
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3-Aminooxetanes: versatile 1,3-amphoteric molecules for intermolecular annulation reactions†
Zengwei
Lai‡
ab,
Renwei
Zhang‡
a,
Qiang
Feng
a and
Jianwei
Sun
 *a
*a
aDepartment of Chemistry, Shenzhen Research Institute, The Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong SAR, Kowloon, China. E-mail: sunjw@ust.hk
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China
First published on 8th September 2020
Abstract
Despite the versatility of amphoteric molecules, stable and easily accessible ones are still limitedly known. As a result, the discovery of new amphoteric reactivity remains highly desirable. Herein we introduce 3-aminooxetanes as a new family of stable and readily available 1,3-amphoteric molecules and systematically demonstrated their amphoteric reactivity toward polarized π-systems in a diverse range of intermolecular [3 + 2] annulations. These reactions not only enrich the reactivity of oxetanes, but also provide convergent access to valuable heterocycles.
Amphoteric molecules, which bear both nucleophilic and electrophilic sites with orthogonal reactivity, represent an attractive platform for the development of chemoselective transformations.1 For example, isocyanides are well-established 1,1-amphoteric molecules, with the terminal carbon being both nucleophilic and electrophilic, and this feature has enabled their exceptional reactivity in numerous multi-component reactions.2 In the past few decades, substantial effort has been devoted to the search for new amphoteric molecules.1–5 Among them, 1,3-amphoteric molecules proved to be versatile. The Yudin and Beauchemin laboratories have independently developed two types of such molecules, α-aziridine aldehydes and amino isocyanates, respectively.4,5 With an electrophilic carbon and a nucleophilic nitrogen in relative 1,3-positions, these molecules are particularly useful for the chemoselective synthesis of heterocycles with high bond-forming efficiency without protective groups (Fig. 1). However, such elegant amphoteric systems still remain scarce. Therefore, the development of new stable amphoteric molecules with easy access remains highly desirable.
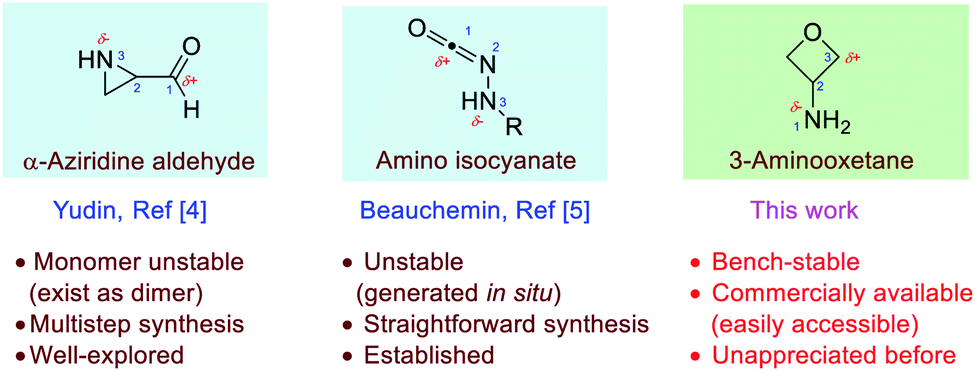 | ||
| Fig. 1 Representative [1,3]-amphoteric molecules versus 3-aminooxetanes. | ||
In this context, herein we introduce 3-aminooxetanes as a new type of 1,3-amphoteric molecules and systematically demonstrate their reactivity in a range of [3 + 2] annulations, providing rapid access to diverse heterocycles. Notably, 3-aminooxetanes are bench-stable and either commercially available or easily accessible. However, their amphoteric reactivity has not been appreciated previously.
Oxetane is a useful functional group in both drug discovery and organic synthesis.6–9 Owing to the ring strain, it is prone to nucleophilic ring-opening, in which it serves as an electrophile (Scheme 1A).6–8 We envisioned that, if a nucleophilic group is installed in the 3-position (e.g., amino group), such molecules should exhibit 1,3-amphoteric reactivity due to the presence of both nucleophilic and electrophilic sites (Scheme 1B). Importantly, the 1,3-relative position is crucial for inhibiting self-destructive intra- or intermolecular ring-opening (i.e. the 3-nucleophilic site attack on oxetane itself) due to high barriers. Thus, such orthogonality is beneficial to their stability. In contrast, the nucleophilic site is expected to react with an external polarized π bond (e.g., X = Y, Scheme 1B), which enables a better-positioned nucleophile (Y) to attack the oxetane and cyclize. Thus, a formal [3 + 2] annulation should be expected. Unlike the well-known SN2 reactivity of oxetanes with simple bond formation, this amphoteric reactivity would greatly enrich the chemistry of oxetanes with multiple bond formations and provide expedient access to various heterocycles. In contrast to the conventional approaches that require presynthesis of advanced intermediates (e.g., intramolecular ring-opening),8 the exploitation of such amphoteric reactivity in an intermolecular convergent manner from simple substrates would be more practically useful. Moreover, more activation modes could be envisioned in addition to oxetane activation. In 2015, Kleij and coworkers reported an example of cyclization between 3-aminooxetane and CO2 in 55% yield, which provided a pioneering precedent.10 However, a systematic study to fully reveal such amphoteric reactivity in a broad context remains unknown in the literature.
