
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe formyloxyl radical: electrophilicity, C–H bond activation and anti-Markovnikov selectivity in the oxidation of aliphatic alkenes†
Miriam
Somekh
a,
Mark A.
Iron
 b,
Alexander M.
Khenkin
a and
Ronny
Neumann
*a
b,
Alexander M.
Khenkin
a and
Ronny
Neumann
*a
aDepartment of Organic Chemistry, Weizmann Institute of Science, Rehovot, 7610001, Israel. E-mail: ronny.neumann@weizmann.ac.il
bComputational Chemistry Unit, Department of Chemical Research Support, Weizmann Institute of Science, Rehovot, 7610001, Israel
First published on 2nd October 2020
Abstract
In the past the formyloxyl radical, HC(O)O˙, had only been rarely experimentally observed, and those studies were theoretical-spectroscopic in the context of electronic structure. The absence of a convenient method for the preparation of the formyloxyl radical has precluded investigations into its reactivity towards organic substrates. Very recently, we discovered that HC(O)O˙ is formed in the anodic electrochemical oxidation of formic acid/lithium formate. Using a [CoIIIW12O40]5− polyanion catalyst, this led to the formation of phenyl formate from benzene. Here, we present our studies into the reactivity of electrochemically in situ generated HC(O)O˙ with organic substrates. Reactions with benzene and a selection of substituted derivatives showed that HC(O)O˙ is mildly electrophilic according to both experimentally and computationally derived Hammett linear free energy relationships. The reactions of HC(O)O˙ with terminal alkenes significantly favor anti-Markovnikov oxidations yielding the corresponding aldehyde as the major product as well as further oxidation products. Analysis of plausible reaction pathways using 1-hexene as a representative substrate favored the likelihood of hydrogen abstraction from the allylic C–H bond forming a hexallyl radical followed by strongly preferred further attack of a second HC(O)O˙ radical at the C1 position. Further oxidation products are surmised to be mostly a result of two consecutive addition reactions of HC(O)O˙ to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. An outer-sphere electron transfer between the formyloxyl radical donor and the [CoIIIW12O40]5− polyanion acceptor forming a donor–acceptor [D+–A−] complex is proposed to induce the observed anti-Markovnikov selectivity. Finally, the overall reactivity of HC(O)O˙ towards hydrogen abstraction was evaluated using additional substrates. Alkanes were only slightly reactive, while the reactions of alkylarenes showed that aromatic substitution on the ring competes with C–H bond activation at the benzylic position. C–H bonds with bond dissociation energies (BDE) ≤ 85 kcal mol−1 are easily attacked by HC(O)O˙ and reactivity appears to be significant for C–H bonds with a BDE of up to 90 kcal mol−1. In summary, this research identifies the reactivity of HC(O)O˙ towards radical electrophilic substitution of arenes, anti-Markovnikov type oxidation of terminal alkenes, and indirectly defines the activity of HC(O)O˙ towards C–H bond activation.
C double bond. An outer-sphere electron transfer between the formyloxyl radical donor and the [CoIIIW12O40]5− polyanion acceptor forming a donor–acceptor [D+–A−] complex is proposed to induce the observed anti-Markovnikov selectivity. Finally, the overall reactivity of HC(O)O˙ towards hydrogen abstraction was evaluated using additional substrates. Alkanes were only slightly reactive, while the reactions of alkylarenes showed that aromatic substitution on the ring competes with C–H bond activation at the benzylic position. C–H bonds with bond dissociation energies (BDE) ≤ 85 kcal mol−1 are easily attacked by HC(O)O˙ and reactivity appears to be significant for C–H bonds with a BDE of up to 90 kcal mol−1. In summary, this research identifies the reactivity of HC(O)O˙ towards radical electrophilic substitution of arenes, anti-Markovnikov type oxidation of terminal alkenes, and indirectly defines the activity of HC(O)O˙ towards C–H bond activation.
Introduction
The formation of the formyloxyl radical, HC(O)O˙, was likely first hypothesized in 1952 as an intermediate in the reaction between a hydroxyl radical and formic acid using ionizing radiation.1 Sometime later in 1960, it was suggested as an intermediate in the electrochemical reduction of CO2 on a dropping mercury electrode.2 The formyloxyl radical was first convincingly identified by analysis of the fluorescence cross sections of the photoexcitation of formic acid in the vacuum-UV region using synchrotron radiation or a pulsed discharge lamp as the light source.3 This research was subsequently revisited in combined experimental–theoretical studies.4 The more recent literature related to the formyloxyl radical involves theory and electronic structure4,5 and its identification as an intermediate in the reduction of CO2 to formic acid and the reverse oxidation of formic acid.6 It is notable that acyloxy radicals typically decarboxylate very quickly with lifetimes of typically less than 1 nanosecond;7 however, it has been inferred that the lifetime of the formyloxyl radical is probably longer due to a somewhat less viable decarboxylation reaction.8 Overall, perusal of the literature reveals that there has been no convenient method to prepare the formyloxyl radical and as a corollary there have not been any in-depth reactivity studies with organic substrates.Recently, we identified the oxygen-centered formyloxyl radical by EPR spectroscopy as an intermediate in the electrochemical oxidation of formic acid/lithium formate on a Pt anode, by forming a spin adduct with a nitrone trap.9 Furthermore, in the presence of the [CoIIIW12O40]5− polyanion as a catalyst, efficient formation of phenyl formate from benzene was observed.9 Presumably the polyanion “stabilizes” the formyloxyl radical by forming an adduct.9 The ability to conveniently prepare the formyloxyl radical by a rather simple electrochemical method, combined with the observed reactivity in the oxidation of some arenes to the corresponding arylformates now sets the stage for a broader investigation into the reactivity of the formyloxyl radical with organic substrates designed to describe its philicity, reactivity toward double bonds and propensity for C–H bond activation – all for the first time. The results show that (i) the formyloxyl radical is mildly electrophilic in substitution reactions with arene substrates. (ii) Reactions with terminal alkenes mainly resulted in the formation anti-Markovnikov oxidative addition products. Such reactions have been an important objective, previously attainable mainly by manipulation of Wacker–Tsuji palladium catalyzed systems10 and by directed evolution of cyctochrome P450 monooxygenase enzymes.11 (iii) The formyloxyl radical is active in hydrogen abstraction reactions only for rather weak benzylic and allylic C–H bonds and shows only slight reactivity toward the C–H bonds in alkanes.
