 for 500 times to receive a stable and reproducible catalytic response.30,31
Scanning transmission electron microscopy (STEM) analyses were carried out using a Cs-corrected Titan Themis 60-300 X-FEG microscope with a high-angle annular dark-field (HAADF) detector (73–352 mrad) using a camera length of 48 mm in combination with energy dispersive X-ray (EDX) chemical mapping. The microscope was optimized with a convergence semi-angle of ∼23.8 mrad, a beam size of ∼0.1 nm and a ∼150 pA current.
Identical location microscopy measurements were performed employing a JEOL microscope (JEM-2800) with a Schottky-type emission source working at 200 kV, equipped with a Gatan OneViewIS camera (4k by 4k, 25 fps). The lattice resolution of the TEM images was 0.09 nm. The point-to-point resolution is 0.20 nm and 0.14 nm for STEM. The spherical aberration and the chromatic aberration were 0.7 mm and 1.3 mm, respectively. EDX mapping was performed with the equipped double silicon drift detectors, with a solid angle of 0.98 steradians with a detection area of 100 mm2, and with 133 eV spectral resolution.
About 3 μL of the nanoparticle suspension was dropcast onto TEM grids for analysis. For the untreated nanoparticles carbon filmed Cu grids (Plano GmbH) were used. For identical location dealloying experiments, the nanoparticles were dropcast onto carbon coated Au grids (Plano GmbH), which were then used as the WE for electrochemical dealloying, since grids from other materials dissolve during the dealloying.
Results and discussion
Dealloying and nanoparticle analyses
Silver–gold nanoparticles were synthesized with a nominal Ag:Au molar ratio of 80:20. The characterization of the nanoparticles by STEM, energy dispersive X-ray (EDX) chemical mapping and electrochemical analysis techniques confirmed this composition, yielding (76 ± 5) and (83 ± 6) mol% Ag, respectively. STEM EDX measurements done on Au grids and on Cu grids showed similar results within the margin of error, confirming that a quantitative analysis of the nanoparticle composition is possible also when using Au grids as a support. This is essential for quantitative analysis of the identical location microscopy data before and after electrochemical dealloying. STEM-HAADF images taken of the untreated AgAu nanoparticles (Fig. 1) show mostly spherical nanoparticles of (30 ± 9) nm in diameter. The average particle size was also determined electrochemically, by measuring the surface area of nanoparticles immobilized on a glassy carbon electrode (GCE) using under potential deposition (UPD) and relating this to the particle volume that was determined afterwards by electrochemical dissolution.27 The determined particle size was (35 ± 7) nm in accordance with the STEM data. The EDX elemental mapping shows nanoparticles with an alloyed Au rich core and an Ag rich shell, as has been reported before.27 The Ag rich shell can also be seen in the CV of the alloy nanoparticles in HCl (Fig. 2). For comparison, pure Ag nanoparticles show a sharp oxidation peak at around 0.1 V in HCl (Fig. 2 green), while pure Au nanoparticles show a broader oxidation peak starting around 0.8 V (Fig. 2 red). Ag forms sparingly soluble AgCl(s)32 and Au forms soluble [AuClx](aq)− complexes,33 according to the following reactions.| Ags + Claq− ⇌ AgCls + e− | (1) |
| Aus + 4Claq− ⇌ [AuCl4]aq− + 3e− | (2) |
| [AuCl4]aq− + 2Aus + 2Claq− ⇌ 3[AuCl2]aq− | (3) |
 | ||
| Fig. 1 STEM-HAADF image of untreated Ag80Au20 nanoparticles (left), EDX elemental mapping of the nanoparticles (right) shows alloy nanoparticles with a Au-rich core. | ||
 | ||
| Fig. 2 CV of pure Ag nanoparticles (green), pure Au nanoparticles (red) and Ag:Au 80:20 nanoparticles (black) in 0.1 M HCl(aq) measured at a scan rate of 0.05 V s−1. | ||
The CV of the AgAu nanoparticles (Fig. 2 black) does not display two completely separate peaks, but an initial silver oxidation peak, followed by a region of increased current due to dealloying of the AgAu alloy merging into an Au oxidation peak. This type of response has been shown to differentiate alloys from core shell nanoparticles before.27,34 The used Ag and Au nanoparticles were found to be (28 ± 3) nm and (61 ± 9) nm in diameter, respectively, using electrochemical methods.
