
3D lithiophilic–lithiophobic–lithiophilic dual-gradient porous skeleton for highly stable lithium metal anode†
Hongfei
Zheng
a,
Qingfei
Zhang
a,
Qiulin
Chen
a,
Wanjie
Xu
a,
Qingshui
Xie
 *a,
Yuxin
Cai
a,
Yating
Ma
a,
Zhensong
Qiao
a,
Qing
Luo
a,
Jie
Lin
a,
Laisen
Wang
a,
Baihua
Qu
b,
Baisheng
Sa
*c and
Dong-Liang
Peng
*a
*a,
Yuxin
Cai
a,
Yating
Ma
a,
Zhensong
Qiao
a,
Qing
Luo
a,
Jie
Lin
a,
Laisen
Wang
a,
Baihua
Qu
b,
Baisheng
Sa
*c and
Dong-Liang
Peng
*a
aDepartment of Materials Science and Engineering, State Key Lab of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, College of Materials, Xiamen University, Xiamen, 361005, China. E-mail: xieqsh@xmu.edu.cn; dlpeng@xmu.edu.cn; Fax: +86-592-2183515; Tel: +86-592-2180155
bPen-Tung Sah Institute of Micro-Nano Science and Technology, Xiamen University, Xiamen, 361005, China
cMultiscale Computational Materials Facility, College of Materials Science and Engineering, Fuzhou University, Fuzhou, 350100, China. E-mail: bssa@fzu.edu.cn
First published on 22nd November 2019
Abstract
The lithium metal anode has been considered the most promising anode in rechargeable batteries to meet the ever-increasing requirements of high energy density. Herein, a 3D porous lithiophilic–lithiophobic–lithiophilic dual-gradient Cu–Au–ZnO–PAN–ZnO (CAZPZ) current collector is fabricated to suppress Li dendrite growth. The lithiophilic Au and ZnO at the bottom are favorable for homogeneous Li nucleation, the ZnO–PAN–ZnO skeleton provides plenty of space to accommodate deposited Li and the lithiated ZnO (Li2O/LixZn) layer can act as an artificial SEI to regulate the well-distributed Li+ flux. As a result, long-term stabilization for 1200 h at 0.5 mA cm−2 and a low overpotential of 22 mV at 3 mA cm−2 are achieved in symmetric cells. Moreover, the CAZPZ–Li hybrid anode exhibits superb electrochemical properties when matched with a LiFePO4 (LFP) cathode. The CAZPZ–Li‖LFP full cells exhibit excellent stabilization for 1000 cycles at 5C with a high capacity retention of 97.3%. The lithiophilic–lithiophobic–lithiophilic dual-gradient design of the 3D porous current collector for the Li metal anode can be a very promising strategy to enable the practical application of Li metal batteries.
Introduction
With the rapid development of consumer electronics, electric vehicles and grid-scale energy storage, the ever-increasing demand for high energy density over 400 W h kg−1 has motivated extensive research on advanced energy storage devices.1–4 With an ultrahigh theoretical specific capacity (3860 mA h g−1) and the lowest electrochemical potential (−3.04 V vs. the standard hydrogen electrode), Li metal is widely deemed to be the most ideal anode.5,6 Fascinatingly, the application of the Li metal anode can not only increase the specific energy of Li metal batteries, but also further expand the choice of cathode material to include lithium-free candidates (such as V2O5, sulfur, and oxygen).7,8Despite great advantages and potential, the current development of the Li metal anode has faced formidable challenges.9,10 Firstly, Li dendrite growth induced by heterogeneous lithium deposition will give rise to dead Li, low coulombic efficiency (CE), and safety issues.11,12 Secondly, the extremely high chemical and electrochemical reactivity of Li leads to severe side reactions with the electrolyte, resulting in a low utilization rate of Li, large polarization and fast capacity fading.13,14 Thirdly, the relatively infinite volume change of Li metal during cycling would induce the cracking of the solid electrolyte interphase (SEI) followed by severe side reactions. Meanwhile, the fresh Li underneath the fractured SEI would result in a large ion flux, inducing non-uniform Li deposition and thus rapid growth of Li dendrites.15
Various superb research studies have been conducted to tackle the aforementioned critical issues.16 Electrolyte optimization (organic solvents, Li salts and functional additives),17–19 fabrication of artificial SEI layers (Li3PO4, Li3N, LiF, etc.)20–22 and designing of solid-state electrolytes (SSEs)23–26 are indeed effective in improving the cycling stability of the Li metal anode and suppressing Li dendrites in previous cycles; however, these methods can't effectively address the hostless nature of Li and uncontrollable Li dendrite growth.27 Thus, designing 3D current collectors is significant to suppress the large volume change, reduce the local current density and develop a stable electrode/electrolyte interface.28,29
The previous reports about 3D current collectors mainly focus on conductive current collectors (carbon-based, metal-based, etc.) to reduce the local current density and nonconductive current collectors (such as glass fiber) with polar groups to regulate homogeneous Li+ flux.30–34 These designs are indeed effective in improving the cycling stability and mitigating the large volume change of Li metal anodes to some extent; however, conductive current collectors can lead to “top-growth” Li deposition and increase the risk of short circuit, while nonconductive current collectors could lose their electric contact easily during plating/stripping.35–37 What's more, most of these current collectors are lithiophobic, leading to a high nucleation overpotential and heterogeneous Li deposition. To overcome these drawbacks, some multifunctional current collectors have been widely researched and several effective approaches have been put forward. Some hosts are modified by lithiophilic materials to reduce the nucleation overpotential and regulate uniform Li nucleation, such as Au NPs@C,38 nitrogen (N) doped graphene,39 Cu@Ni nanowires,40 and G-CNF with gradient-distributed ZnO.41 An interfacial layer with a lithiophilic–lithiophobic gradient is also reported to provide better control of Li deposition and suppress dendrite growth.42 Besides, Luo et al. reported a mixed ion- and electron-conducting (MIEC) network to regulate homogeneous Li plating/stripping behavior.36 These commendable reports pointed out that multifunctional composite current collectors are extraordinarily promising. Nevertheless, homogeneous Li nucleation and growth are still hard to control effectively. Thus, it is of great importance to develop novel current collectors with elaborately regulated lithiophilic affinity and conductive properties.