Results and discussion
The formyloxyl radical was electrochemically generated by a one-electron oxidation of a formate ion adsorbed on a Pt anode in a HCOOH/LiOOCH solution catalyzed by K5[CoW12O40]. Hydrogen gas is formed at the cathode. Typical reaction conditions involved dissolving 10 μmol K5Co(III)W12O40, 1 mmol substrate, and 0.5 mmol LiOOCH in 3 mL HCOOH. Reactions were carried out in an undivided cell configuration at a potential of 1.8 V versus SHE using a Pt gauze anode, a Pt wire cathode and a Pt reference electrode at room temperature. Under the same conditions but without an applied electrochemical potential, no reaction of any substrates was observed. HC(O)O˙ was previously shown to react with arenes to yield the corresponding phenyl formates via a proposed cyclohexadienyl intermediate.9 An alternative possibility of a substrate electrochemical oxidation followed a nucleophilic substitution reaction was discounted since no reactions occur in acetic acid/acetate. It is now possible to estimate the philicity of HC(O)O˙ using a Hammett linear free energy relationship (LFER), log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kX/kH = ρσ. Since these are electrochemical reactions, the reaction rate was measured as a function of the charge transferred (Q) rather than reaction time. Extrinsic factors such as the distance between the electrodes and their positioning in the cell vary somewhat from experiment to experiment and affect the current. The precision of measurements based on time was limited but rate kinetics as a function of Q gave very good precision and repeatability.
kX/kH = ρσ. Since these are electrochemical reactions, the reaction rate was measured as a function of the charge transferred (Q) rather than reaction time. Extrinsic factors such as the distance between the electrodes and their positioning in the cell vary somewhat from experiment to experiment and affect the current. The precision of measurements based on time was limited but rate kinetics as a function of Q gave very good precision and repeatability.
The reaction of benzene is hypothesized to follow a pseudo-first-order rate law because HC(O)O˙ is continuously generated on the anode and its concentration can be considered constant during the reaction. This gives the rate law in eqn (1), which can be plotted as shown in eqn (2).
| d[PhO(O)CH]/dQ = kobs[HC(O)O˙]0[PhH]1 | (1) |
| ln(X) = kobsQ | (2) |
Fig. 1 indeed shows a good pseudo-first-order behavior with a measured value of kobs = 0.28 (mA h)−1 with r2 = 0.96. Note that there is a lag (almost no reaction of benzene with HC(O)O˙) up to 4 mA h at the beginning of reaction, associated with the charge needed to attain a low but steady-state concentration of HC(O)O˙.
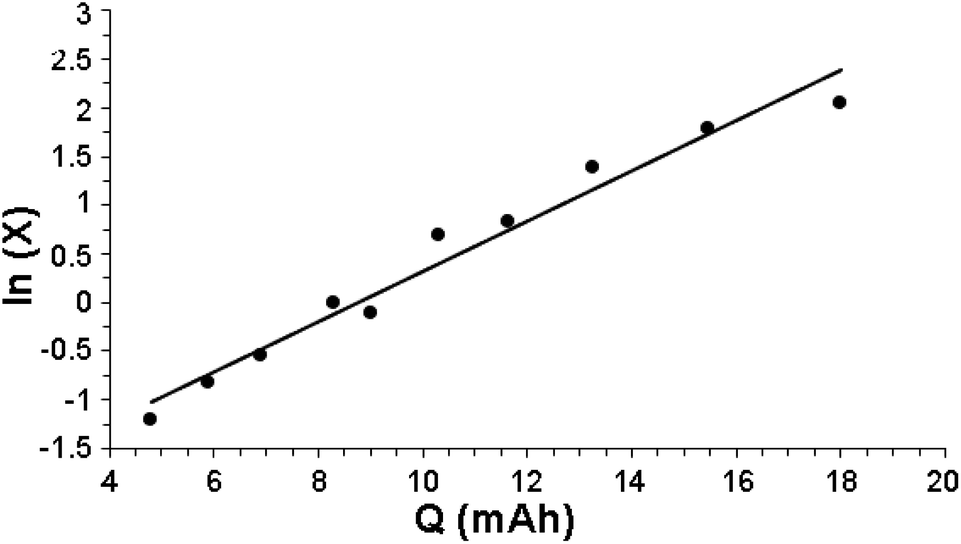 | ||
| Fig. 1 Pseudo-first-order kinetics for the reaction of benzene with the formyloxyl radical. | ||
The rate constants for substituted arenes, PhX (X = tert-Bu, F, Cl and Br), are best determined by competitive reactions of equimolar mixtures of PhX and PhH. For electron donating substituents such as OMe and Me the reactions were not selective (see also below for reactions at benzylic C–H bonds) while for electron withdrawing substituents such as CF3 and NO2 the reactions were too slow to obtain accurate results. Thus, the rate constants were derived from the ratio of the two products, PhO(O)CH/XPhO(O)CH, eqn (3).
