DOI:
10.1039/D0SC04036C
(Edge Article)
Chem. Sci., 2020,
11, 9937-9944
Bouncing off walls – widths of exit channels from shallow minima can dominate selectivity control†
Received
23rd July 2020
, Accepted 28th August 2020
First published on 31st August 2020
Abstract
A selectivity model based on the widths of pathways to competing products, rather than barrier heights, is formulated for the butadiene + allyl cation reaction. This model was arrived at via analysis of stationary points, intrinsic reaction coordinates, potential energy surface shapes and direct dynamics trajectories, all determined using quantum chemical methods.
Introduction
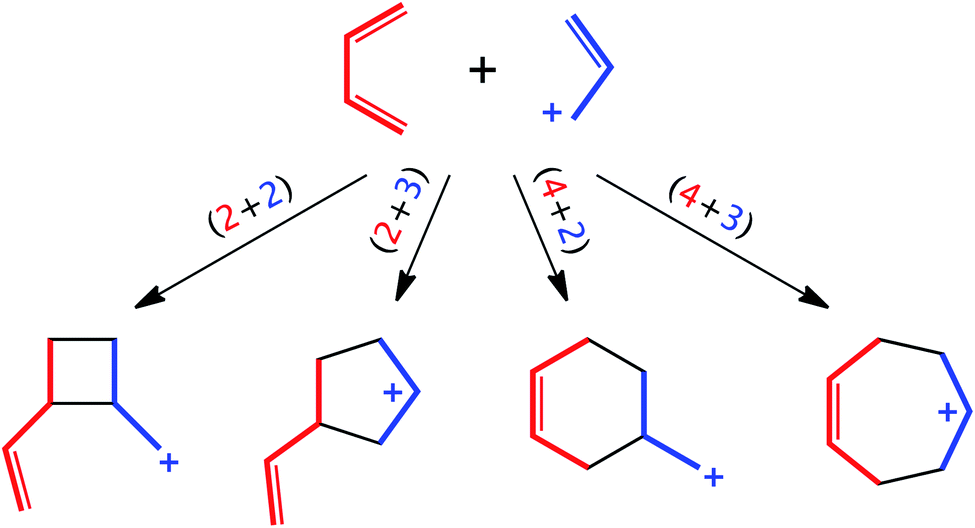
Reactions between butadienes and allyl cations have been studied extensively from both mechanistic and synthetic perspectives. Reactions of these two types of components can lead to 4-, 5-, 6- or 7-membered rings via concerted or stepwise (formal) (2 + 2)/[π2 + π2],1,2 (2 + 3)/[π2 + π2],3,4 (4 + 2)/[π4 + π2]5–9 (the so-called “ionic Diels–Alder reaction”) or (4 + 3)/[π4 + π2]3,4,10–12 cycloaddition reactions, respectively (Scheme 1). Which products are observed can be influenced by which substituents are attached to each component and the environment in which the reaction is run. Here we reinvestigate the parent reaction of butadiene + allyl cation (in the gas phase and DMSO), adding results from density functional theory (DFT) calculations to Pascual-Teresa and Houk's previously reported HF and MP2 results,13 and including an expanded view of the butadiene + allyl cation potential energy surface (PES). In addition, we have examined variational transition states (VTSs)14 and subjected this reaction to ab initio molecular dynamics (AIMD) simulations, of both downhill and uphill varieties,15 to assess whether or not non-statistical dynamic effects play a role in determining product selectivity.16–19 Our results have led to the conclusion that the widths of pathways to products can sometimes be more important than barrier heights—an entropic effect that has often been overlooked but has potential utility in constructing useful models of selectivity and in designing new selective reactions.
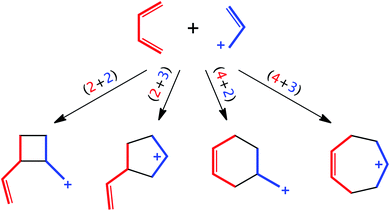 |
| | Scheme 1 Possible modes of cycloaddition for butadiene + allyl cation. | |
Computational methods
The Gaussian 09 software suite20 was used for all DFT calculations. The geometries and frequencies of minima and transition-state structures (TSSs) were optimized at the B3LYP-D3/6-311+G(d,p),21–25 M06-2X/6-311+G(d,p),26–28 M06-2X/6-311G(d), and ωB97X-D/6-311G(d)29 levels of theory. TSSs were defined as such by frequency calculations that predicted exactly one imaginary frequency,30 while minima showed no imaginary frequencies. Molecular structures were visualized using CYLview.31 Intrinsic reaction coordinate (IRC)32–34 calculations were performed to confirm that minimum energy pathways (MEPs) connected TSSs to the reactants and the products indicated. To explore the effect of solvation, calculations were also performed with M06-2X/6-311G(d) and the PCM solvation model for DMSO.35 In addition, single-point energies were computed at the CCSD(T)-F12a/AVDZ36 level using the MOLPRO 2015 package.37 As shown in Fig. 1, the relative energies of stationary points are similar among different levels. VTSs and associated reaction paths were determined using Gaussrate38/Polyrate39 with application of the Reorientation Of the Dividing Surface (RODS) algorithm at the M06-2X/6-311G(d) level of theory.14,40
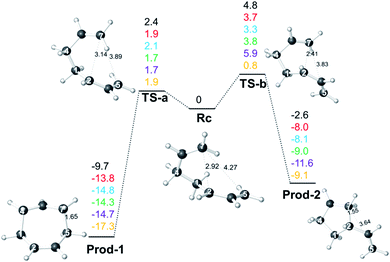 |
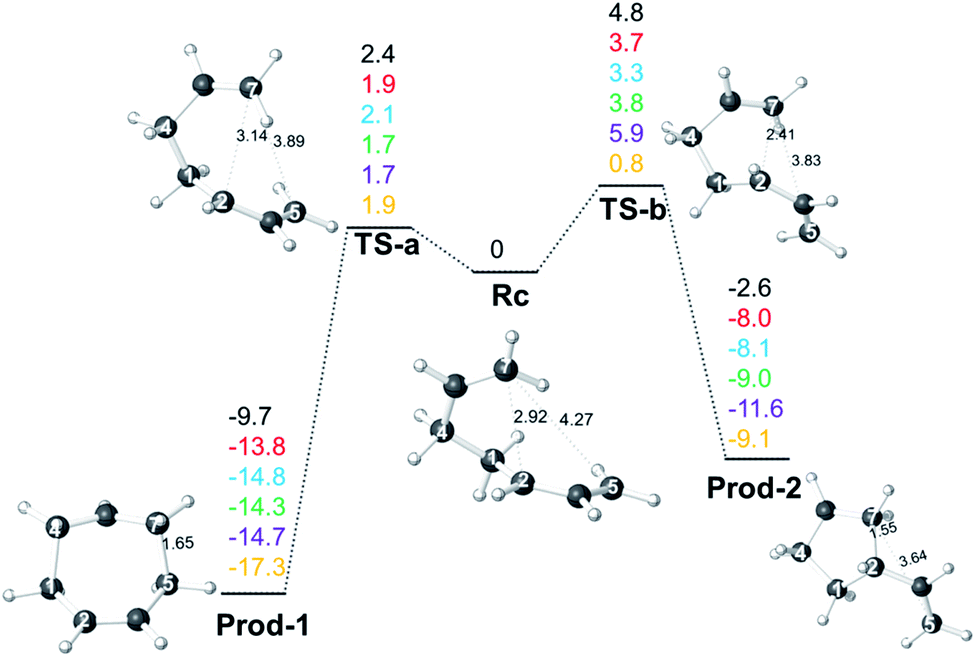
| | Fig. 1 Relative free energies (single point electronic energies for CCSD(T) results) of stationary points on the allyl cation + butadiene PES (in kcal mol−1 relative to Rc) at 298.15 K. The values shown are from top to bottom: B3LYP-D3/6-311+G (d,p) (black), M06-2X/6-311+G(d,p) (red), M06-2X/6-311G(d) (blue), ωB97X-D/6-311G(d) (green), PCM(DMSO)-M06-2X/6-311G(d) (purple), CCSD(T)-F12a/AVDZ//M06-2X/6-311G(d) (yellow). Distances shown (Å) are from structures optimized with M06-2X/6-311G(d). Structure labels are defined in the text below. | |