Herein, a 3D porous lithiophilic–lithiophobic–lithiophilic dual-gradient ZnO–PAN–ZnO skeleton is covered on a Au-modified Cu substrate (Cu–Au) to develop a Cu–Au–ZnO–PAN–ZnO (denoted as CAZPZ) current collector for the Li metal anode. The Cu–Au substrate is conductive while the conductivity of the ZnO–PAN–ZnO skeleton is poor; Li+ ions are inclined to homogeneously nucleate on the Au surface because Li+ ions can easily gain electrons from the Cu–Au substrate. In CAZPZ, lithiophilic Au and the bottom ZnO can provide a lot of nucleation sites and decrease the Li nucleation overpotential greatly, causing homogeneous Li nucleation.43,44 Further Li deposition takes place between the lithiophilic Cu–Au substrate and the bottom of the ZnO–PAN–ZnO skeleton. The deposited Li will exhibit strong affinity to lithiophilic Au and ZnO, and as a result, the deposited Li can be endowed with the function of a “glue” to integrate the Cu–Au substrate and ZnO–PAN–ZnO skeleton tightly, preventing their separation and strengthening their electric contact. Subsequently deposited Li will compactly wrap the PAN fibers with lithiophilic ZnO modification and then gradually fill into the porous space of the ZnO–PAN–ZnO skeleton from the bottom to the top. The porous space of the ZnO–PAN–ZnO skeleton can mitigate the large volume change. Furthermore, the upper ZnO layer can react with Li+ ions and electrons during the initial Li plating stage to form a highly ion-conductive and lithiophilic artificial SEI layer of Li2O/LixZn, which can regulate the well-distributed Li+ flux.45 What's more, the much higher mechanical strength of Li2O (141 GPa) can avoid direct penetration of Li (4.9 GPa).46 As a result, the dual-gradient structural design of the CAZPZ current collector can guide bottom-to-top Li nucleation/deposition and suppress Li dendrites. Long-term symmetrical cycling for 1200 h at 0.5 mA cm−2 and a low overpotential of 22 mV at 3 mA cm−2 are achieved in symmetric cells. Moreover, the CAZPZ–Li‖LiFePO4 full cell exhibits excellent stabilization for 1000 cycles with a high capacity retention of 97.3% at 5C.
Results and discussion
The preparation process of the 3D lithiophilic–lithiophobic–lithiophilic dual-gradient CAZPZ current collector is illustrated in Fig. S1.† The SEM images (Fig. 1a–c) show that the electrospun PAN fibers are substantially uniform with an average diameter of about 500 nm and are randomly intertwined, and the as-formed pure PAN skeleton possesses a porous structure. A similar morphology can be found in the designed ZnO–PAN–ZnO skeleton (Fig. 1d–f) obtained by sputtering ZnO on both sides of a porous PAN skeleton. However, the surface of fibers becomes rough due to the modification with ZnO particles (Fig. 1f). When used as the current collector for a lithium metal anode, the porous structure of the skeleton can effectively reduce the local flux of Li+ but cannot hinder the transfer of Li+ and provide enough space for Li plating. The EDS maps in Fig. 1g–j confirm that ZnO is successfully coated on the surface of PAN fibers facing the magnetron sputtering target and the thickness of the ZnO layer is about 15 nm. As shown in Fig. S2,† the thickness of the ZnO–PAN–ZnO skeleton is about 80 μm and the corresponding Zn elemental EDS data reveal that the content of ZnO on the top and bottom is higher than that in the middle (almost no ZnO), which evidences the successful fabrication of the 3D porous lithiophilic–lithiophobic–lithiophilic dual-gradient skeleton. In order to further decrease the overpotential of Li deposition, Au is uniformly sputtered on the surface of Cu foil (Fig. S3 and S4†). | ||
| Fig. 1 Characterization of the PAN and ZnO–PAN–ZnO skeletons. SEM images of the (a–c) PAN and (d–f) ZnO–PAN–ZnO skeletons. (g–j) EDS maps of Zn, O, C and N distribution in a single ZnO-modified PAN fiber. | ||
The Li plating/stripping behavior on the Cu and 3D dual-gradient CAZPZ current collectors is illustrated in Fig. 2. In the initial plating process on Cu, the plated Li is inclined to form isolated Li domains due to the lithiophobicity and defective surface of Cu foil, resulting in dendrite formation on these formed Li domains.47 Dead Li forms while stripping, leading to large polarization and fast capacity fading.36 Before Li deposition on CAZPZ, ZnO first interacts with Li+ ions and electrons to form highly ion-conductive LixZn alloys and Li2O, which is helpful in homogeneous Li+ transport (Fig. 2b).48,49 Li+ is introduced at the bottom and nucleates on the Au surface with a low nucleation overpotential due to the lithiophilicity of Au and ZnO. Further Li is plated on deposited Li and fills into the porous space of the ZnO–PAN–ZnO skeleton from the bottom to the top. The deposited Li can homogeneously strip from the CAZPZ current collector and no dendrites form.