The electrochemical dealloying of the AgAu nanoparticles is schematically shown in Fig. 3. The Ag will be leached out of the alloy and a porous or hollow Au rich structure is supposed to remain.26 The dealloying is performed using various numbers of cycles in 1.0 M HClO4. In the first cycle, the dealloying CVs of AgAu nanoparticles show a distinct Ag oxidation peak, starting at around 0.3 V (Fig. 4a). This peak is followed by a region of increased current, compared to the base line current before the peak. This current is due to Ag, which has been stabilized by alloying with Au, being leached out of the alloy (dealloying).34 At around 1.1 V, a second peak is observed, which is associated with surface oxidation of Au. In agreement with the literature, no dissolution of Au was observed in HClO4.35,36 The cathodic (negative) peak detected at around 0.3 V on the reverse scan is associated with a partial reduction of Ag+(aq) ions back to Ag(s) at the electrode. In the second cycle, a slightly shifted Ag oxidation peak is observed between 0.3 V and 0.4 V but no dealloying region is visible in the CV, in agreement with previous findings.34 Starting between the 70th and 100th cycle, peaks are no longer observable in the CV and subsequent cycles are nearly identical. This may be interpreted to indicate that no further dealloying occurs. Under this assumption, the number of cycles for dealloying has been reported as the minimum number necessary to receive a stable response in a CV, in previous work.37 Indeed, the overall amount of Ag found in the nanoparticles, using electrochemical methods, after dealloying (see Fig. 4b) does not significantly change anymore after 100 cycles. This shows that the nanoparticle composition remains unaltered during further cycling.
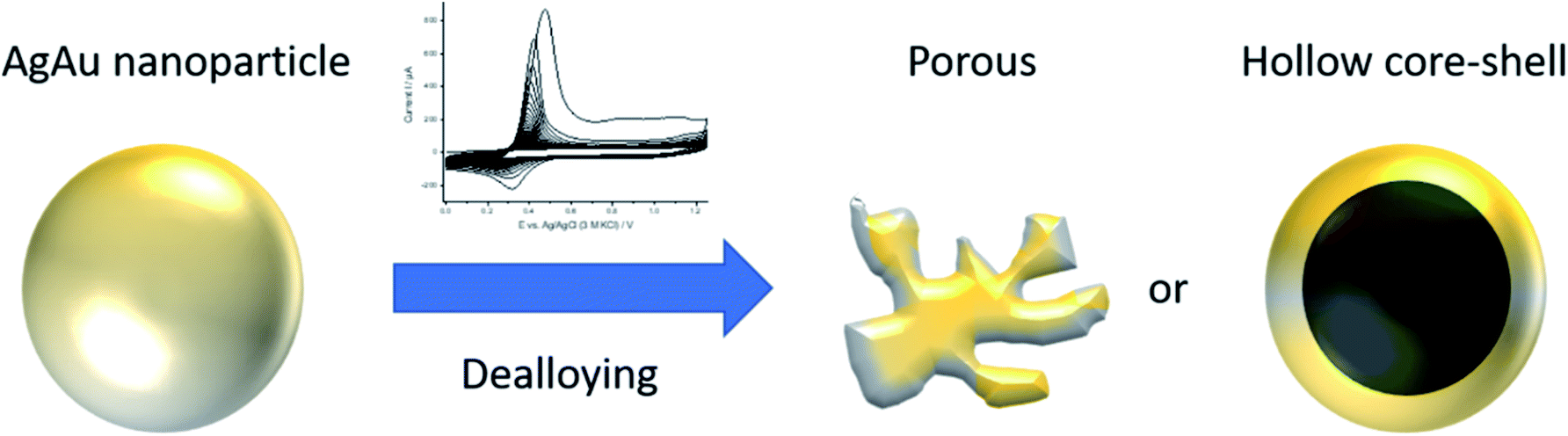 | ||
| Fig. 3 Schematic representation of a silver–gold nanoparticle with a gold rich alloyed core and a silver rich shell before dealloying (left) and the possible porous or hollow structure resulting after dealloying (right). | ||
 | ||
| Fig. 4 (a) Cyclic-voltammetric dealloying of AgAu alloy nanoparticles in 1.0 M HClO4 at 1 V s−1, showing the first, second, 100th, 500th, 1000th, 2000th and 4000th cycle of 4000 cycle dealloying measurements, (b) relative molar amount of Ag remaining in the nanoparticles after dealloying, determined by electrochemical dissolution (coulometry) in HCl(aq), (c) nanoparticle size measured by TEM (number of nanoparticles analyzed n = 96, 136, 188 and 183, respectively; histograms in Fig. S1 in the ESI†) and (d) averaged measured catalytic current density at −0.3 V vs. RHE in 0.5 M H2SO4. | ||
Complementary microscopic analysis is performed next, to identify if the nanoparticle morphology and porosity also remain constant afterwards. As visible in Fig. 5, STEM analysis of the dealloyed nanoparticles shows significant morphology changes throughout, for increasing cycle numbers. Additionally, EDX analysis reveals that the individual composition of the nanoparticles also still changes slightly. After one dealloying cycle, the nanoparticles show only slight changes in their morphology, despite leaching about 20% of the Ag out of the nanoparticles (see Fig. 4b and Table S1 in the ESI†). The formed nanoparticles generally appear less spherical and exhibit some rough edges. EDX mapping of the nanoparticles hints towards the presence