Both quasi-classical and classical AIMD simulations were performed using M06-2X/6-311G(d). For the former, zero-point energies are included in initial sampling while for the latter, they are not. Quasi-classical AIMD simulations also were performed using ωB97X-D/6-311G(d). All trajectories were propagated using the Progdyn41 script. To explore the effect of continuum solvent on dynamics, the PCM(DMSO)-M06-2X/6-311G(d) level of theory was employed. Uphill (initiated in the Rc region) and downhill (initiated in transition state regions) dynamics simulations were both carried out. For downhill dynamics, starting geometries were generated from a Boltzmann distribution of vibrations at 298 K and trajectories were propagated in both product and reactant directions with the Verlet algorithm, using Gaussian 09 to calculate forces at each time step (the time step was set as 1 fs).42 For uphill dynamics simulations, starting geometries were generated from a Boltzmann sampling of vibrations at 298 K then propagated towards the product direction until one or the other of the products is formed. Starting points for uphill dynamics are overlaid in Fig. 2. Note that our analysis here is for a “relaxed” Rc; i.e., momentum for its formation is not included.18,19 The following geometric criteria (atom numbers shown in Fig. 1) were employed to halt trajectories: when the C1–C4 and C5–C7 distances both dropped below 1.70 Å, a trajectory was labeled as the (4 + 3) adduct (7-membered ring product, Prod-1); when the C1–C4 and C2–C7 distances both dropped below 1.60 Å, a trajectory was labeled as the (2 + 3) adduct (5-membered ring product, Prod-2).
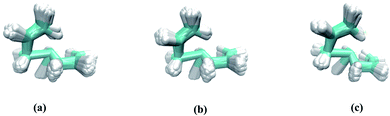 |
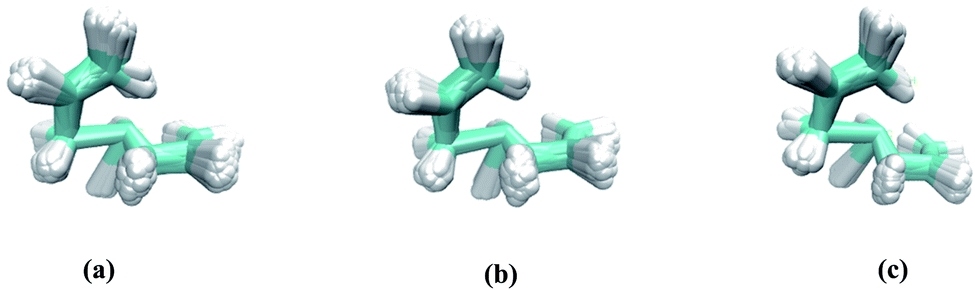
| | Fig. 2 Overlays of all starting geometries consistent with a Boltzmann distribution at 298 K in the region of Rc: (a) M06-2X/6-311G(d), (b) ωB97X-D/6-311G(d), (c) PCM(DMSO)-M06-2X/6-311G(d). | |
Results and discussion
Reaction coordinates
Energy profiles from different levels of theory for the allyl cation + butadiene reaction are shown in Fig. 1. No TSSs for concerted cycloaddition or formation of Rc, the product of addition of one end of the butadiene π-system to one end of the allyl π-system, were found; it appears that adduct formation is barrierless, at least in the absence of explicit solvent (appropriate modeling of which is beyond our current capabilities). This conclusion is consistent with previous studies on related reactions.11c,12,13 Cation Rc can then be converted to 7- or 5-membered ring-containing products via concerted processes involving TS-a and TS-b, respectively. Note that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group involved in bond formation (that which originated in the allyl fragment) rotates in opposite directions to form the 7- and 5-membered ring-containing products (i.e., for an overall pathway connecting Prod-1 to Prod-2, the migrating CH2 group interacts antarafacially with the allyl group over which it migrates). While tempting to consider this observation a result of “vestiges of orbital symmetry,”43 that cannot be done with confidence without more detailed examination of the PES preceding Rc. At all DFT levels of theory (but not CCSD(T), for which the data correspond to single-point electronic energies), TS-b is predicted to be higher in energy than TS-a and Prod-2 was predicted to be higher in energy than Prod-1. Namely, in terms of either kinetic or thermodynamic control, formation of the 7-membered ring product, Prod-1, is favored. However, Rc is a shallow minimum, which can increase the lifetime and play a significant role in the dynamics, particularly at low temperatures.