 | ||
| Fig. 2 Schematic diagram of Li plating/stripping behavior on (a) Cu and (b) Cu–Au–ZnO–PAN–ZnO current collectors. | ||
To illustrate the evolution of Li plating/stripping, the Cu, Au and CAZPZ current collectors were galvanostatically plated at a fixed Li capacity (from 0.25 to 1 mA h cm−2) and stripped to 0.5 V at a current density of 0.05 mA cm−2. The voltage profiles of Cu, Au and CAZPZ during 1 mA h cm−2 Li plating are illustrated in Fig. S5,† indicating that the nucleation overpotentials of both Au and CAZPZ current collectors are very low but that of Cu is large. Fig. 3a–c show the typical morphology of Li plating/stripping on a Cu current collector. Li is heterogeneously distributed on the surface of Cu foil to form island-like domains which would grow and sparsely disperse with further Li plating. After stripping, the Cu surface is foamed. This phenomenon is caused by heterogeneous nucleation and subsequent Li growth from Li crystal nuclei.39,50 Unlike Cu, more uniform Li plating is observed on the Cu–Au current collector and the surface is flat after Li stripping (Fig. 3d–f) because the good lithiophilicity of Au has a positive effect on regulating homogeneous Li plating/stripping. Fig. 3g–i show that the top surface of the ZnO–PAN–ZnO skeleton maintains its porous structure and no lithium deposition occurs on the top even after plating 1 mA h cm−1 of lithium. The initial deposition of Li (0.25 mA h cm−2) takes place on the surface of the bottom ZnO–PAN fibers while the fibers still maintain a porous morphology (Fig. 3j–l). When Li plating increases to 1 mA h cm−2, the Li would wrap the bottom lithiophilic ZnO–PAN fibers first and then uniformly deposit into the porous space. Fig. 3m–o suggest that the Li-plated Au surface of the CAZPZ collector exhibits similar behavior to a single Au collector but is more homogeneous due to the effect of the ZnO–PAN–ZnO skeleton in homogenizing the Li+ flux. Besides, the Au surface of CAZPZ is also flat after stripping. From the SEM images in Fig. 3g–o and cross-sectional SEM images in Fig. S6,† during the Li plating process on the CAZPZ current collector, the well-distributed Li+ flux is guided to the bottom to easily gain electrons and then it preferentially nucleates on the surface of Au. Then lithium deposition occurs between the Cu–Au and the bottom ZnO–PAN–ZnO skeleton. The deposited Li will generate strong affinity with lithiophilic Au and ZnO; as a result, it can be endowed with the function of a “glue” to integrate the Cu–Au substrate and ZnO–PAN–ZnO skeleton tightly (Fig. S6a†), preventing their separation and strengthening their electric contact to avoid the formation of dead Li. Subsequently, Li+ ions will unceasingly be plated on deposited Li and compactly wrap the PAN fibers. The thickness of the Li layer gradually increases from the bottom to the top until it uniformly fills into the whole porous space of the ZnO–PAN–ZnO skeleton (Fig. S6b–e†). An SEI layer would form on the Li frontier surface, embedding the PAN skeleton. When charged to 0.5 V, the plated Li on the CAZPZ current collector can be homogeneously stripped thoroughly (Fig. S6f†). After 5 mA h cm−2 Li plating, the top surface of the CAZPZ current collector maintains its porous structure, while Li grows outside the surface partially, but no pointed Li dendrites occur after plating 10 mA h cm−2 of Li (Fig. S6g–j†). This phenomenon indicates that the largest Li storage capacity of the designed porous skeleton is very close to 10 mA h cm−2. The bottom-to-top plating behavior of Li is in accordance with the above-mentioned schematic diagram.
 | ||
| Fig. 3 Morphological observations of the Li plating/stripping processes. SEM images after plating 0.25 mA h cm−2 and 1 mA h cm−2 and stripping 1 mA h cm−2 (that is, recharged to 0.5 V) on the (a–c) Cu collector, (d–f) Au collector, (g–i) top ZnO–PAN–ZnO surface of CAZPZ, (j–l) bottom ZnO–PAN–ZnO surface of CAZPZ and (m–o) Au surface of CAZPZ, respectively. The current density is 0.05 mA cm−2. | ||
To demonstrate the effect of the dual-gradient CAZPZ current collector in improving the cycling performance of the Li anode, Cu, Au–Cu, Cu–PAN, Cu–Au–PAN and Cu–Au–ZnO–PAN–ZnO working electrodes were paired with pure lithium foil as the counter electrode for cycling tests. To remove the surface contamination and stabilize the SEI layer, the galvanostatic process was performed first at a current density of 50 μA cm−2 from 0.01 V to 0.5 V (vs. Li+/Li) for 5 cycles (Fig. S7†). Fig. S8 and S9† indicate that ZnO first interacts with Li+ ions and electrons under large voltage polarization before Li plating due to the limited electrochemical reaction kinetics caused by the poor electronic conductivity of the ZnO–PAN–ZnO skeleton, and the formed Li2O/LixZn layer can regulate the well-distributed Li+ flux.51–53 The DFT calculations also show the same results, that is, the adsorbed Li atoms would interact with ZnO spontaneously with a reaction energy Er of −3.04 eV before Li plating during the first discharge process (Computational details in the ESI†). After the lithiation reaction of ZnO, Li+ ions would be guided to preferentially plate on the surface of the Cu–Au substrate because of the lower nucleation overpotential and abundant electrons on the Au surface. When testing the CEs, 1 mA h cm−2 of Li was plated on the working electrodes at 0.5 mA cm−2, and then stripped from the working electrodes until a cut-off potential of 0.5 V (vs. Li+/Li) was reached. As depicted in Fig. 4a, the initial coulombic efficiency (CE) of Cu–Au–ZnO–PAN–ZnO (98.24%) is higher than those of pure Cu (97.15%) and Cu–PAN (97.48%) and is close to those of Cu–Au–PAN (98.01%) and Cu–Au (98.47%). From Fig. 4a and b, it can be observed that the mass-transfer overpotentials (voltage plateau in the black dashed rectangle in Fig. 4a) of all the working electrodes are closed to 20 mV, indicating that the introduction of the ZnO–PAN–ZnO skeleton does not increase the transfer resistance due to its porous structure. In contrast, the nucleation overpotentials of the Cu–Au, Cu–Au–ZnO–PAN–ZnO, Cu–Au–PAN, Cu and Cu–PAN current collectors are 13.79, 14.44, 20.49, 32.96 and 52.83 mV, respectively. Undoubtedly, both Au and ZnO have an obvious effect on reducing the Li nucleation overpotential owing to their lithiophilic properties. Benefitting from the effective synergistic effect between lithiophilic Au and the porous ZnO–PAN–ZnO skeleton, the average CE of Cu–Au–ZnO–PAN–ZnO is maintained stably above 99% (Fig. 4c). By contrast, the Cu–Au–PAN, Cu–Au, Cu–PAN and Cu current collectors display lower average CEs of ∼95%, ∼94%, ∼93% and ∼79% for only 100 cycles, respectively. The low CE is mainly induced by dendrite growth during cycling. In Fig. S10 and S11,† it is clearly observed that no dendrites grow on the top surface of the Cu–Au–ZnO–PAN–ZnO electrode after different cycles and its original porous structure is well maintained. However, severe dendrite growth can be observed on other electrodes after 100 cycles, accounting for their low CEs. As shown in Fig. S12 and S13,† the CAZPZ‖Li asymmetric cell exhibits better CEs than the Cu–Au–ZnO–PAN‖Li and Cu–Au–PAN–ZnO‖Li asymmetric cells, illustrating the merits of the design of the lithiophilic–lithiophobic–lithiophilic dual-gradient porous skeleton, and the CAZPZ‖Li asymmetric cell also exhibits a very steady cycling performance for more than 500 h and its average CE reaches 98.7% even at a high capacity of 5 mA h cm−2.
