
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of pentasubstituted 1,3-dienes via Ni-catalyzed reductive coupling of unsymmetrical internal alkynes†
Zhijun
Zhou
,
Jiachang
Chen
,
Herong
Chen
and
Wangqing
Kong
 *
*
Institute for Advanced Studies (IAS), Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: wqkong@whu.edu.cn
First published on 10th September 2020
Abstract
The reductive coupling of alkynes represents a powerful strategy for the rapid synthesis of highly substituted 1,3-dienes. This method has the advantages of high atom and step economy, and readily available substrates. Unfortunately, the intermolecular coupling of unsymmetrical internal alkynes remains extremely challenging due to the difficulty in controlling self-dimerization and cross-coupling, as well as stereo- and regioselectivity. Previous reports are still limited to intramolecular processes or the use of stoichiometric amounts of metal catalyst. Herein, we report that nickel-catalyzed reductive coupling of two unsymmetrical internal alkynes can overcome the above-mentioned limitations by using a hemilabile directing group strategy to control the regioselectivity. A series of synthetically challenging penta-substituted 1,3-dienes are obtained in good yields with high regio- and enantioselectivity (mostly > 20/1 rr, >90% ee).
Introduction
The 1,3-dienes have been recognized to be among the most useful building blocks for the construction of diverse carbo- and heterocycles by Diels–Alder or pericyclic reactions.1 They are also important industrial chemicals in the production of rubbers and adiponitrile, an intermediate in nylon production.2 In addition, 1,3-dienes are ubiquitous structural units in many biologically active natural products such as callystatin A and arenicolide C (Fig. 1).3 | ||
| Fig. 1 Conjugated 1.3-dienes. | ||
Not surprisingly, many synthetic approaches have been developed for the synthesis of 1,3-dienes.4 Less-substituted 1,3-dienes can be easily prepared via olefination of unsaturated carbonyl compounds5 or ene-yne metathesis.6 However, when considering the stereo- and regioselective alignment of those substituents on the 1,3-diene skeleton, the synthesis of highly substituted 1,3-dienes is very challenging, and the synthetic difficulty increases with a growing number of substituents (Scheme 1a).
 | ||
| Scheme 1 Challenges in reductive coupling of internal alkynes for the stereoselective synthesis of pentasubstituted 1,3-dienes. | ||
Traditional cross-coupling reaction of alkenyl halides with alkenyl metals or alkynes is effective for the synthesis of highly substituted 1,3-dienes.7 However, this approach usually requires many steps to pre-prepare two stereodefined coupling partners. So far, the rapid synthesis of highly substituted 1,3-diene is still rare.8 The direct reductive coupling of two alkynes constitutes an efficient and atom-efficient methodology for the rapidly synthesis of highly substituted 1,3-dienes. Unfortunately, the intermolecular coupling of unsymmetrical internal alkynes is extremely challenging due to the difficulty in controlling homo-dimerization and cross-coupling, as well as the stereo- and regioselectivity in the carbon–carbon forming process (Scheme 1b). The vast majority of examples describe the coupling of alkynes is the self-dimerization of diarylacetylenes, or intramolecular diyne cyclization,9 where regioselectivity is determined by the enforced proximity of these two triple bonds.
The reductive coupling of alkynes with active carbonyl-based systems has made great progress,10 while there are only few reports on regioselective coupling of relatively low reactivate internal alkynes. In 1989, Buchwald pioneered a Zr-mediated coupling reaction of internal alkynes with TMS-substituted alkynes.11 Subsequently, Sato and co-workers reported a Ti-mediated coupling of terminal alkynes with TMS-substituted alkynes or conjugated 2-alkynoates (Scheme 1c).12 Micalizio's group further developed the regioselective coupling of two internal alkynes through Ti-mediated alkoxide-directed strategy,13 which has been used to the synthesis of many bioactive natural products containing 1,3-diene moiety (Scheme 1d).14 The main limitation of this method is that it requires cumbersome operations, using stoichiometric amounts of catalyst and excess organometallic reagents, such as Grignard reagents or organolithium reagents, which preclude the incorporation of sensitive functional groups. To the best of our knowledge, the catalytic, regio- and stereoselective reductive coupling of two unsymmetrical internal alkynes has never been reported.15
Herein, we report Ni-catalyzed reductive self- and cross-coupling of two unsymmetrical internal alkynes using hemilabile directing group strategy to control the regioselectivity, providing the direct access to pentasubstituted 1,3-dienes in good yields with high enantioselectivity. The present protocol features high efficiency and atom-economy, simple operation, and is tolerant of a variety of functional groups (Scheme 1e).