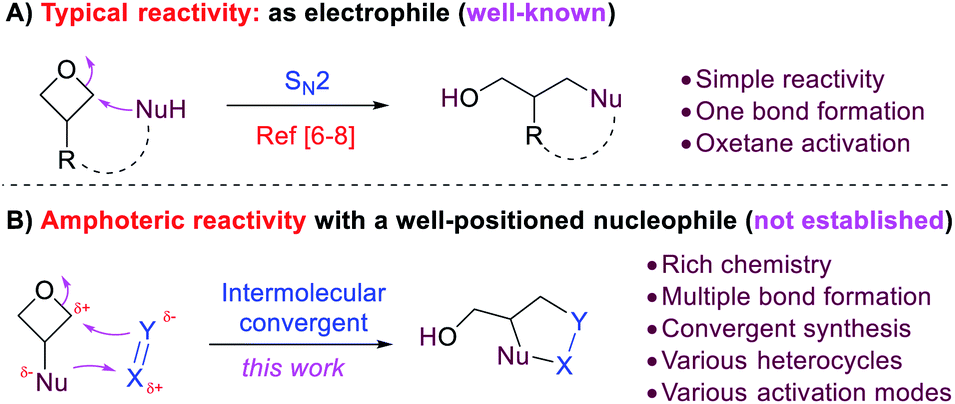 | ||
| Scheme 1 Typical oxetane reactivity and the new amphoteric reactivity. | ||
To test our hypothesis, we began with the commercially available 3-aminooxetanes 1a and 1b as the model substrates. Phenyl thioisocyanate 2a and CS2 were initially employed as reaction partners, as they both have a polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond as well as a relatively strong sulfur nucleophilic motif. Moreover, the resulting desired products, iminothiazolidines and mercaptothiazolidines, are both heterocycles with important biological applications (Fig. 2).11 To our delight, simple mixing these two types of reactants in DCM resulted in spontaneous reactions at room temperature without any catalyst. The corresponding [3 + 2] annulation products iminothiazolidine 3a and mercaptothiazolidine 4a were both formed with excellent efficiency (Scheme 2). It is worth mentioning that catalyst-free ring-opening of an oxetane ring is rarely known, particularly for intermolecular reactions.6–9 In this case, the high efficiency is likely attributed to the suitable choice and perfect position of the in situ generated sulfur nucleophile.
S bond as well as a relatively strong sulfur nucleophilic motif. Moreover, the resulting desired products, iminothiazolidines and mercaptothiazolidines, are both heterocycles with important biological applications (Fig. 2).11 To our delight, simple mixing these two types of reactants in DCM resulted in spontaneous reactions at room temperature without any catalyst. The corresponding [3 + 2] annulation products iminothiazolidine 3a and mercaptothiazolidine 4a were both formed with excellent efficiency (Scheme 2). It is worth mentioning that catalyst-free ring-opening of an oxetane ring is rarely known, particularly for intermolecular reactions.6–9 In this case, the high efficiency is likely attributed to the suitable choice and perfect position of the in situ generated sulfur nucleophile.
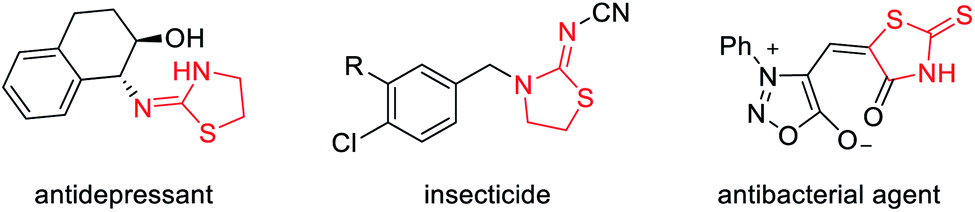 | ||
| Fig. 2 Selected bioactive molecules containing iminothiazolidine and mercaptothiazolidine motifs. | ||
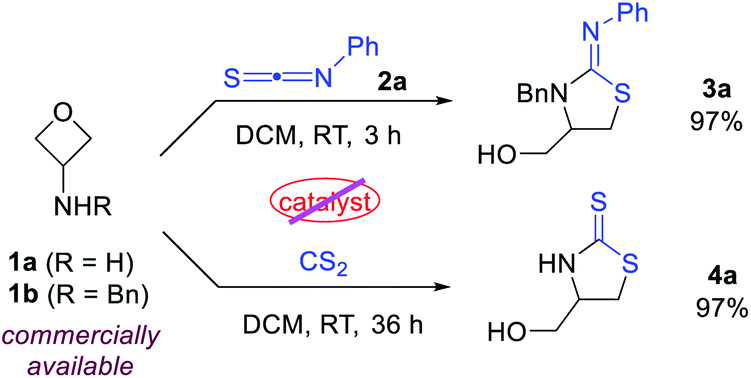 | ||
| Scheme 2 Initial results between 3-aminooxetanes and thiocarbonyl compounds. | ||
The catalyst-free annulation protocol is general with respect to various 3-aminooxetanes and isothiocyanates. A range of iminothiazolidines and mercaptothiazolidines were synthesized with high efficiency under mild conditions (Scheme 3). Many of them were obtained in quantitative yield. Quaternary carbon centers could also be generated from 3-substituted 3-aminooxetanes (e.g., 3j). The structure of product 3b was unambiguously confirmed by X-ray crystallography.