 | ||
| Fig. 4 Electrochemical characterization comparison. (a) Voltage profiles and (b) nucleation overpotentials of Cu, Cu–Au, Cu–PAN, Cu–Au–PAN and Cu–Au–ZnO–PAN–ZnO (red dashed rectangle in (a)) in the 1st cycle. (c) Comparison of the coulombic efficiency of 1 mA h cm−2 Li plating/stripping at 0.5 mA cm−2. Electrochemical impedance spectra (EIS) of (d) Cu–Au–ZnO–PAN–ZnO||Li and (e) Cu||Li cells after the activation process at 50 μA cm−2 and after 50 cycles at 0.5 mA cm−2. | ||
Electrochemical impendence spectroscopy (EIS) measurements were conducted and the corresponding Nyquist plots at different cycles are illustrated in Fig. 4d and e. The semicircle in the high frequency region is related to interfacial charge transfer resistance (Rct).54,55 After the galvanostatic activation process, the Cu‖Li cell reveals a large interfacial resistance (118.3 Ω) which is much higher than that of Cu–Au–ZnO–PAN–ZnO‖Li (42.4 Ω). Similarly, the interfacial resistance of the Cu‖Li cell (150.9 Ω) is also much higher than that of Cu–Au–ZnO–PAN–ZnO‖Li (18.3 Ω) after 50 cycles (Table S1†). Distinctly, the Cu–Au–ZnO–PAN–ZnO current collector is advantageous in stabilizing the SEI layer and reducing the interfacial charge transfer resistance.
Simulation of the electric field distributions of the CAZPZ–Li and Cu–Li anodes after Li nucleus formation was performed and is shown in Fig. 5a and b (simulation of the electric field distribution in the ESI†). This shows that the high electric field intensity around the Li nuclei on the Cu current collector dramatically increases due to tip effects (clearly visible in red), causing uneven Li+ flux distribution and subsequent heterogeneous growth of Li dendrites.56 In contrast, the CAZPZ current collector exhibits a well-distributed electric field, promoting uniform Li+ flux and heterogeneous Li deposition on the CAZPZ current collector.57 The simulation results agree with the Li plating process revealed in Fig. 3.
 | ||
| Fig. 5 Simulation models of electric field values of the plated Li on the (a) CAZPZ and (b) Cu current collectors after Li nucleation. The calculated binding energies between Li atoms and (c) Cu, (d) Au, and (e) Li2O substrates. The charge density differences of the adsorption of one Li atom on the (f) Cu, (g) Au, and (h) Li2O surfaces. The loss of electrons is indicated in yellow and gain of electrons is indicated in blue. | ||
To further evaluate the Li adsorption ability, DFT calculations are carried out to determine the binding energies between a Li atom and different substrates (Computational details in the ESI†). As indicated in Fig. 5c–e, the Au surface shows a more negative binding energy of −3.28 eV than the Cu surface (−2.98 eV), indicating that Au is more lithiophilic and benefits the decrease of the nucleation overpotential of Li. Surprisingly, the Li2O surface exhibits the most negative binding energy of −6.56 eV; the strong binding energy between Li and Li2O demonstrates that Li2O is much more lithiophilic than both Cu and Au, which can further reduce the nucleation overpotential of Li effectively. Thus, the nucleation overpotential of the Cu–Au–ZnO–PAN–ZnO collector (14.44 mV) is much less than that of the Cu–PAN collector (52.83 mV) as shown in Fig. 4b. To understand the chemical origin of the above different binding energies, the charge density differences of lithiated Cu, Au and Li2O surfaces are illustrated in Fig. 5f–h, respectively.58 For these three cases, a net loss of electronic charge could be found above the Li atom, and a net gain of electronic charge is found on the adjacent substrate surfaces as well. For Cu (or Au), metallic Li–Cu (or Li–Au) bonding is characterized by the accumulated near free electron gas between the adsorbed Li atom and the Cu (or Au) surface. Since the Li–Au charge transfer is stronger than the Li–Cu one, its corresponding energy is more negative. It is worth noting that Li–O ionic bonding is observed between the adsorbed Li atom and the Li2O surface in Fig. 5h, resulting in the most negative binding energy.
