
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Forwards and backwards – synthesis of Laurencia natural products using a biomimetic and retrobiomimetic strategy incorporating structural reassignment of laurefurenynes C–F†
Hau Sun Sam
Chan
 ,
Amber L.
Thompson‡
,
Kirsten E.
Christensen‡
and
Jonathan W.
Burton
*
,
Amber L.
Thompson‡
,
Kirsten E.
Christensen‡
and
Jonathan W.
Burton
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: jonathan.burton@chem.ox.ac.uk
First published on 8th October 2020
Abstract
Laurefurenynes C–F are four natural products isolated from Laurencia species whose structures were originally determined on the basis of extensive nuclear magnetic resonance experiments. On the basis of a proposed biogenesis, involving a tricyclic oxonium ion as a key intermediate, we have reassigned the structures of these four natural products and synthesized the four reassigned structures using a biomimetic approach demonstrating that they are the actual structures of the natural products. In addition, we have developed a synthesis of the enantiomers of the natural products laurencin and deacetyllaurencin from the enantiomer of (E)-laurefucin using an unusual retrobiomimetic strategy. All of these syntheses have been enabled by the use of tricyclic oxonium ions as pivotal synthetic intermediates.
Introduction
The C15-acetogenin natural products isolated from Laurencia species display both wide structural diversity and wide ranging biological activity which have piqued the interest of synthetic organic chemists since the initial isolation of (+)-laurencin in 1965.1–3 These natural products generally contain cyclic ethers, and frequently medium-ring ethers, and have served as a test-bed for the development of numerous new and efficient methodologies to allow access to complex cyclic ethers.4–7 The structural assignment of many of these acetogenic natural products has come primarily from high-field NMR experiments with single crystal X-ray diffraction being used where possible. The unambiguous assignment of natural products structures with compounds not suitable for single crystal X-ray analysis8 is a challenging task and it is inevitable that incorrect structures are reported in the literature with these structural misassignments frequently being uncovered through total synthesis.9–11 Within the C15 acetogenic natural products isolated from Laurencia species a number of structural misassignments have also occurred that have varied from gross structural reassignments,12–15 through to atom transpositions16–19 and stereochemical reassignment.20–22 Key to correcting many of these structural misassignments have been postulates regarding the biosyntheses of acetogenic Laurencia natural products, many of which have been proposed to proceed through complex oxonium ion intermediates.15,23–33 These biogenetic postulates have allowed rational prediction of the likely structures of a number of the natural products15,21,22,26,34 and in a number of cases the biogenetic arguments have been augmented by DFT calculations of both proton and carbon NMR chemical shifts14,22,34 for a range of candidate structures with ultimately the structure of the natural products being established through total synthesis.35 Among the acetogenic Laurencia natural products whose structures have been reassigned are laurefucin,36,37 obtusallenes V, VI and VII,16,19,26 elatenyne,12–15,38 laurendecumenyne B,15,39,40 aplysiallene,41,42 a chloroenyne from Laurencia majuscula13,34,38,43 and laurefurenynes A and B,20–22 additionally the structures of the ocellenynes have been tentatively reassigned on the basis of biogenetic considerations and DFT calculations of NMR chemical shifts.24,44,45The laurefurenynes are a series of six acetogenic natural products which were isolated and characterized by Jaspars and co-workers from a sample of Laurencia sp. collected in the Philippines in 1991.20 The structures of these six natural products ((Z/E)-1, (Z/E)-2 and (Z/E)-3) were assigned through extensive 1D and 2D NMR experiments (Fig. 1).46 In 2013 the Britton group21 and our own group22 published structural reassignments of laurefurenynes A and B from (Z/E)-1 to (Z/E)-4 based on total synthesis and DFT calculations of NMR chemical shifts. In the original paper of Jaspars, a plausible biogenesis of the laurefurenynes (((Z/E)-1, (Z/E)-2 and (Z/E)-3) was proposed based on epoxide and/or bromonium ion cyclizations. However, the reassigned structures of laurefurenynes A and B (Z/E)-4 fit with a proposed biosynthesis involving the previously characterized tricyclic oxonium ions 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47 which likely arise via transannular displacement of bromide or chloride from the halofucins 5 and 6 (Scheme 1),15,48,49 followed by opening of the oxonium ions 7 at C-7 with water and then displacement of the C-12 bromine atom with water with inversion of configuration.21,22 If it is indeed the case that laurefurenynes A and B (Z/E)-4 are biosynthesized from the halofucins 5 and 6 then it would be reasonable to postulate that laurefurenynes C–F are produced on the same biosynthetic pathway.21 Here, opening of the oxonium ions 7 with water, or a water equivalent,26,34 at C-10 would produce the reassigned structures of laurefurenynes E and F (Z/E)-950 and displacement of the bromine substituent with inversion of configuration by water or a water equivalent would yield the reassigned structures of laurefurenynes C and D (Z/E)-10.