| ln[PhO(O)CH]Q/[XPhO(O)CH]Q = −(kHobs − kXobs)Q | (3) |
The data are presented in Fig. S1–S4,† and the resulting Hammett plot that was obtained from the measured log(kXobs/kHobs) values and σpara12 gives ρ = −1.5 (r2 = 0.96) as shown in Fig. 2. The kinetic results and ρ value obtained show that HC(O)O˙ is electrophilic. This is expected based on the general classification of radicals as being electrophilic if the radical is centered on an atom (here oxygen) that is more electrophilic than carbon, as can be seen from the computed spin density shown in Fig. 3. A single literature example of the formation of aryl esters from RC(O)O˙ radicals showed that electrophilicity of three ArC(O)O˙ radicals, measured in a similar manner, yielded similar negative ρ values.13
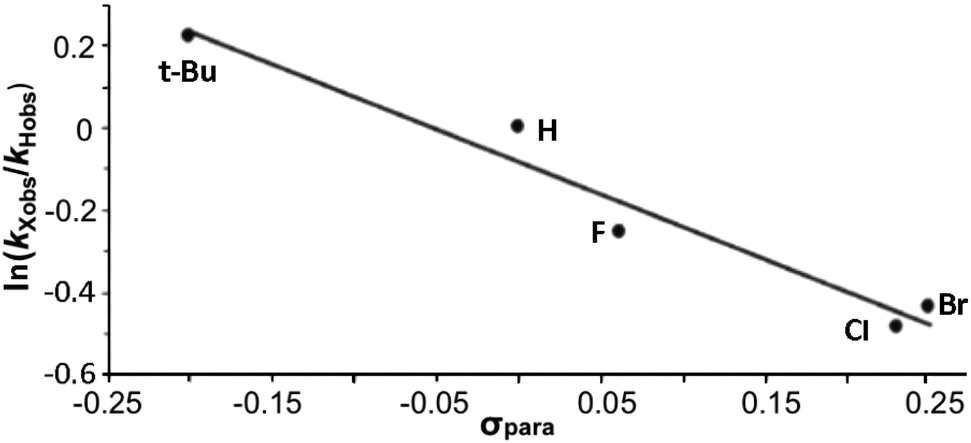 | ||
| Fig. 2 Hammett plot for the reaction of PhX with the formyloxyl radical. | ||
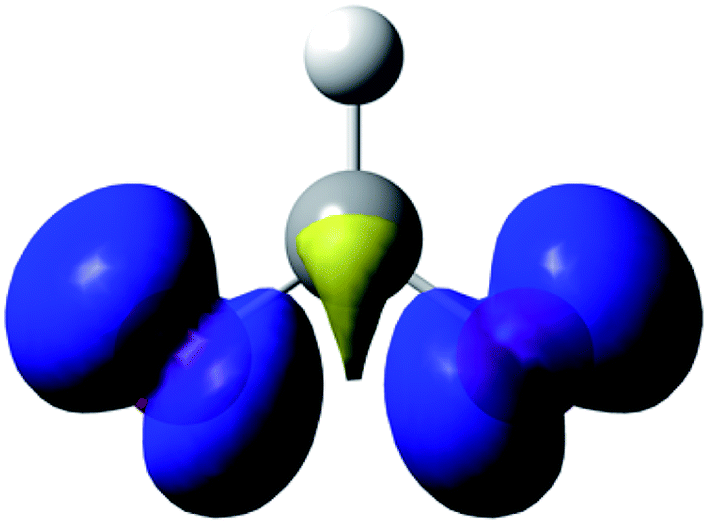 | ||
| Fig. 3 Spin density on the formyloxy radical (0.004 a.u. isosurface). | ||
The presumptive reaction mechanism (Scheme 1) was evaluated computationally using density functional theory (at the SMD(HCOOH)-xrev-DSD-PBEP86-D4/ma-def2-QZVPP//SMD(HCOOH)-PW6B95D3BJ/def2-SVP level of theory, see Computational Details section). Overall the calculations show a low barrier for the radical substitution reaction, which is very exergonic, Table 1. Based on a substitution reaction at the para position (lowest barriers), the calculated 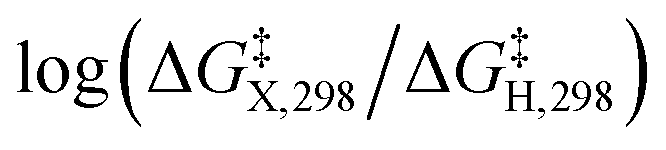 values plotted versus σpara yield ρ = −1.02 (r2 = 0.99), Fig. S5.† There is a very good correlation between the experimental and calculated results supporting the conclusion that the formyloxyl radical is electrophilic, and as such the reactivity is dominated by polar effects following the Bronsted–Evans–Polanyi formalism. It should be noted that arene oxyesterifications as well as alkene dihydroxylations were also reported using substituted malonyl peroxides in acidic, hydrogen-bonding perfluoro alcohols as solvents where different electrophilic ionic mechanisms have been proposed.15
values plotted versus σpara yield ρ = −1.02 (r2 = 0.99), Fig. S5.† There is a very good correlation between the experimental and calculated results supporting the conclusion that the formyloxyl radical is electrophilic, and as such the reactivity is dominated by polar effects following the Bronsted–Evans–Polanyi formalism. It should be noted that arene oxyesterifications as well as alkene dihydroxylations were also reported using substituted malonyl peroxides in acidic, hydrogen-bonding perfluoro alcohols as solvents where different electrophilic ionic mechanisms have been proposed.15
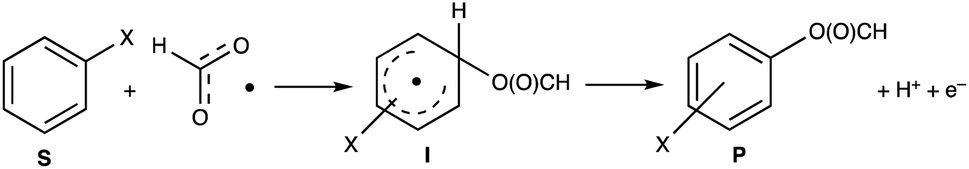 | ||
| Scheme 1 Proposed reaction pathway for reaction of arenes with the formyloxy radical. | ||
|
|
S + HCOO˙ → I, ΔG298 |
|
||
|---|---|---|---|---|
| H | — | 2.2 | −5.1 | −39.2 |
| F | o | 4.5 | −3.7 | −34.7 |
| m | 5.0 | −3.7 | −37.2 | |
| p | 2.6 | −3.4 | −37.2 | |
| Cl | o | 4.8 | −4.9 | −35.7 |
| m | 5.9 | −2.8 | −36.9 | |
| p | 4.1 | −4.0 | −36.6 | |
| Br | o | 4.6 | −4.1 | −36.2 |
| m | 5.7 | −3.4 | −37.2 | |
| p | 4.3 | −4.3 | −37.1 | |
| t Bu | o | 9.0 | −3.2 | −36.2 |
| m | 2.6 | −4.1 | −37.8 | |
| p | 1.5 | −4.3 | −37.8 |
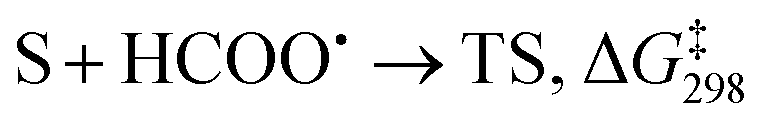
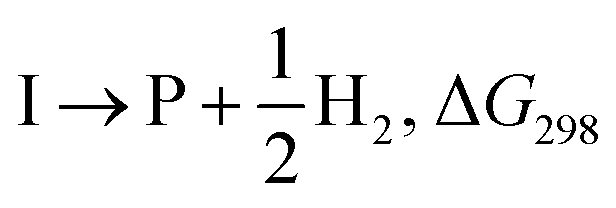
The reaction of alkenes, and especially terminal alkenes, with the formyloxyl radical is also of significant interest. Thus, reactions with1-hexene, 1-heptene and 1-octene as representative substrates yielded mixtures of products as shown in Chart 1. Three classes of products were identified. The major products (∼60–80%) are the linear aldehydes (1-3a) with the same number of carbon atoms and the primary allylic alcohols (1-3b). Both products are formally the result of an anti-Markovnikov oxidative addition reaction. It is probable that the initial products are allylic formate esters that undergo fast hydrolysis14 to yield the allylic alcohol, which are isomerized to yield the linear aldehyde. The second set of products (20–25%) are the result of the further oxidation of an initially formed, unobserved intermediate to yield the terminal vicinal diol (1-3e) that can then be oxidatively cleaved to yield the corresponding linear aldehyde with one less carbon atom (1-3f). The third set of minor products are ketones resulting from overall Markovnikov oxidative addition to the terminal alkene. The overall anti-Markovnikov oxidative addition selectivity is quite striking, typically ∼10:1.