C group involved in bond formation (that which originated in the allyl fragment) rotates in opposite directions to form the 7- and 5-membered ring-containing products (i.e., for an overall pathway connecting Prod-1 to Prod-2, the migrating CH2 group interacts antarafacially with the allyl group over which it migrates). While tempting to consider this observation a result of “vestiges of orbital symmetry,”43 that cannot be done with confidence without more detailed examination of the PES preceding Rc. At all DFT levels of theory (but not CCSD(T), for which the data correspond to single-point electronic energies), TS-b is predicted to be higher in energy than TS-a and Prod-2 was predicted to be higher in energy than Prod-1. Namely, in terms of either kinetic or thermodynamic control, formation of the 7-membered ring product, Prod-1, is favored. However, Rc is a shallow minimum, which can increase the lifetime and play a significant role in the dynamics, particularly at low temperatures.
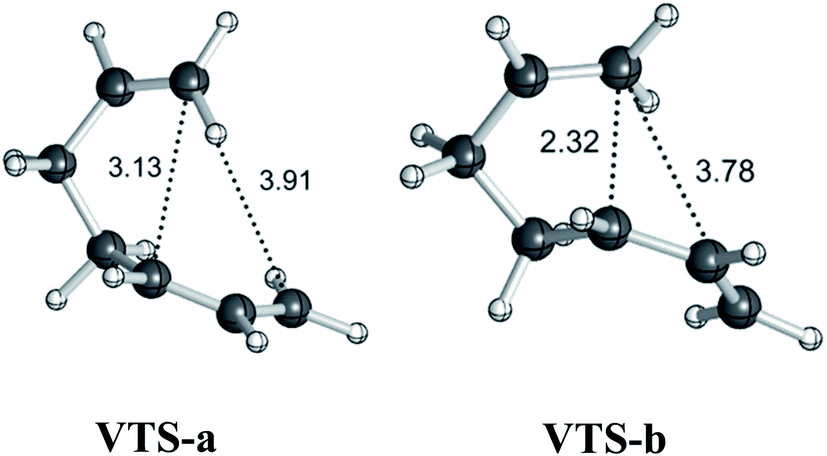
Variational transition states
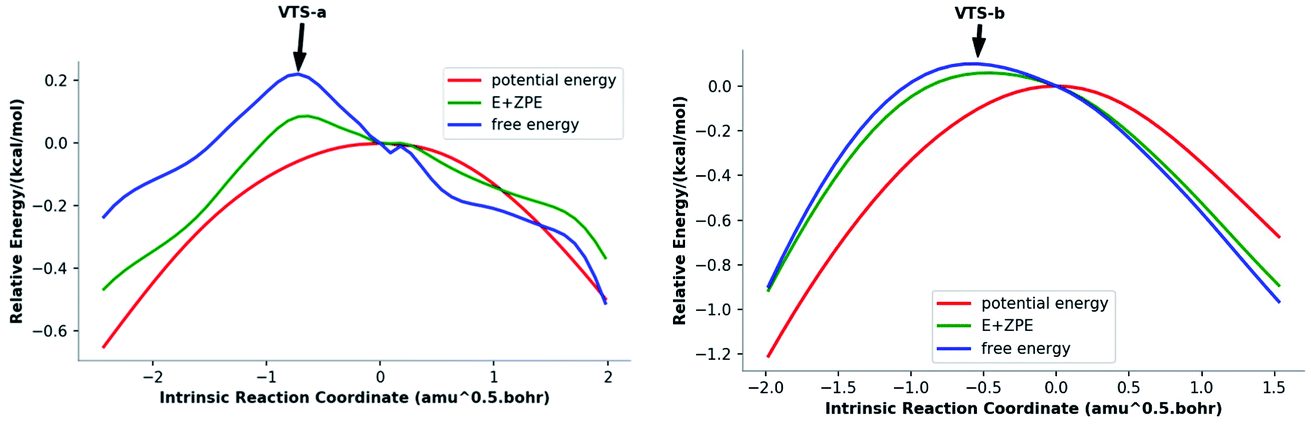
To address issues with standard computations of entropies, reaction pathways derived using variational transition state theory (VTST) calculations (using M06-2X/6-311G(d)) were computed; these are shown in Fig. 3. For both pathways, VTSs differ from the PES TSSs (compare the geometries of the VTSs in Fig. 4 with those of the TSSs in Fig. 1). The C5–C7 distance in VTS-a is 0.02 Å longer than that in TS-a, while the C2–C7 distance in VTS-a is 0.01 Å shorter than that in TS-a. The C5–C7 and C2–C7 distances in VTS-b are both shorter than those in TS-b, by 0.05 Å and 0.09 Å, respectively. Both VTSs are higher in free energy than the corresponding TSSs. As shown in Fig. 3, this effect is not simply an entropy effect, but also an effect of zero point energy. Note, however, that the difference in energy between the peaks of the free energy and potential energy curves is small—tenths of a kcal mol−1—consistent with flat surfaces, in terms of both potential energy and free energy, near the transition states. No evidence for “entropic intermediates” is found here.44,45
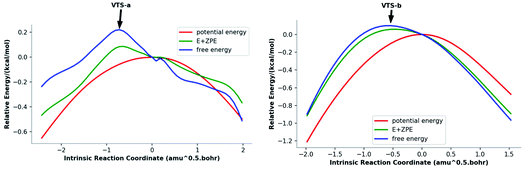 |
| | Fig. 3 Reaction coordinate plots (M06-2X/6-311G(d)) for pathways a (left) and b (right). The locations of VTS-a and VTS-b are highlighted. | |
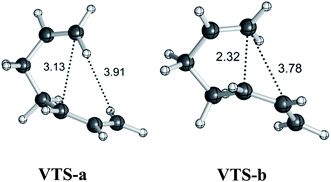 |
| | Fig. 4 Geometries (M06-2X/6-311G(d)) of variational transition states. Selected distances are shown in Å. | |
Direct dynamics trajectories
Given the flatness of the PES near Rc and the transition states for cyclization, AIMD trajectory simulations were employed to evaluate (1) whether either transition state can lead to both products and (2) the factors influencing product selectivity.
Downhill trajectories initiated from TS-a did in fact lead to both products, but just barely (Table 1), while trajectories initiated from TS-b did not. In both cases, however, a large amount of recrossing (here including trajectories connecting the reactant to the reactant, one product to the same product, or one product to the other product) was observed, consistent with a flat region of the PES.
Table 1 Results of downhill dynamics simulations initiated from TS-a and TS-b (M06-2X/6-311G(d))
|
|
Total trajectories |
Trajectories that form Prod-1 |
Trajectories that form Prod-2 |
Trajectories that recross |
|
TS-a
|
702 |
403 |
3 |
296 |
|
TS-b
|
699 |
0 |
451 |
248 |
Downhill trajectories were also initiated from VTS-a and VTS-b. Only Prod-1 was formed from VTS-a and only Prod-2 was formed from VTS-b (Table 2). While recrossing trajectories were much reduced for VTS-b, compared to TS-b, a high percentage of recrossing was still observed for VTS-a.