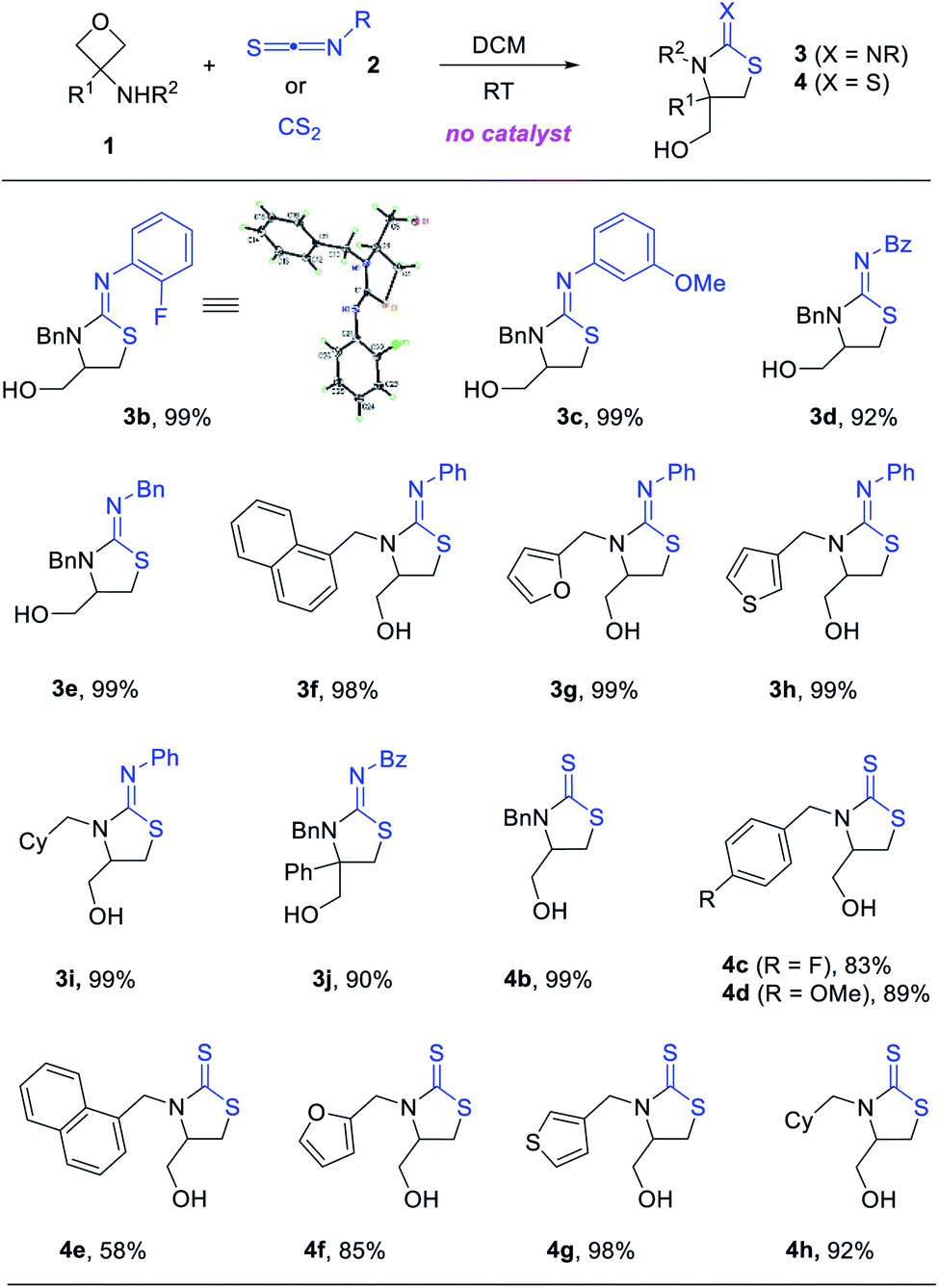 | ||
| Scheme 3 Formal [3 + 2] annulation with isothiocyanates and CS2. Reaction conditions: 1 (0.3–0.4 mmol), 2 (1.1 equiv.) or CS2 (1.5 equiv.), DCM (2 mL), RT, 3 h for 3 and 36 h for 4. Yields are for the isolated products. | ||
With the initial success of thiocarbonyl partners, we next turned our attention to isocyanates, in which the carbonyl group serves as the [3 + 2] annulation motif. Compared with sulfur as the nucleophilic site in the above cases, the oxygen atom is less nucleophilic. As expected, initial tests of the reactivity by mixing 1b and 5a resulted in no desired annulation product 6a in the absence of a catalyst (Table 1, entry 1). Next, Brønsted acids, including TsOH and the super acid HNTf2, were examined as catalysts, but with no success (entries 2 and 3). We then resorted to various Lewis acids, particularly those oxophilic ones, in hope of activating the oxetane unit. Unfortunately, many of them still remained ineffective (e.g., ZnCl2, AuCl, and FeCl3). However, to our delight, further screening of stronger Lewis acids helped identify Sc(OTf)3, Zn(OTf)2, and In(OTf)3 to be effective at room temperature, leading to the desired iminooxazolidine product 6a in good yield (entries 7–9). Its structure was confirmed by X-ray crystallography. Nevertheless, aiming to search for a cheaper catalyst, we continued to optimize this reaction at a higher temperature using previous ineffective catalysts. Indeed, FeCl3 was found to be effective at 80 °C (61% yield, entry 10), while Brønsted acid TsOH remained ineffective at this temperature (entry 11). Notably, decreasing the loading of FeCl3 to 1 mol% led to a higher yield (89% yield, entry 12). However, further decreasing to 0.5 mol% resulted in slightly diminished efficiency (entry 13).