Results and discussion
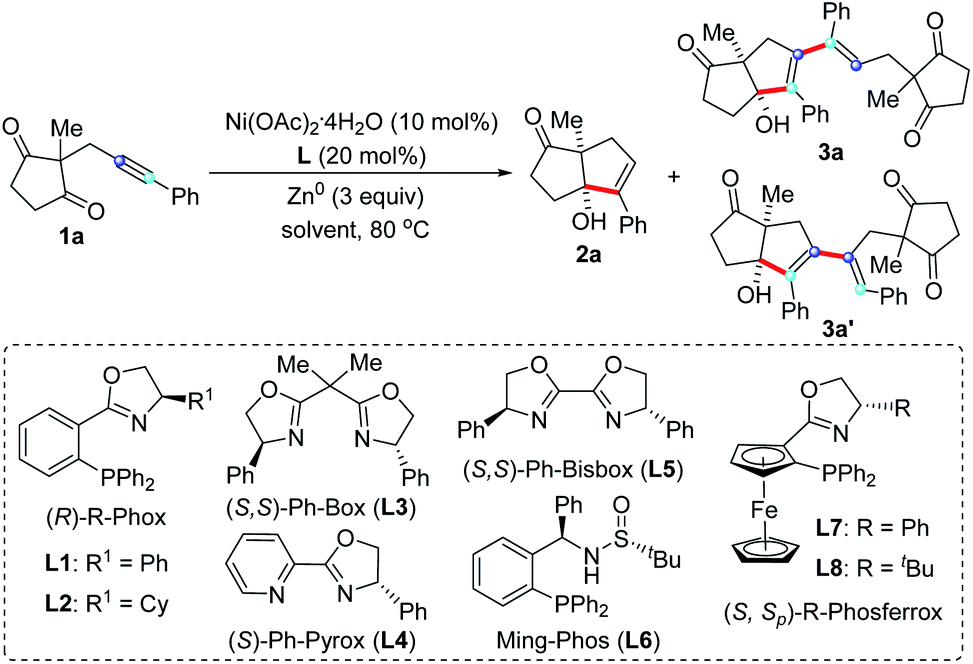
Based on the hypothesis that we proposed in Scheme 1e, we started to explore the nickel-catalyzed reductive coupling reaction of unsymmetrical internal alkynes using alkynone 1a as model substrate (Table 1). After carefully evaluating many reaction parameters, we are delight to find that a combination of Ni(OAc)2·4H2O/Phox ligand (L1) as catalyst that was found to be effective in alkynone cyclization,16 Zn0 as reducing agent and MeOH as solvent, provided pentasubstituted 1,3-diene 3a in 44% yield with 91% ee, along with the cyclization side product 2a in 12% yield (entry 1). Various solvents were tested (entries 2–6), revealing that TFE is the most suitable, providing 3a in 56% yield with 95% ee (entry 6). Reducing the reaction temperature to 40 °C resulted in a significant deterioration of the regioselectivity (entry 7, 3a/3a′ = 2/1). We also found that the reaction concentration had a pronounced influence on the reaction outcome. A low concentration slowed down the reaction; while high concentration resulted in poor regioselectivity (entry 8, 3a/3a′ = 2/1). To further improve the yield and enantioselectivity of 3a, different types of chiral ligands L3–L9 were investigated (entries 9–15). L9 gave the highest enantioselectivity (98% ee), but the chemoselectivity was poor (entry 15). The best result was achieved using TFE/HFIP (4/1) as solvent, providing 3a in 85% yield and 95% ee with high chemo- and regioselectivity (3a/3a′ > 20/1) (entry 16). Unsurprisingly, the reaction did not proceed in the absence of Ni(OAc)2·4H2O or Zn0 (entries 17 and 18). Finally, the absolute configuration of pentasubstituted 1,3-diene 3a was determined by X-ray crystallography (Fig. 2).17|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Yield of 2ab (%) | Yield of 3ab (%) | ee of 3ac (%) | rrd |