of Ag on the surface (Fig. 5a). It is therefore likely that incomplete dissolution of Ag occurs on the nanoparticles' surface during the first scan, despite the high positive potentials applied during dealloying. Cycling for 100 cycles reduces the Ag content to about 20 mol% in the nanoparticles according to the CV analysis. While the EDX mappings show less Ag in the outer parts of particles, there are still nanoparticles found with an almost unaltered composition. The nanoparticles show no porosity and did not significantly shrink compared to their original size, despite losing a high amount of Ag (Fig. 5b). A size comparison after dealloying (Fig. 4c) reveals that even after 500 cycles the nanoparticles only shrink slightly (by about 17%). After longer cycling, the fraction of fused/aggregated nanoparticles gets so high that average particle sizes can no longer be assigned from TEM imaging, due to the small number of individual nanoparticles detected (Fig. 5e and f).
 | ||
| Fig. 5 STEM images and STEM EDX mappings of dealloyed AgAu nanoparticles from different samples using 0.5 (a), 100 (b), 500 (c), 1000 (d), 2000 (e) and 4000 cycles (f), respectively. | ||
After 500 cycles of dealloying, no further decrease in particle size compared to 100 cycles is observable, but porosity can be detected in a significant portion of the nanoparticles (Fig. 5c). Identical location microscopy measurements were performed prior to dealloying and after 100 cycles (Fig. S2 in the ESI†) and 500 cycles (Fig. 6) of dealloying, to directly observe the changes in the nanoparticles. After 500 cycles, some nanoparticles shrink to about half their original size, while others remain almost unchanged (Fig. 6). These nanoparticles exhibit visible porosity and do not seem to be hollow core–shell like, which was hypothesized for AgAu nanoparticles being dealloyed using fast cycling.26 The EDX mappings of the identical location STEM (Fig. 6) show that the nanoparticles lose most of their Ag content. It can also be seen that three nanoparticles appear in a sintered like state. A growing fraction of aggregated nanoparticles is detected for samples with increasing cycling numbers, for instance, after 1000 or 2000 cycles (Fig. 5d, e and 7). This aggregation reduces the overall surface area, while the formation of pores increases the surface area, as shown in Fig. 3. The UPD of Pb2+ to Pb on Ag and Au allows us to electrochemically determine the surface area of the nanoparticles and thus to identify the dominating influence for the different number of dealloying cycles. This revealed an initial decrease in the surface area compared to the untreated nanoparticles, but no further reproducible change could be measured for larger cycling numbers. It is therefore evident that the porosity cannot be easily detected using UPD and the aggregation of nanoparticles during the dealloying process strongly depends on the starting configuration of the nanoparticles. This leads to a high variability in the measured surface area. In the images taken before the dealloying, very small pure Ag nanoparticles can be seen at the very right nanoparticle (Fig. 6), which completely vanish after the dealloying. These pure Ag nanoparticles might be responsible for the overall reduction in Ag content seen in the ensemble measurements using CV, with respect to the determined individual particle composition.