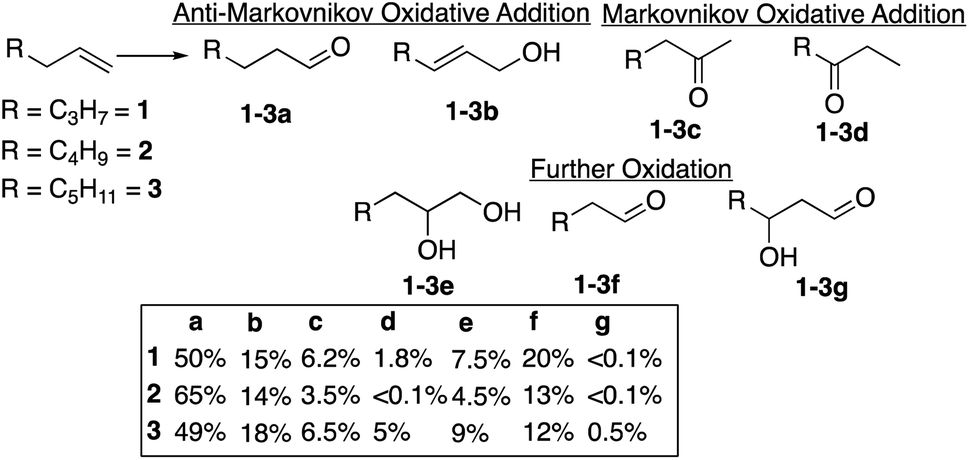 | ||
| Chart 1 Oxidation of terminal alkenes with formyloxyl radicals. Reaction conditions: 1 mmol 1-alkene, 3 mL 1:1 HCOOH:CH3CN, 70 mg LiOCOH, 35 mg K5CoIIIW12O40. Working electrode – Pt net, reference electrode – Pt wire, counter electrode Pt; 1.8 V vs. SHE in an undivided cell; t = 1.5 h; RT. Product analyses were carried out as described in the Experimental section. Small amounts of the allylic formate esters were identified as unhydrolyzed precursors of the allylic alcohols 1-3b. | ||
Using 1-hexene as a standard substrate, the effects of variations in reaction temperature, potential and addition of water were examined. Reaction carried out between −10 °C and 22 °C showed little influence of the temperature on the overall yield and the ratio between the products, Table S1.† On the other hand, increasing the potential, maintained the ratio of the anti-Markovnikov/Markovnikov oxidation products but showed a clear inhibition of the formation of further oxidation products, Table S2.† Especially notable is the strong proportional decrease in the amount of 1,2-hexanediol formed going from 1.4 V to 2.0 V versus SHE. The addition of water combined with a slight increase in temperature to 40 °C had a similar effect on the reaction selectivity vis-à-vis the influence on the formation of 1,2-hexanediol, Table S3.†
The formation of a ketone at the 3-position of the linear alkenes is unusual. Therefore, it was of interest to observe product distribution in reactions of β-substituted terminal alkenes with tertiary and quaternary carbon centers where such ketone formation is not possible. The reaction of the formyloxyl radical with vinylcyclohexane (4), Scheme 2, yielded anti-Markovnikov oxidative addition products, 4a and 4b, as well as the further oxidation products, the vicinal diol (4e) and the C–C bond cleavage product (4f). Interestingly, no oxidation products were formed by a Markovnikov oxidative addition, rather double bond isomerization yielded some ethylidenecyclohexane (4c) and then cyclohexanone (4d).
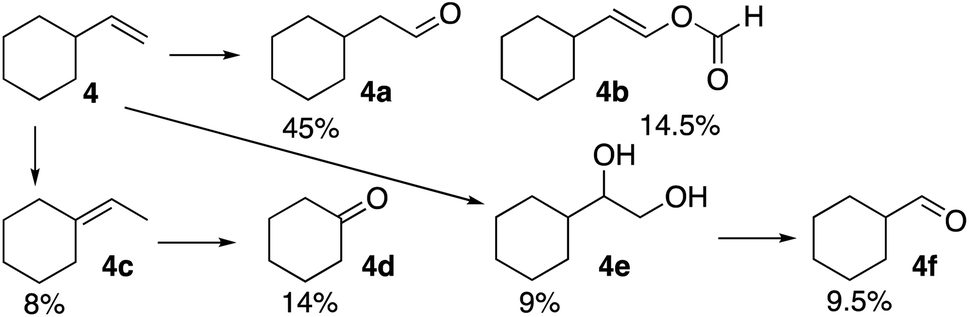 | ||
| Scheme 2 Oxidation of vinylcyclohexane. Reaction conditions: 1 mmol 1-vinylcyclohexene, 3 mL 1:1 HCOOH:CH3CN, 70 mg LiOCOH, 35 mg K5CoIIIW12O40. Working electrode – Pt net, reference electrode – Pt wire, counter electrode Pt; 1.8 V vs. SHE in an undivided cell; t = 1.5 h; RT. Product analyses were carried out as described in the Experimental section. | ||
The reaction of the formyloxyl radical with 3,3-dimethyl-1-butene (5), Scheme 3, also yielded the anti-Markovnikov oxidative addition product 5a as well as the further oxidation products, the vicinal diol (5e) and the C–C bond cleavage product (5f). Here, the formation of a radical on the quaternary carbon is not possible. Only, a minor amount of oxidative Markovnikov products, 5c and 5d were formed.