| a Reactions were carried out with 1a (0.2 mmol), Ni(OAc)2·4H2O (10 mol%), ligand (20 mol%), reducing agent (0.6 mmol) in 2 mL solvent (0.1 M) at 80 °C, unless noted otherwise. b Isolated yields. c Determined by HPLC analysis with a chiral column. d rr is regioisomeric ratio of 3a/3a′. e 40 °C. f Reaction concentration: 0.2 M. g TFE/HFIP = 4/1. h Without Ni(OAc)2·4H2O. i Without Zn0. TFE = trifluoroethanol. HFIP = hexafluoroisopropanol. | ||||||
| 1 | L1 | MeOH | 12 | 44 | 91 | 20/1 |
| 2 | L1 | MeCN | 18 | — | — | — |
| 3 | L1 | THF | 40 | — | — | — |
| 4 | L1 | EtOH | 31 | 29 | 85 | 20/1 |
| 5 | L1 | iPrOH | 31 | 26 | 78 | 20/1 |
| 6 | L1 | TFE | <2 | 56 | 95 | 20/1 |
| 7e | L1 | TFE | <2 | 42 | 94 | 2/1 |
| 8f | L1 | TFE | <2 | 35 | 95 | 2/1 |
| 9 | L2 | TFE | <2 | 57 | 86 | 20/1 |
| 10 | L3 | TFE | No reaction | — | — | — |
| 11 | L4 | TFE | No reaction | — | — | — |
| 12 | L5 | TFE | No reaction | — | — | — |
| 13 | L6 | TFE | 4 | 59 | 33 | 20/1 |
| 14 | L7 | TFE | 18 | 62 | 88 | 20/1 |
| 15 | L8 | TFE | 39 | 14 | 98 | 10/1 |
| 16 | L1 | TFE/HFIPg | <2 | 87 | 95 | 20/1 |
| 17h | L1 | TFE/HFIPg | No reaction | — | — | — |
| 18i | L1 | TFE/HFIPg | No reaction | — | — | — |
 | ||
| Fig. 2 ORTEP representation of the product 3a. | ||
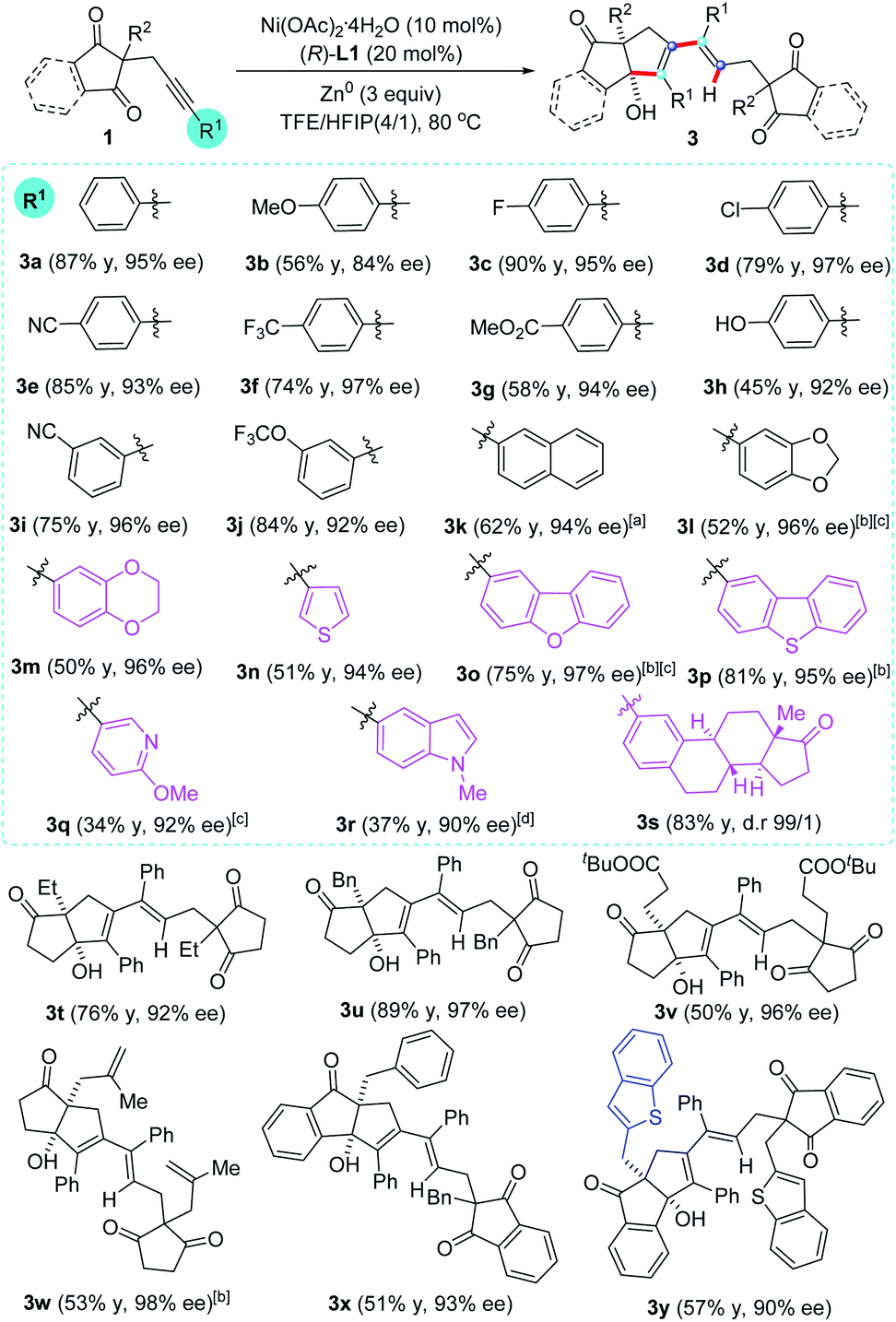
With the optimized reaction conditions on hand (Table 1, entry 16), we sought to investigate the generality of regio- and enantioselective reductive self-coupling of alkynones. As shown in Scheme 2, a wide range of pentasubstituted 1,3-dienes were synthesized in good yields and excellent enantioselectivity. Whether