CAZPZ‖Li 2025 coin cells were assembled, and then 5 mA h cm−2 of Li was pre-electroplated onto the CAZPZ current collector to construct a CAZPZ–Li hybrid anode and then CAZPZ–Li‖Li cells. Cu–Li‖Li cells were also constructed for comparison. At a current density of 0.5 mA cm−2, as shown in Fig. 6a, the Cu–Li‖Li cell exhibits the same stripping/plating overpotential (∼8 mV) as the CAZPZ–Li‖Li cell; however, its overpotential drops abruptly after 387 h of cycling, representing short circuit due to Li dendrite growth, while the CAZPZ–Li‖Li cell exhibits very high stability up to 1200 h without any sign of short circuit or apparent overpotential increase, indicating better cyclability. When tested at 1 mA cm−2, the CAZPZ–Li‖Li cell can continuously work for up to 600 h (300 cycles) with a small and stable overpotential of ∼18 mA, whereas the Cu–Li‖Li cell exhibits higher overpotential throughout the entire cycling test, unstable overpotential around 200 h and a huge overpotential increase after cycling for 450 h (Fig. 6b). Though steady cycling is achieved at 3 mA cm−2 in the CAZPZ–Li‖Li cell, the Li plating overpotential on the pure Li side sharply increases after 95 h (Fig. S14a†), which is induced by the severe dendrite formation on the pure Li electrode (Fig. S15†). To avoid this issue, a CAZPZ–Li‖CAZPZ–Li symmetric cell was assembled, and a pure Li‖Li symmetric cell was assembled for comparison. As illustrated in Fig. 6c, when cycled at 3 mA cm−2, the voltage hysteresis of the pure Li symmetric cell is very unstable and oscillates drastically. In contrast, the voltage hysteresis of the CAZPZ–Li symmetric cell is small and stable (∼22 mV) for 200 h (300 cycles) of cycling. As observed in the Fig. 6c inset, the voltage–time profile of the CAZPZ–Li symmetric cell remains flat and constant, indicating its high cycling stability. However, the pure Li symmetric cell exhibits not only a larger Li stripping/plating overpotential (∼100 mV) but also a necking behavior, which is a characteristic sign of dendrite formation in the early stage and SEI accumulation later.43Fig. 6d shows that the rate performance of the CAZPZ–Li symmetric cell again surpasses that of the pure Li symmetric cell at various current densities of 0.25, 0.5, 1, 3 and 5 mA cm−2. Even at 5 mA cm−2 for 3 mA h cm−2, the Li stripping/plating overpotential of the CAZPZ–Li symmetric cell is only around 50 mV for stable cycling over 60 h, whereas that of the pure Li symmetric cell is too large and unstable (Fig. S14b†). The above contrasting results demonstrate that the CAZPZ current collector can improve the stability and high-rate performance of the Li metal anode.
 | ||
| Fig. 6 Voltage–time curves of Li plating/stripping. (a) The voltage–time curves of the Cu–Li‖Li and Cu–Au–ZnO–PAN–ZnO–Li‖Li cells at 0.5 mA cm−2; the insets in (a) show the detailed voltage profiles from the dashed blue and purple rectangles. (b) The voltage–time curves at 1 mA cm−2. (c) The voltage–time curves at 3 mA cm−2 and (d) rate performances of Cu–Au–ZnO–PAN–ZnO–Li and pure Li symmetric cells. The plating/stripping capacity for each cycle is set to 1 mA h cm−2. | ||
To illustrate the practical applications of the self-fabricated 3D dual-gradient CAZPZ current collector, CAZPZ–Li‖LiFePO4 full cells were assembled by using commercial LiFePO4 (LFP, 2 mg cm−2) as the cathode and the CAZPZ–Li as the anode, and pure-Li‖LFP cells were assembled for comparison. Fig. 7a illustrates representative charge and discharge profiles from 0.1C to 5C between 2.5–4.0 V (1C = 170 mA g−1) for the CAZPZ–Li‖LFP full cell. The CAZPZ–Li‖LFP full cell exhibits high initial specific discharge capacities of 163.3, 158.1, 152.8, 145.3, 134.0 and 121.1 mA h g−1 at 0.1, 0.2, 0.5, 1, 2 and 5C rates, respectively, and low voltage hysteresis, evidencing outstanding electrochemical reaction kinetics and rate performance.59 The cycling stability was tested at 1C and 5C as shown in Fig. 7b and c. The initial discharge capacities of the CAZPZ–Li‖LFP cell are 144.0 and 116.5 mA h g−1 at 1C and 5C, respectively. The discharge capacities of the CAZPZ–Li‖LFP full cell are 141.8 mA h g−1 after 200 cycles at 1C with a large capacity retention of 98.5% and 113.3 mA h g−1 after 1000 cycles at 5C with a high capacity retention of 97.3%. However, the Li‖LFP cell exhibits discharge capacities of only 110.3 mA h g−1 after 200 cycles at 1C (77.8% capacity retention) and 55.8 mA h g−1 after 1000 cycles at 5C (47.7% capacity retention). The surface SEM images of the pure Li and CAZPZ–Li anodes after 1000 cycles at 5C are illustrated in Fig. 7d and e. Foamed Li dendrites are formed on the surface of the pure Li anode; however, no dendrites can be found on the surface of the CAZPZ–Li anode. The growth of Li dendrites mainly contributes to the difference in the stability performance of full cells. These results evidence that the design of the 3D dual-gradient CAZPZ current collector can enhance the structural stability of the Li metal anode effectively and improve its electrochemical properties.