Table 2 Results of downhill dynamics simulations initiated from VTSs (M06-2X/6-311G(d))
|
|
Total trajectories |
Trajectories that form product |
Trajectories that recross |
|
VTS-a
|
139 |
64 |
75 |
|
VTS-b
|
103 |
93 |
10 |
Results for uphill AIMD trajectories initiated at Rc are shown in Table 3. At all levels of theory, formation of the 7-membered ring-containing product, Prod-1, is much preferred. However, the predicted Prod-1/Prod-2 ratio is lower than expected based on differences in TSS free energies (Fig. 1). For example, at ωB97-XD/6-311G(d), the ratio from dynamics simulation is 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, while that from free energy differences for TSSs is 97:3. As described above, VTS free energies differ only slightly from TSS free energies. With PCM(DMSO)-M06-2X/6-311G(d), free energy differences predict essentially no Prod-2, but our dynamics simulations predict that 16.5% of trajectories lead to this product. What is the origin of this discrepancy? Is it technical or chemical in nature?
:10, while that from free energy differences for TSSs is 97:3. As described above, VTS free energies differ only slightly from TSS free energies. With PCM(DMSO)-M06-2X/6-311G(d), free energy differences predict essentially no Prod-2, but our dynamics simulations predict that 16.5% of trajectories lead to this product. What is the origin of this discrepancy? Is it technical or chemical in nature?
Table 3 Results of uphill dynamics simulations initiated from Rc
| Method |
Total trajectories |
Trajectories that form Prod-1 |
Trajectories that form Prod-2 |
| M06-2X/6-311G(d) |
206 |
171 (83%) |
35 (17%) |
| ωB97X-D/6-311G(d) |
154 |
138 (90%) |
16 (10%) |
| PCM(DMSO)-M06-2X/6-311G(d) |
91 |
76 (84%) |
15 (16%) |
Since our trajectories often involved thousands of fs to form products (see ESI†), we were concerned about ZPE leakage (generally a worry past ∼0.5 ps).46–49 To address this issue, we also carried out classical AIMD simulations. Results (using M06-2X/6-311G(d)) for uphill AIMD trajectories initiated at Rc and for downhill AIMD trajectories initiated at VTS-a are shown in Tables 4 and 5, respectively. For uphill simulations, the predicted Prod-1/Prod-2 ratio does not change significantly between classical and quasi-classical simulations (compare Tables 3 and 4). For downhill simulations, a significantly smaller proportion of trajectories recrossed for classical vs. quasi-classical dynamics (compare Tables 2 and 5), but a large proportion of recrossing was still observed.
Table 4 Classical dynamics results of uphill dynamics simulations initiated from Rc (M06-2X/6-311G(d))
|
|
Total trajectories |
Trajectories that form Prod-1 |
Trajectories that form Prod-2 |
|
Rc
|
90 |
75 (83%) |
15 (17%) |
Table 5 Classical dynamics results of downhill dynamics simulations initiated from VTS-a (M06-2X/6-311G(d))
|
|
Total trajectories |
Trajectories that form product |
Trajectories that recross |
|
VTS-a
|
130 |
91 |
39 |
Potential energy surfaces
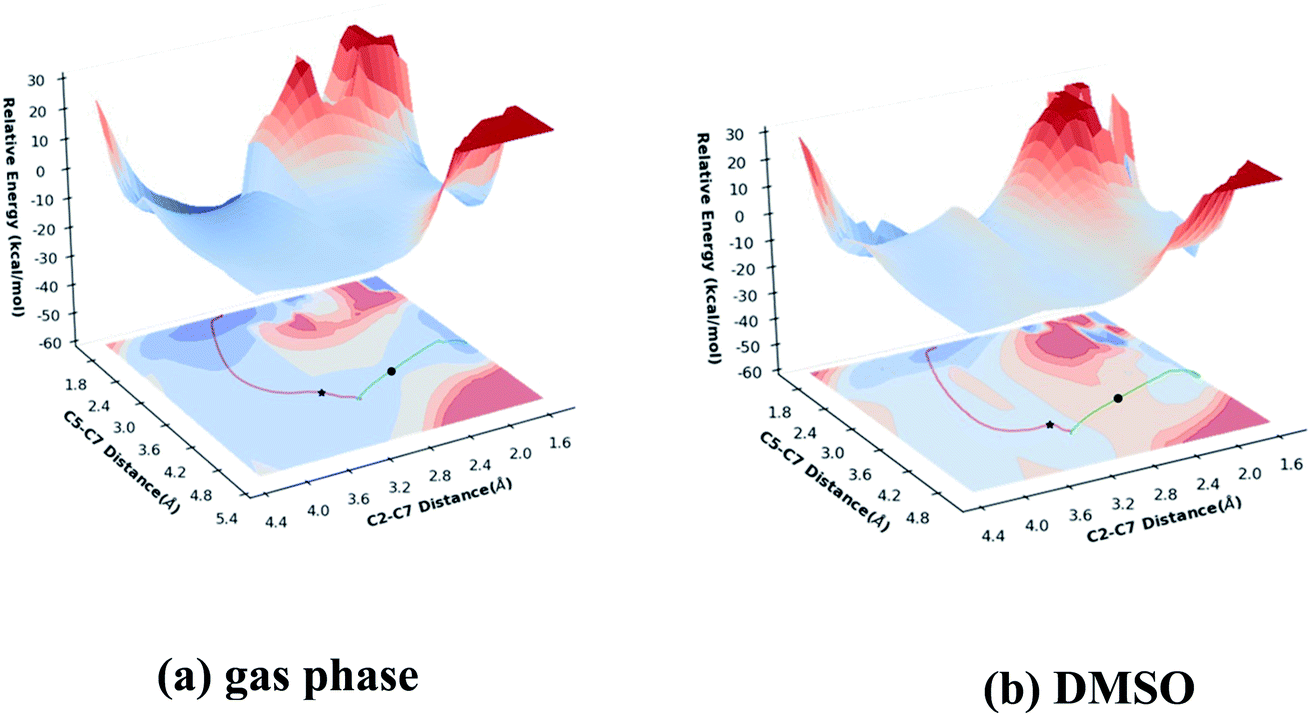
To explore how shape of the PES influences the selectivity of the reaction, PESs for gas phase (M06-2X/6-311G(d)) and DMSO (PCM-M06-2X/6-311G(d)) reactions were built (Fig. 5) using relaxed scans along C5–C7 and C2–C7 distances (see Fig. 1 for atom labels) as x- and y-axes and energy displayed as height and color (top surfaces) or color alone (bottom surfaces, projections of top surfaces into the x–y plane). While these are reduced-dimension PESs, the geometric parameters used correspond to those of the forming bonds.50,51 Small C5–C7 distances correspond to the Prod-1 region, while small C2–C7 distances correspond to the Prod-2 region. The locations of the two TSSs are marked, as are the IRC paths. One observation is immediately apparent from these surfaces: the pathway toward Prod-1 is wider than the pathway toward Prod-2.