the terminal of the alkyne is an electron-rich or electron-deficient aryl group (3a–3l), high yields and ee values were observed. Useful functional groups such as methoxyl (3b), fluoro (3c), chloro (3d), cyano (3e and 3i), trifluoromethyl (3f), ester (3g), free hydroxyl (3h) and trifluoromethoxyl (3j) are well compatible. A range of heterocycles such as benzodioxan (3l and 3m), thiophene (3n), dibenzofuran (3o), dibenzothiophene (3p), pyridine (3q), and indole (3r) could be successfully embedded into the target products in good yields with high enantioselectivity (90–97% ee). The reaction of estrone-substituted alkynone 1s under standard conditions produced the desired product 3s in 83% yield with complete diastereospecificity (>99/1 d.r.), which demonstrated the practicability of our method. However, complex mixture was observed with alkyl-substituted alkynone.
 | ||
| Scheme 2 Substrate scope for the reductive self-coupling reaction of alkynones. | ||
The influence of the substituents (R2) at the 2-position of the cyclopentane-1,3-diones on the reaction outcome was also studied. Ethyl, benzyl, including those functionalized with 3-(tert-butoxy)-3-oxopropyl, were all well tolerated leading to the corresponding 1,3-dienes 3t–3v in good yields with excellent enantioselectivity. Notably, the allyl substituted 1,3-dione proceeded efficiently to provide 3w in 98% ee, providing an opportunity to further functionalize these obtained products. It is remarkable that the indan-1,3-dione-containing substrates, which usually resulted in very low enantioselectivity in previous report,16 were also found to be compatible with this transformation, affording the desired 1,3-dienes 3x and 3y in 93% ee and 90%, respectively.