47 which likely arise via transannular displacement of bromide or chloride from the halofucins 5 and 6 (Scheme 1),15,48,49 followed by opening of the oxonium ions 7 at C-7 with water and then displacement of the C-12 bromine atom with water with inversion of configuration.21,22 If it is indeed the case that laurefurenynes A and B (Z/E)-4 are biosynthesized from the halofucins 5 and 6 then it would be reasonable to postulate that laurefurenynes C–F are produced on the same biosynthetic pathway.21 Here, opening of the oxonium ions 7 with water, or a water equivalent,26,34 at C-10 would produce the reassigned structures of laurefurenynes E and F (Z/E)-950 and displacement of the bromine substituent with inversion of configuration by water or a water equivalent would yield the reassigned structures of laurefurenynes C and D (Z/E)-10.
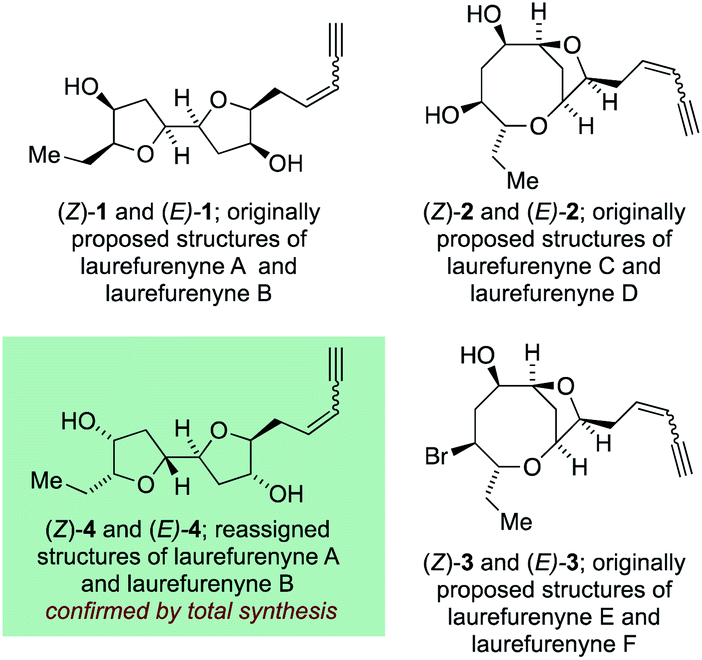 | ||
| Fig. 1 Originally proposed structures of laurefurenynes A–F along with reassigned structures of laurefurenynes A and B. | ||
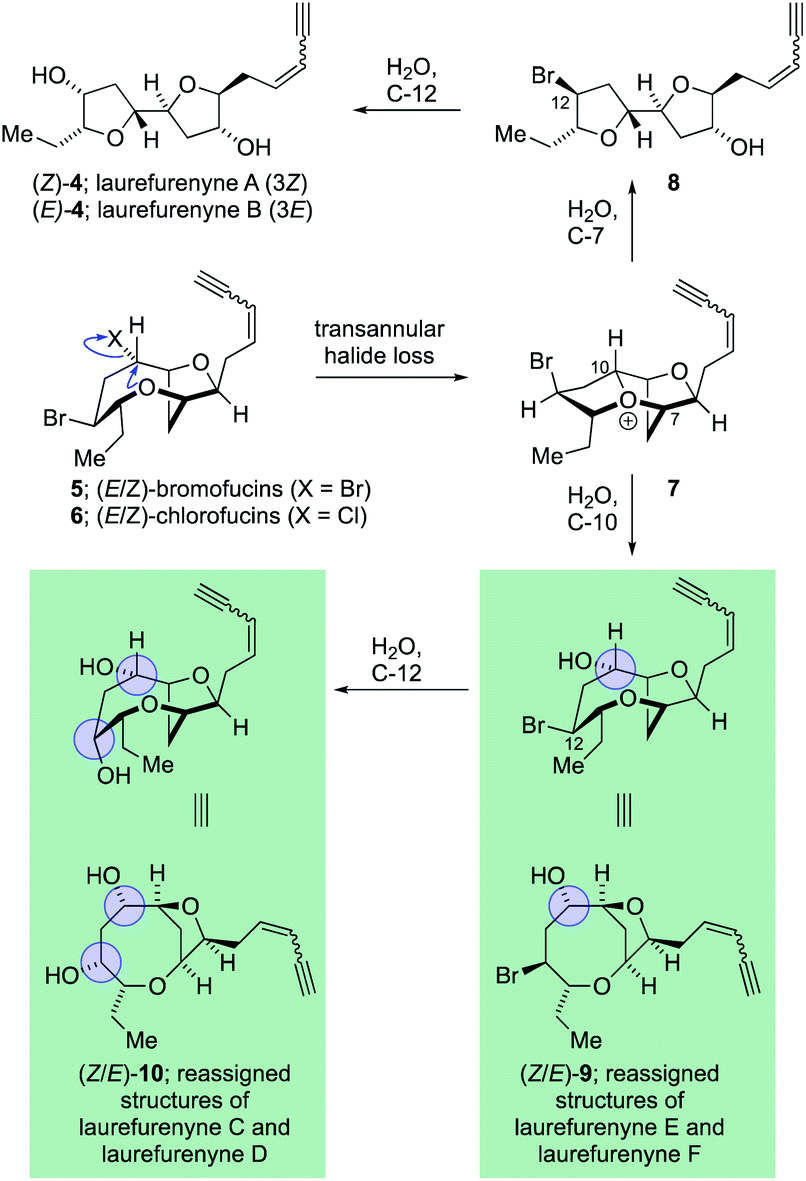 | ||
| Scheme 1 Proposed biosynthesis of the reassigned structures of laurefurenynes A–F from the halofucins. | ||
Based on the above analysis, we report the first synthesis and the first biomimetic synthesis of the reassigned structures of laurefurenynes C–F (Z/E)-10 and (Z/E)-9. Confirmation that the structures of the actual natural products are represented by (Z/E)-10 and (Z/E)-9 comes through comparative 1H and 13C nuclear magnetic resonance data for our synthetic material with that of the natural material,20 Mosher's ester analysis to establish the absolute configuration of a newly installed secondary alcohol,51,52 single crystal X-ray diffraction data giving the structure of the reassigned natural product laurefurenyne D (E)-10 and the reassigned dioxabicyclo[5.2.1]decane cores 12 of laurefurenynes C–F.53 In combination with the reported reassigned structures of laurefurenynes A and B (Z/E)-4 by Britton and our own group,21,22 reassignment of the structures of the remaining laurefurenynes (Z/E)-9 and (Z/E)-10 offers chemical evidence to prompt reevaluation of the initially proposed biosynthesis of these natural products.20 In addition, the development of methods necessary for the total synthesis of the reassigned structures of laurefurenynes C–F (Z/E)-9 and (Z/E)-10 enabled the total synthesis of the enantiomers of the natural products laurencin1–3 and deacetyllaurencin54,55 namely ent-laurencin and ent-deacetyllaurencin using an unusual retrobiomimetic approach.56
Results and discussion
The synthesis of the reassigned structures of laurefurenynes C–F (Z/E)-10 and (Z/E)-9 began with studies concerning the expansion of the scope of our previously reported bromocyclization–nucleophilic quenching reaction of known enantiopure bromomesylates 1157 to include oxygen nucleophiles (Fig. 2). In our previous report, only chloride and bromide had been used as nucleophiles in this transformation and extra steps were required for the introduction of oxygen substituents at the desired C-10 position.47 The expansion of the scope of nucleophiles would allow a direct bromocyclization–oxygenation reaction of 11 to provide the dioxabicyclo[5.2.1]decane core of the reassigned structures of laurefurenynes E–F (12) more efficiently, obviating the need for the intermediacy of 13 (Nu = Cl, Br) as a synthetic intermediate. We conducted the initial studies on expanding the scope of the bromocyclization/nucleophilic quenching reaction with the known enantiopure bromomesylate 1457 instead of 11, as the reaction with 14 was consistently found to give exclusive C-10 opening products with halide nucleophiles,47 therefore simplifying reaction analysis. Thus, exposure of the bromomesylate 14 to our previously described conditions (AgAl(pftb)4·CH2Cl2 (pftb = perfluoro-t-butoxy) and TiCl4) at low temperature which likely generates the oxonium ion 15, followed by addition of saturated aqueous NaHCO3 solution, unexpectedly gave what appeared to be the previously reported chloride 16 as the major product by TLC analysis versus an authentic sample (Scheme 2).47 However, using excess AgOBz as the oxygen nucleophile in the place of saturated aqueous NaHCO3, gave the C-10 alcohol 17 in 74% overall yield after methanolysis of the crude benzoate ester; 17 has previously been reported by both Kim and Snyder.58–60 The formation of 17 with AgOBz rather than aqueous NaHCO3 solution is most likely due to the chloride anion scavenging ability of Ag+, rendering chloride anions unable to compete with benzoate anions for nucleophilic attack at the oxonium ion intermediate 15.61 Following Kim's two step procedure58 compound 17 was readily transformed into ent-(E)-laurefucin 18.36,37,58,59,62,63 Hence, cross-metathesis of 17 with crotonaldehyde using catalyst 19 and copper(I) iodide,64 followed by the Colvin–Ohira reaction using trimethylsilyldiazomethane and lithium diisopropylamide installed the (E)-enyne in 41% overall yield giving ent-(E)-laurefucin 18 that had identical 1H NMR data to a sample we had prepared previously. The matching spectroscopic data for our synthetic 17 and 18 with material prepared by Kim and Snyder confirmed that the C-10 alcohol had the configuration shown.65
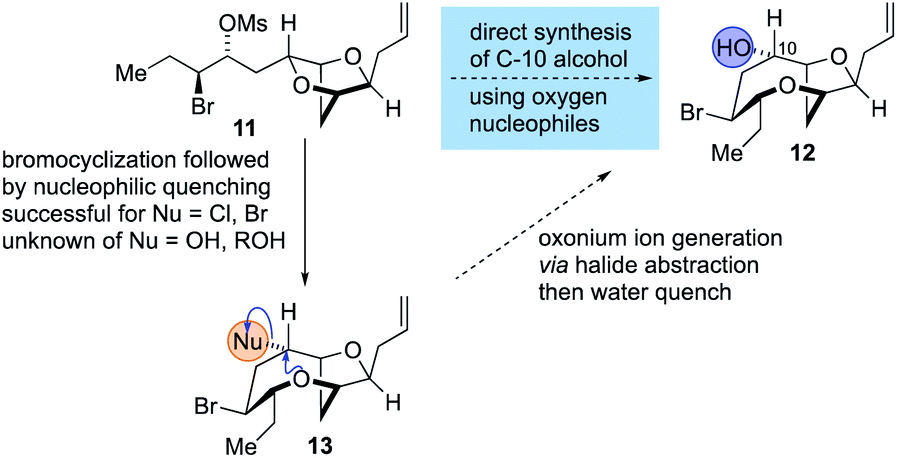 | ||
| Fig. 2 Proposed strategy for the synthesis of 12 by the direct use of oxygen nucleophiles in the bromocyclization–nucleophilic quenching reaction. | ||