 | ||
| Scheme 3 Oxidation of 3,3-dimethyl-1-butene. Reaction conditions: 1 mmol 3,3-dimethyl-1-butene, 3 mL 1:1 HCOOH:CH3CN, 70 mg LiOCOH, 35 mg K5CoIIIW12O40. Working electrode – Pt net, reference electrode – Pt wire, counter electrode Pt; 1.8 V vs. SHE in an undivided cell; t = 1.5 h; RT. Product analyses were carried out as described in the Experimental section. nd-not detected. | ||
The reaction that most resembles that of the presently reported reaction of the formyloxyl radical with alkenes is the well-known Kharasch–Sosnovsky reaction. This is a copper-catalyzed oxidation of alkenes with peresters that, however, yields allylic oxidation products with preferred substitution of terminal alkenes at the C3 rather than C1 position.16 It is generally thought that the mechanism of the Kharasch–Sosnovsky reaction, using RC(O)OO-tBu as an oxidant, is cleavage of the O–O peroxide bond leading to formation of the tert-butoxy radical and CuIIO(O)CR.16b The radical then reacts with the alkene to form an allyl radical that then reacts with the CuIIO(O)CR to give the allylic ester product. Although the reaction of the allyl radical with CuIIO(O)CR was originally suggested to take place either via an allyl cation16c or is ligand controlled,17 it would appear that a pericyclic mechanism best explains the observed reactivity that includes double bond isomerization.18
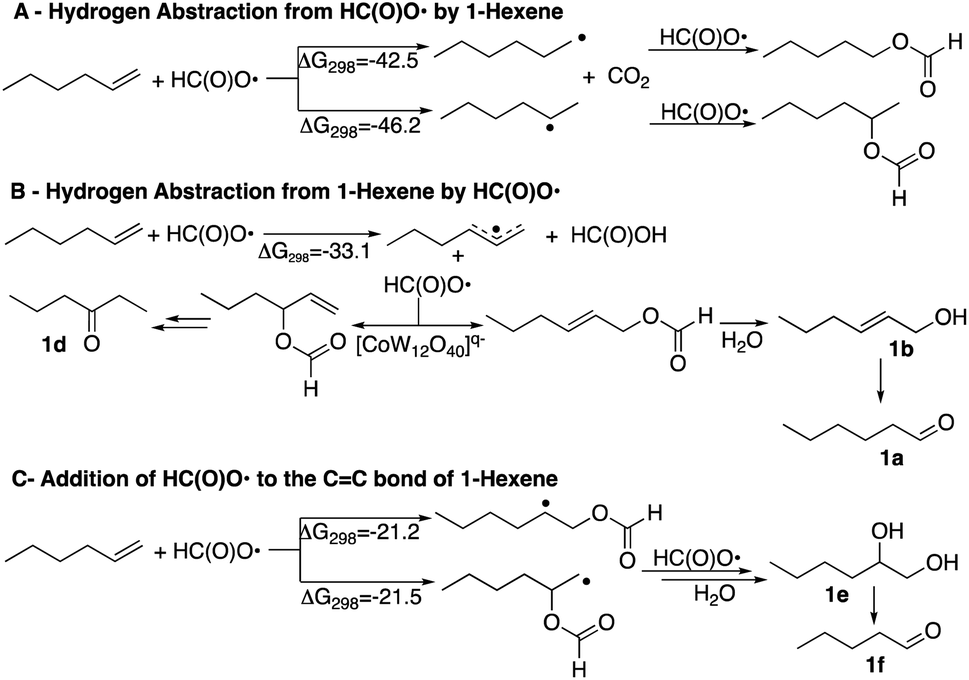
It is therefore of interest to survey possible reaction pathways to explain the reaction selectivity that was observed using 1-hexene as a representative substrate. Three pathways for the initial reaction of HC(O)O˙ with 1-hexene were considered, Scheme 4. DFT calculations show that hydrogen abstraction from the formyloxyl radical by 1-hexene (A) is the most exergonic of all the pathways considered. As would be expected the 2-hexyl radical was calculated to be more stable than the 1-hexyl radical by ΔG298 = −3.7 kcal mol−1. Further radical–radical heterocoupling reactions would yield 2-hexylformate and 1-hexylformate in very exergonic reactions (∼−84 kcal mol−1). 2-Hexylformate and 1-hexylformate could conceivably react with additional formyloxyl radicals to yield the corresponding alcohols and carbonyl products by a subsequent hydrolysis/oxidation sequence. However, (i) neither 2-hexylformate and 1-hexylformate nor their hydrolysis products 2-hexanol and 1-hexanol were observed as products, (ii) the C1 experimentally observed selectivity is not verified by the relative stabilities of the hexyl radicals where oxidation at C2 rather than C1 would be expected, and (iii) the oxidation of 1-hexanol under the same reactions conditions used for the oxidation of 1-hexene (see caption to Scheme 1) revealed that 1-hexanol is an order of magnitude less reactive than 1-hexene. 2-Hexanol was more reactive than 1-hexanol and considering that 2-hexanone was a minor product, pathway A could explain its formation.
 | ||
| Scheme 4 Reaction pathways for the reaction of 1-hexene. | ||
A second pathway (B) is the reaction of the formyloxyl radical with 1-hexene to give the hexallyl radical in an exergonic, barrierless reaction, Scheme 4. The spin density on the hexallyl radical is evenly distributed between C1 and C3, Fig. S6.† Radical–radical heterocoupling reactions, which are also very exergonic (∼71 kcal mol−1) could yield the various allylic formate esters as shown in Scheme 4. Only hex-2-en-1-yl formate was experimentally observed, but in small amounts. Hydrolysis of hex-2-en-1-yl formate would yield 2-hexen-1-ol that was observed (15 mol%, Chart 1). Isomerization of 2-hexen-1-ol would yield hexanal. This isomerization reaction was verified in a control reaction using 2-hexen-1-ol as substrate, where some of the further oxidation products were also obtained in minor amounts. The product of the reaction of the hexallyl radical with HC(O)O˙ at the C3 position to yield 1-hexen-3-formate or the hydrolysis product, 1-hexen-3-ol was not observed, but the formation of 3-hexanone can be also be inferred via an isomerization reaction. It should be noted that hexanal, in principle could be formed by the formation of vinylic formates via a [1,3]H-sigmatropic shift that may be catalyzed by [CoIIIW12O40]5−. However, calculations show that such a reaction is endergonic, where the resulting vinylic formate is less stable than the allylic formate by 3.8 kcal mol−1. A [1,3]H-sigmatropic shift is more likely in reactions of cyclic alkenes with HC(O)O˙ since the shift is going to give a more stable secondary radical. Thus, cyclohexene yielded also cyclohexanone and 3-cyclohexen-1-ol and 1-methyl cyclohexene yielded 2-methyl cyclohexanone and 3-methyl-3-cyclohexen-1-ol, Scheme S1.† The observed formation of n-hexanal (1a) as the major product as well as the primary allylic alcohol (1b), its ester, and 3-hexanone (1d) is apt for a reaction initiated by the formation of a hexaallyl intermediate, but is not an obvious explanation for the formation of 2-hexanone (1c) and the further oxidation products, 1,2-hexanediol (1e) and n-pentanal (1f).