Based on the successful realization of the self-coupling of internal alkynes, we further explored the regio- and enantioselective cross-coupling of two unsymmetrical internal alkynes. Alkynone 1b and propargyl amide 4a were chosen as model substrates to test our reaction design (Table 2). Under the previously established self-coupling reaction conditions (Table 1, entry 6 or entry 16), only a trace amount of cross-coupling product 5ba was detected (Table 2, entry 1), which reinforces the concept that the catalytic cross-coupling of two unsymmetrical internal alkynes is far from easy.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Additive | Yield of 5bab (%) | ee of 5bac (%) | rrd |
| a Reactions were carried out with 1b (0.2 mmol), 4a (0.1 mmol), Ni(OAc)2·4H2O (10 mol%), ligand (20 mol%), Zn0 (0.3 mmol) and additive (0.1 mmol) in 1 mL TFE (0.1 M) at 80 °C, unless noted otherwise. b Yields are of isolated products. c Determined by HPLC analysis on a chiral stationary phase. d 5ab/5ab′. e Using TFE/HFIP (4/1) or TFE as solvent. f 4b was used instead of 4a. g 4c was used instead of 4a. | |||||
| 1e | (S)-L1 | — | <5 | — | — |
| 2 | (S)-L1 | A1 | 64 | 92 | 20/1 |
| 3 | (S)-L1 | A2 | 29 | 92 | 20/1 |
| 4 | (S)-L1 | A3 | 31 | 92 | 20/1 |
| 5 | (S)-L1 | A4 | 58 | 92 | 20/1 |
| 6 | (S)-L1 | NEt3 | 0 | — | — |
| 7 | (S)-L1 | PPh3 | 0 | — | — |
| 8 | (S)-L1 | P(OEt)3 | 0 | — | — |
| 9 | (S)-L2 | A1 | 62 | 88 | 20/1 |
| 10 | (S)-BINAP | A1 | 0 | — | — |
| 11 | L6 | A1 | 24 | 35 | 20/1 |
| 12f | (S)-L1 | A1 | No reaction | — | — |
| 13g | (S)-L1 | A1 | Complex mixture of products | — | — |

Montgomery et al. found that the introduction of electron-deficient olefins to NHC–Ni(0) complexes can significantly enhance their stability and catalytic performance.18 Inspired by this discovery, various π-acidic additives such as acrylates A1–A3 and fumarate A4, were evaluated (entries 2–5). To our delight, the addition of methyl acrylate (A1) indeed led to a dramatic improvement of the reaction efficiency, delivering cross-coupling product 5ba in 64% yield with 92% ee and a more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of regioisomers (entry 2). The structure of π-acidic additives has a great influence on the reaction outcome (entries 3–5), indicating that they may be used as additional ligands coordinated with the nickel catalyst, thereby enhancing the catalytic activity. Lewis base additives (Et3N, PPh3 or P(OEt)3) were also tested, but they completely suppressed the transformation (entries 6–8). (S)-Cy-PHOX was also effective, giving similar result to L1 (entry 9). Other chiral ligands such as (S)-BINAP and (R,S)-Mingphos (L6) were less effective and afforded lower yields of 5ba (entries 10 and 11).
:1 ratio of regioisomers (entry 2). The structure of π-acidic additives has a great influence on the reaction outcome (entries 3–5), indicating that they may be used as additional ligands coordinated with the nickel catalyst, thereby enhancing the catalytic activity. Lewis base additives (Et3N, PPh3 or P(OEt)3) were also tested, but they completely suppressed the transformation (entries 6–8). (S)-Cy-PHOX was also effective, giving similar result to L1 (entry 9). Other chiral ligands such as (S)-BINAP and (R,S)-Mingphos (L6) were less effective and afforded lower yields of 5ba (entries 10 and 11).
Interestingly, in a comparative experiment, the reductive coupling reaction of alkynone 1b with an unsymmetrical internal alkyne lacking a carboxyl group (4b or 4c) resulted in a complex mixture of products (entries 12 and 13). These experiments clearly show that the carboxyl group plays a crucial role in determining the regioselectivity in C–C bond formation process and achieving a productive mode of reactivity.
After identifying suitable reaction conditions for regio- and enantioselective cross-coupling of unsymmetrical alkynes, we decided to investigate the substrate compatibility of the reaction with propargyl amides, esters, ketones, and alcohols (Scheme 3). Both the propargyl amide 4a, imides 4d–4e and carbamates 4f–4h were found to be compatible with the cross-coupling reaction with alkynone 1b, providing the corresponding 1,3-dienes 5ba–5bh in 94–97% ee and more than 20/1 ratio of regioisomers. As the N-Boc is easily removed, it constitutes a way to generate 1,3-diene bearing a free amine group. Propargyl sulfonamide 4i also gave the desired product 5bi in 92% yield with 97% ee, but the regioselectivity is slightly reduced (4/1). The homopropargyl ester 4j–4l were all viable substrates for this transformation and gave the desired products 5bj–5bl in 92–95% ee with more than 20/1 regioselectivity. Moreover, we also found that two structurally distinguishable alkynones could also undergo reductive cross-coupling, affording the pentasubstituted 1,3-dienes 5bm–5bo in good yields with high regio- and enantioselectivity. To further extend the synthetic applications of the reductive cross-coupling, we found that internal alkyne 4p bearing a free hydroxyl group was also tolerated, providing the desired 1,3-diene 5bp in 80% yield with 94% ee and a more than 20:1 ratio of regioisomers. These results indicate that either the carbonyl group or the free hydroxyl group can be used as a hemilabile directing group to control regioselectivity.19
 | ||
| Scheme 3 Substrate scope for the cross-coupling of unsymmetrical internal alkynes. | ||
Under slightly modified reaction conditions, the reactions of alkynones 1a with symmetrical diarylacetylenes bearing fluorine (4r and 4t) and chlorine (4s), or dithiopheneacetylene (4u) proceeded smoothly to afford the conjugated dienes 5aq–5au in synthetically useful yields with 82–90% ee. Remarkably, unsymmetrical diarylacetylenes 4v–4w were also effective coupling partners, providing the stereodefined 1,3-dienes 5av–5aw with highly regioselectivity (10/1), where the carbon–carbon bond was formed on the alkyne carbon with an electron-deficient aryl group. These results show that the electronic properties of the substituents also have a great influence on the regioselectivity of the reaction. However, no reaction was observed using a dialkylacetylene.