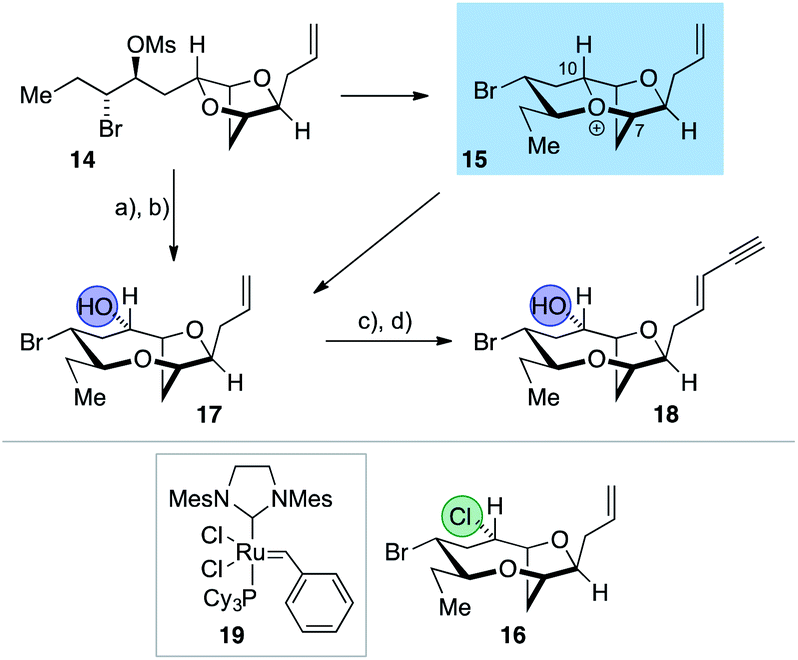 | ||
| Scheme 2 Synthesis of 17 by the direct use of oxygen nucleophile (AgOBz) and its conversion to ent-(E)-laurefucin 18. Reagents and conditions: (a) AgAl(pftb)4·CH2Cl2, TiCl4, CH2Cl2, −40 °C, 2 h, then AgOBz, −78 °C, 1 h, then work-up with sat. NaHCO3 and excess Bu4N+I−; (b) K2CO3, MeOH, 74% (2 steps); (c) crotonaldehyde, cat. 19, cat. CuI, Et2O, RT, overnight; (d) TMSCHN2, lithium diisopropylamide, −78 °C, 1 h, then 0 °C, 1 h, 18 41% E:Z > 20:1 (two steps). | ||
The use of AgOBz as the oxygen nucleophile was then applied to the bromocyclization–nucleophilic quenching reaction with known enantiopure bromomesylate 11 (Scheme 3). The desired C-10 oxygenated compound 12 was obtained in 63% overall yield after methanolysis of the crude benzoate ester, without any product from C-7 oxygenation being observed. The structure of alcohol 12 was confirmed by single crystal X-ray diffraction studies53 (Flack x absolute structure parameter = −0.034(5); shown in Scheme 3).66 Alcohol 12 was then transformed into the reassigned structures of laurefurenyne E and F (Z)-9 and (E)-9 using known procedures for the stereoselective synthesis of (Z)- and (E)-enynes. Thus, oxidative cleavage of the terminal olefin in 12,67 followed by Stork–Zhao olefination gave the corresponding (Z)-vinyl iodide,68 followed by Sonogashira coupling with trimethylsilylacetylene and TMS deprotection gave (Z)-9. In a related manner, the reassigned structure of laurefurenyne F (E)-9 was synthesized from 12 by a cross-metathesis, Colvin–Ohira sequence.58 We were unable to obtain X-ray crystal structures of (E)- and (Z)-9, however, (E)- and (Z)-9 were also synthesized from (E)- and (Z)-bromofucin (E)- and (Z)-5via the previously characterized oxonium ions 747 suggesting that the synthetic route from 12 to 9 had not resulted in undesired alterations of stereocenters. Hence, the oxonium ions (Z)- and (E)-7, readily formed from the corresponding bromofucins on treatment with a silver(I) salt followed by low temperature NMR analysis, were quenched with water to give (Z)- and (E)-9 respectively.
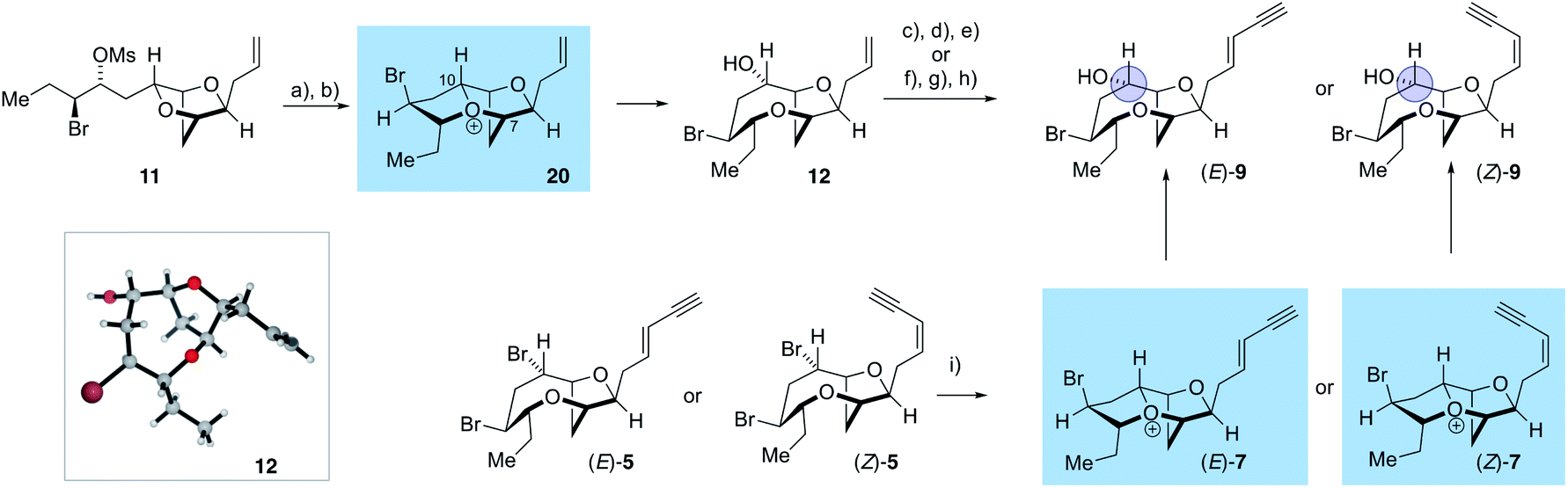 | ||
Scheme 3 Synthesis of the reassigned structures of laurefurenynes E–F 9via12. (a) TiCl4, AgAl(pftb)4·CH2Cl2, CH2Cl2, −40 °C, 2 h then add AgOBz, −78 °C, 1 h, then work-up with sat. NaHCO3 and excess Bu4NI; (b) K2CO3, MeOH, 63% (2 steps); (c) OsO4, NaIO4, 2,6-lutidine, dioxane, water; (d) ICH2PPh3I, NaHMDS, HMPA, THF, −78 °C to r.t.; (e) Me3SiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, Pd(PPh3)4, CuI, Et3N, then K2CO3, MeOH, (Z)-9 35% Z:E > 10:1 (four steps); (f) crotonaldehyde, cat. 19, cat. CuI, Et2O, r.t., overnight; (g) TMSCHN2, lithium diisopropylamide, −78 °C, 1 h, then 0 °C, 1 h; (h) crude treated with K2CO3, MeOH, (E)-9 29% E:Z > 20:1 (three steps); (i) AgAl(pftb)4·CH2Cl2, CD2Cl2, −40 °C, 1.5 h, then filter at −78 °C and NMR analysis of 7, then retrieve NMR sample, excess H2O followed by NaHCO3(s), −78 °C, 15 min then warm to RT (Z)-9 39%; (E)-9 47%. CH, Pd(PPh3)4, CuI, Et3N, then K2CO3, MeOH, (Z)-9 35% Z:E > 10:1 (four steps); (f) crotonaldehyde, cat. 19, cat. CuI, Et2O, r.t., overnight; (g) TMSCHN2, lithium diisopropylamide, −78 °C, 1 h, then 0 °C, 1 h; (h) crude treated with K2CO3, MeOH, (E)-9 29% E:Z > 20:1 (three steps); (i) AgAl(pftb)4·CH2Cl2, CD2Cl2, −40 °C, 1.5 h, then filter at −78 °C and NMR analysis of 7, then retrieve NMR sample, excess H2O followed by NaHCO3(s), −78 °C, 15 min then warm to RT (Z)-9 39%; (E)-9 47%. | ||
The stereochemical assignment at C-10 of laurefurenynes E and F 9 was further supported by Mosher ester analysis of the C-10 secondary alcohol (blue circle) (see ESI†),51,52 providing further evidence that the absolute configuration of C-10 is (S), in keeping with the X-ray crystal structure of 12. The spectroscopic data of (E)-9 were in accordance with the data for the natural product laurefurenyne F ([α]D25 = +17.5 (c = 0.12, MeOH)lit.20 [α]D25 = +17.0 (c = 0.10, MeOH)) for (Z)-9, the NMR spectroscopic data are in accordance with the published data for the natural product laurefurenyne E; however, the specific rotation of natural laurefurenyne E was reported to be [α]D25 = +11.0 (c = 0.10, MeOH) whereas that of our synthetic material was [α]D25 = −5.9 (c = 0.12, MeOH) for a sample that has a Z:E ratio of > 15:1. Measurement of the specific rotation of a sample with a lower Z:E ratio of 7.8:1 gave [α]D25 = −4.7 (c = 0.17, MeOH) which is still of a lower magnitude and of opposite sign compared to that reported in the isolation paper.20