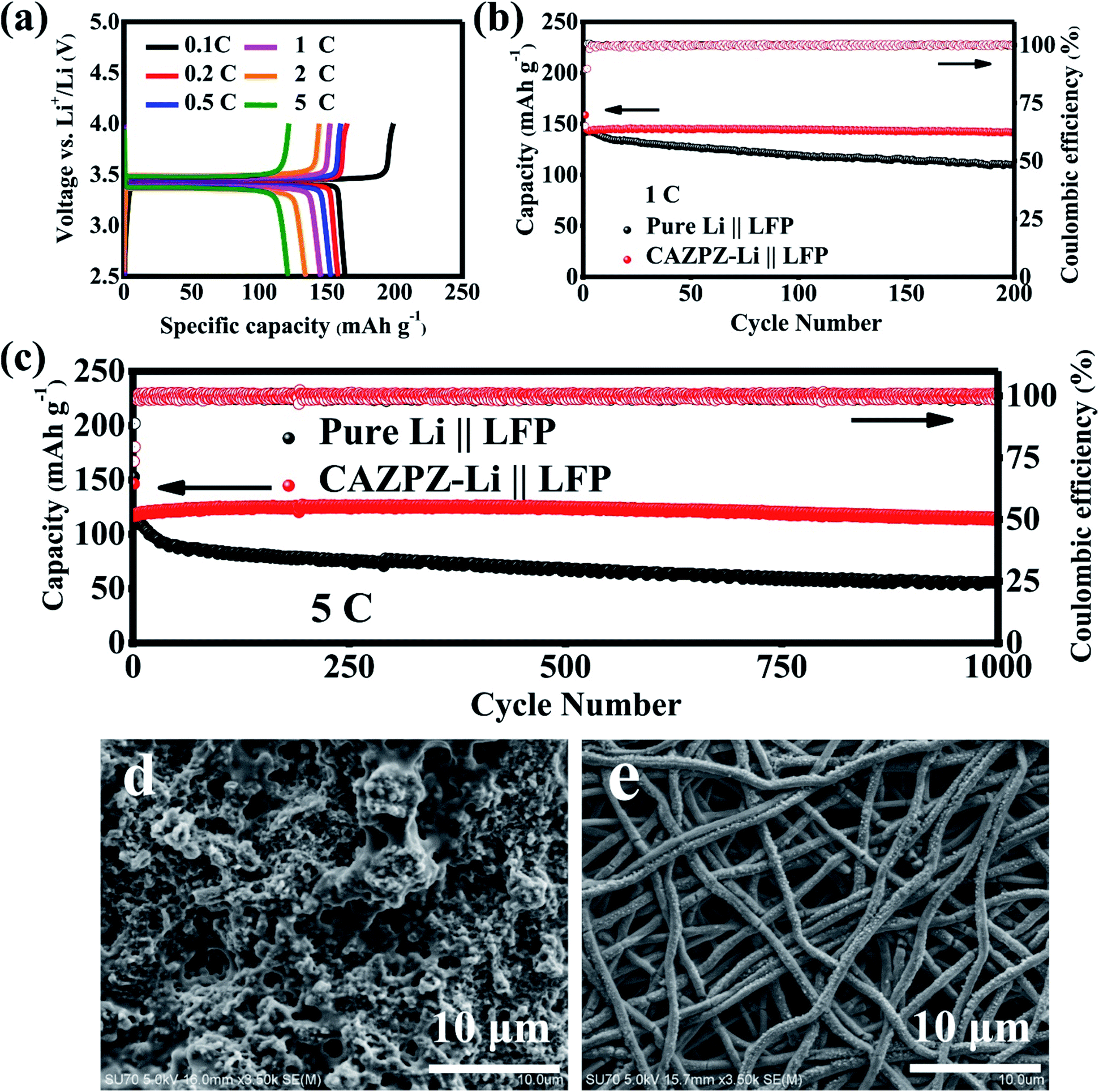 | ||
| Fig. 7 Comparison between CAZPZ–Li‖LFP and pure-Li‖LFP full cells. (a) Galvanostatic charge–discharge profiles of the CAZPZ–Li‖LFP full cell at different rates, cycling performances (b) at 1C and (c) 5C, and SEM images of (d) pure-Li and (e) CAZPZ–Li after 1000 cycles at 5C. | ||
The self-synthesized Li1.2Mn0.54Ni0.13Co0.13O2 (MNC-411) and the commercial Li4Ti5O12 (LTO) are also used as the cathodes to assemble full cells. The structure and morphology characterization of the used cathode materials are revealed in Fig. S16 and S17.† In this work, the electrochemical performances of the CAZPZ–Li‖MNC-411 full cell were tested from 2.0 V to 4.6 V. As shown in Fig. 8a and b, when cycled at 5C (1C = 250 mA g−1), the CAZPZ–Li‖MNC-411 full cell exhibits outstanding stability and high voltage retention with an almost 100% CE, and a high capacity of 141.7 mA h g−1 can be achieved after 200 cycles. Even when tested at a large current density of 10C for 400 cycles, the CAZPZ–Li‖MNC-411 full cell still maintains a decent reversible capacity of 121.9 mA h g−1 with a capacity retention over 80% and a high voltage retention of 96.4%. The CAZPZ–Li‖LTO full cell was tested from 1.0 V to 3.0 V. As shown in Fig. 8c and d, its reversible capacity remains 128.9 mA h g−1 after 1000 cycles at 5C (1C = 175 mA g−1) and 110.7 mA h g−1 after steady cycling for 2000 cycles at 10C (Fig. 8c and d). The stability of the CAZPZ–Li‖LTO full cell is higher than that of the pure-Li‖LTO cell (Fig. S18†).