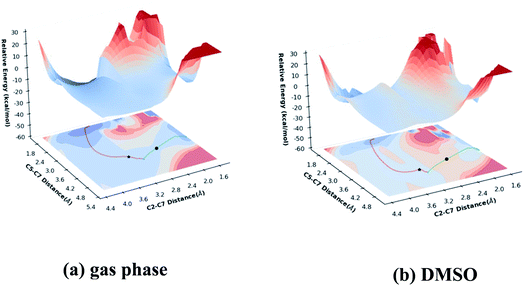 |
| | Fig. 5 Potential energy surfaces: (a) M06-2X/6-311G(d), (b) PCM(DMSO)-M06-2X/6-311G(d). IRCs for TS-a are shown as red lines and IRCs for TS-b are shown as green lines. TS-a and TS-b are indicated as stars and circles, respectively. | |
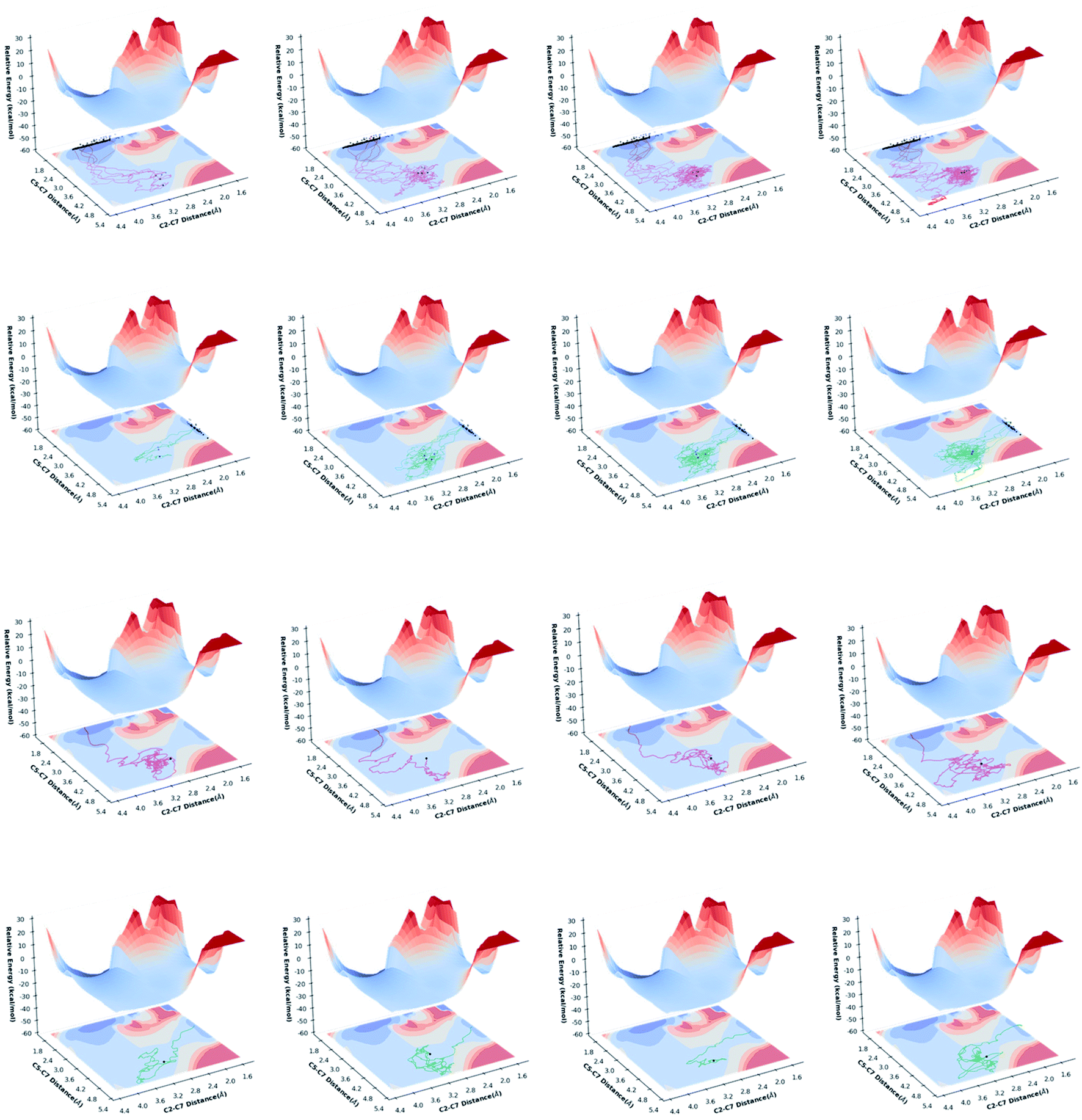
Representative gas phase trajectories are plotted in Fig. 6. As expected, long-time trajectories sample more of the Rc region on the PES before forming products. Importantly, some trajectories heading toward product “turn back” before crossing the dividing surface that separates reactant from products, i.e., they miss the exit channel and instead “bounce off the wall” flanking it. This is distinct from recrossing, where the dividing surface is passed before a trajectory turns back (at least for the dimensions we have plotted here).16–19 This is also distinct from bouncing off the side walls of a valley en route to product(s), i.e., where deviations from an IRC still lead to product(s), not back to reactant(s).16 The proportion of Prod-1 forming trajectories that arose from “turning back” (62/171 or 36%) is larger than the proportion of Prod-2 forming trajectories that arose from “turning back” (7/35 or 20%), i.e., more Prod-1 is formed from trajectories that initially miss the exit channel toward which they started, presumably because the exit channel toward Prod-1 is wider. The product ratio predicted on the basis of “turn back” trajectories alone is 90:10 (62 vs. 7 trajectories). The ratio predicted on the basis of direct trajectories (here there are 137, making for a total of 206 as shown in Table 3) is 80:20, and the overall ratio accounting for all product-forming trajectories is 83:17 (Table 3). Thus, the ratio predicted from direct uphill trajectories does not reflect the predicted free energies of transition states, be they TSSs or VTSs, and the “turning back” phenomenon increases selectivity for the product formed via the wider pathway. A similar effect was recently discussed for a different carbocation reaction, but from the perspective of downhill trajectories and post-transition state bifurcations.52
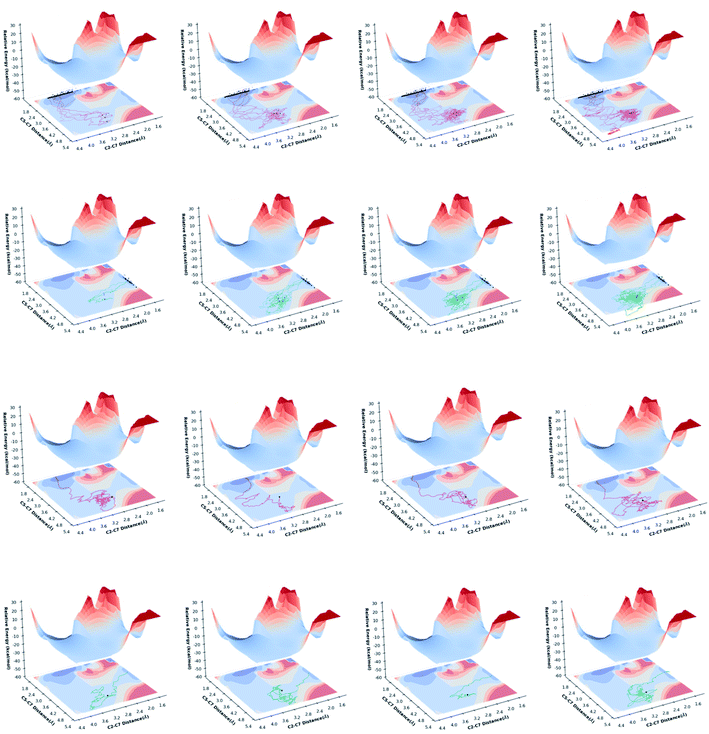 |
| | Fig. 6 (1) Projections of trajectories onto the M06-2X/6-311G(d) PES, based on trajectory length (time). Left to right: 0–500 fs, 500–1000 fs, 1000–1500 fs, 1500–2000 fs. The first row corresponds to Prod-1-forming trajectories. The second row corresponds to Prod-2-forming trajectories. (2) Projections of representative “turn back” trajectories onto the M06-2X/6-311G(d) PES. The third row shows representative Prod-1-forming “turn back” trajectories. The bottom row shows representative Prod-2-forming “turn back” trajectories. | |
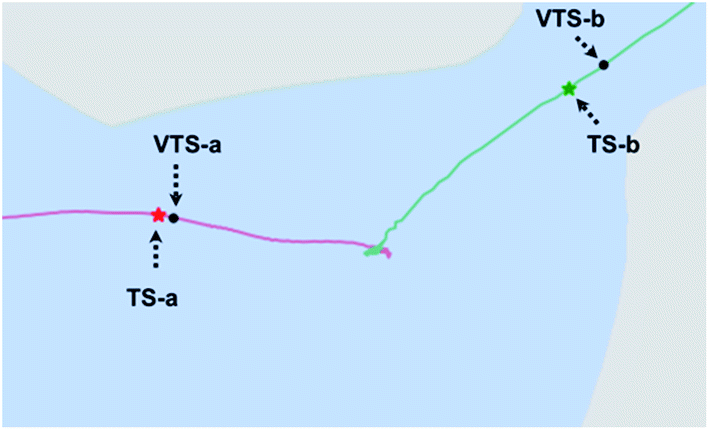