The successful generation of laurefurenynes E and F 9 from the bromofucins 5 provides chemical evidence to support their potential biosynthesis via the oxonium ions 7 as with other related natural products (Scheme 1).47 As noted above laurefurenynes C and D may well therefore arise biosynthetically from laurefurenynes E and F via an SN2 reaction at C-12 with an oxygen nucleophile. However, this transformation was expected to be challenging due to the neopentyl-like nature of C-12 coupled with the β-oxygen substituent which is known to retard the rates of nucleophilic substitution.69 Under a range of conditions reported for substitution at neopentyl-like centres with oxygen nucleophiles,70–72 substrate 12 was either unreactive, underwent low conversion or yielded the elimination product 21 (Scheme 4, see ESI† for details). Ultimately, using Fleet's method (cesium trifluoroacetate in DMF at 120 °C for 12 hours)73 gave the desired substitution product 22 in 62% yield along with a trace of elimination product 21. Previously, Fujiwara and Murai have reported that bromination of alcohols with a β-oxygen that is part of a medium ring, can occur with inversion or retention of configuration depending on reaction conditions with retention of configuration being ascribed to neighboring group participation by the medium ring oxygen atom.74 We therefore needed to confirm the relative configuration of 22. Pleasingly, 22 was isolated as a white crystalline solid which allowed single crystal X-ray diffraction analysis (absolute structure parameter of 22: −0.01(9))53,66 which demonstrated that the C-12 alcohol in 22 was formed with inversion of configuration and confirmed that the relative configuration was as expected (Scheme 4). Analogous to the synthesis of the reassigned structures of laurefurenynes E and F 9, two separate protocols were used for the synthesis of laurefurenynes C and D 10, with the first synthesis starting from diol 22. Thus, diol 22 was readily converted into laurefurenyne C by modified Lemieux–Johnson oxidation67 of the terminal alkene in 22 followed by Stork–Zhao olefination and Sonogashira coupling followed by deprotection. This provided the (Z)-enyne (Z)-10 in 31% yield over the 4 steps with >10:1 Z:E selectivity. Alternatively, the (E)-enyne 10 was readily formed by Kim's procedure58 involving cross metathesis of 22 with crotonaldehyde followed by Colvin–Ohira reaction giving laurefurenyne D (E)-10. Laurefurenynes C and D 10 were also synthesized from laurefurenynes E and F 9 by displacement of the secondary bromides with cesium trifluoroacetate (Scheme 4).73 The NMR spectroscopic data for the reassigned structures of laurefurenynes C and D 10 was identical with that of the natural products. Furthermore, the optical rotations of laurefurenyne C (Z)-10 ([α]D25 = + 10.8 (c = 0.13, MeOH), lit.20 [α]D25 = +20.0 (c = 0.10, MeOH)) and laurefurenyne D (E)-10 ([α]D25 = +14.0 (c = 0.10, MeOH), lit.20 [α]D25 = +32.0 (c = 0.10, MeOH)) were of the same sign but of reduced magnitude compared to those of the isolated natural products indicating that the absolute configuration of the natural products is likely as shown in Schemes 1 and 4 in keeping with the proposed biosynthesis (Scheme 1). Synthetic laurefurenyne D (E)-10 was initially isolated as a colorless oil. However, a single crystal suitable for single crystal X-ray diffraction analysis was obtained by slow diffusion of pentane into an ethyl acetate solution of (E)-10. The crystal structure of (E)-10 (shown in Scheme 4) was obtained with an absolute structure parameter of −0.07(9), providing further confirmation of its structure.53
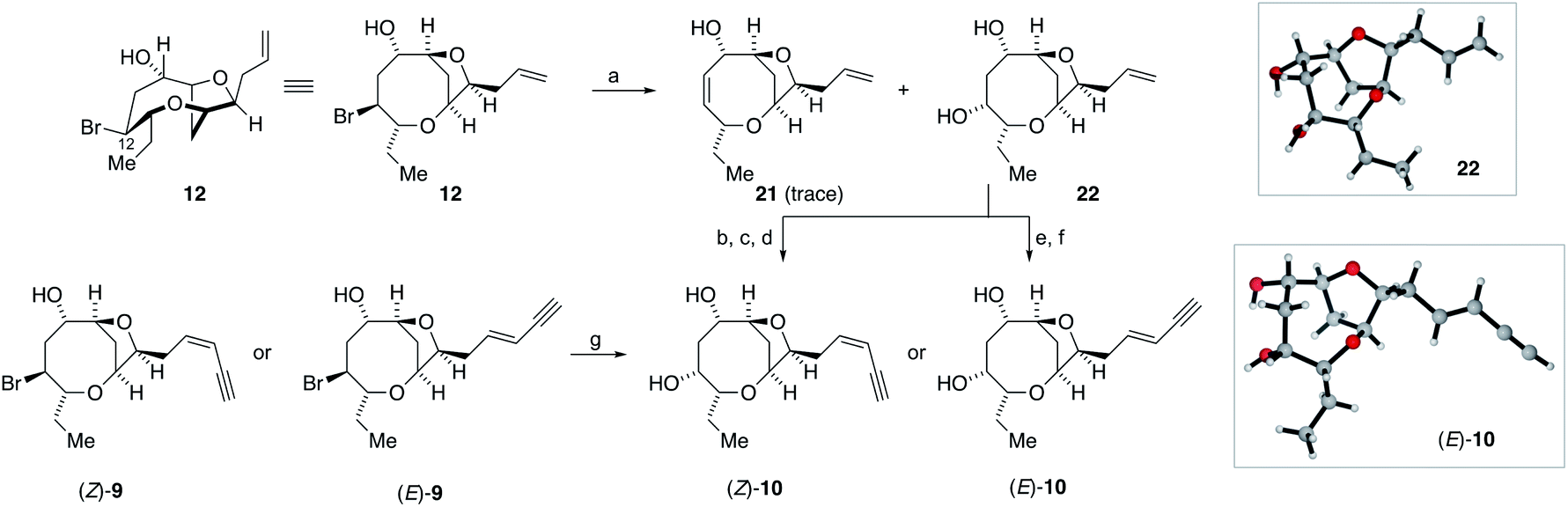 | ||
Scheme 4 Synthesis of the reassigned structures of laurefurenynes C-D 10via12. (a) CF3CO2Cs, Me2NCHO, 120 °C, 12 h, 22 62%, 21, trace; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, water; (c) ICH2PPh3I, NaHMDS, HMPA, THF, −78 °C to RT; (d) Me3SiCCH, Pd(PPh3)4, CuI, Et3N, then K2CO3, MeOH, (Z)-10 32% Z:E > 10:1 (four steps); (e) CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO, cat. 19, cat. CuI, Et2O, RT; (f) TMSCHN2, nBuLi, −78 °C, 1 h, then 0 °C, 30 min, then 1.0 M HCl (E)-10 81% E:Z > 20:1 (two steps); (g) CF3CO2Cs, Me2NCHO, 120 °C, 12 h, (Z)-10 23%, (E)-10 38%. CHCHO, cat. 19, cat. CuI, Et2O, RT; (f) TMSCHN2, nBuLi, −78 °C, 1 h, then 0 °C, 30 min, then 1.0 M HCl (E)-10 81% E:Z > 20:1 (two steps); (g) CF3CO2Cs, Me2NCHO, 120 °C, 12 h, (Z)-10 23%, (E)-10 38%. | ||
The success of the synthesis of ent-(E)-laurefucin 18, and the reassigned structures of the laurefurenynes C–F 9 and 10, that were enabled by the direct C-10 oxygenation of bromomesylates 11 and 14 in the bromocyclization–nucleophilic quenching reaction, prompted us to further investigate the scope of this reaction. We proposed that the enantiomer of the Δ4-oxocene natural products laurencin and deacetyllaurencin,75–78 namely ent-laurencin 24 and ent-deacetyllaurencin 23 could be synthesized via an unusual retrobiomimetic C9–O bond cleavage from ent-(E)-prelaurefucin 25 (Scheme 5). This proposed C9–O bond cleavage requires the formation of a formal anion at C-10 (26) with subsequent E1cB elimination. Reduction of ent-(E)-prelaurefucin 25 with Zn was unlikely to be selective for formation of ent-deacetyllaurencin given the previous precedent in a related system37,79 and we therefore elected to investigate two elimination reactions for the formation of 23 and 24, namely a Kishner–Leonard elimination (27)80,81 and elimination to form a vinyl sulfone (28 to 29) which could undergo subsequent selective reduction with the mild reducing agent sodium dithionite.82 In order to put this plan into practice we needed to further expand the scope of the bromocyclization–nucleophilic quenching reaction to include sulfur nucleophiles for the synthesis of sulfones. The requisite sulfone 30 was readily prepared from enantiopure bromomesylate 14. Oxonium ion formation from 14 was achieved as before followed by the addition of 1-phenyl-1H-tetrazole-5-thiol 31 as the sulfur nucleophile to give corresponding C-10 sulfide in 76% yield.83,84 The sulfide was then oxidized to the desired sulfone 30 in 77% yield by cat. Mo7O24(NH4)6·4H2O and 30% aqueous H2O2 in 77% yield (Scheme 6).