 | ||
| Fig. 6 Identical location TEM with STEM images and EDX mapping, before and after dealloying for 500 cycles in 1 M HClO4. | ||
 | ||
| Fig. 7 Zoomed in STEM images from different samples of (a) untreated Ag80Au20 nanoparticles, (b) after 0.5 cycles, (c) after 100 cycles, (d) after 500 cycles, (e) after 1000 cycles, (f) after 2000 cycles, and (g) after 4000 cycles, the scale bar is the same for all images. | ||
Extended cycling for 1000, 2000 and 4000 cycles follows the trends observed for 500 cycles. More porous but also more aggregated nanoparticles are observed. Using lead UPD, it was found that the surface of the nanoparticles is composed of Au for cycle numbers of 100 cycles or more. In a previous UPD study, we detected a surface termination by Ag for the as-synthesized AgAu particles, even for high Au contents, in agreement with theoretical predictions.27,38 Since no aggregation of AgAu nanoparticles was found by Sieradzki et al.26 during dealloying at constant potentials, we attribute this to the repetitive formation and dissolution of a Au oxide layer on the nanoparticle surface upon potentiodynamic dealloying. This repetitive alteration of the surfaces is suggested to favour the motion of Au atoms, which promotes the dealloying process, but also the aggregation of particles that are in contact with each other. Interestingly, some nanoparticles seem completely unchanged in the dealloying process, even though they are in direct contact with a different particle which is almost completely dealloyed and porous. This was also noticed after 4000 cycles (Fig. 5f). A similar phenomenon of silver enriched clusters has been observed in nanoporous gold.39 It was explained by Monte Carlo simulations, which suggest that the formation of at least a monolayer of Au on top of the AgAu alloy may occur already at early stages of dealloying. This passivating layer prevents further dealloying. Usually, researchers seek to prevent this passivation behavior by having a high amount of Ag in the starting alloy (in this case 80 mol%) and a particle size above 25 nm for AgAu, as stated by Li et al.26 However, our results unambiguously show that individual nanoparticles passivate, revealing that individual particle reactivities can differ from the integral average behavior. This is attributable to the statistical nature of both the elemental distribution of Ag and Au in the nanoparticle after synthesis and the surface diffusion of Au during the dealloying. It was found that about 10% of the nanoparticles passivate at an early stage and maintain their composition, independent of the number of dealloying cycles. Insufficient electrical contact as the reason for low dealloying is less likely an explanation, as the nanoparticles show a change in their outer layer (see lower nanoparticle in Fig. 6) and are directly touching regularly dealloyed nanoparticles in some cases (see Fig. 5f).