To access the synthetic potential of the reductive coupling of internal alkynes, we are pleased to find that the resulting penta-substituted 1,3-dienes can be converted to various complex polycyclic molecular architectures containing multiple stereogenic centers (Scheme 4). Heating of 3a with DMAD (dimethyl but-2-ynedioate) under reflux in toluene afforded the [5,5,6]-tricyclic product 6 as a single diastereoisomer in 53% yield. To our surprise, treatment of 3a with maleic anhydride in toluene under reflux furnished the tetracyclic diene 8 bearing five stereocenters in 71% yield and high diastereoselectivity (>99/1). We speculate that product 8 is formed by the Diels–Alder reaction of maleic anhydride with diene intermediate 7, which was generated in situ from 3avia protonation of the hydroxyl group followed by elimination of H2O. The structure of 8 was confirmed by X-ray crystallographic analysis.20 Moreover, the tetracyclic compound 9 was obtained in 70% yield with high diastereoselectivity (>99/1) by reacting enantiopure diene 5bm (92% ee) with PTAD (4-phenyl-1,2,4-triazoline-3,5-dione) at room temperature.21
 | ||
| Scheme 4 Synthetic transformations. | ||
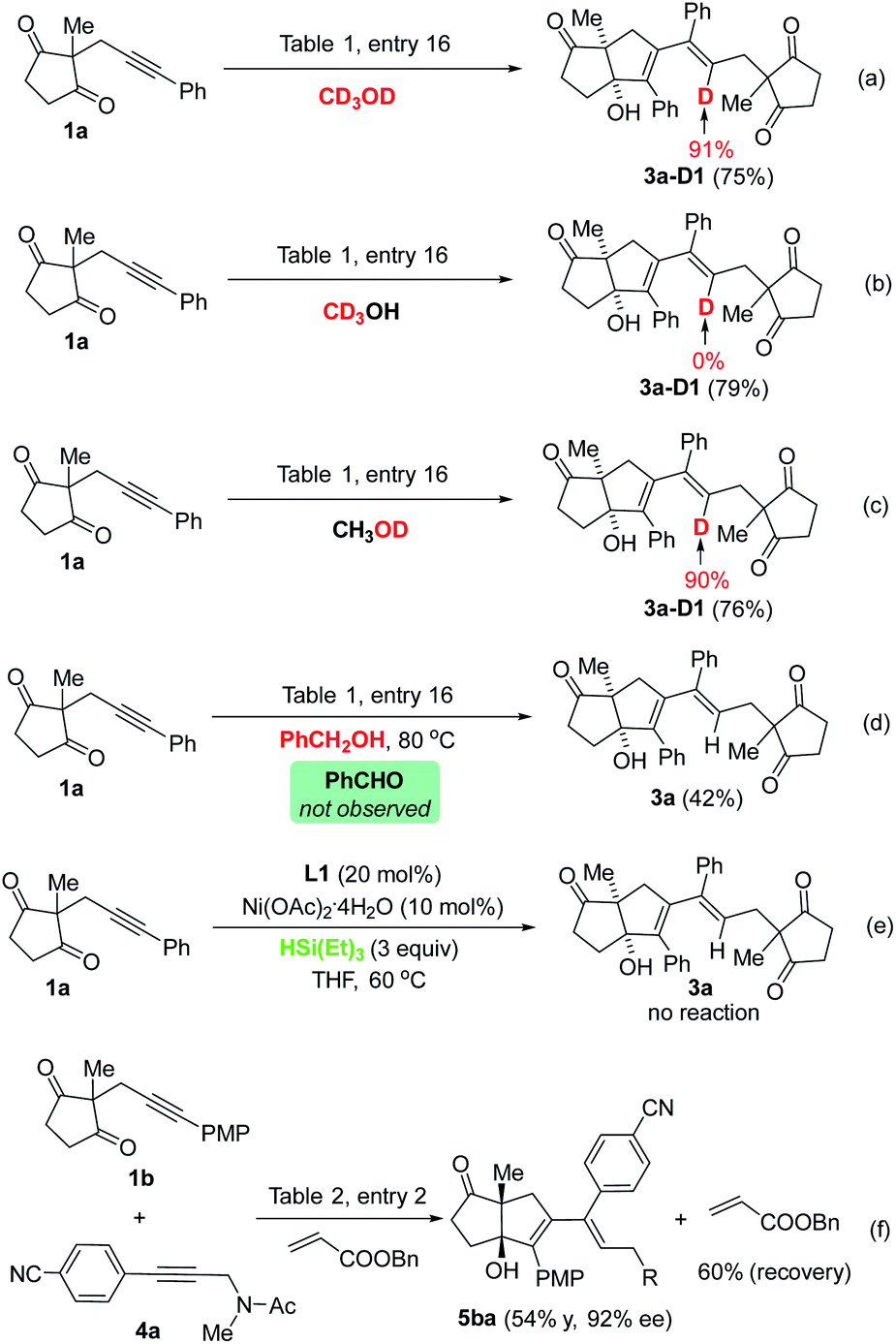
To provide some mechanistic insight for the cross-coupling reaction, isotope labelling by deuterium was performed to determine the origin of H. As shown in Scheme 5, the conversion of 1a to 3a-D1 was firstly conducted under our standard reaction conditions using deuterium-labelled CD3OD as the solvent, the desired product 3a-D1 was isolated in 75% yield with 91% deuteration rate. This result clearly shows that methanol instead of water is the proton source for this transformation (Scheme 5a). When CD3OH was used as solvent, no incorporation of deuterium was detected in the product 3a (Scheme 5b), suggesting that the methyl moiety of MeOH is not the proton source. Further investigation with CH3OD as solvent furnished 3a-D1 in 76% yield with 91% deuterium incorporation (Scheme 5c), revealing that the hydroxyl of methanol is the proton source of the reaction.22
 | ||
| Scheme 5 Mechanistic study. | ||
When the reaction was conducted in benzyl alcohol, not even traces of benzaldehyde were detected in the crude reaction mixture (Scheme 5d). This result further confirms that the alcohol solvent is not a reducing agent for this transformation. Alcohols with different structures have a great influence on the reaction outcome (Table 1), which may suggests that alcohol is not only hydrogen source, but also interacts with the reaction intermediate in the catalytical cycle. To our surprise, under previously reported reaction conditions for intramolecular reductive cyclization of alkynones using HSiEt3 as both the hydride source and reductant,23 the reaction virtually stopped (Scheme 5e). Using benzyl acrylate instead of methyl acrylate (A1) as π-acidic additive, the target product 5ba was obtained in 54% yield and 92% ee, while more than 60% of benzyl acrylate could be recovered (Scheme 5f). This result further confirms that acrylate may serves as additional ligand to activate nickel catalyst rather than reactant.
To further verify the catalytic model, Ph-PHOX ligand L2 was prepared in six different levels of enantiopurity and used in the reductive coupling and cyclization reaction of alkynone 1a. As shown in Fig. 3, the ee value of the product 3a was linearly correlated to the ee value of the chiral ligand (R2 = 0.99), which supports that the active catalytic species was composed of a nickel complex with a single Ph-PHOX ligand.
 | ||
| Fig. 3 Relationship between the ee values of the ligand Ph-PHOX (L2) and product 3a. | ||
Although a detailed mechanism for this transformation requires further investigation, based on the above-mentioned experimental observations, a possible catalytical cycle for the reductive cross coupling is depicted in Scheme 6. Initially, reduction of NiII precatalyst by Zn0 gave a catalytically active Ni0 species. Oxidative cycloaddition of alkynone 1 with unsymmetrical internal alkyne 2 afforded a five-membered nickelacycle A. The tethered carbonyl or hydroxyl group may serve as a hemilabile directing group to control the regioselectivity.24 Selective protonation of intermediate A by alcohol25 would then produce the conjugated dienylnickel species B. The crowded environment promotes the reversible cis–trans isomerization of nickel intermediate B,16a,26 thereby forming a new dienylnickel intermediate C. Finally, the target pentasubstituted 1,3-diene 3 was formed through the nucleophilic attack of the dienylnickel C to one of the ketone carbonyls, and the subsequent protonation.