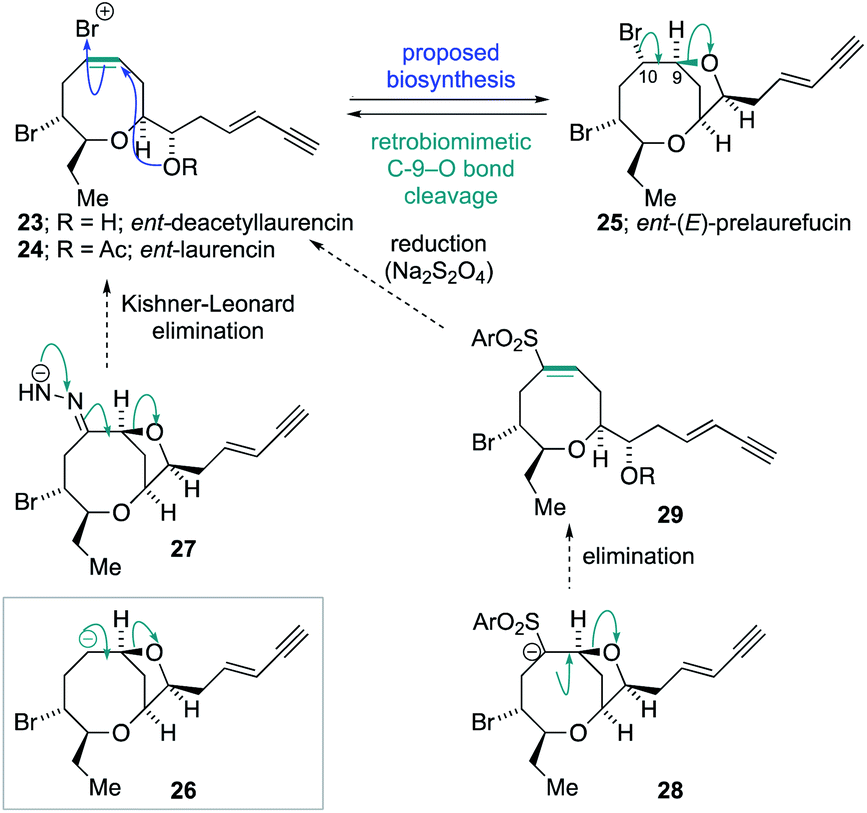 | ||
| Scheme 5 Proposed routes to ent-laurencin by a retrobiomimetic strategy. | ||
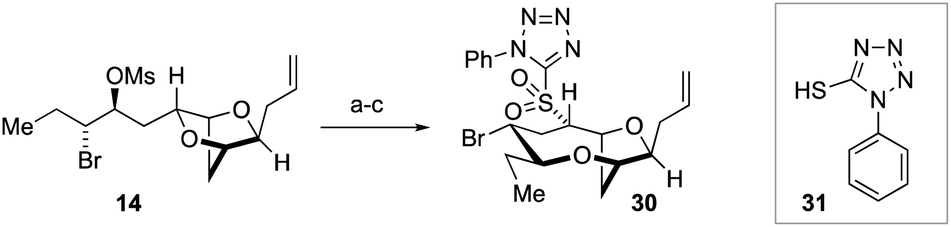 | ||
| Scheme 6 Synthesis of sulfone 30. Reagents and conditions: (a) TiCl4, AgAl(pftb)4·CH2Cl2, CH2Cl2, −40 °C, 2 h; (b) then add 31, −78 °C, 1 h; (c) 30% aq. H2O2, cat. Mo7O24(NH4)6·4H2O, EtOH, 0 °C to RT overnight, 59% from 14. | ||
Treatment of 30 with 1.1 equivalents of strong base (NaHMDS) at −78 °C resulted in no conversion of starting material but on warming to ambient temperature, complete conversion of the sulfone was observed giving the two cyclopropane-containing products 32 and 33 in a 1:2.2 ratio in 90% combined yield (Scheme 7). The structures of the sulfones were assigned by extensive NMR experiments including 1H–1H NOE experiments (see ESI†). A plausible mechanism for the formation of 32 and 33 is shown below. Thus, deprotonation the acidic proton adjacent to the sulfone in 30 would give the anion 34 from which direct cyclopropanation can occur giving 32 or E1cB elimination can occur giving alkoxide 35. Alkoxide 35 can then undergo intramolecular proton transfer to provide the α-sulfonyl anion 36 from which cyclopropanation can readily occur giving 33. The generation of product 33 indicated that the desired E1cB fragmentation was possible although the anion stabilizing properties of the sulfone promoted further reactions.