The third pathway (C) involves addition of the formyloxyl radical to the CC bond as shown in Scheme 4. Further coupling reactions of the radical species with formyloxyl radicals would lead to the formation of 1,2-hexanediol. A control reaction using 1,2-hexanediol as substrate under the common reaction conditions (see caption of Scheme 1) shows that it was efficiently converted only to n-pentanal commensurate with the amount of n-pentanal formed in the oxidation of 1-hexene.
Since the oxidation of alkenes plausibly involves hydrogen abstraction at the allylic position (Scheme 4, B) where the bond dissociation energy (BDE) for 1-hexene is 83.4 kcal mol−1,19a it was of interest to evaluate the potential of the formyloxyl radical for activation of stronger C–H bonds. Cyclohexane (BDE = 99.5 kcal mol−1) showed only very low reactivity, but a series of alkyl arenes – toluene, p-xylene, ethyl benzene and cumene with BDEs of 89.7, 87.7, 85.4 and 84.4 kcal mol−1 respectively19 all reacted at the benzylic position as well as on the ring. The ratios of benzylic versus ring reactivity (mol%) was 75:25, 69:31, 78:22, 98:2, respectively, and provides a measure of C–H activation versus aromatic substitution reactions.
In the absence of K5[CoIIIW12O40] only trace amounts of products are formed. Thus, the role of the K5[CoIIIW12O40] as catalyst requires consideration. Previously, we observed that the EPR spectrum associated with the spin adduct formed by the reaction of HC(O)O˙ with the BMPO (5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide) spin trap is quenched in the presence of [CoIIIW12O40]5−, which during the reaction is reduced to [CoIIW12O40]6−.9 This indicates that [CoIIIW12O40]5− reacted very fast with a radical species, either HC(O)O˙ and/or the BMPO spin adduct. Considering that the “resting state” of the polyanion is reduced despite the strongly oxidizing reaction (anodic) conditions and that the CoII/CoIII/CoIV redox metal is coordinatively and sterically inaccessible, it is reasonable to assume that an outer-sphere electron transfer forms a donor–acceptor [D+–A−] complex between the formyloxyl radical donor and the polyanion acceptor.20 In the copper catalyzed Kharasch–Sosnovsky reaction, it is generally accepted that an intermediate complex is formed between the very short-lived acyloxyl radical and the Cu(II) catalyst.16–18 Considering the steric bulk of the polyanion, whose hydrodynamic diameter is >1 nm, and its redox potentials versus SHE as measured in acetonitrile, Scheme 5, the existence of a [D+–A−] complex is probably related to the anti-Markovnikov selectivity and the isomerization reactions observed in the formyloxylation of terminal alkenes.
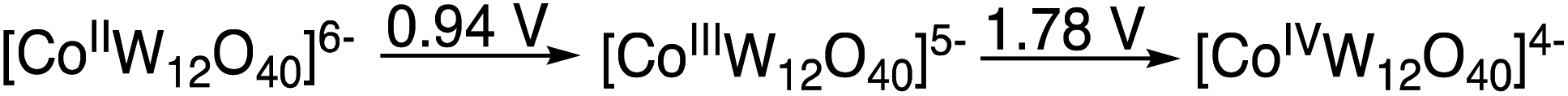 | ||
| Scheme 5 Redox potentials of [CoW12O40]q− in acetonitrile versus SHE. | ||
Experimental
Materials
All chemicals were reagent grade and used as supplied. Alkene substrates were purified on alumina column and analyzed by GC-MSD prior to use. Formic acid was 98+% from Acros Organic. The K5CoIIIW12O40·16H2O polyoxometalate was prepared following a literature method.21Reaction analysis
Combined gas chromatography-flame ionization (GC-FID) and gas chromatography-mass selective (GC-MSD) measurements were carried out to identify and quantify reactions products using a HP 6890 instrument with a flame ionization detector and a HP 5973 instrument with a mass selective detector. Separations were carried out on a 30 m column (Restek 5 MS, 0.32 mm internal diameter) with a 0.25 μm thick coating of 5% phenylmethylsilicone with helium as the carrier gas.Electrochemistry
The electrocatalytical experiments were performed unless stated otherwise noted at room temperature in a glass three-electrode cell equipped with a platinum gauze anode working electrode (3 cm2 area), a platinum cathode, and a Pt wire quasi reference electrode. Typically, a magnetically stirred solution of substrate (1 mmol), lithium formate (0.25 M) and K5CoIIIW12O40 (10 mmol) in 3 mL of a 1:1 mixture of HCOOH:acetonitrile was electrolyzed at constant potential of 1.8 V versus SHE until transfer of a known amount of charge. Identification and quantification of products were determined by GC-MSD and GC-FID using also reference standards as described above.
Kinetics of benzene formyloxylation
Benzene (1 mmol) was electrolyzed under the conditions described above until transfer of 20 mA h charge (about 90 minutes). During electrolysis, ten aliquots (about 1.5 μL each) were collected over time from the reaction mixture without pausing the electrolysis. The syringe was washed with a 1:1 mixture of HCOOH and CH3CN between collections of the samples flushed twice with the reaction mixture just before sampling. The samples were analyzed by GC-FID.
Kinetics of arene formyloxylation
An equimolar mixture of benzene and the substituted arene (1 mmol in total) was electrolyzed under the conditions described above. The collection of sample aliquots was performed as described above.Electrochemical formyloxylation of alkenes
The substrate of interest was electrolyzed under the conditions described in the Electrochemistry subsection. Typically, a magnetically stirred solution of substrate (1 mmol), lithium formate (0.25 M) and K5CoIIIW12Ou (10 mmol) in 3 mL of a 1:1 mixture of HCOOH:acetonitrile was electrolyzed at constant potential of 1.8 V versus SHE. Products were analyzed by GC-MSD and GC-FID.