 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have demonstrated a Ni-catalyzed reductive self-coupling and cross-coupling of two unsymmetrical internal alkynes using a hemilabile directing group strategy to control the regioselectivity, enabling the rapidly synthesis of pentasubstituted conjugated dienes with diverse functional groups in good yields with high regio- and enantioselectivity. The reaction features high atom- and step-economy, without requiring the use of prepared stereodefined coupling partners (vinyl halide or vinyl organometallic) compared to traditional cross-coupling reactions. The penta-substituted 1,3-dienes are very useful and can be converted to various complex polycyclic molecular architectures containing multiple stereogenic centers. Further synthetic applications and mechanistic studies are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Wuhan University, the “1000-Youth Talents Plan” and NSFC (21702149) for financial support.Notes and references
- For selected reviews, see:
(a) J.-E. Bäckvall, R. Chinchilla, C. Nájera and M. Yus, Chem. Rev., 1998, 98, 2291–2312 CrossRef
; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS
- C. A. Tolman, J. Chem. Educ., 1986, 63, 199–201 CrossRef CAS
-
(a)
G. Mehta and H. S. P. Rao, Synthesis of Conjugated Dienes and Polyenes, in The Chemistry of Dienes and Polyenes, ed. Z. Rappoport, Wiley, New York, 1997, Vol. 1, p. 359 Search PubMed
- For a recent review on 1,3-diene synthesis, see: M. De Paolis, I. Chataigner and J. Maddaluno, Top. Curr. Chem., 2012, 327, 87–146 CrossRef CAS
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS
-
(a) S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317–1382 CrossRef CAS
- For selected reviews and examples, see:
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS
- Selected examples for the synthesis of pentasubstituted 1,3-dienes, see:
(a) X. Zhang and R. C. Larock, Org. Lett., 2003, 5, 2993–2996 CrossRef CAS
- For selected review, see:
(a) K. P. C. Vollhardt, Angew. Chem., Int. Ed., 1984, 23, 539–644 CrossRef
- For selected reviews and examples, see:
(a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2916 CrossRef CAS
-
(a) S. L. Buchwald and B. T. Watson, J. Am. Chem. Soc., 1987, 109, 2544–2546 CrossRef CAS
-
(a) T. Hamada, D. Suzuki, H. Urabe and F. Sato, J. Am. Chem. Soc., 1999, 121, 7342–7344 CrossRef CAS
-
(a) J. Ryan and G. C. Micalizio, J. Am. Chem. Soc., 2006, 128, 2764–2765 CrossRef CAS
- Selected examples, see:
(a) H. L. Shimp and G. C. Micalizio, Org. Lett., 2005, 7, 5111–5114 CrossRef CAS
- References on Ni-catalyzed reductive coupling of symmetric diarylacetylenes, see:
(a) T. Wu, J. Chen and Y. Wu, Org. Lett., 2011, 13, 4794–4797 CrossRef CAS
-
(a) C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS
- CCDC 1907444 (3a) contain the supplementary crystallographic data for this paper.†.
-
(a) D. P. Todd, B. B. Thompson, A. J. Nett and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 12788–12791 CrossRef CAS
-
(a) L. Xiao, L. Cheng, W. Feng, M. Li, J. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461–464 CrossRef CAS
- CCDC 1946921 (8) contain the supplementary crystallographic data for this paper.†.
- CCDC 2009296 (9) contain the supplementary crystallographic data for this paper.†.
- K. Nakai, Y. Yoshida, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2014, 136, 7797–7800 CrossRef CAS
- W. Fu, M. Nie, A. Wang, Z. Cao and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 2520–2524 CrossRef CAS
-
(a) K. M. Miller, T. Luanphaisarnnont, C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2004, 126, 4130–4131 CrossRef CAS
-
(a) J. Marco-Martínez, V. López-Carrillo, E. Buñuel, R. Simancas and D. J. Cárdenas, J. Am. Chem. Soc., 2007, 129, 1874–1875 CrossRef
- For recent examples on cis–trans isomerization of alkenylnickel species, see:
(a) X. Zhang, X. Xie and Y. Liu, Chem. Sci., 2016, 7, 5815–5820 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, and NMR spectra. CCDC 1907444, 1946921 and 2009296. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04173d |
| This journal is © The Royal Society of Chemistry 2020 |