 | ||
| Fig. 8 Electrochemical properties of CAZPZ–Li‖MNC-411/LTO. Cycle performance at (a) 5C and (b) 10C for the CAZPZ–Li‖MNC-411 full cells. Cycle performance at (c) 5C and (d) 10C for the CAZPZ–Li‖LTO full cells. | ||
Conclusions
To summarize, we use a facile strategy combining electrostatic spinning and magnetron sputtering to produce a 3D porous dual-gradient (lithiophilic–lithiophobic–lithiophilic) Cu–Au–ZnO–PAN–ZnO current collector for the Li metal anode. With the synergistic effect of lithiophilic Au and dual-gradient ZnO, it is able to reduce the nucleation overpotential greatly and regulate homogeneous Li plating. As a result, long-term cyclability for 1200 h at 0.5 mA cm−2 and for 200 h at 3 mA cm−2 can be achieved in symmetric cells. Furthermore, the effectiveness and practicability of the dual-gradient CAZPZ–Li hybrid anode in improving electrochemical performance are further demonstrated to be successful in different full cell systems matched with LTO, LFP and MNC-411 cathode materials. Specifically, excellent stabilization for 1000 cycles at 5C with a high capacity retention of 97.3% in the CAZPZ–Li‖LFP full cell and for 2000 cycles at 10C with a capacity retention of 87.1% in the CAZPZ–Li‖LTO full cell is achieved. A layered lithium-rich cathode (MNC-411) is introduced into the Li metal full cell, and a high capacity retention of over 80% and good voltage retention of 96.3% can be achieved after 400 cycles at 10C. The design of the 3D porous lithiophilic–lithiophobic–lithiophilic dual-gradient current collector plays a positive role in accommodating the large volume change of Li, accelerating the good distribution of Li+ flux and developing a stable SEI layer, finally leading to a greatly enhanced electrochemical performance of the lithium metal anode.Author contributions
H. Z., Q. X., B. S. and D. P. designed the research and co-wrote the paper. H. Z., Q. L., J. L., Q. Z. and Q. C. performed the electrochemical tests and analyzed the data. W. X. and B. Q. drew the schematic diagrams using 3D Max. Y. C., Y. M. and Z. Q. performed XRD and SEM characterization. L. W. and B. S. carried out the DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 51701169 and 51871188), the National Key R&D Program of China (Grant No. 2016YFA0202602), the Key Projects of Youth Natural Foundation for the Universities of the Fujian Province of China (No. JZ160397), the Natural Science Foundation of Fujian Province of China (No. 2019J06003 and 2017J05087), the Fundamental Research Funds for the Central Universities of China (Xiamen University: No. 20720160082 and 20720190013) and the “Double-First Class” Foundation of Materials and Intelligent Manufacturing Discipline of Xiamen University.References
- B. Liu, Y. Sun, L. Liu, S. Xu and X. Yan, Adv. Funct. Mater., 2018, 28, 1704973 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- S. Hy, H. Liu, M. Zhang, D. Qian, B.-J. Hwang and Y. S. Meng, Energy Environ. Sci., 2016, 9, 1931–1954 RSC.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- M. S. Whittingham, Proc. IEEE, 2012, 100, 1518–1534 CAS.
- W. Tang, X. Yin, S. Kang, Z. Chen, B. Tian, S. L. Teo, X. Wang, X. Chi, K. P. Loh, H. W. Lee and G. W. Zheng, Adv. Mater., 2018, 30, 1801745 CrossRef PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- C. Yang, K. Fu, Y. Zhang, E. Hitz and L. Hu, Adv. Mater., 2017, 29, 1701169 CrossRef PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- Y. Guo, H. Li and T. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- W. Li, H. Zheng, G. Chu, F. Luo, J. Zheng, D. Xiao, X. Li, L. Gu, H. Li, X. Wei, Q. Chen and L. Chen, Faraday Discuss., 2014, 176, 109–124 RSC.
- Y. Liu, P. He and H. Zhou, Adv. Energy Mater., 2018, 8, 1701602 CrossRef.
- X.-Q. Zhang, X.-B. Cheng and Q. Zhang, Adv. Mater. Interfaces, 2018, 5, 1701097 CrossRef.
- Z. Liang, D. Lin, J. Zhao, Z. Lu, Y. Liu, C. Liu, Y. Lu, H. Wang, K. Yan, X. Tao and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2862–2867 CrossRef CAS PubMed.
- X. Wang, W. Zeng, L. Hong, W. Xu, H. Yang, F. Wang, H. Duan, M. Tang and H. Jiang, Nat. Energy, 2018, 3, 227–235 CrossRef CAS.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 5301–5305 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- L. Suo, Y. S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- Y. Gao, Y. Zhao, Y. C. Li, Q. Huang, T. E. Mallouk and D. Wang, J. Am. Chem. Soc., 2017, 139, 15288–15291 CrossRef CAS PubMed.
- X. Liang, Q. Pang, I. R. Kochetkov, M. S. Sempere, H. Huang, X. Sun and L. F. Nazar, Nat. Energy, 2017, 2, 17119 CrossRef CAS.
- S. Choudhury, R. Mangal, A. Agrawal and L. A. Archer, Nat. Commun., 2015, 6, 10101 CrossRef CAS PubMed.
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- S. Zekoll, C. Marriner-Edwards, A. K. O. Hekselman, J. Kasemchainan, C. Kuss, D. E. J. Armstrong, D. Cai, R. J. Wallace, F. H. Richter, J. H. J. Thijssen and P. G. Bruce, Energy Environ. Sci., 2018, 11, 185–201 RSC.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- H. Wang, Y. Li, Y. Li, Y. Liu, D. Lin, C. Zhu, G. Chen, A. Yang, K. Yan, H. Chen, Y. Zhu, J. Li, J. Xie, J. Xu, Z. Zhang, R. Vila, A. Pei, K. Wang and Y. Cui, Nano Lett., 2019, 19, 1326–1335 CrossRef PubMed.
- J. Lang, Y. Jin, X. Luo, Z. Liu, J. Song, Y. Long, L. Qi, M. Fang, Z. Li and H. Wu, J. Mater. Chem. A, 2017, 5, 19168–19174 RSC.
- J. W. Jin Xie, H. Ryoung Lee, K. Yan, Y. Li, F. Shi, W. Huang, A. Pei, G. Chen, R. Subbaraman, J. Christensen and Y. Cui, Sci. Adv., 2018, 4, eaat5168 CrossRef PubMed.
- R. Zhang, X. B. Cheng, C. Z. Zhao, H. J. Peng, J. L. Shi, J. Q. Huang, J. Wang, F. Wei and Q. Zhang, Adv. Mater., 2016, 28, 2155–2162 CrossRef CAS PubMed.