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
| a Reaction scale: 1b (0.1 mmol), 5a (0.1 mmol), catalyst (10 mol%), toluene (1 mL). b Yield based on analysis of the 1H NMR spectra of the crude reaction mixture using trichloroethylene as an internal reference. For all the entries, the urea product from simple amine addition to isocyanate 5a accounts for the mass balance. c Run at 80 °C. d Isolated yield. | ||
| 1 | — | 0 |
| 2 | TsOH·H2O | 0 |
| 3 | HNTf2 | 0 |
| 4 | ZnCl2 | 0 |
| 5 | AuCl | 0 |
| 6 | FeCl3 | 0 |
| 7 | Sc(OTf)3 | 74 |
| 8 | Zn(OTf)2 | 78 |
| 9 | In(OTf)3 | 90 |
| 10 | FeCl3c | 61 |
| 11 | TsOH·H2Oc | 0 |
| 12 | FeCl3c (1 mol%) | 89(84)d |
| 13 | FeCl3c (0.5 mol%) | 85 |
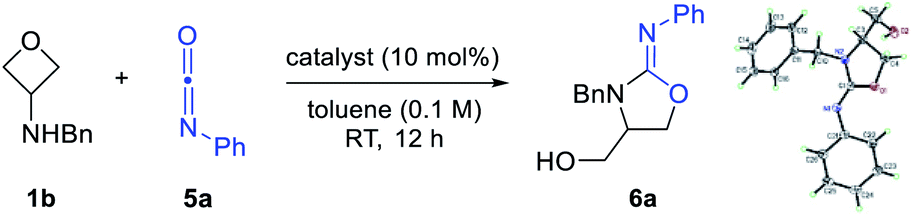
While there are multiple effective catalysts, FeCl3 was selected for the scope study in view of its low price. Various substituted 3-aminooxetanes and isocyanates were subjected to this annulation protocol (Scheme 4). The corresponding iminooxazolidine products were all obtained in good to excellent yields. Isocyanates containing an electron-donating or electron-withdrawing group were both suitable reaction partners. Remarkably, a 1.5 mmol scale reaction of 6a also worked efficiently.
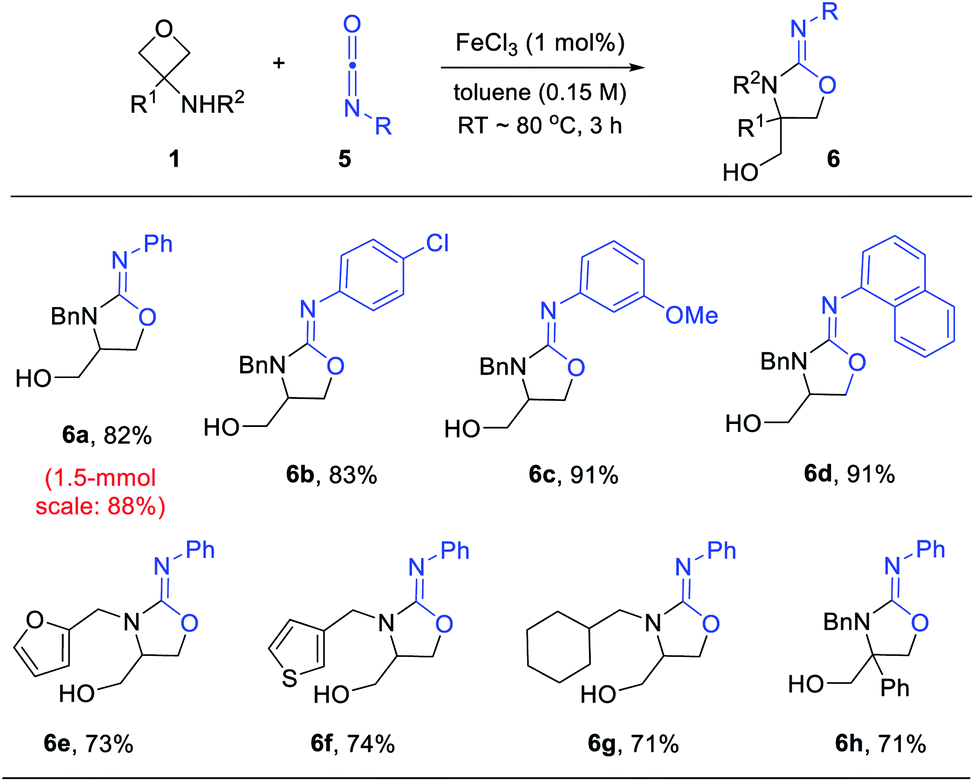 | ||
| Scheme 4 Formal [3 + 2] annulation between 3-aminooxetanes and isocyanates. Reaction scale: 1 (0.3 mmol), 5 (0.3 mmol), FeCl3 (1 mol%), toluene (2 mL). | ||
Although (thio)isocyanates and CS2 have been successfully utilized in the formal [3 + 2] annulation with 3-aminooxetanes, these partners are relatively reactive. We were curious about whether the CO bond in relatively inert molecules could react in a similar manner. For example, the CO bond in CO2 is both thermodynamically and kinetically inert relative to typical organic carbonyl groups. However, as a cheap, abundant and green one-carbon source, CO2 has been a subject of persistent investigations owing to its versatility in various transformations leading to valuable materials.12 Specifically, if CO2 could be employed as a partner for the [3 + 2] annulation with 3-aminooxetanes, it would represent an attractive synthesis of oxazolidinones, a well-known heterocycle with applications in both organic synthesis and medicinal chemistry.13 In this context, we next studied the possibility of utilizing CO2 in our annulation.
As expected, the reaction between 1b and CO2 at 1 atmospheric pressure did not proceed without a catalyst (Table 2, entry 1). Next, we examined representative Lewis acids, such as Sc(OTf)3, In(OTf)3 and FeCl3. Among them, Sc(OTf)3 exhibited the highest catalytic activity at room temperature (22% yield, entry 2). The reaction efficiency could be improved at 80 °C (65% yield, entry 6), but no further improvement could be made at a higher temperature or with other solvents. Next, we resorted to organic nitrogen bases, as they were known as effective activators of CO2.14 While Et3N and DABCO were completely ineffective for the reaction in MeCN at 80 °C, fortunately, TMG, TBD, and DBU were