Catalysis
Next, the electrocatalytic properties of nanoparticles were analyzed, using the HER as a model reaction. It was used to probe whether continued dealloying treatment further increases the catalytic activity or if there is an optimum dealloying time. Each catalytic curve displays the averages of at least three independent experiments (Fig. 8). Pure Au nanoparticles were also analyzed to test if the activity of the dealloyed nanoparticles is merely moving towards the activity of pure Au, or if a further increased electrocatalytic activity can be achieved. Fig. 8 clearly confirms the latter case, as dealloyed AgAu nanoparticles show much higher activity than pure Au nanoparticles which have comparable activity to untreated Ag80Au20 nanoparticles, with respect to their determined surface area. | ||
| Fig. 8 Averaged catalytic LSV curves from multiple independent experiments at 2 mV s−1 in 0.5 M H2SO4 using pure silver and pure gold; 100 cycles “dealloyed” gold, non-dealloyed AgAu 80:20 and dealloyed AgAu nanoparticles after different numbers of dealloying cycles. The plotted currents are normalized to the electrochemically determined surface area of the nanoparticles. | ||
Notably, the dealloying treatment does not increase the activity of pure Au nanoparticles irrespective of the cycling numbers. This verifies that Ag leaching from the AgAu nanoparticles causes the strong increase of their electrocatalytic activity. Untreated AgAu nanoparticles reach a current density of about 10 mA cm−2 at −1.1 V (see Fig. S3 in the ESI†). The overall current is very low, and the nanoparticles show almost no catalytic activity in the chosen potential window for the HER. Dealloying of the nanoparticles greatly improved their activity. Already a dealloying treatment of only 0.5 cycles resulted in a ten-fold increase in current at −1.0 V. Overall, the highest current and current density were measured after 100 cycles of dealloying (see Fig. 4d). Notably, this is very close to the region of around 70 to 100 cycles at which the dealloying CV no longer shows any features and does not change anymore afterwards. Cycling for longer than 100 cycles starts to decrease the activity again, but the current density does not decrease below about 75 mA cm−2. This reveals that even after long treatment under harsh conditions dealloyed AgAu nanoparticles still show a much higher catalytic activity than untreated alloy and monometallic nanoparticles. After 500 cycles, the activity is very similar to the activity after 4000 cycles, but it is higher after 1000 and 2000 cycles. The very high activity after 100 cycles with a steep decrease after 500 cycles clearly suggests that there is an optimal amount of cycles for dealloying.
An increase in catalytic activity after chemical40 and electrochemical41 dealloying has also been observed for reduction catalysis before. This fits well with our results and highlights the fact that the activity of various alloy systems can be improved through dealloying. While we found that extensive cycling leads to a decrease in catalytic activity compared to shorter cycling times, which hints toward an optimal dealloying time, Wang et al.42 found that longer cycling (100000 cycles) still improved the catalytic activity. While these results seem to contradict each other at first glance, a deeper comparison reveals that both results hint at a common factor. We observed the highest catalytic activity after 100 cycles, while the dealloying CV did not show any changes anymore after 70 to 100 cycles. Further cycling decreased the activity. In the case observed by Wang et al.,42 the CV still changed between 5000 and 100000 cycles, which indicates they did not reach a stable response for their system after 5000 cycles and, thus, did not reach the highest activity yet. Accordingly, further cycling still increased their activity, which is in line with our findings.
Conclusions
In this work we demonstrated that the loss of features in successive CVs during the cyclic-voltammetric dealloying of alloy nanoparticles does not imply a stable or persistent morphology or activity of the dealloyed nanoparticles. The morphology of the nanoparticles, their individual composition and the catalytic activity still change significantly. For the used AgAu nanoparticles, the highest activity towards the hydrogen evolution reaction was observed after 100 cycles of dealloying, which is close to the start of a constant CV response. At this stage, the nanoparticles show almost no porosity, but the particle surface appears mostly dealloyed in STEM EDX mappings. Longer cycling of the nanoparticles leads to an increase in their porosity. However, this turned out to negatively affect the activity towards the HER. Hence, we conclude that the optimum dealloying extent is reached when the CV response of the nanoparticles does not change anymore in subsequent cycles. This “abort criterion” for electrochemical dealloying is readily identifiable in practice, in contrast to having to control the