Computational methods
All calculations were performed using Gaussian16 Revisions B.01 and C.01 (ref. 22) and Orca versions 4.2.0 and 4.2.1;23 geometries were optimized with the former and accurate double-hybrid energies were calculated with the latter. Geometries were optimized using Zhao and Truhlar's PW6B95D3BJ exchange–correlation functional,24 which includes the third version of Grimme's dispersion25 with the Becke–Johnson dampening function.26 With this functional the double-ζ version of the second revision (def2) of Ahlrichs and coworkers' basis sets (def2-SVP)27 was used. Accurate energies were calculated using Santra et al.‘s revised version of the older DSD-PBEP86 double-hybrid functional, specifically xrevDSD-PBEP86-D4;28 this functional was shown to be more accurate, especially for radical systems.23 The functional includes the fourth generation of Grimme's dispersion correction (i.e., D4).29 With this functional, the quadruple-ζ with two sets of polarization functions of the second revision (def2) of Ahlrichs and coworkers' basis sets (def2-QZVPP)27 minimally-augmented with sp diffuse functions only as per Zheng et al. (i.e., ma-def2-QZVPP)30 was used. With Orca, the resolution-of-the-identity (RI) with the def2-QZVPPD/C31 and def2/J32 auxiliary basis sets was used in conjunction with the resolution of identity–chain of spheres exchange (RIJCOSX) approximation33 to improve the performance of all energy calculations. Bulk solvent effects were approximated by using a polarizable continuum model (PCM), specifically the integral equation formalism model (IEF-PCM)34 with formic acid as the solvent as in the experiments and in particular Truhlar's empirically parameterized version Solvation Model with Dispersion (SMD) was used.35 Solvation was used for all calculations, including geometry optimizations. The connectivity of all transition states was confirmed by intrinsic reaction coordinate (IRC) calculations using the default Hessian-based projector-corrector integrator scheme,36 or where the IRCs were inconclusive (e.g., when the surface around the transition state is too flat) by distorting the transition state along its imaginary frequency in each direction and following it downhill to the reactant(s) and product(s).Conclusions
Linear free energy relationships (LFERs) are typically derived from kinetic measurements carried out as a function of time. In these reactions, where the formyloxyl oxidant is formed in situ electrochemically, we found higher precision and repeatability for kinetic measurements in an undivided cell configuration as a function of charge transferred (Q). In this way the philicity of the formyloxyl radical was measured by carrying out aromatic substitution reactions using a Hammett LFER. The experimental results yielded a ρ value of −1.5 with a very good correlation with σpara values indicating that HC(O)O˙ can be considered electrophilic. A Hammett LFER using DFT-calculated free energies of activation,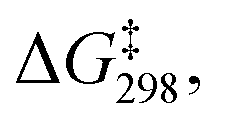 supports this conclusion.
supports this conclusion.
The reaction of HC(O)O˙ with five terminal alkenes revealed the preference for the formation of the anti-Markovnikov linear aldehyde and the 2-en-1-ol/formate over the formation of the Markovnikov ketone, typically in 10:1 or greater ratios. Major by-products, predominantly the aldehyde formed via the cleavage of the C1–C2 bond, are the result of further oxidation. Three plausible reaction pathways, based on the oxidation of 1-hexene, supported by “control” oxidation reactions with 1-hexanol, 2-hexanol, 2-hexen-1-ol and 1,2-hexanediol as substrates, are considered to explain the results. All the reaction pathways considered are initiated by very exergonic reactions, Scheme 4. It would appear that the most straight-forward pathway to the anti-Markovnikov linear aldehyde is a reaction involving hydrogen atom abstraction by HC(O)O˙ at the allylic position (C–H BDE ∼83–84 kcal mol−1) to form an allylic radical (Scheme 4, pathway B). A heteroradical coupling reaction between HC(O)O˙ and the allylic radical to yield the primary allylic formate followed hydrolysis yields 2-hexen-1-ol, which was shown to undergo an isomerization reaction to yield n-hexanal as the major product from 1-hexene. A similar pathway but with a different regioselectivity in the heteroradical coupling step also explains the formation of 3-hexanone from 1-hexene as a minor product. It is proposed that the [CoIIIW12O40]5− catalyst reversibly forms donor–acceptor [D+–A−], complexes with radical species. Thus, catalysis is thought to be related to (1) the inhibition of HC(O)O˙ radical decomposition, for example by anodic one-electron oxidation or unimolecular decarboxylation, (2) catalysis of isomerization reactions and (3) the steric bulk of the [CoIIIW12O40]5− polyanion that may direct reactions toward anti-Markovnikov oxidative addition reactions.
A different reaction pathway (Scheme 4, pathway C) is proposed for the formation of the further oxidation products, whereby addition of HC(O)O˙ to the CC alkene double bond would result in the formation of a radical-ester intermediate. A further radical heterocoupling would yield the vicinal diol after hydrolysis. Such diols are susceptible to C–C bond cleavage, as also demonstrated here, yielding the aldehyde with one less carbon atom as a major by-product. It is worthwhile noting that the reaction pathway (Scheme 4, pathway A) that has the most exergonic first step does not appear to be relevant for the formation of most of the products, except perhaps for the formation of the minor Markovnikov oxidative addition product (i.e., 2-hexanone from 1-hexene).
The reactivity of HC(O)O˙ toward hydrogen abstraction was further investigated in order to obtain an empirical limit for C–H bond activation. It was found that cyclohexane (BDE = 99.5 kcal mol−1) was only marginally reactive. However, the reaction of HC(O)O˙ with a series of primary, secondary and tertiary alkylated benzene derivatives with BDEs ranging from 84.4 to 89.7 kcal mol−1 demonstrated that benzylic C–H bond activation occurs in all cases in competition with electrophilic aromatic substitution reactions. These results would indicate that reactions with HC(O)O˙ could be rather efficient for the activation of C–H bonds that have BDEs of up to ∼90 kcal mol−1. Thus, the formyloxyl radical is apparently “weaker” than the more commonly used hydroxyl and tert-butoxyl oxygen centered radicals, which in the future may translate into higher selectivity for C–H bond activation. Given the unique reactivity of HC(O)O˙, the objective of ongoing research is to minimize the further too fast oxidation of HC(O)O˙ to CO2 that limits the faradaic efficiency and leads to only low yields of these reactions as also previously noted.9
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Israel Science Foundation grant 1237/18. R. N. is the Rebecca and Israel Sieff Professor of Organic Chemistry.Notes and references
- (a) E. J. Hart, J. Phys. Chem., 1952, 56, 594–599 CrossRef CAS; (b) W. M. Garrison and J. K. Rollefson, Discuss. Faraday Soc., 1952, 12, 155–161 RSC.
- J. Jordan and P. T. Smith, Proc. Chem. Soc., 1960, 546–547 Search PubMed.
- M. Suto, X. Wang and L. C. Lee, J. Phys. Chem., 1988, 92, 3764–3768 CrossRef CAS.