competent for the desired process (entries 7–11). Among them, DBU exhibited the best performance, leading to the desired product 7a in 89% yield (entry 11). It is worth noting that the polar solvent MeCN was found to be crucial for the base-catalyzed reactivity. Less polar solvents, such as toluene, DCE or THF, completely shut down the reaction. We believe that effective stabilization of certain polar intermediates involved here is critically beneficial to decreasing the reaction barrier. Finally, unlike the previous Lewis acid-catalyzed annulation with isocyanates, this base-catalyzed [3 + 2] annulation with CO2 proceeds via a different activation mode (i.e., to activate CO2 rather than oxetane). We believe that expansion of possible activation modes in this type of amphoteric reactivity will enrich the chemistry of oxetanes.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | T | Conv. (%) | Yield (%) |
| a Reaction scale: 1b (0.1 mmol), CO2 (1 atm), solvent (0.5 mL). Yields based on analysis of the 1H NMR spectra of the crude reaction mixture using CH2Br2 as an internal standard. | ||||
| 1 | — | RT | 0 | 0 |
| 2 | Sc(OTf)3 | RT | 48 | 22 |
| 3 | In(OTf)3 | RT | 33 | 9 |
| 4 | Zn(OTf)2 | RT | 7 | 0 |
| 5 | Sc(OTf)3 | 60 °C | 100 | 61 |
| 6 | Sc(OTf)3 | 80 °C | 100 | 65 |
| 7 | Et3N | 80 °C | 0 | 0 |
| 8 | DABCO | 80 °C | 5 | 0 |
| 9 | TMG | 80 °C | 72 | 54 |
| 10 | TBD | 80 °C | 100 | 88 |
| 11 | DBU | 80 °C | 100 | 89 |
|
||||
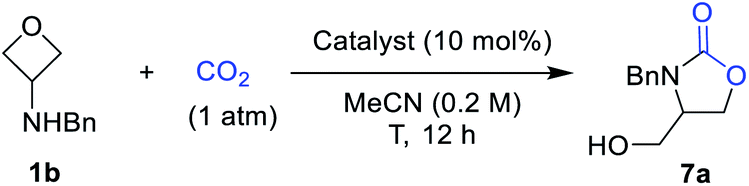
We next examined the scope of this CO2-fixation process. Unfortunately, at a larger scale (0.5 mmol), the same condition (entry 11, Table 2) could not lead to complete conversion within 12 h. Therefore, further optimization aiming to accelerate the reaction was performed. Indeed, a higher concentration (1.0 M) resulted in a higher rate without affecting the yield. As shown in Scheme 5, a wide variety of 3-aminooxetanes were smoothly converted to the corresponding oxazolidinones in high yields. Both electron-donating and electron-withdrawing substituents on the N-benzyl group did not affect the efficiency. Heterocycle-based N-benzyl or N-allylic substituents are all suitable substrates. However, for regular alkyl substituents, such as homobenzyl (7h) or n-butyl (7j), the stronger base catalyst TBD was needed to achieve good efficiency. Furthermore, this reaction can tolerate steric hindrance in the 3-position of the oxetane (7k), where a quaternary carbon center could be incorporated. However, increasing the size of the N-substituent, such as the secondary alkyl groups in 7i and 7l, did influence the reactivity, thus requiring a higher temperature (100 °C). This process exhibited good compatibility with diverse functional groups, such as ethers, pyridines, aryl halides, olefins, silyl-protected alcohols, and phthalimides. Finally, this protocol is also capable of generating various oxazolidinones embedded in a different structural context, such as chiral oxazaolidinone 7l, bis(oxazolidinone) 7m, and polyheterocycle-fused oxazolidinone 7o.