A close-up view of the reactant region of the PES is shown in Fig. 7, which also shows the locations of the VTSs. TS-a and VTS-a occupy similar positions in this plot, consistent with similar amounts of recrossing in downhill AIMD. VTS-b is noticeably later (closer to product in terms of the geometric parameters plotted here) than is TS-b, consistent with reduced recrossing. Being later, it is further into the least wide region of the path to products, consistent with it being even more difficult for uphill trajectories to reach Prod-2 than if they only had to reach TS-b. This region of decreased width indicates that varying the C5–C7 distance happens to be particularly difficult near the transition state for formation of the C2–C7 bond. This phenomenon was unexpected, but is perhaps related to the fact that the forming σ-bond is much shorter in TS-b and VTS-b than in TS-a and VTS-a and conformational restrictions due to hyperconjugation with the forming σ-bond are expected, therefore, to be more severe.
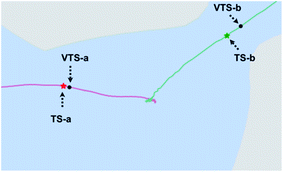 |
| | Fig. 7 Close-up view of the reactant region of the surface from Fig. 5a. | |
This pathway width effect is clearly connected to entropy, i.e., a wider path is related to more flexibility and more possible ways to cross through a dividing surface.40,45,53–55 This effect appears not to be captured fully in VTS free energies, however, at least for the system examined here, which involves a flat energy surface, although we cannot definitively rule out the possibility that the apparent discrepancy is due to the particulars of running each type of calculation. Nonetheless, interpreting results of this type in terms of pathway widths allows one to better tie specific structural features to entropy changes along reaction coordinates.
Conclusions
We have explored the mechanism of the reaction between butadiene and allyl cation using quantum chemical computations on PESs and direct AIMD simulations (quasi-classical and classical). Consistent with results from previous calculations at other levels of theory,13 concerted cycloaddition pathways are not found. Instead, stepwise pathways leading to products of formal (4 + 3) and (2 + 3) cycloaddition are present, but these involve a shallow intermediate with low barrier exit channels. As for other reactions with shallow intermediates—twixtyls, calderas, mesas, para-intermediates19,56–58—non-traditional effects appear to play a key role in product formation. We propose that the product selectivity in the system examined here—and likely others52 —is determined by dynamic effects related to the width of pathways to products. In particular, there is an entropy effect—transition states with wider energy wells orthogonal to the reaction coordinate have more possible paths through them—which is modulated, enhanced in this case, by an unequal probability of trajectories turning back before reaching transition states. We are currently expanding on this work to see if models based on pathway width can rationalize product selectivity for cases with existing experimental evidence for entropy-controlled selectivity where the origins of that entropy control are not known.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
Support from the National Science Foundation (CHE-1856416 and supercomputing resources from the XSEDE program via CHE-030089) and the National Natural Science Foundation of China (contract no. 21573027 and 21973009) is gratefully acknowledged. Mengna Bai thanks the fund from the China Scholarship Council (CSC).