- (a) E. H. Kim, S. E. Bradforth, D. W. Arnold, R. B. Metz and D. M. Neumark, J. Chem. Phys., 1995, 103, 7801–7814 CrossRef CAS; (b) E. Garand, K. Klein, J. F. Stanton, J. Zhou, T. I. Yacovitch and D. M. Neumark, J. Phys. Chem. A, 2010, 114, 1374–1383 CrossRef CAS.
- (a) A. Rauk, D. Yu, P. Borowski and B. Roos, Chem. Phys., 1995, 1971, 73–80 CrossRef; (b) P. Y. Ayala and H. B. Schlegel, J. Chem. Phys., 1998, 108, 7560–7567 CrossRef CAS; (c) D. Feller, D. A. Dixon and J. S. Francisco, J. Phys. Chem. A, 2003, 107, 1604–1617 CrossRef CAS; (d) W. Sun and M. Saeys, J. Phys. Chem. A, 2008, 112, 6918–6928 CrossRef CAS.
- (a) J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Noerskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS; (b) S. Back, J.-H. Kim, Y.-T. Kim and Y. Jung, Phys. Chem. Chem. Phys., 2016, 18, 9652–9657 RSC; (c) J. M. Hull, M. R. Provorse and C. M. Aikens, J. Phys. Chem. A, 2012, 116, 5445–5452 CrossRef CAS; (d) J. Scaranto and M. Mavrikakis, Surf. Sci., 2016, 648, 201–211 CrossRef CAS; (e) I. Palacio, J. M. Rojo and O. Rodríguez de la Fuente, ChemPhysChem, 2012, 13, 2354–2369 CrossRef CAS.
- A. Fraind, R. Turncliff, T. Fox, J. Sodano and L. R. Ryzhkov, J. Phys. Org. Chem., 2011, 24, 809–820 CrossRef CAS.
- A. Rauk, D. Yu and D. A. Armstrong, J. Am. Chem. Soc., 1994, 116, 8222–8228 CrossRef CAS.
- A. M. Khenkin, M. Somekh, R. Carmieli and R. Neumann, Angew. Chem., Int. Ed., 2018, 57, 5403–5407 CrossRef CAS.
- (a) J. J. Dong, W. R. Browneand and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 734–744 CrossRef CAS; (b) Z. K. Wickens, K. Skakuj, B. Morandi and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 890–893 CrossRef CAS; (c) Z. K. Wickens, B. Morandi and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 11257–11260 CrossRef CAS; (d) X.-S. Ning, M.-M. Wang, C.-Z. Yao, X.-M. Chen and Y.-B. Kang, Org. Lett., 2016, 18, 2700–2703 CrossRef CAS; (e) K.-F. Hu, X.-S. Ning, J.-P. Qu and Y.-B. Kang, J. Org. Chem., 2018, 83, 11327–11332 CrossRef CAS.
- S. C. Hammer, G. Kubik, E. Watkins, S. Huang, H. Minges and F. H. Arnold, Science, 2017, 358, 215–218 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. E. Kurz and M. Pellegrini, J. Org. Chem., 1970, 35, 990–992 CrossRef CAS.
- H. W. Gibson, Chem. Rev., 1969, 69, 673–692 CrossRef CAS.
- (a) A. Dragan, T. M. Kubczyk, J. H. Rowley, S. Sproules and N. C. O. Tomkinson, Org. Lett., 2015, 17, 2618–2621 CrossRef CAS; (b) A. Pilevar, A. Hosseini, M. Šekutor, H. Hausmann, J. Becker, K. Turke and P. R. Schreiner, J. Org. Chem., 2018, 83, 10070–10079 CrossRef CAS; (c) J. C. Griffith, K. M. Jones, S. Picon, M. J. Rawling, B. M. Kariuki, M. Campbell and N. C. O. Tomkinson, J. Am. Chem. Soc., 2010, 132, 14409–14411 CrossRef CAS; (d) A. Pilevar, A. Hosseini, J. Becker and P. R. Schreiner, J. Org. Chem., 2019, 84, 12377–12386 CrossRef CAS.
- (a) M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS; (b) M. B. Andrus and J. C. Lashley, Tetrahedron, 2002, 58, 845–866 CrossRef CAS; (c) C. Walling and A. A. Zavitsas, J. Am. Chem. Soc., 1963, 85, 2084–2090 CrossRef CAS.
- J. K. Kochi and H. E. Mains, J. Org. Chem., 1965, 30, 1862–1872 CrossRef CAS.
- A. L. J. Beckwith and A. A. Zavitsas, J. Am. Chem. Soc., 1986, 108, 8230–8234 CrossRef CAS.
- (a) Y.-R. Luo, BDEs of C–H bonds, in Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007, pp. 19–145 CrossRef; (b) D. F. McMillen and D. M. Golden, Ann. Rev. Phys. Chem., 1982, 33, 493–532 CrossRef CAS.
- L. Eberson, J. Am. Chem. Soc., 1983, 105, 3192–3199 CrossRef CAS.
- L. C. W. Baker and T. P. McCutcheon, J. Am. Chem. Soc., 1956, 78, 4503–4506 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed; (c) F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. A. Kreig, J. Chem. Phys., 2010, 132, 154104 CrossRef; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- (a) A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154104 CrossRef; (b) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef; (c) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) G. Santra, N. Sylvetsky and J. M. L. Martin, J. Phys. Chem. A, 2019, 123, 5129–5143 CrossRef CAS; (b) J. M. L. Martin and G. Santra, Isr. J. Chem., 2020, 60, 787–804 CrossRef CAS.
- (a) E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef; (b) E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- A. Hellwig, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS; (b) S. Kossmann and F. Neese, Chem. Phys. Lett., 2009, 481, 240–243 CrossRef CAS; (c) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS; (d) S. Kossmann and F. Neese, J. Chem. Theory Comput., 2010, 6, 2325–2338 CrossRef CAS; (e) R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef.
- (a) B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS; (b) E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef; (c) B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS; (d) J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct., 1999, 464, 211–226 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- (a) H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS; (b) H. P. Hratchian and H. B. Schlegel, in Theory and Applications of Computational Chemistry: The First 40 Years, ed. C. E. Dykstra, G. Frenking, K. S. Kim, and G. E. Scuseria, Elsevier, Amsterdam, 2004, pp. 194–249 Search PubMed; (c) H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetic data, computational data, tables, and schemes of results of additional reactions. See DOI: 10.1039/d0sc04936k |
| This journal is © The Royal Society of Chemistry 2020 |