- L. L. Lu, J. Ge, J. N. Yang, S. M. Chen, H. B. Yao, F. Zhou and S. H. Yu, Nano Lett., 2016, 16, 4431–4437 CrossRef CAS PubMed.
- S. H. Wang, Y. X. Yin, T. T. Zuo, W. Dong, J. Y. Li, J. L. Shi, C. H. Zhang, N. W. Li, C. J. Li and Y. G. Guo, Adv. Mater., 2017, 29, 1703729 CrossRef PubMed.
- X. B. Cheng, T. Z. Hou, R. Zhang, H. J. Peng, C. Z. Zhao, J. Q. Huang and Q. Zhang, Adv. Mater., 2016, 28, 2888–2895 CrossRef CAS PubMed.
- D. Lin, Y. Liu, Z. Liang, H.-W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef CAS PubMed.
- J. Li, P. Zou, S. W. Chiang, W. Yao, Y. Wang, P. Liu, C. Liang, F. Kang and C. Yang, Energy Storage Materials, 2019 DOI:10.1016/j.ensm.2019.06.019.
- C. Zhang, S. Liu, G. Li, C. Zhang, X. Liu and J. Luo, Adv. Mater., 2018, 30, 1801328 CrossRef PubMed.
- P. Zou, S.-W. Chiang, J. Li, Y. Wang, X. Wang, D. Wu, A. Nairan, F. Kang and C. Yang, Energy Storage Materials, 2019, 18, 155–164 CrossRef.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS.
- R. Zhang, X. R. Chen, X. Chen, X. B. Cheng, X. Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7764–7768 CrossRef CAS PubMed.
- L.-L. Lu, Y. Zhang, Z. Pan, H.-B. Yao, F. Zhou and S.-H. Yu, Energy Storage Materials, 2017, 9, 31–38 CrossRef.
- Y. Nan, S. Li, Y. Shi, S. Yang and B. Li, Small, 2019, e1903520, DOI:10.1002/smll.201903520.
- H. Zhang, X. Liao, Y. Guan, Y. Xiang, M. Li, W. Zhang, X. Zhu, H. Ming, L. Lu, J. Qiu, Y. Huang, G. Cao, Y. Yang, L. Mai, Y. Zhao and H. Zhang, Nat. Commun., 2018, 9, 3729 CrossRef PubMed.
- Y. Liu, D. Lin, Z. Liang, J. Zhao, K. Yan and Y. Cui, Nat. Commun., 2016, 7, 10992 CrossRef CAS PubMed.
- B. Hong, H. Fan, X.-B. Cheng, X. Yan, S. Hong, Q. Dong, C. Gao, Z. Zhang, Y. Lai and Q. Zhang, Energy Storage Materials, 2019, 16, 259–266 CrossRef.
- C. Wang, A. Wang, L. Ren, X. Guan, D. Wang, A. Dong, C. Zhang, G. Li and J. Luo, Adv. Funct. Mater., 2019, 1905940, DOI:10.1002/adfm.201905940.
- K. Yan, H. W. Lee, T. Gao, G. Zheng, H. Yao, H. Wang, Z. Lu, Y. Zhou, Z. Liang, Z. Liu, S. Chu and Y. Cui, Nano Lett., 2014, 14, 6016–6022 CrossRef CAS PubMed.
- H. Dai, K. Xi, X. Liu, C. Lai and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17515–17521 CrossRef CAS PubMed.
- L. Wang, X. Zhu, Y. Guan, J. Zhang, F. Ai, W. Zhang, Y. Xiang, S. Vijayan, G. Li, Y. Huang, G. Cao, Y. Yang and H. Zhang, Energy Storage Materials, 2018, 11, 191–196 CrossRef.
- K.-H. Chen, A. J. Sanchez, E. Kazyak, A. L. Davis and N. P. Dasgupta, Adv. Energy Mater., 2019, 9, 1802534 CrossRef.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- G. Zhang, S. Hou, H. Zhang, W. Zeng, F. Yan, C. C. Li and H. Duan, Adv. Mater. Interfaces, 2015, 27, 2400–2405 CrossRef CAS PubMed.
- B. Zhu, N. Liu, M. McDowell, Y. Jin, Y. Cui and J. Zhu, Nano Energy, 2015, 13, 620–625 CrossRef CAS.
- C. Wang, Y. Gong, B. Liu, K. Fu, Y. Yao, E. Hitz, Y. Li, J. Dai, S. Xu, W. Luo, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 565–571 CrossRef CAS PubMed.
- Y. An, H. Fei, G. Zeng, X. Xu, L. Ci, B. Xi, S. Xiong, J. Feng and Y. Qian, Nano Energy, 2018, 47, 503–511 CrossRef CAS.
- Q. Li, S. Zhu and Y. Lu, Adv. Funct. Mater., 2017, 27, 1606422 CrossRef.
- L. Ye, M. Liao, H. Sun, Y. Yang, C. Tang, Y. Zhao, L. Wang, Y. Xu, L. Zhang, B. Wang, F. Xu, X. Sun, Y. Zhang, H. Dai, P. G. Bruce and H. Peng, Angew. Chem., Int. Ed., 2019, 58, 2437–2442 CrossRef CAS PubMed.
- S. Liu, X. Xia, Y. Zhong, S. Deng, Z. Yao, L. Zhang, X.-B. Cheng, X. Wang, Q. Zhang and J. Tu, Adv. Energy Mater., 2018, 8, 1702322 CrossRef.
- Q. Peng, K. Hu, B. Sa, J. Zhou, B. Wu, X. Hou and Z. Sun, Nano Res., 2017, 10, 3136–3150 CrossRef CAS.
- Y. Deng, C. Yang, K. Zou, X. Qin, Z. Zhao and G. Chen, Adv. Energy Mater., 2017, 7, 1601958 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, SEM images, EDS results, EIS results, XRD patterns and electrochemical performance. See DOI: 10.1039/c9ta09505e |
| This journal is © The Royal Society of Chemistry 2020 |