References
- Y. J. Hong and D. J. Tantillo, How Cyclobutanes are Assembled in Nature - Insights from Quantum Chemistry, Chem. Soc. Rev., 2014, 43, 5042–5050 RSC.
- P. G. Gassman and A. C. Lottes, Cyclobutane Formation in the 2π+2π Cycloaddition of Allyl and Related Cations to Unactivated Olefins. Evidence for the 2nd Step in the Proposed Mechanism of the Ionic Diels-Alder Reaction, Tetrahedron Lett., 1992, 33, 157–160 CrossRef CAS.
- H. M. R. Hoffmann, The Cycloaddition of Allyl Cations to 1,3-Dienes - General-Method for the Synthesis of 7-Membered Carbocycles, Angew. Chem. Int. Ed., 1984, 23, 1–19 CrossRef.
- H. M. Hoffmann, Syntheses of 7-Membered and 5-Membered Rings from Allyl Cations, Angew. Chem. Int. Ed., 1973, 12, 819–835 CrossRef.
- R. K. Haynes, G. R. King and S. C. Vonwiller, Preparation of a Bicyclic Analog of Qinghao (Artemisinic) Acid via a Lewis-Acid Catalyzed Ionic Diels-Alder Reaction Involving a Hydroxy Diene and Cyclic Enone and Facile Conversion into (+/-)-6,9-Desdimethylqinghaosu, J. Org. Chem., 1994, 59, 4743–4748 CrossRef CAS.
- P. G. Gassman and D. B. Gorman, Stepwise Mechanism for the Formation of 2π+ 4π Cycloadducts in the Ionic Diels-Alder Reaction, J. Am. Chem. Soc., 1990, 112, 8624–8626 CrossRef CAS.
- P. G. Gassman and D. A. Singleton, Acid-Catalyzed Intramolecular Diels-Alder Reactions - the Cycloaddition of Allyl Cations to 1,3-Dienes, J. Am. Chem. Soc., 1984, 106, 6085–6086 CrossRef CAS.
- P. G. Gassman, D. A. Singleton, J. J. Wilwerding and S. P. Chavan, Acrolein Acetals as Allyl Cation Precursors in the Ionic Diels-Alder Reaction, J. Am. Chem. Soc., 1987, 109, 2182–2184 CrossRef CAS.
- Y. J. Ko, S. B. Shim and J. H. Shin, A Mechanistic Study on the Intramolecular Ionic Diels-Alder Reaction of 2-Methyl-3,9,11-Tridecatriene-2-ol and 2,11-Dimethyl-1,3,9,11-Dodecatetraene, Tetrahedron Lett., 2007, 48, 863–867 CrossRef CAS.
-
R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Elsevier, 2013 Search PubMed.
-
(a) M. Harmata, The (4+3)-Cycloaddition Reaction: Simple Allylic Cations as Dienophiles, Chem. Commun, 2010, 46, 8886–8903 RSC;
(b) M. Harmata, Intramolecular Cycloaddition Reactions of Allylic Cations, Tetrahedron, 1997, 53, 6235–6280 CrossRef CAS;
(c) M. Harmata and P. R. Schreiner, Intramolecular 4 + 3 Cycloadditions. A Theoretical Analysis of Simple Diastereoselectivity in Reactions of Alkoxyallylic Cations and Furans, Org. Lett., 2001, 3, 3663–3665 CrossRef CAS.
- C. J. Cramer and S. E. Barrows, Quantum chemical characterization of cycloaddition reactions between 1,3-butadiene and oxyallyl cations of varying electrophilicity, J. Phys. Org. Chem., 2000, 13, 176–186 CrossRef CAS.
- B. de Pascual Teresa and K. N. Houk, The Ionic Diels-Alder Reaction of the Allyl Cation and Butadiene: Theoretical Investigation of the Mechanism, Tetrahedron Lett., 1996, 37, 1759–1762 CrossRef CAS.
- J. L. Bao and D. G. Truhlar, Variational Transition State Theory: Theoretical Framework and Recent Developments., Chem. Soc. Rev., 2017, 46, 7548–7596 RSC.
- R. Pradhan and U. Lourderaj, Can Reactions Follow Non-Traditional Second-Order Saddle Pathways Avoiding Transition States?, Phys. Chem. Chem. Phys., 2019, 21, 12837–12842 RSC.
- X. Y. Ma and W. L. Hase, Perspective: Chemical Dynamics Simulations of Non-Statistical Reaction Dynamics, Philos. Trans. R. Soc., A, 2017, 375, 20160204 CrossRef.
- U. Lourderaj, K. Park and W. L. Hase, Classical Trajectory Simulations of Post-Transition State Dynamics, Int. Rev. Phys. Chem., 2008, 27, 361–403 Search PubMed.
- B. K. Carpenter, Intramolecular Dynamics for the Organic Chemist, Acc. Chem. Res., 1992, 25, 520–528 CrossRef CAS.
-
B. K. Carpenter, Nonstatistical Dynamics in Thermal Reactions of Polyatomic Molecules, in Ann. Rev. Phys. Chem.; Annual Review of Physical Chemistry, Annual Reviews, Palo Alto, 2005, vol. 56, pp. 57–89 Search PubMed.
-
M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, A New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, Density-Functional Thermochemistry .3. The Role of exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- J. Tirado-Rives and W. L. Jorgensen, Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements?, J. Chem. Theory Comput., 2016, 12, 4303–4325 CrossRef CAS.
- A. Sengupta and K. Raghavachari, Solving the Density Functional Conundrum: Elimination of Systematic Errors to Derive Accurate Reaction Enthalpies of Complex Organic Reactions, Org. Lett., 2017, 19, 2576–2579 CrossRef CAS.
- D. J. Tantillo, Biosynthesis via Carbocations: Theoretical Studies on Terpene Formation, Nat. Prod. Rep., 2011, 28, 1035–1053 RSC.
-
C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009 Search PubMed.