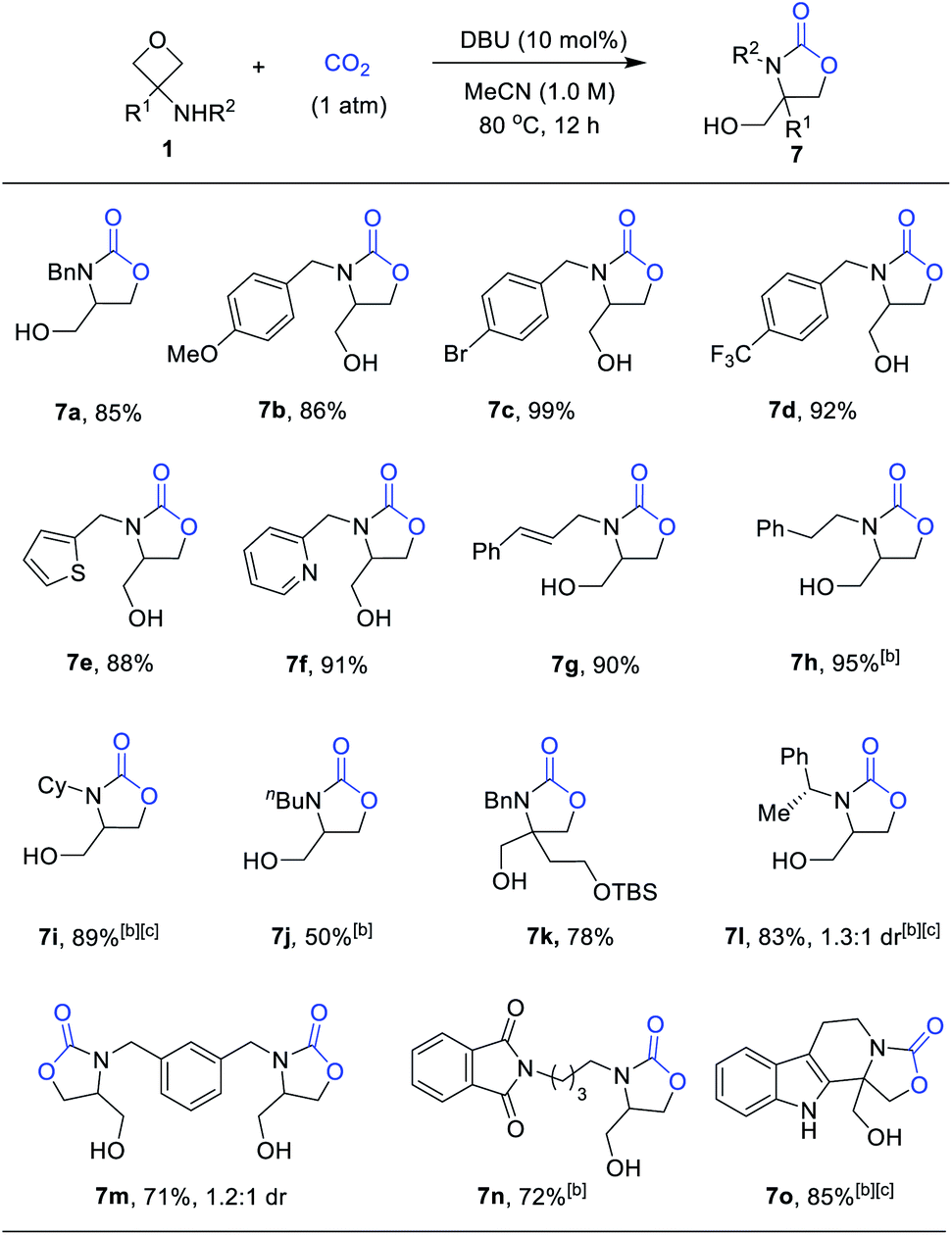 | ||
| Scheme 5 Formal [3 + 2] annulation between 3-aminooxetanes and CO2. aReaction scale: 1 (0.5 mmol), CO2 (1 atm), DBU (10 mol%), MeCN (0.5 mL). Isolated yield. bRun with TBD as the catalyst. cRun with DMF as solvent at 100 °C. | ||
In summary, 3-aminooxetanes have been systematically demonstrated, for the first time, as versatile 1,3-amphoteric molecules. They are a new addition to the limited family of amphoteric molecules. Though previously unappreciated, these molecules exhibited various advantages over the related known 1,3-amphotric molecules (e.g., α-aziridine aldehydes and amino isocyanates), including easy access and extraordinary stability. The perfect position of the nucleophilic nitrogen together with the orthogonal electrophilic carbon allowed them to participate in a diverse range of intermolecular formal [3 + 2] annulations with polarized π-systems, leading to rapid access to various valuable nitrogen heterocycles. Different types of polarized double bonds, from reactive (thio)isocyanates to inert CO2, all participated efficiently in these highly selective annulations with or without a suitable catalyst. Furthermore, the involvement of more functional groups in such amphoteric reactivity allowed manifold activation modes, thereby greatly enriching the reactivity of the already versatile oxetane unit to a new dimension. These reactions, proceeding in an intermolecular convergent manner from readily available substrates, provide expedient access to various valuable nitrogen heterocycles, thus being complementary to those traditional methods that either required multiple steps or less available substrates. More studies on the 1,3-amphoteric reactivity of 3-oxetanes, particularly those with other partners as well as their asymmetric variants, are ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by NSFC (21572192, 81703341 and 91956114) and Hong Kong RGC (16302318 and 16302617). We thank Zhiyang Li for repeating the results.References