morphological changes for any specific nanocatalyst. The fact that this is in line with previously reported dealloying conditions of Pt-based alloy nanoparticles towards maximum activity in the oxygen evolution reaction37 suggests that this “abort criterion”-guideline may be generally applicable to different nanocatalysts and reactions. Noteworthily, cycling of pure gold nanoparticles is in agreement with that, as it yields a stable CV response directly after the first cycle and the highest activity of these particles was indeed detected for the untreated nanoparticles.The present findings provide an excellent starting point to establish dealloying as an effective tool to tune nanoparticle properties and provide general guidance to identify optimum dealloying conditions. Typically, higher porosity leads to higher catalytic activity, while in the case presented herein, higher particle porosity did not necessarily result in a higher activity. The particle aggregation occurring in line with porosity formation during extensive cycling in our study caused a loss of intrinsic catalytic activity. This outweighed the benefits of the higher porosity. This shows that while increased material porosity is beneficial in general, the presence of pores alone is not sufficient to make a good nano-electrocatalyst. Additional essential material properties, like the pore size, connectivity, structural integrity and local composition of the catalyst should be considered as design factors. We showed that smaller cycling numbers result in higher activity, even before significant pore formation is observed. Consequently, it should be aimed at keeping nanoparticle dealloying times low and close to the beginning of stable CV responses. It is often extremely difficult to change the particle size or shape, for bi- or multimetallic nanoparticles during bottom-up synthesis, independently from their composition. Thus, applying a small number of dealloying cycles may be used as an activation post-treatment of nanoparticles to fine-tune their composition without greatly influencing their morphology. Alternatively, more extended dealloying may be used to tune nanoparticle morphology to a larger extent. This makes electrochemical dealloying a versatile and efficient tool to fine-tune the properties and activity of nano- and micro-scale materials, which may help researchers to markedly speed up the development of improved electrocatalysts in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2033 – Projektnummer 390677874. It was also funded by the Ministry of Innovation, Science and Research of North Rhine-Westphalia (“NRW Rückkehrerprogramm”). C. S. and A. G. M acknowledge financial support from the German Science Foundation SCHE 634/21-1. We would also like to thank Mathies V. Evers for his help with the EDX mappings.Notes and references
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Evolution of nanoporosity in dealloying, Nature, 2001, 410, 450–453 CrossRef CAS.
- K. M. Kosuda, A. Wittstock, C. M. Friend and M. Bäumer, Oxygen-mediated coupling of alcohols over nanoporous gold catalysts at ambient pressures, Angew. Chem., Int. Ed. Engl., 2012, 51, 1698–1701 CrossRef CAS.
- K. M. Kosuda, A. Wittstock, C. M. Friend and M. Bäumer, Sauerstoffinduzierte Kupplung und Oxidation von Alkoholen über nanoporösem Gold, Angew. Chem., 2012, 124, 1730–1733 CrossRef.
- A. Wittstock, A. Wichmann and M. Bäumer, Nanoporous Gold as a Platform for a Building Block Catalyst, ACS Catal., 2012, 2, 2199–2215 CrossRef CAS.
- M. Graf, M. Haensch, J. Carstens, G. Wittstock and J. Weissmüller, Electrocatalytic methanol oxidation with nanoporous gold: microstructure and selectivity, Nanoscale, 2017, 9, 17839–17848 RSC.
- X. Xiao, C. Engelbrekt, Z. Li and P. Si, Hydrogen evolution at nanoporous gold/tungsten sulfide composite film and its optimization, Electrochim. Acta, 2015, 173, 393–398 CrossRef CAS.
- G. G. Li, Y. Lin and H. Wang, Residual Silver Remarkably Enhances Electrocatalytic Activity and Durability of Dealloyed Gold Nanosponge Particles, Nano Lett., 2016, 16, 7248–7253 CrossRef CAS.
- J. Snyder, I. McCue, K. Livi and J. Erlebacher, Structure/processing/properties relationships in nanoporous nanoparticles as applied to catalysis of the cathodic oxygen reduction reaction, J. Am. Chem. Soc., 2012, 134, 8633–8645 CrossRef CAS.
- B. E. Peecher and J. R. Hampton, Dealloying Behavior of NiCo and NiCoCu Thin Films, Int. J. Electrochem., 2016, 2016, 1–10 CrossRef.
- X.-L. Wu, H. Wu, Z.-M. Wang, H. Aizawa, J. Guo and Y.-H. Chu, Nanoporous Microsphere Assembly of Iodine-Functionalised Silver Nanoparticles as a Novel Mini-Substrate for Enriching and Sensing, Sci. Rep., 2017, 7, 1–8 CrossRef.
- Z. Yang, X. Wang, X. Kang, S. Zhang and Y. Guo, The PtPdAg/C electrocatalyst with Pt-rich surfaces via electrochemical dealloying of Ag and Pd for ethanol oxidation, Electrochim. Acta, 2017, 236, 72–81 CrossRef CAS.
- J. Erlebacher and R. Seshadri, Hard Materials with Tunable Porosity, MRS Bull., 2009, 34, 561–568 CrossRef CAS.
- Y. Ding, Y.-J. Kim and J. Erlebacher, Nanoporous Gold Leaf: “Ancient Technology”/Advanced Material, Adv. Mater., 2004, 16, 1897–1900 CrossRef CAS.
- J. Erlebacher, An Atomistic Description of Dealloying, J. Electrochem. Soc., 2004, 151, C614 CrossRef CAS.
- L. H. Qian and M. W. Chen, Ultrafine nanoporous gold by low-temperature dealloying and kinetics of nanopore formation, Appl. Phys. Lett., 2007, 91, 83105 CrossRef.
- J. Snyder and J. Erlebacher, Kinetics of Crystal Etching Limited by Terrace Dissolution, J. Electrochem. Soc., 2010, 157, C125 CrossRef CAS.
- S. Koh and P. Strasser, Electrocatalysis on bimetallic surfaces: modifying catalytic reactivity for oxygen reduction by voltammetric surface dealloying, J. Am. Chem. Soc., 2007, 129, 12624–12625 CrossRef CAS.
- I. McCue, E. Benn, B. Gaskey and J. Erlebacher, Dealloying and Dealloyed Materials, Annu. Rev. Mater. Res., 2016, 46, 263–286 CrossRef CAS.
- J. F. Gomes, A. C. Garcia, L. H. S. Gasparotto, N. E. de Souza, E. B. Ferreira, C. Pires and G. Tremiliosi-Filho, Influence of silver on the glycerol electro-oxidation over AuAg/C catalysts in alkaline medium: a cyclic voltammetry and in situ FTIR spectroscopy study, Electrochim. Acta, 2014, 144, 361–368 CrossRef CAS.
- C. Poochai, W. Veerasai, E. Somsook and S. Dangtip, Dealloyed ternary Cu@Pt-Ru core-shell electrocatalysts supported on carbon paper for methanol electrooxidation catalytic activity, Electrochim. Acta, 2016, 222, 1243–1256 CrossRef CAS.
- S. Rudi, L. Gan, C. Cui and P. Strasser, Dealloying of Pt Bimetallic Catalysts at Constant Electrode Potentials, ECS Trans., 2013, 58, 581–586 CrossRef.
- F. Zhou, J. Chen, Y. Wang, J. Zhang, R. Luo, X. Wei, G. Wang and R. Wang, The study of platinum-tellurium intermetallic nanoparticles for formic acid electro-oxidation, Electrochim. Acta, 2017, 248, 307–312 CrossRef CAS.
- S. Ju, J. Feng, P. Zou, W. Xu, S. Wang, W. Gao, H.-J. Qiu, J. Huo and J.-Q. Wang, A robust self-stabilized electrode based on Al-based metallic glasses for a highly efficient hydrogen evolution reaction, J. Mater. Chem. A, 2020, 8, 3246–3251 RSC.
- H.-J. Qiu, J. J. Gao, F.-K. Chiang, Y. R. Wen, A. Y. Yao, P. Du, G. Fang, J. Q. Wang and X. J. Liu, A general and scalable approach to produce nanoporous alloy nanowires with rugged ligaments for enhanced electrocatalysis, J. Mater. Chem. A, 2018, 6, 12541–12550 RSC.
- D. M. Artymowicz, J. Erlebacher and R. C. Newman, Relationship between the parting limit for de-alloying and a particular geometric high-density site percolation threshold, Philos. Mag., 2009, 89, 1663–1693 CrossRef CAS.
- X. Li, Q. Chen, I. McCue, J. Snyder, P. Crozier, J. Erlebacher and K. Sieradzki, Dealloying of noble-metal alloy nanoparticles, Nano Lett., 2014, 14, 2569–2577 CrossRef CAS.
- V. Grasmik, C. Rurainsky, K. Loza, M. V. Evers, O. Prymak, M. Heggen, K. Tschulik and M. Epple, Deciphering the Surface Composition and the Internal Structure of Alloyed Silver-Gold Nanoparticles, Chemistry, 2018, 24, 9051–9060 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, A study of the nucleation and growth processes in the synthesis of colloidal gold, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- D. T. Sawyer, A. Sobkowiac and J. L. Roberts, Electrochemistry for Chemists, John Wiley & Sons, New York, 2nd edn, 1995 Search PubMed.
- L. Wan, Y. Qin and J. Xiang, Rapid Electrochemical Fabrication of Porous Gold Nanoparticles for High-performance Electrocatalysis towards Oxygen Reduction, Electrochim. Acta, 2017, 238, 220–226 CrossRef CAS.
- M. Gatalo, P. Jovanovič, F. Ruiz-Zepeda, A. Pavlišič, A. Robba, M. Bale, G. Dražić, M. Gaberšček and N. Hodnik, Insights into electrochemical dealloying of Cu out of Au-doped Pt-alloy nanoparticles at the sub-nano-scale, J. Electrochem. Sci. Eng., 2018, 8, 87 CrossRef CAS.
- C. Batchelor-McAuley, A. Martinez-Marrades, K. Tschulik, A. N. Patel, C. Combellas, F. Kanoufi, G. Tessier and R. G. Compton, Simultaneous electrochemical and 3D optical imaging of silver nanoparticle oxidation, Chem. Phys. Lett., 2014, 597, 20–25 CrossRef CAS.
- Y.-G. Zhou, N. V. Rees, J. Pillay, R. Tshikhudo, S. Vilakazi and R. G. Compton, Gold nanoparticles show electroactivity: counting and sorting nanoparticles upon impact with electrodes, Chem. Commun., 2012, 48, 224–226 RSC.
- E. N. Saw, V. Grasmik, C. Rurainsky, M. Epple and K. Tschulik, Electrochemistry at single bimetallic nanoparticles - using nano impacts for sizing and compositional analysis of individual AgAu alloy nanoparticles, Faraday Discuss., 2016, 193, 327–338 RSC.
- S. Bruckenstein and M. Shay, An in situ weighing study of the mechanism for the formation of the adsorbed oxygen monolayer at a gold electrode, J. Electroanal. Chem. Interfacial Electrochem., 1985, 188, 131–136 CrossRef CAS.
- J. S. Gordon and D. C. Johnson, Application of an electrochemical quartz crystal microbalance to a study of water adsorption at gold surfaces in acidic media, J. Electroanal. Chem., 1994, 365, 267–274 CrossRef CAS.
- P. Strasser, S. Koh and J. Greeley, Voltammetric surface dealloying of Pt bimetallic nanoparticles: an experimental and DFT computational analysis, Phys. Chem. Chem. Phys., 2008, 10, 3670–3683 RSC.
- G. Guisbiers, R. Mendoza-Cruz, L. Bazán-Díaz, J. J. Velázquez-Salazar, R. Mendoza-Perez, J. A. Robledo-Torres, J.-L. Rodriguez-Lopez, J. M. Montejano-Carrizales, R. L. Whetten and M. José-Yacamán, Electrum, the Gold-Silver Alloy, from the Bulk Scale to the Nanoscale: Synthesis, Properties, and Segregation Rules, ACS Nano, 2016, 10, 188–198 CrossRef CAS.
- T. Krekeler, A. V. Straßer, M. Graf, K. Wang, C. Hartig, M. Ritter and J. Weissmüller, Silver-rich clusters in nanoporous gold, Mater. Res. Lett., 2017, 5, 314–321 CrossRef CAS.
- F.-J. Lai, W.-N. Su, L. S. Sarma, D.-G. Liu, C.-A. Hsieh, J.-F. Lee and B.-J. Hwang, Chemical dealloying mechanism of bimetallic Pt-Co nanoparticles and enhancement of catalytic activity toward oxygen reduction, Chemistry, 2010, 16, 4602–4611 CrossRef CAS.
- J. Snyder and J. Erlebacher, ECS Trans., 2011, 1021–1030 CAS.
- D. Wang, Y. Yu, J. Zhu, S. Liu, D. A. Muller and H. D. Abruña, Morphology and activity tuning of Cu3Pt/C ordered intermetallic nanoparticles by selective electrochemical dealloying, Nano Lett., 2015, 15, 1343–1348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Nanoparticle characterization, electrocatalytic response. See DOI: 10.1039/d0ta04880a |
| This journal is © The Royal Society of Chemistry 2020 |