- C. Gonzalez and H. B. Schlegel, Reaction-Path Following in Mass-Weighted Internal Coordinates, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- K. Fukui, The Path of Chemical-Reactions - the IRC Approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- S. Maeda, Y. Harabuchi, Y. Ono, T. Taketsugu and K. Morokuma, Intrinsic Reaction Coordinate: Calculation, Bifurcation, and Automated Search, Int. J. Quant. Chem., 2015, 115, 258–269 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- L. G. Kong, F. A. Bischoff and E. F. Valeev, Explicitly Correlated R12/F12 Methods for Electronic Structure, Chem. Rev., 2012, 112, 75–107 CrossRef CAS.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schutz, Molpro: a General-Purpose Quantum Chemistry Program Package. WIREs, Comput. Mol. Biosci., 2012, 2, 242–253 CrossRef CAS.
-
J. Zheng, S. Zhang, J. C. Corchado, Y.-Y. Chuang, E. L. Coitino, B. A. Ellingson and D. G. Truhlar, GAUSSRATE, version 2009-A, University of Minnesota, Minneapolis, MN, 2010 Search PubMed.
-
J. Zheng, et al, POLYRATE-version 2010, University of Minnesota, Minneapolis, MN, 2010 Search PubMed.
- A. Gonzalez-Lafont, J. Villa, J. M. Lluch, J. Bertran, R. Steckler and D. G. Truhlar, Variational Transition State Theory and Tunneling Calculations with Reorientation of the Generalized Transition States for Methyl Cation Transfer, J. Phys. Chem. A, 1998, 102, 3420–3428 CrossRef CAS.
- B. R. Ussing, C. Hang and D. A. Singleton, Dynamic Effects on the Periselectivity, Rate, Isotope Effects, and Mechanism of Cycloadditions of Ketenes with Cyclopentadiene, J. Am. Chem.Soc., 2006, 128, 7594–7607 CrossRef CAS.
-
D. Marx and J. Hutter, Ab initio molecular dynamics: basic theory and advanced methods, Cambridge University Press, 2009 Search PubMed.
- A. G. Leach, S. Catak and K. N. Houk, Mechanism of the Forbidden 3s,5s-Sigmatropic Shift: Orbital Symmetry Influences Stepwise Mechanisms Involving Diradical Intermediates, Chem.–Eur. J., 2002, 8, 1290–1299 CrossRef CAS.
- Z. Y. Yang, C. S. Jamieson, X. S. Xue, M. Garcia-Borras, T. Benton, X. F. Dong, F. Liu and K. N. Houk, Mechanisms and Dynamics of Reactions Involving Entropic Intermediates, Trends Chem., 2019, 1, 22–34 CrossRef.
- O. M. Gonzalez-James, E. E. Kwan and D. A. Singleton, Entropic Intermediates and Hidden Rate-Limiting Steps in Seemingly Concerted Cycloadditions. Observation, Prediction, and Origin of an Isotope Effect on Recrossing, J. Am. Chem. Soc., 2012, 134, 1914–1917 CrossRef CAS.
- L. Bonnet and J. C. Rayez, Quasiclassical Trajectory Method for Molecular Scattering Processes: Necessity of a Weighted Binning Approach, Chem. Phys. Lett., 1997, 277, 183–190 CrossRef CAS.
- C. Doubleday, M. Boguslav, C. Howell, S. D. Korotkin and D. Shaked, Trajectory Calculations for Bergman Cyclization Predict H/D Kinetic Isotope Effects Due to Nonstatistical Dynamics in the Product, J. Am. Chem. Soc., 2016, 138, 7476–7479 CrossRef CAS.
- M. Bennun and R. D. Levine, Conservation of Zero-point Energy in Classical Trajectory Computations by a Simple Semiclassical Correspondence, J. Chem. Phys., 1994, 101, 8768–8783 CrossRef.
- M. C. Reis, C. S. Lopez, O. N. Faza and D. J. Tantillo, Pushing the Limits of Concertedness. A Waltz of Wandering Carbocations, Chem. Sci., 2019, 10, 2159–2170 RSC.
- H.-H. Chuang, D. J. Tantillo and C.-P. Hsu, Construction of Two-Dimensional Potential Energy Surfaces of Reactions with Post-Transition-State Bifurcations, J. Chem. Theory Comput., 2020, 16, 4050–4060 CrossRef CAS.
-
B. K. Carpenter, Understanding Chemical Reaction Mechanisms, in Of Minds and Molecules, ed. N. Bhushan and S. Rosenfeld, Oxford University Press New York, 2000, pp. 223–224 Search PubMed.
- S. R. Hare, R. P. Pemberton and D. J. Tantillo, Navigating Past a Fork in the Road: Carbocation-π Interactions Can Manipulate Dynamic Behavior of Reactions Facing Post-Transition-State Bifurcations, J. Am. Chem. Soc., 2017, 139, 7485–7493 CrossRef CAS.
-
(a)
E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006, vol. 369, pp. 412–413 Search PubMed;
(b) C. Stegelmann, A. Andreasen and C. T. Campbell, Degree of Rate Control: How Much the Energies of Intermediates and Transition States Control Rates, J. Am. Chem. Soc., 2009, 131, 8077–8082 CrossRef CAS.
- Z. H. Wang, J. S. Hirschi and D. A. Singleton, Recrossing and Dynamic Matching Effects on Selectivity in a Diels-Alder Reaction, Angew. Chem. Int. Ed., 2009, 48, 9156–9159 CrossRef CAS.
- J. N. Harvey, Understanding the Kinetics of Spin-forbidden Chemical Reactions, Phys. Chem. Chem. Phys., 2007, 9, 331–343 RSC.
- R. Hoffmann, S. Swaminathan, B. G. Odell and R. Gleiter, Potential Surface for a Nonconcerted Reaction-Tetramethylene, J. Am. Chem. Soc., 1970, 92, 7091–7097 CrossRef CAS.
- W. V. Doering, J. L. Ekmanis, K. D. Belfield, F. G. Klarner and B. Krawczyk, Thermal Reactions of Anti- and Syn-Dispiro 5.0.5.2 Tetradeca-1,8-Dienes: Stereomutation and Fragmentation to 3-Methylenecyclohexenes. Entropy-Dictated Product Ratios from Diradical Intermediates?, J. Am. Chem. Soc., 2001, 123, 5532–5541 CrossRef.
- B. H. Northrop, D. P. O'Malley, A. L. Zografos, P. S. Baran and K. N. Houk, Mechanism of the Vinylcyclobutane Rearrangement of Sceptrin to Ageliferin and Nagelamide E., Angew. Chem. Int. Ed., 2006, 45, 4126–4130 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details on computations, including coordinates and energies for computed structures and IRC plots (PDF). See DOI: 10.1039/d0sc04036c |
| ‡ M. B. and Z. F. contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Jun
Li
b,
Jun
Li