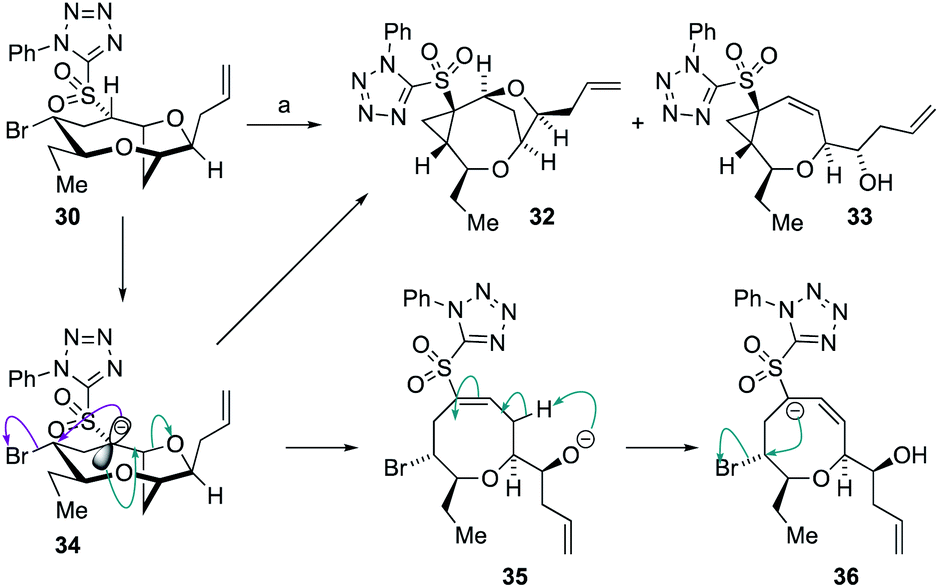 | ||
| Scheme 7 Formation of cyclopropanes 32 and 33 from 30, and a proposed mechanism for product generation. Reagents and conditions: (a) 1.1 eq. NaHMDS, THF, −78 °C to RT, 15 min 32:33 = 1:2.2 90%. | ||
Our attention therefore turned to investigate the Kishner–Leonard elimination from the C-10 hydrazone 38 (Scheme 8). Oxidation of the previously synthesized alcohol 17 using Dess–Martin periodinane gave ketone 37, which on treatment with hydrazine hydrate gave the requisite hydrazone 38 (not isolated). Following a literature procedure,85 treatment of the crude hydrazone 38 with dimsyl sodium on a small scale (0.7 mmol NaH – caution – sodium hydride in DMSO is a known explosion hazard)86 gave an inseparable mixture of the cyclopropane 39, as the major product, along with the diene 40 (39:40, 4.5:1); the structures of 39 and 40 were assigned by extensive 1H and 13C NMR experiments (Scheme 8). Both the cyclopropane 39 and the diene 40 most likely arise from the hydrazone anion corresponding to 27 with formation of the diene requiring a further elimination of HBr. We were disappointed that the cyclopropane 39 was the major product of this reaction. However, when the crude hydrazone 38 was treated with 1.1 equivalents of NaHMDS in THF at −78 °C with gradual warming to room temperature, during which gas evolution was observed, the desired Δ4-oxocene 41 was isolated along with a trace of the 1,3-diene 40 as a side product (42% combined yield over 2 steps). This procedure was then applied to the natural product ent-(E)-laurefucin 18 for the synthesis of ent-laurencin 24. Thus, oxidation of alcohol 18 gave the ketone 42 from which the corresponding hydrazone was prepared. In order to effect the Kishner–Leonard elimination on the hydrazone derived from ketone 42 it was necessary to increase in the equivalents of NaHMDS from 1.1 to 2.0 equivalents presumably due to the deprotonation of the acidic acetylene C–H. Using this procedure gave ent-deacetyllaurencin 23 and diene 43 in a 3:1 ratio in 43% combined yield over 2 steps. The natural product ent-laurencin 24 was obtained by acetylation of ent-deacetyllaurencin 23 in 93% yield. The spectroscopic data of 23 and 24 are in accordance with published data2,55 (synthetic ent-laurencin: [α]D25 = −61.0 (c = 0.10, CHCl3), lit.2 natural (+)-laurencin [α]D23 = +70.2 (c = 1.0, CHCl3); synthetic ent-deacetyllaurencin [α]D25 = −34.7 (c = 0.05, CHCl3), lit.55 natural deacetyllaurencin [α]D17 = +46.1 (c = 1.15, CHCl3)).
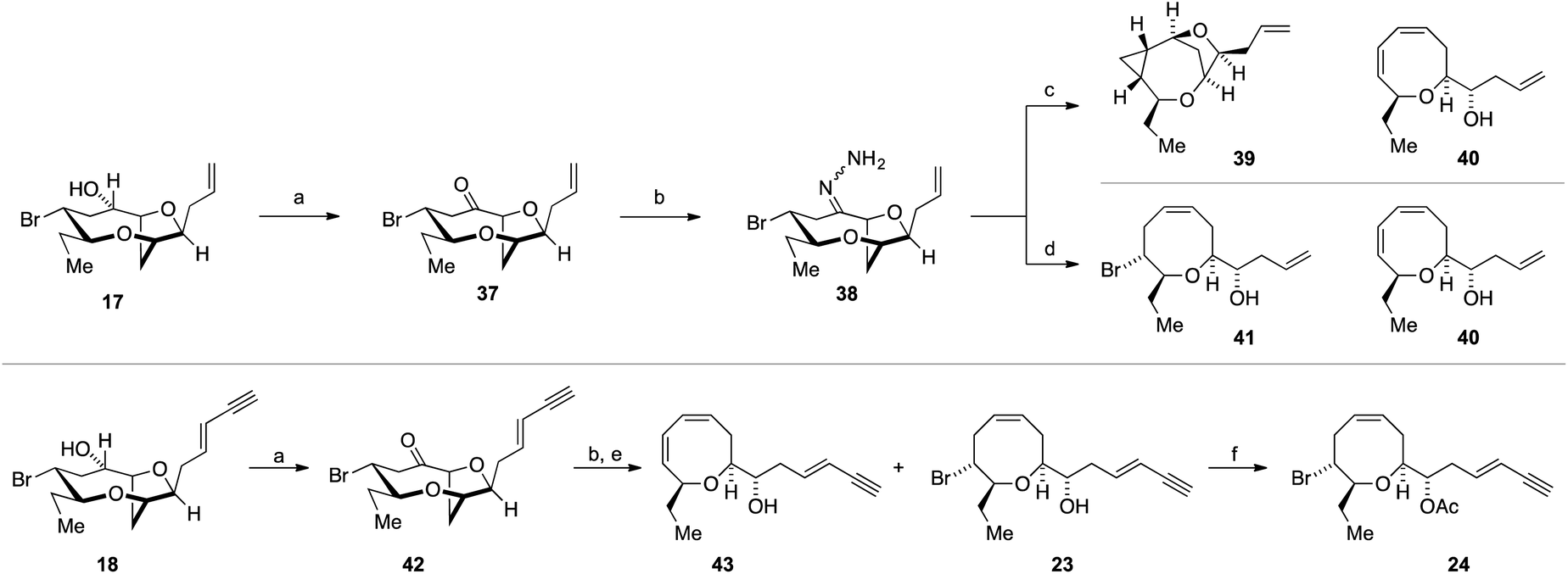 | ||
| Scheme 8 Kishner–Leonard fragmentation for Δ4-oxocene synthesis and the synthesis of ent-deacetyllaurencin 23 and ent-laurencin 24. Reagents and conditions: (a) Dess–Martin periodinane, CH2Cl2, RT, 1 h, 37 85%; 42 quant.; (b) N2H4·H2O, EtOH, RT 2 h; (c) 3.5 eq. NaH, DMSO, then add crude 38 RT, 60% 4.5:1 mixture of 39 and 40 (from 37); (d) 1.1 eq. NaHMDS, THF, −78 °C to RT, 41 42% and 40 trace (two steps); (e) 2.0 eq. NaHMDS, THF, −78 °C to RT, 23 32% and 43 11% (two steps); (f) Ac2O, DMAP, CH2Cl2, r.t., 1 h, 91%. | ||
Conclusions
In conclusion, we have reported structural reassignment of laurefurenynes C–F 9–10 confirmed through the first synthesis and the first biomimetic synthesis of the natural products based on their proposed biosynthesis. Confirmation of the reassigned natural product structures came from extensive spectroscopic and crystallographic evidence. The structural reassignment and biomimetic generation of the laurefurenynes enables them to be considered in line with a series of structurally analogous Laurencia natural products, which further lend weight to their proposed biosynthesis from the bromofucins via complex oxonium ions such as 7. In addition, the expansion of the scope of nucleophiles compatible with the bromocyclization–nucleophilic quenching reactions enabled the preparation of a series of substrates for the investigations that led to the total synthesis of ent-deacetyllaurencin 23 and ent-laurencin 24 through a retrobiomimetic C9–O cleavage, which was ultimately achieved using the Kishner–Leonard elimination reaction. The use of a broad two directional biomimetic and retrobiomimetic strategy has facilitated the synthesis of six natural products. Further development of oxonium ion chemistry in general with applications in natural product synthesis are ongoing and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Croucher Foundation for the award of a University of Oxford Croucher Scholarship to HSSC and we are very grateful to Prof. Marcel Jaspars for providing the primary NMR data of natural laurefurenynes C, D, E and F.Notes and references
- T. Irie, M. Suzuki and T. Masamune, Tetrahedron Lett., 1965, 6, 1091–1099 CrossRef.
- T. Irie, M. Suzuki and T. Masamune, Tetrahedron, 1968, 24, 4193–4205 CrossRef CAS.
- For the X-ray crystal structure of (+)-laurencin see: (a) A. F. Cameron, K. K. Cheung, G. Ferguson and J. M. Robertson, Chem. Commun., 1965, 638 RSC; (b) A. F. Cameron, K. K. Cheung, G. Ferguson and J. M. Robertson, J. Chem. Soc. B, 1969, 559–564 RSC.
- B. G. Wang, J. B. Gloer, N. Y. Ji and J. C. Zhao, Chem. Rev., 2013, 113, 3632–3685 CrossRef CAS.
- Z. F. Zhou, M. Menna, Y. S. Cai and Y. W. Guo, Chem. Rev., 2015, 115, 1543–1596 CrossRef CAS.
- T. Wanke, A. C. Philippus, G. A. Zatelli, L. F. O. Vieira, C. Lhullier and M. Falkenberg, Rev. Bras. Farmacogn., 2015, 25, 569–587 CrossRef CAS.
- K. Fujiwara, in Marine Natural Products, ed. H. Kiyota, Springer-Verlag, Berlin, Heildelberg, 2006, vol. 5, pp. 97–148 Search PubMed.
- The crystalline sponge method and the CryoEM/MicroED method allow the determination of structures of small organic molecules that are not suitable for single crystal X-ray diffraction. Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS; C. G. Jones, M. W. Martynowycz, J. Hattne, T. J. Fulton, B. M. Stoltz, J. A. Rodriguez, H. M. Nelson and T. Gonen, ACS Cent. Sci., 2018, 4, 1587–1592 CrossRef.
- K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 2050 CrossRef.
- M. E. Maier, Nat. Prod. Rep., 2009, 26, 1105–1124 RSC.
- T. L. Suyama, W. H. Gerwick and K. L. McPhail, Bioorg. Med. Chem., 2011, 19, 6675–6701 CrossRef CAS.
- J. G. Hall and J. A. Reiss, Aust. J. Chem., 1986, 39, 1401–1409 CrossRef CAS.
- H. M. Sheldrake, C. Jamieson and J. W. Burton, Angew. Chem., Int. Ed., 2006, 45, 7199–7202 CrossRef CAS.
- S. G. Smith, R. S. Paton, J. W. Burton and J. M. Goodman, J. Org. Chem., 2008, 73, 4053–4062 CrossRef CAS.
- B. S. Dyson, J. W. Burton, T. I. Sohn, B. Kim, H. Bae and D. Kim, J. Am. Chem. Soc., 2012, 134, 11781–11790 CrossRef CAS.
- G. Guella, I. Mancini, A. Öztunç and F. Pietra, Helv. Chim. Acta, 2000, 83, 336–348 CrossRef CAS.
- D. C. Braddock, R. Bhuva, D. S. Millan, Y. Perez-Fuertes, C. A. Roberts, R. N. Sheppard, S. Solanki, E. S. E. Stokes and A. J. P. White, Org. Lett., 2007, 9, 445–448 CrossRef CAS.
- D. C. Braddock and H. S. Rzepa, J. Nat. Prod., 2008, 71, 728–730 CrossRef CAS.
- D. C. Braddock, D. S. Millan, Y. Perez-Fuertes, R. H. Pouwer, R. N. Sheppard, S. Solanki and A. J. P. White, J. Org. Chem., 2009, 74, 1835–1841 CrossRef CAS.
- W. M. Abdel-Mageed, R. Ebel, F. A. Valeriote and M. Jaspars, Tetrahedron, 2010, 66, 2855–2862 CrossRef CAS.
- M. T. Holmes and R. Britton, Chem.–Eur. J., 2013, 19, 12649–12652 CrossRef CAS.
- D. J. Shepherd, P. A. Broadwith, B. S. Dyson, R. S. Paton and J. W. Burton, Chem.–Eur. J., 2013, 19, 12644–12648 CrossRef CAS.
- A. Fukuzawa, M. Aye, M. Nakamura, M. Tamura and A. Murai, Tetrahedron Lett., 1990, 31, 4895–4898 CrossRef CAS.
- H. Kikuchi, T. Suzuki, E. Kurosawa and M. Suzuki, Bull. Chem. Soc. Jpn., 1991, 64, 1763–1775 CrossRef CAS.
- A. Murai, in Comprehensive Natural Products Chemistry, ed. D. H. R. Barton, O. Meth-Cohn and K. Nakinishi, Elsevier, Oxford, 1999, vol. 1, pp. 303–324 Search PubMed.
- D. C. Braddock, Org. Lett., 2006, 8, 6055–6058 CrossRef CAS.
- K. J. Bonney, D. C. Braddock, A. J. White and M. Yaqoob, J. Org. Chem., 2011, 76, 97–104 CrossRef CAS.
- D. C. Braddock and D. T. Sbircea, Chem. Commun., 2014, 50, 12691–12693 RSC.
- S. A. Snyder, D. S. Treitler, A. P. Brucks and W. Sattler, J. Am. Chem. Soc., 2011, 133, 15898–15901 CrossRef CAS.
- Y. A. Zhang, N. Yaw and S. A. Snyder, J. Am. Chem. Soc., 2019, 141, 7776–7788 CrossRef CAS.
- S. Keshipeddy, I. Martinez, B. F. Castillo 2nd, M. D. Morton and A. R. Howell, J. Org. Chem., 2012, 77, 7883–7890 CrossRef CAS.
- M. T. Taylor and J. M. Fox, Tetrahedron Lett., 2015, 56, 3560–3563 CrossRef CAS.
- C. A. Taylor, Y.-A. Zhang and S. A. Snyder, Chem. Sci., 2020, 11, 3036–3041 RSC.
- E. D. Shepherd, B. S. Dyson, W. E. Hak, Q. N. N. Nguyen, M. Lee, M. J. Kim, T. I. Sohn, D. Kim, J. W. Burton and R. S. Paton, J. Org. Chem., 2019, 84, 4971–4991 CrossRef CAS.
- For a very recent example of where total synthesis has so far not uncovered the structure of a misassigned natural product see: M. Heinrich, J. J. Murphy, M. K. Ilg, A. Letort, J. T. Flasz, P. Philipps and A. Furstner, J. Am. Chem. Soc., 2020, 142, 6409–6422 CrossRef CAS.
- A. Fukuzawa, E. Kurosawa and T. Irie, Tetrahedron Lett., 1972, 13, 3–6 CrossRef.
- A. Furusaki, E. Kurosawa, A. Fukuzawa and T. Irie, Tetrahedron Lett., 1973, 4579–4582 CrossRef CAS.
- H. M. Sheldrake, C. Jamieson, S. I. Pascu and J. W. Burton, Org. Biomol. Chem., 2009, 7, 238–252 RSC.
- N. Y. Ji, X. M. Li, K. Li and B. G. Wang, J. Nat. Prod., 2007, 70, 1499–1502 CrossRef CAS.
- N. Y. Ji, X. M. Li, K. Li and B. G. Wang, J. Nat. Prod., 2010, 73, 1192 CrossRef CAS.
- M. Suzuki and E. Kurosawa, Phytochemistry, 1985, 24, 1999–2002 CrossRef CAS.
- J. Wang and B. L. Pagenkopf, Org. Lett., 2007, 9, 3703–3706 CrossRef CAS.
- A. D. Wright, G. M. Konig, R. Denys and O. Sticher, J. Nat. Prod., 1993, 56, 394–401 CrossRef CAS.
- G. R. Schulte, M. C. H. Chung and P. J. Scheuer, J. Org. Chem., 1981, 46, 3870–3873 CrossRef CAS.
- D. Jeong, T. I. Sohn, J. Y. Kim, G. Kim, D. Kim and R. S. Paton, Org. Lett., 2017, 19, 6252–6255 CrossRef CAS.
- The absolute configurations of the laurefurenynes A-F was not assigned by the isolation chemists and the configurations of 2 and 3 were depicted as enantiomeric to that shown in Fig. 1.
- H. S. S. Chan, Q. N. N. Nguyen, R. S. Paton and J. W. Burton, J. Am. Chem. Soc., 2019, 141, 15951–15962 CrossRef.
- B. Kim, T. I. Sohn, D. Kim and R. S. Paton, Chem.–Eur. J., 2018, 24, 2634–2642 CrossRef CAS.
- D. Kim, T.-i. Sohn, B. Kim and R. S. Paton, Heterocycles, 2018, 97, 179–191 CrossRef.
- The natural product isolaurefucin methyl ether would correspond to the methyl ether of the reassigned structure of laurefurenyne F. Isolation and structure determination of isolaurefucin methyl ether see: (a) R. Denys, J. C. Coll, A. R. Carroll and B. F. Bowden, Aust. J. Chem., 1993, 46, 1073–1077 CrossRef CAS ; synthesis of isolaurefucin methyl ether see:; (b) Ref. 49 .
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS.
- Low temperature data were collected using a (Rigaku) Oxford Diffraction Supernova A diffractometer (λ = 1.54184 Å) and reduced using CrysAlisPro. The structures were solved using SuperFlip and refined using CRYSTALS. J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105–107 CrossRef CAS; L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef; P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef; P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef; R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef . Full details can be found in the ESI† (CIF); crystallographic data have also been deposited with the Cambridge Crystallographic Data Centre and copies of these data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/data_request/cif.
- E. Kurosawa, T. Irie and A. Fukuzawa, Tetrahedron Lett., 1972, 2121–2124 CrossRef CAS.
- G. M. König and A. D. Wright, J. Nat. Prod., 1994, 57, 477–485 CrossRef.
- N. Ungur, V. Kulciţki, O. Chetraru, M. Grinco and P. F. Vlad, Helv. Chim. Acta, 2013, 96, 864–871 CrossRef CAS.
- The enantiopure bromomesylates 11 and 14 are readily available from diacetone-D-glucose see ref. 47.
- B. Kim, M. Lee, M. J. Kim, H. Lee, S. Kim, D. Kim, M. Koh, S. B. Park and K. J. Shin, J. Am. Chem. Soc., 2008, 130, 16807–16811 CrossRef CAS.
- S. A. Snyder, A. P. Brucks, D. S. Treitler and I. Moga, J. Am. Chem. Soc., 2012, 134, 17714–17721 CrossRef CAS.
- The spectroscopic data of our synthetic 17 matched that of Kim and Snyder with the optical rotation of our synthetic material being equal in magnitude but opposite in sign to that reported by Kim.
- Addition of excess tetrabutylammonium iodide during workup prevented the formation of baseline species.
- S. J. Wratten and D. J. Faulkner, J. Org. Chem., 1977, 42, 3343–3349 CrossRef CAS.
- For the total synthesis of the natural enantiomer of laurefucin see ref. 58. For synthesis of laurefucin in racemic form see ref. 59. For the synthesis of the unnatural enantiomer of laurefucin see ref. 47.
- K. Voigtritter, S. Ghorai and B. H. Lipshutz, J. Org. Chem., 2011, 76, 4697–4702 CrossRef CAS.
- We have previously synthesized ent-(E)-laurefucin and our synthetic material provided crystals suitable for single crystal X-ray diffraction studies (see ESI† CIF).
- The Flack x parameter was determined by full-matrix least squares refinement. H. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876–881 CrossRef CAS; H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CrossRef; A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2011, 44, 1017–1022 CrossRef.
- W. Yu, Y. Mei, Y. Kang, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217–3219 CrossRef CAS.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 2173–2174 CrossRef CAS.
- S. S. Shaik, J. Am. Chem. Soc., 1983, 105, 4359–4367 CrossRef CAS.
- B. Radüchel, Synthesis, 1980, 292–295 CrossRef.
- S. Raghavan and V. Krishnaiah, Tetrahedron Lett., 2006, 47, 7611–7614 CrossRef CAS.
- R. M. Moriarty, H. Zhuang, R. Penmasta, K. Liu, A. K. Awasthi, S. M. Tuladhar, M. S. C. Rao and V. K. Singf, Tetrahedron Lett., 1993, 34, 8029–8032 CrossRef CAS.
- A. A. Bell, L. Pickering, M. Finn, C. delaFuente, T. M. Krulle, B. G. Davis and G. W. J. Fleet, Synlett, 1997, 1077–1078 CrossRef CAS.
- K. Fujiwara, M. Kobayashi, D. Awakura and A. Murai, Synlett, 2000, 1187–1189 CAS.
- For the synthesis of laurencin in racemic form see: (a) T. Masamune and H. Matsue, Chem. Lett., 1975, 4, 895–898 CrossRef; (b) A. Murai, H. Murase, H. Matsue and T. Masamune, Tetrahedron Lett., 1977, 18, 2507–2510 CrossRef; (c) T. Masamune, H. Matsue and H. Murase, Bull. Chem. Soc. Jpn., 1979, 52, 127–134 CrossRef CAS; (d) T. Masamune, H. Murase, H. Matsue and A. Murai, Bull. Chem. Soc. Jpn., 1979, 52, 135–141 CrossRef CAS.
- For previous total syntheses of (+)-laurencin see: (a) K. Tsushima and A. Murai, Tetrahedron Lett., 1992, 33, 4345–4348 CrossRef CAS; (b) A. Fukuzawa, M. Aye and A. Murai, Chem. Lett., 1990, 1579–1580 CrossRef CAS; (c) M. Bratz, W. H. Bullock, L. E. Overman and T. Takemoto, J. Am. Chem. Soc., 1995, 117, 5958–5966 CrossRef CAS; (d) J. W. Burton, J. S. Clark, S. Derrer, T. C. Stork, J. G. Bendall and A. B. Holmes, J. Am. Chem. Soc., 1997, 119, 7483–7498 CrossRef CAS; (e) M. T. Crimmins and K. A. Emmitte, Org. Lett., 1999, 1, 2029–2032 CrossRef CAS; (f) S. Baek, H. Jo, H. Kim, H. Kim, S. Kim and D. Kim, Org. Lett., 2005, 7, 75–77 CrossRef CAS; (g) K. Fujiwara, S. Yoshimoto, A. Takizawa, S.-i. Souma, H. Mishima, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2005, 46, 6819–6822 CrossRef CAS.
- For a formal synthesis of laurencin and ent-laurencin see: M. T. Mujica, M. M. Afonso, A. Galindo and J. A. Palenzuela, Synlett, 1996, 983–984 CrossRef CAS.
- For the synthesis of deacetyllaurencin from laurencin see: (a) T. Irie, M. Suzuki and T. Masamune, Tetrahedron Lett., 1965, 6, 1091–1099 CrossRef; (b) T. Irie, M. Suzuki and T. Masamune, Tetrahedron, 1968, 24, 4193–4205 CrossRef CAS; (c) A. Fukuzawa, M. Aye and A. Murai, Chem. Lett., 1990, 1579–1580 CrossRef CAS . For a synthesis of deacetyllaurencin from D-galactose pentaacetate see: ref. 76g.
- S. Teruaki, K. Kuniko, S. Minoru and K. Etsuro, Chem. Lett., 1983, 12, 1639–1642 CrossRef.
- N. J. Leonard and S. Gelfand, J. Am. Chem. Soc., 1955, 77, 3272–3278 CrossRef CAS.
- N. J. Leonard and S. Gelfand, J. Am. Chem. Soc., 1955, 77, 3269–3271 CrossRef CAS.
- M. Julia, H. Lauron, J.-P. Stacino, J.-N. Verpeaux, Y. Jeannin and Y. Dromzee, Tetrahedron, 1986, 42, 2475–2484 CrossRef CAS.
- It was also possible to open the oxonium 15, prepared from 14, with tosylhydrazine to give the corresponing C-10 tosylhydrazine S6 (see ESI†).
- The configuration of the C-10 sulfide was assigned by analogy with the corresponding previously prepared C-10 halides (ref. 47).
- J. Jiang, Y. Ichikawa and M. Isobe, Tetrahedron, 1997, 53, 5103–5122 CrossRef.
- Q. Yang, M. Sheng, J. J. Henkelis, S. Tu, E. Wiensch, H. Zhang, Y. Zhang, C. Tucker and D. E. Ejeh, Org. Process Res. Dev., 2019, 23, 2210–2217 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2005091–2005094. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04120c |
| ‡ Authors to whom correspondence regarding X-ray crystallography should be addressed. |
| This journal is © The Royal Society of Chemistry 2020 |