
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining alkali metals and zinc to harness heterometallic cooperativity in cyclic ester ring-opening polymerisation†
Weronika
Gruszka
a,
Anna
Lykkeberg
a,
Gary S.
Nichol
 a,
Michael P.
Shaver
b,
Antoine
Buchard
c and
Jennifer A.
Garden
*a
a,
Michael P.
Shaver
b,
Antoine
Buchard
c and
Jennifer A.
Garden
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
bSchool of Natural Sciences, Department of Materials, Henry Royce Institute, University of Manchester, Manchester, M13 9PL, UK
cDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 12th October 2020
Abstract
Heterometallic cooperativity is an emerging strategy to elevate polymerisation catalyst performance. Here, we report the first heterotrimetallic Na/Zn2 and K/Zn2 complexes supported by a ProPhenol ligand, which deliver “best of both” in cyclic ester ring-opening polymerisation, combining the outstanding activity (Na/K) and good control (Zn2) of homometallic analogues. Detailed NMR studies and density-functional theory calculations suggest that the Na/Zn2 and K/Zn2 complexes retain their heterometallic structures in the solution-state. To the best of our knowledge, the K/Zn2 analogue is the most active heterometallic catalyst reported for rac-lactide polymerisation (kobs = 1.7 × 10−2 s−1), giving activities five times faster than the Na/Zn2 complex. These versatile catalysts also display outstanding performance in ε-caprolatone and δ-valerolactone ring-opening polymerisation. These studies provide underpinning methodologies for future heterometallic polymerisation catalyst design, both in cyclic ester polymerisation and other ring-opening (co)polymerisation reactions.
Introduction
Cyclic ester ring-opening polymerisation (ROP) is an efficient method for producing aliphatic polyesters such as poly(lactic acid) (PLA), poly(ε-caprolactone) (PCL) and poly(δ-valerolactone) (PVL).1–3 The degradable and biocompatible properties of these polyesters have led to applications across packaging,4 electronics and medicine.5 ROP is dependent on the catalyst, and well-defined organometallic complexes have exerted excellent activities, selectivities and control over the polymer microstructure. The first examples comprised homoleptic homometallic alkoxides, e.g. Al(OiPr)3, however bimetallic catalysts have recently gathered increased attention,6 with most examples based on biocompatible Al, Ca, Fe, K, Na, Ti and Zn metals.7 Despite the high activity of alkali metal catalysts,8 bimetallic zinc catalysts are generally more efficient at combining high activities with polymerisation control in ROP.9–11 We recently reported a highly active bimetallic zinc-benzoxide catalyst ([LZn2OBn]),12 based on the Trost ProPhenol ligand (LH3), for the controlled homo- and co-polymerisation of rac-lactide (rac-LA), ε-caprolactone (ε-CL) and rac-β-butyrolactone.The activity and selectivity of homometallic species can be enhanced by introducing a heterometal into the same complex, which can result in heterometallic cooperativity. Inspired by nature, which has long utilised heterometallic metalloenzymes in biological transformations,13,14 chemists have observed unprecedented activity and selectivity enhancements with heterometallic complexes in metal–halogen exchange,15 C–H bond activation16 and olefin polymerisation.17,18 However, the concept remains underexplored in cyclic ester ROP despite heterometallic complexes with a M–O–M′ (M ≠ M′) framework (and thus intermetallic electronic communication via the O heteroatom) having the potential to enhance monomer coordination by increasing the metal Lewis acidity and accelerate propagation by enhancing the metal-alkoxide nucleophilicity.17,19 To date, the best performing heterometallic catalysts for LA and ε-CL ROP have generally comprised metals with large ionic radii and significant electronegativity differences between the metals, e.g. Al/Zn,20 La/Mg,21 Li/In,22 Li/Mg and Li/Zn,23 Li/Sm,24 Na/Sm,25 Sm/Al26 and Ti/Zn27 (Fig. 1). Combining Zn with electropositive alkali metals, which are highly active, inexpensive, earth abundant and non-toxic, is thus attractive from scientific, economic and environmental perspectives, yet remains underexplored.7 Herein, the synthesis and activity of novel heterometallic complexes [LMZn2Et2(THF)2] (Fig. 1, where M = Na or K) are reported for cyclic ester ROP.
 | ||
| Fig. 1 Heterometallic M/Zn complexes reported for cyclic ester ROP. | ||
Results and discussion
Homo- and hetero-metallic complex synthesis
The heterometallic precursors [LH2Na(THF)2] (1) and [LH2K(THF)2] (2) were synthesised via mono-deprotonation of the phenolic-OH of LH3 with NaH or KH, respectively (Scheme 1), and characterised by NMR spectroscopy, mass spectrometry and elemental analysis (Fig. S1–S6†). 1H and DOSY NMR analysis indicated that 1 and 2 are centrosymmetric and mononuclear in THF-d8 solution (Fig. S2 and S5†). Crystals of 1 were obtained by cooling a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF:toluene mixture to −34 °C, yet despite multiple attempts, the crystals of 2 obtained were unsuitable for X-ray crystallographic analysis. 1 was mononuclear in the solid-state, with pentacoordinate Na displaying a low τ value (τ = 0.41, Fig. 2a)28 suggesting that the structure is closer to a square pyramidal geometry than a trigonal bipyramidal geometry. The Na coordination sphere comprises a longer central Na1–O2 bond (2.355(5) Å) and four shorter dative bonds, two from the benzylic-OH [Na1–O1 (2.258(6) Å) and Na1–O3 (2.254(6) Å)] and two from THF [Na1–O4 (2.278(7) Å) and Na1–O5 (2.258(7) Å)]. In contrast to the solution-state, 1 was non-centrosymmetric in the solid-state, as evidenced by the tetragonal space group (P43).
:1 THF:toluene mixture to −34 °C, yet despite multiple attempts, the crystals of 2 obtained were unsuitable for X-ray crystallographic analysis. 1 was mononuclear in the solid-state, with pentacoordinate Na displaying a low τ value (τ = 0.41, Fig. 2a)28 suggesting that the structure is closer to a square pyramidal geometry than a trigonal bipyramidal geometry. The Na coordination sphere comprises a longer central Na1–O2 bond (2.355(5) Å) and four shorter dative bonds, two from the benzylic-OH [Na1–O1 (2.258(6) Å) and Na1–O3 (2.254(6) Å)] and two from THF [Na1–O4 (2.278(7) Å) and Na1–O5 (2.258(7) Å)]. In contrast to the solution-state, 1 was non-centrosymmetric in the solid-state, as evidenced by the tetragonal space group (P43).
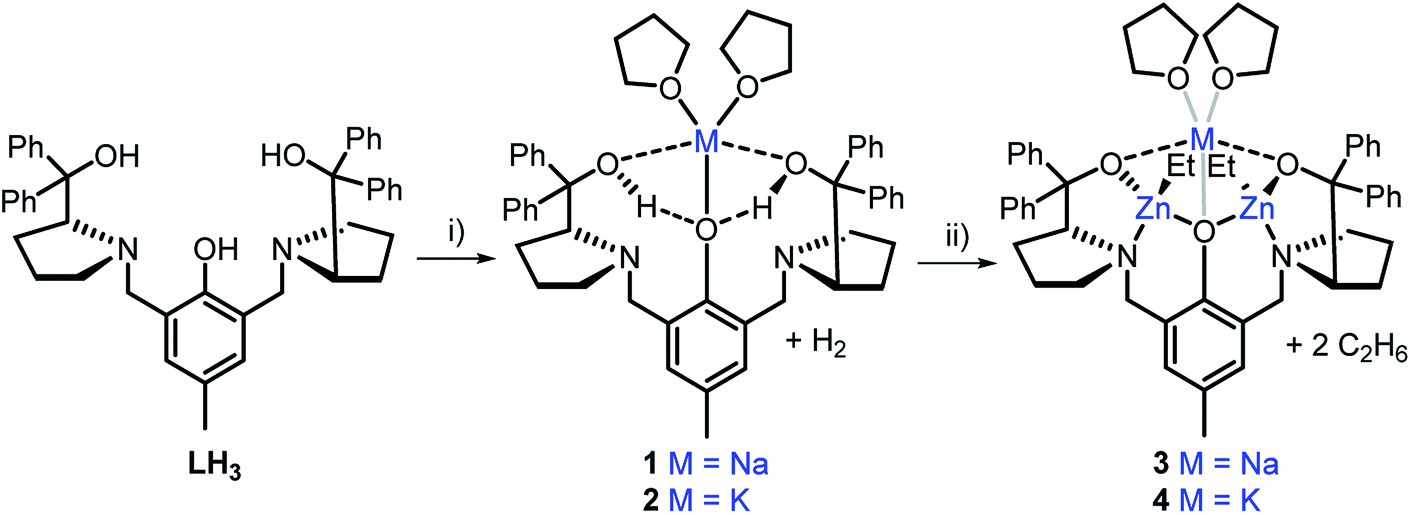 | ||
| Scheme 1 Synthesis of monometallic complexes 1 and 2 and heterometallic complexes 3 and 4. Reagents and conditions: (i) 1.1 eq. MH, THF, R.T., 2 h; (ii) 2 eq. ZnEt2, THF, R.T., 1 h. | ||
 | ||
| Fig. 2 (a) Molecular structure of 1. Ellipsoids set at 50% probability level. H atoms and co-crystallised THF have been omitted for clarity. Selected bond lengths (Å): Na1–O1 2.258(6), Na1–O2 2.355(5), Na1–O3 2.254(6), Na1–O4 2.278 (7), Na1–O5 2.258(7). Selected bond angles (°): O1–Na1–O2 70.6(2), O2–Na1–O3 73.7(2), O1–Na1–O4 90.2(3), O3–Na1–O5 96.1(2), O4–Na1–O5 119.6(3). (b) Molecular structures of 3′ and 4′ with the lowest free enthalpies computed by DFT (refer to Tables S6 and S13†). | ||
Although complex 1 features two benzylic OH groups that are acidified through hydrogen bonding to the phenolic O, these groups were not deprotonated with further equivalents of NaH (≤3 eq. in total). This suggests that the product selectivity is influenced by both the pKa of the OH groups and the ionic radii of the alkali metals. Notably, Na+ and K+ are significantly larger (102 and 138 pm, respectively) than Li+ (76 pm),29 and indeed, metalation of LH3 with nBuLi was less selective. NMR spectroscopic studies revealed two products, one symmetric (attributed to lithiation of the phenol-OH) and one asymmetric (lithiation of the benzylic OH). However, 1 and 2 were selectively deprotonated with 2 eq. of ZnEt2, yielding complexes [LNaZn2Et2(THF)2] (3) and [LKZn2Et2(THF)2] (4) (Scheme 1), which were characterised by NMR spectroscopy, mass spectrometry and elemental analysis (Fig. S7–S12†). The centrosymmetric solution-state structure of 1 and 2 was also prominent in 3 and 4, suggesting that each Et2Zn deprotonates one benzylic OH and retains one ethyl group (Fig. S7 and S10†). DOSY NMR analysis suggests that 3 and 4 are both monomeric in the solution state (Fig. S8 and S11†). Unfortunately, attempts to isolate single crystals of 3 and 4 suitable for X-ray diffraction studies proved unsuccessful. However, density-functional theory (DFT) calculations confirmed the heterometallic structures and stability of 3 and 4 (refer to ESI†). The calculations suggest that the R,R configuration of the N atoms observed in the molecular structure of 1 is likely retained with 3′ and 4′ (′ denotes computationally modelled structures, see ESI†), resulting in the two Zn-Et moieties facing in opposite directions relative to the phenol ring plane (Fig. 2b). However, ligand rearrangement to a meso (R,S) configuration at the N atoms, with Zn-Et groups facing in the same direction, was found to be only slightly endergonic for both 3′ (+2.0 kcal mol−1) and 4′ (+7.7 kcal mol−1) (Tables S6 and S13†).
Rac-LA polymerisation: heterometallic vs. homometallic catalysis
Complexes 3 and 4 were found to be highly efficient initiators for rac-LA ROP with 2 eq. of benzyl alcohol (BnOH, Table 1). Complex 4 exhibited an exceptional polymerisation rate of kobs = 1.7 × 10−2 s−1 in THF solvent at room temperature (R.T., Fig. S17†), converting 60 eq. of rac-LA in just 20 seconds. Not only is 4 five times faster than 3 (kobs = 3.2 × 10−3 s−1) but it is, to the best of our knowledge, the most active heterometallic catalyst system reported for LA ROP and the first heterometallic K/Zn catalyst reported for cyclic ester ROP. Previously, some of the most active heterometallic catalysts for LA ROP were Li/Zn and Li/Mg complexes supported by a bis(phenol) ligand,23 which respectively converted 62 eq. and 88 eq. rac-LA in 15 min at R.T. in the presence of 1 eq. neopentyl alcohol. Complexes 3 and 4 (with 2 eq. of BnOH) also displayed activity for rac-LA ROP at 0.2–0.5 mol% catalyst loadings, generating PLA with Mn of up to 12100 g mol−1 (Table S1†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Time (min) | Conv.b (%) | M n,obs (Da) | M n,calc (Da) | Đ |
|
a 100:1:2 LA:cat:BnOH, [LA] = 1 M in THF.
b Calculated using 1H NMR spectroscopy.
c Determined by GPC using polystyrene standards in THF. Values corrected by Mark–Houwink factor (0.58).31
d Calculated from the monomer conversion Mn,calc = M0 × ([M]/[I]) × conversion assuming 2 chains per catalyst.
e No BnOH used.
f Polymerisations run at 60 °C.
g Calculated from the monomer conversion Mn,calc = M0 × ([M]/[I]) × conversion assuming 1 chain per catalyst.
h
[BnOK] generated in situ from KH and BnOH before adding [LZn2OBn].
|
||||||
| 1e,f | 3 | 2.5 | 12 | — | — | — |
| 2 | 3 | 0.33 | 47 | 2100 | 3400 | 1.3 |
| 3 | 3 | 2.5 | 71 | 3000 | 5100 | 1.2 |
| 4 | 3 | 7.5 | 86 | 3900 | 6200 | 1.4 |
| 5ef | 4 | 2.5 | 13 | — | — | — |
| 6 | 4 | 0.08 | 45 | 1900 | 3300 | 1.7 |
| 7 | 4 | 0.33 | 60 | 2500 | 4300 | 1.4 |
| 8 | 4 | 1.25 | 81 | 3900 | 5800 | 1.4 |
| 9 | 4 | 2 | 93 | 4300 | 6700 | 1.4 |
| 10e | 1 | 1.25 | 79 | 14800 |
11400g |
4.1 |
| 11e | [BnONa] | 1.25 | 88 | 20400 |
12700g |
2.7 |
| 12e | 2 | 0.33 | 72 | 7300 | 10400g |
4.3 |
| 13e | [BnOK] | 0.25 | 94 | 13100 |
13600g |
1.9 |
| 14e | [LZn2OBn] | 7.5 | 17 | — | — | — |
| 15e,h | [BnOK] + [LZn2OBn] | 0.25 | 99 | 8300 | 7100 | 1.6 |
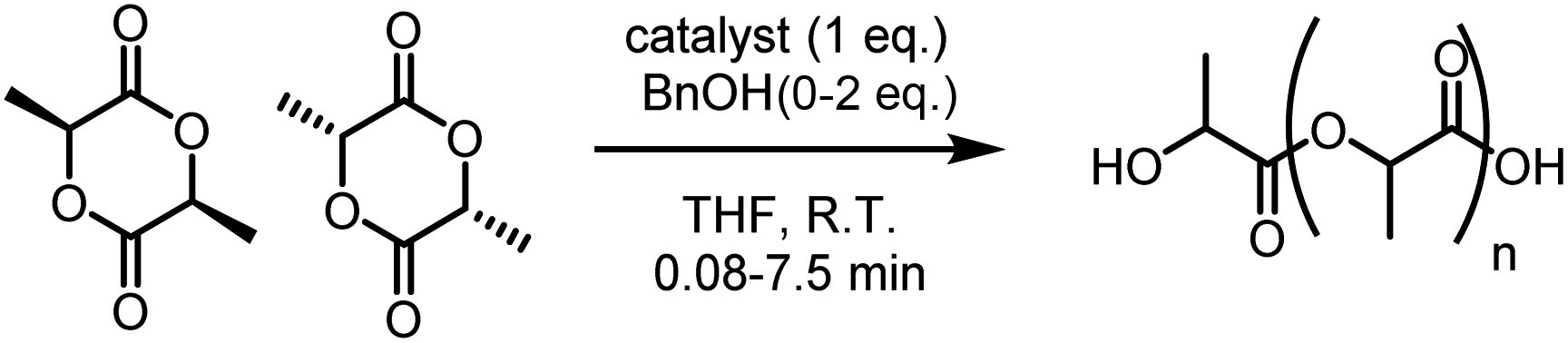
The polymerisations with 3 or 4 (and 2 eq. BnOH) were controlled with a linear relationship between Mn and monomer conversion (Table 1, Fig. S18 and S19†). The discrepancy between the observed and calculated Mn values was attributed to transesterification reactions, as evidenced by MALDI-ToF analysis (refer to ESI†). End-group analysis revealed the expected α-benzoxy, ω-hydroxy end-capped polymer chains. However, unlike related homometallic Trost ProPhenol catalysts ([(LH)2Zr]30 and [LZn2OBn]12) no ligand end groups were detected with complexes 3 and 4; this improved control may arise from the increased chelate stability and steric congestion of 3 and 4. Similarly to homometallic [LZn2OBn],12 the PLA generated from rac-LA was either atactic (maximum Pi = 0.53 with 3, Table S1†) or showed a modest isotactic bias (maximum Pi = 0.62 with 4). Kinetic studies of L-LA ROP (Fig. S34†) indicated that 4 is twice as active in rac-LA ROP (kobs = 1.7 × 10−2 s−1) than L-LA ROP (kobs = 7.8 × 10−3 s−1), whereas 3 displays similar polymerisation rates for rac- and L-LA (kobs = 3.2 × 10−3 and 2.7 × 10−3 s−1, respectively). These results suggest that while 3 likely has a similar degree of preference for D- and L-LA enchainment, 4 might display a slight preference for D-LA coordination and insertion, resulting in a modest isotactic bias. Notably, only trace rac-LA (<13%) was converted in the absence of BnOH co-initiator (Table 1, entries 1 and 5); these conversions were only mildly improved by the addition of 1 eq. BnOH to give 15% conversion with 3 after 5 min, and 20% conversion with 4 after 1.25 min (THF at R.T, Table S1,† entries 12 and 25). The dramatically reduced activity in the presence of 1 eq. BnOH suggests that 3 and 4 are unlikely to operate via an activated monomer mechanism as 2 eq. BnOH are required to efficiently initiate the proposed coordination-insertion mechanism. Complexes 3 and 4 also remained active under immortal polymerisation conditions (10 eq. BnOH, Table S1,† entries 13 and 26).
Complexes 3 and 4 were benchmarked against homometallic complexes 1–2, [BnONa], [BnOK] and [LZn2OBn] in THF (Table 1). The alkali metal analogues were highly active but poorly controlled; the MALDI-ToF data shows transesterified ω-hydroxy end-capped and cyclic PLA (see ESI†). [BnONa] and [BnOK] also displayed poor solubility in THF and toluene, emphasising a potential benefit of heterometallic initiators, which are often more soluble than the homometallic counterparts. Although [LZn2OBn] displays good activities in toluene,12 the activity is diminished in THF (entry 14, Table 1). In contrast, 3 and 4 gave improved activities in THF, which was attributed to the Lewis acidic alkali metals, particularly the larger K+ in 4 (vs. Na+ in 3), providing additional coordination sites, thus preventing competitive THF/LA coordination. Indeed, DFT calculations suggest a slight preference for coordination of 2 eq. THF to 4′vs. 1 eq. THF to 3′ (Fig. 2, Tables S6 and S13†), even if coordination of 2 eq. THF to both 3 and 4 was observed by NMR analysis. Complexes 3 and 4 were also significantly faster than in situ generated [LZn2OBn] in rac-LA ROP in toluene at 60 °C (Table S3†), with 3 and 4 converting 89 eq. and 86 eq. rac-LA in 2.5 and 1 min, respectively (vs. 87 eq. in 10 min with in situ generated [LZn2OBn]). The activity and control differences between 3–4 and their homometallic analogues suggest cooperative interactions between Na/K and Zn2.
Reactivity insights: experimental and computational studies
The in situ generation of [LNaZn2(OBn)2(THF)2] (5) and [LKZn2(OBn)2(THF)2] (6) from 3 or 4 and 2 eq. BnOH was investigated by NMR analysis in THF-d8, which indicated the rapid loss of BnOH and the formation of ethane (0.85 ppm) and new centrosymmetric complexes with OBn co-ligands (Fig. S44 and S45†). Notably, the Zn-Et groups of 3 remained intact in the presence of 10 eq. rac-LA until the addition of 2 eq. BnOH whereupon the Zn-Et groups disappeared and PLA was rapidly formed (Fig. S46†). DOSY NMR analysis of in situ generated 5 and 6 confirmed that the OBn co-ligands and L were part of the same complex (Fig. S47 and S48†). DFT calculations suggested 5′ and 6′ conserve the R,R ligand stereochemistry at the N atoms (vide supra) but with ligand rearrangement to a meso (R,S) configuration also possible under the polymerisation conditions (Tables S9 and S14†). The in situ dissociation of 5 and 6 in THF to [LZn2OBn] and [BnONa] or [BnOK], respectively, was deemed unlikely based on NMR and DFT calculations (Fig. S49, S50 and Table S10†). The reaction of 1 and 2 with 1 eq. BnOH was also investigated but gave no reaction i.e. no [BnONa] or [BnOK] was formed (Fig. S51†). These findings suggest that the rearrangement of 3 and 4 to homometallic species is unlikely under the polymerisation conditions. This was further supported by monitoring the reaction of [LZn2OBn] with 1 eq. of in situ generated [BnOK] in THF-d8 by 1H NMR, which also generated 6 (Fig. S52†). Testing a 1:1 [BnOK]:[LZn2OBn] mixture in rac-LA ROP gave excellent activity in both THF and toluene (Tables 1 and S3†), albeit with reduced polymerisation control (Đ = 1.6–2.0) vs.[LZn2OBn] and 4 (Đ < 1.5). Similarly to LiCl addition boosting the activity of conventional Grignard reagents by forming heterometallic Turbo-Grignard reagents,32 our findings suggest that addition of an alkali metal alkoxide to a bis-Zn complex may provide a simple yet effective strategy for improving the performance of homometallic ROP initiators. However, in this case, the optimal balance between the polymerisation activity and control was achieved with in situ generation of 5 and 6via alcoholysis of 3 and 4 (Table 1, vide infra).
Coordination of 1–2 eq. L-LA to 5′ and 6′ was modelled by DFT (see ESI†); these reactions were either neutral or slightly exergonic. The most stable structures feature the R,R ligand configuration at the N atoms and one L-LA coordinated to the alkali metal, although coordination of two L-LA may also be accessible under polymerisation conditions. While no significant differences in structural and L-LA coordination preferences were found between 5′ and 6′, the data suggests that coordination of two L-LA is more accessible for 6′ (vs.5′), in line with the increased ionic radius of K+. The activity differences between 3 and 4 (with 2 eq. BnOH) may thus have a kinetic origin; this requires modelling of ROP transition states which are currently under investigation in our laboratories.
Previous studies showed that isolated [LZn2OBn] gave a ten-fold activity increase vs. the in situ generated complex,12 and so the isolation of 5 and 6 was investigated. However, the isolated heterometallic species showed reduced activity in rac-LA ROP compared to the in situ generated analogues (with 2 eq. BnOH); isolated 5 was approximately six times slower than 3, and 6 gave half the rate of 4 (THF, R.T.). In contrast to in situ generated 5 and 6, DOSY NMR analysis of isolated 5 and 6 (Fig. S54 and S55†) suggested formation of a two-component mixture involving higher MW species (approx. 826 Da in both cases) and lower MW species comprising OBn anions (252 Da for 5 and 298 Da for 6). This may explain the reduced activity of the isolated species, as steric congestion around the metals in 5 and 6 could decrease the stability over time in THF (1 h at R.T.), leading to the formation of (mixed) metal-OBn aggregates. It is also plausible that increased concentration upon solvent removal for product isolation leads to formation of different structures, as Lewis donor solvents are well-known to influence aggregation states of organometallic complexes. Importantly, on the timescale of the in situ generated polymerisations (<7.5 min at R.T. in THF), there was no evidence of decomposition by NMR analysis.
Catalyst scope
Complex 3 (with 2 eq. BnOH) is also extremely active in ε-CL and δ-valerolactone (δ-VL) ROP (Table S4†), converting 53 eq. ε-CL and 99 eq. δ-VL in just 5 s at R.T. Notably, 3 converts up to 760 eq. ε-CL in 4 min at R.T., producing PCL with Mn up to 19600 g mol−1. While complex 4 converted 94 eq. δ-VL in 5 s, it was less active than 3 in ε-CL ROP (Table S4†), which contrasts with the higher activity of 4 (vs.3) in rac-LA ROP. The more Lewis basic character of ε-CL vs. δ-VL and LA (based on the FT-IR carbonyl shifts: ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) = 1732 cm−1 for ε-CL, 1747 cm−1 for δ-VL and 1770 cm−1 for LA)33 may promote decomposition/rearrangement of 4; this may be more likely with 4 than 3 due to the larger and more electropositive K+ centre facilitating ε-CL coordination.
O) = 1732 cm−1 for ε-CL, 1747 cm−1 for δ-VL and 1770 cm−1 for LA)33 may promote decomposition/rearrangement of 4; this may be more likely with 4 than 3 due to the larger and more electropositive K+ centre facilitating ε-CL coordination.
Conclusions
In summary, two novel heterometallic complexes 3 and 4 were reported to be highly active in rac-LA, ε-CL and δ-VL ROP with 2 eq. BnOH. Complexes 3 and 4 outperform their homometallic counterparts, combining high activities with good polymerisation control. To the best of our knowledge, 4/BnOH (2 eq.) is the fastest heterometallic catalytic system for rac-LA ROP reported to date. These rate enhancements demonstrate the benefit of combining metals known to be highly active in cyclic ester ROP (Zn) with abundant, inexpensive and non-toxic alkali metals (Na and K). When bridged through a heteroatom, electronic communication between heterometals can alter the properties of each metal through an “ate” activation. While alkali metals are known to boost the reactivity of Zn towards C–H activation,34 our studies suggest that this concept can also be translated to cyclic ester ROP. The activity enhancements observed may arise from increased Lewis acidity of the more electropositive metal (Na/K) and labilisation of the M–OR bonds around the more electrophilic metal(s) (Zn). Heterometallic catalysts remain underexplored in ROP and offer a promising area for further investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the CRITICAT Centre for Doctoral Training and EPSRC (W. G., EP\L016419\1; M. P. S.; EP\S025200\1 and EP\P026095\2), Royal Society (J. A. G., RSG\R1\180101; A. B., UF\160021 fellowship), British Ramsay Memorial Trust (J. A. G.) and L’Oréal-UNESCO For Women in Science (J. A. G.) for funding. We thank the University of Bath HPC for computing resources.Notes and references
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- C. Ha and J. A. Gardella, Chem. Rev., 2005, 105, 4205–4232 CrossRef CAS.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- R. Platel, L. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS.
- C. Wombwell and E. Reisner, Dalton Trans., 2014, 43, 4483–4493 RSC.
- T. D. Bluemke, W. Clegg, P. Garcia-Alvarez, A. R. Kennedy, K. Koszinowski, M. D. McCall, L. Russo and E. Hevia, Chem. Sci., 2014, 5, 3552–3562 RSC.
- A. J. Martinez-Martinez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef CAS.
- S. K. Mandal and H. Roesky, Inorg. Chem., 2007, 46, 10158–10167 CrossRef CAS.
- J. P. Mcinnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 13, 2545–2557 CrossRef.
- Z. Cai and D. Xiao, Comments Inorg. Chem., 2019, 39, 27–50 CrossRef CAS.
- A. H. Gao, W. Yao, Y. Mu, W. Gao, M.-T. Sun and Q. Su, Polyhedron, 2009, 28, 2605–2610 CrossRef CAS.
- L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2006, 25, 1012–1020 CrossRef.
- M. Normand, E. Kirillov, T. Roisnel and J.-F. Carpentier, Organometallics, 2012, 31, 1448–1457 CrossRef CAS.
- J. Char, E. Brule, P. C. Gros, M.-N. Rager, V. Guerineau and C. M. Thomas, J. Organomet. Chem., 2015, 796, 47–52 CrossRef CAS.
- W. Li, Z. Zhang, Y. Yao, Y. Zhang and Q. Shen, Organometallics, 2012, 31, 3499–3511 CrossRef CAS.
- H. T. Sheng, J. M. Li, Y. Zhang, Y. M. Yao and Q. Shen, Polyhedron, 2008, 27, 1665–1672 CrossRef CAS.
- J. Hao, J. Li, C. Cui and H. W. Roesky, Inorg. Chem., 2011, 50, 7453–7459 CrossRef CAS.
- H.-Y. Chen, M.-Y. Liu, A. K. Sutar and C.-C. Lin, Inorg. Chem., 2010, 49, 665–674 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- B. Rajashekhar, S. K. Roymuhury, D. Chakraborty and V. Ramkumar, Dalton Trans., 2015, 44, 16280–16293 RSC.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333–3336 CrossRef CAS.
- P. Dubois, C. Jacobs, R. Jerome and P. Teyssie, Macromolecules, 1991, 24, 2266–2270 CrossRef CAS.
- R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743–755 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, EA, MS characterisation data, polymer MALDI-ToF, GPC, kinetic and DOSY studies and DFT calculations. CCDC 2002496. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04705h |
| This journal is © The Royal Society of Chemistry 2020 |