- Z. He, A. Zajdlik and A. K. Yudin, Acc. Chem. Res., 2014, 47, 1029–1040 CrossRef CAS.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- (a) J. D. St. Dennis, Z. He and A. K. Yudin, ACS Catal., 2015, 5, 5373–5379 CrossRef; (b) P. Trinchera, V. B. Corless and A. K. Yudin, Angew. Chem., Int. Ed., 2015, 54, 9038–9041 CrossRef CAS; (c) D. B. Diaz, C. C. G. Scully, S. K. Liew, S. Adachi, P. Trinchera, J. D. St. Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2016, 55, 12659–12663 CrossRef CAS.
- (a) R. Hili and A. K. Yudin, J. Am. Chem. Soc., 2006, 128, 14772–14773 CrossRef CAS; (b) R. Hili and A. K. Yudin, Angew. Chem., Int. Ed., 2008, 47, 4188–44191 CrossRef CAS; (c) R. Hili and A. K. Yudin, J. Am. Chem. Soc., 2009, 131, 16404–16406 CrossRef CAS; (d) R. Hili, V. Rai and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 2889–2891 CrossRef CAS; (e) B. H. Rotstein, V. Rai, R. Hili and A. K. Yudin, Nat. Protoc., 2010, 5, 1813–1822 CrossRef CAS; (f) S. Baktharaman, N. A. Afagh, A. Vandersteen and A. K. Yudin, Org. Lett., 2010, 12, 240–243 CrossRef CAS; (g) L. L. W. Cheung, Z. He, S. M. Decker and A. K. Yudin, Angew. Chem., Int. Ed., 2011, 50, 11798–11802 CrossRef CAS.
- (a) J.-G. Roveda, C. Clavette, A. D. Hunt, S. I. Gorelsky, C. J. Whipp and A. M. Beauchemin, J. Am. Chem. Soc., 2009, 131, 8740–8741 CrossRef CAS; (b) J.-G. Roveda, C. Clavette, W. Gan, A. Bongers, T. Markiewicz, A. B. Toderian, S. I. Gorelsky and A. M. Bearchemin, J. Am. Chem. Soc., 2012, 134, 16111–16114 CrossRef; (c) C. Clavette, J.-F. Vincent Rocan and A. M. Beauchemin, Angew. Chem., Int. Ed., 2013, 52, 12705–12708 CrossRef CAS; (d) J.-F. Vincent-Rocan, C. Clavetter, K. Leckett and A. M. Beauchemin, Chem.–Eur. J., 2015, 21, 3886–3890 CrossRef CAS.
- For leading reviews on oxetane chemistry, see: (a) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS; (b) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272–286 CrossRef CAS; (c) Z. Wang, Z. Chen and J. Sun, Org. Biomol. Chem., 2014, 12, 6028–6032 RSC; (d) C. A. Malapit and A. R. Howell, J. Org. Chem., 2015, 80, 8489–8495 CrossRef CAS; (e) S. Li and J. Xu, Prog. Chem., 2016, 28, 1798–1810 CAS; (f) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS.
- Selected examples of intermolecular nucleophilic opening of oxetanes: (a) Y. Wang, H. Bekolo and A. R. Howell, Tetrahedron, 2002, 58, 7101–7107 CrossRef CAS; (b) H. Yoshida, Y. Asatsu, Y. Mimura, Y. Ito, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2011, 50, 9676–9679 CrossRef CAS; (c) N. Ishida, Y. Nakanishi and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 11875–11878 CrossRef CAS; (d) Z. Wang, Z. Chen and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 6685–6688 CrossRef CAS; (e) W. Yang, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 6954–6958 CrossRef CAS.
- Selected examples of intramolecular nucleophilic opening of oxetanes: (a) T. Bach and J. Shröder, Tetrahedron Lett., 1997, 38, 3707–3710 CrossRef CAS; (b) T. Bach, K. Kather and O. Krämer, J. Org. Chem., 1998, 63, 1910–1918 CrossRef CAS; (c) T. Bach and J. Shröder, J. Org. Chem., 1999, 64, 1265–1273 CrossRef CAS; (d) T. Bach, Synlett, 2000, 1699–1707 CAS; (e) R. N. Loy and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 2786–2787 CrossRef CAS; (f) J. A. Burkhard, B. H. Tchitchanov and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5379–5382 CrossRef CAS; (g) W. Zhao, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 6209–6213 CrossRef CAS; (h) Z. Chen, B. Wang, Z. Wang, G. Zhu and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 2027–2031 CrossRef CAS; (i) S. A. Ruider, S. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11908–11911 CrossRef CAS; (j) R. A. Croft, J. J. Mousseau, C. Choi and J. A. Bull, Chem.–Eur. J., 2016, 22, 16271–16276 CrossRef CAS; (k) W. Yang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 1868–1871 CrossRef CAS; (l) A. R. White, R. A. Kozlowski, S.-C. Tsai and C. D. Vanderwal, Angew. Chem., Int. Ed., 2017, 56, 10525–10529 CrossRef CAS; (m) D. Xu, H. Wei, Y. Zhen, Y.-Q. Gao, R. Li, X. Li, Y. He, Z. Zhang and W. Xie, Org. Chem. Front., 2019, 6, 1681–1685 RSC; (n) L. G. DeRatt, E. C. Lawson, C.-Y. Wang and S. D. Kuduk, Org. Lett., 2019, 21, 9642–9645 CrossRef CAS; (o) R. Zhang, W. Guo, M. Duan, K. N. Houk and J. Sun, Angew. Chem., Int. Ed., 2019, 58, 18055–18060 CrossRef CAS; (p) H. Huang, W. Yang, Z. Chen, Z. Lai and J. Sun, Chem. Sci., 2019, 10, 9586–9590 RSC.
- Selected recent examples of oxetane ring expansion: (a) D. Rix, R. Ballesteros-Garrido, W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2011, 50, 7308–7311 CrossRef CAS; (b) C. Gronnier, S. Kramer, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2012, 134, 828–831 CrossRef CAS; (c) B. Guo, G. Schwarzwalder and J. T. Njardarson, Angew. Chem., Int. Ed., 2012, 51, 5675–5678 CrossRef CAS; (d) Q. Yin and S.-L. You, Org. Lett., 2014, 16, 1810–1813 CrossRef CAS; (e) Y.-N. Wang, L.-C. Yang, Z.-Q. Rong, T.-L. Liu, R. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1596–1600 CrossRef CAS; (f) S. Jana, Z. Yang, C. P. X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129–10134 RSC.
- J. Rintjema, W. Guo, E. Martin, E. C. Escudero-Adan and A. W. Kleij, Chem.–Eur. J., 2015, 21, 10754–10762 CrossRef CAS.
- (a) M.-H. Shih, Y.-Y. Xu, Y.-S. Yang and G.-L. Lin, Molecules, 2015, 20, 6520–6532 CrossRef CAS; (b) U. K. Shukla, R. Singh, J. M. Khanna, A. K. Saxena, H. K. Singh, R. N. Sur, B. N. Dhawan and N. Anand, Collect. Czech. Chem. Commun., 1992, 57, 415–424 CrossRef CAS; (c) A. Hense, R. Fischer, E.-R. Gesing, S. Herrmann, K. Kather, S. Lehr, K. Voigt, H.-J. Riebel, P. Jeschke, C. Erdelen, W. Andersch, P. Lösel and U. Reckmann, PCT Int. Appl., 2002, 96872 Search PubMed; (d) M. D’hooghe and N. De Kimpe, Tetrahedron, 2006, 62, 513–535 CrossRef.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS; (b) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644–659 CrossRef CAS.
- (a) M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010–2023 CrossRef CAS; (b) T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529–542 CrossRef CAS; (c) K. Michalska, I. Karpiuk, M. Krul and S. Tyski, Bioorg. Med. Chem., 2013, 21, 577–591 CrossRef CAS; (d) M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef CAS.
- DBU and other organic bases have been known to activate CO2: (a) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382–404 RSC; (b) C. Villiers, J.-P. Dognon, R. Pollet, P. Thuéry and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1987318 and 1987319. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04254d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |