
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Divergent synthesis of 5-substituted pyrimidine 2′-deoxynucleosides and their incorporation into oligodeoxynucleotides for the survey of uracil DNA glycosylases†
Ai
Tran
a,
Song
Zheng
a,
Dawanna S.
White
b,
Alyson M.
Curry
b and
Yana
Cen
 *bc
*bc
aDepartment of Pharmaceutical Sciences, Albany College of Pharmacy and Health Sciences, Colchester, VT 05446, USA
bDepartment of Medicinal Chemistry, Virginia Commonwealth University, Richmond, VA 23219, USA. E-mail: ceny2@vcu.edu; Tel: +1-804-828-7405
cInstitute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA
First published on 7th October 2020
Abstract
Recent studies have indicated that 5-methylcytosine (5mC) residues in DNA can be oxidized and potentially deaminated to the corresponding thymine analogs. Some of these oxidative DNA damages have been implicated as new epigenetic markers that could have profound influences on chromatin function as well as disease pathology. In response to oxidative damage, the cells have a complex network of repair systems that recognize, remove and rebuild the lesions. However, how the modified nucleobases are detected and repaired remains elusive, largely due to the limited availability of synthetic oligodeoxynucleotides (ODNs) containing these novel DNA modifications. A concise and divergent synthetic strategy to 5mC derivatives has been developed. These derivatives were further elaborated to the corresponding phosphoramidites to enable the site-specific incorporation of modified nucleobases into ODNs using standard solid-phase DNA synthesis. The synthetic methodology, along with the panel of ODNs, is of great value to investigate the biological functions of epigenetically important nucleobases, and to elucidate the diversity in chemical lesion repair.
Introduction
5-Methylcytosine (5mC), also known as the fifth base, is both an endogenous DNA damage product and an intermediate in a recently identified epigenetic reprogramming pathway.1 Accumulating evidence suggests that active demethylation could involve iterative oxidation of 5mC to 5-hydroxymethylcytosine (5hmC),2 and then to 5-formylcytosine (5foC) and 5-carboxylcytosine (5caC),3,4 mediated by ten-eleven translocation (TET) enzymes, with the conversion of 5caC to C by a putative decarboxylase5 (Scheme 1). Direct deformylation of 5foC can also lead to the restoration of cytosine (C).6 An alternative proposal involves 5mC deamination by activation-induced deaminase (AID),7,8 followed by thymine DNA glycosylase (TDG)-initiated base excision repair (BER).9 Recent findings also implicate the removal of modified C or uracil (U) derivatives by DNA glycosylase/BER/nucleotide excision repair (NER) as another potential demethylation mechanism.10–14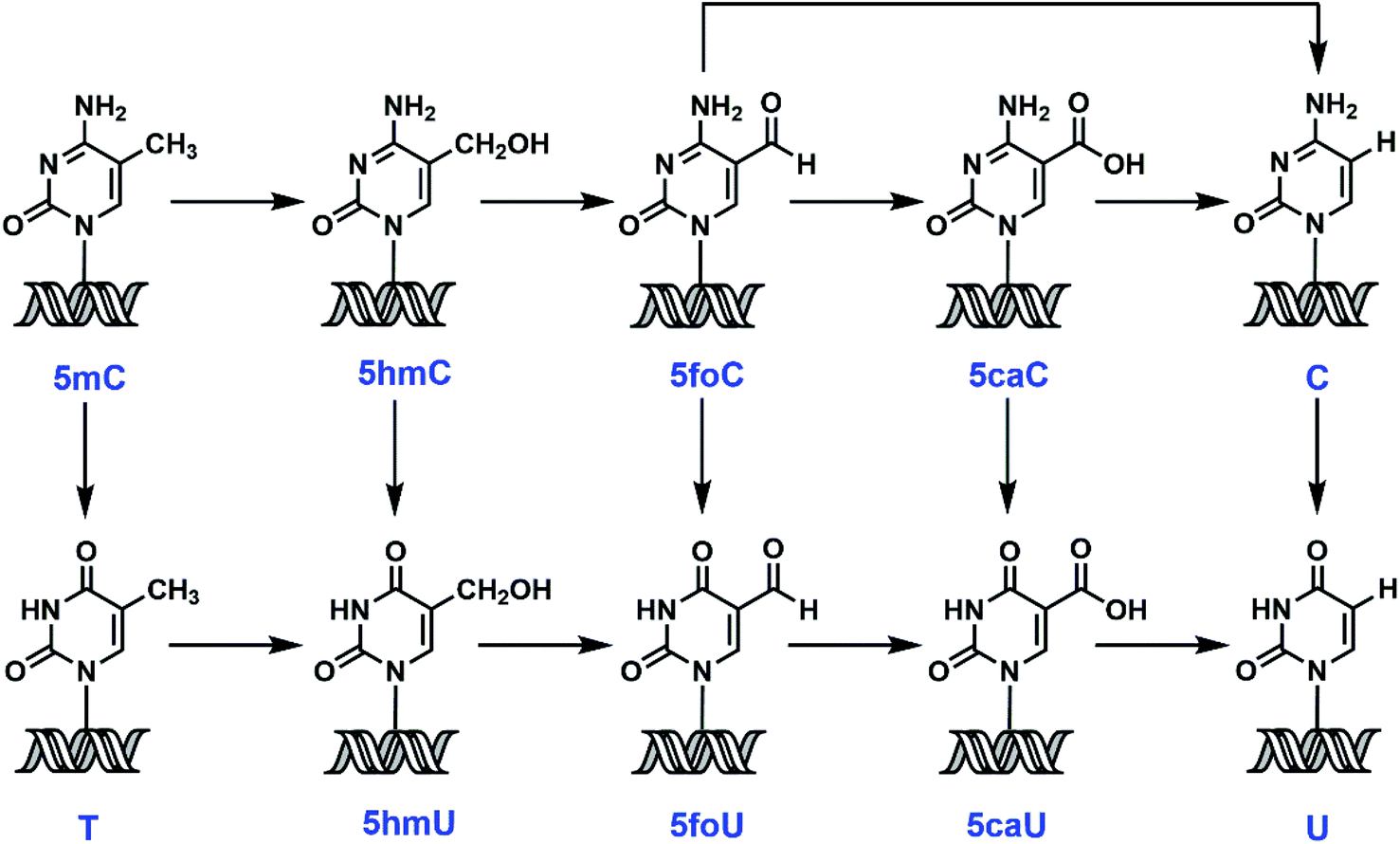 | ||
| Scheme 1 Potential pathways for the oxidation, deamination and demethylation of 5mC and derivatives. 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; 5foC, 5-formylcytosine; 5caC, 5-carboxylcytosine; 5hmU, 5-hydroxymethyluracil; 5foU, 5-formyluracil; 5-caU, 5-carboxyluracil. | ||
The significance of 5mC in transcription silencing, genomic imprinting and cellular differentiation has been well established.15,16 However, the same cannot be said for its chemical relatives. Much has been debated about whether these 5mC derivatives are of physiological relevance or just transient intermediates along the above-mentioned pathways.17 For example, great progress has been made to map and sequence 5-hydroxymethyluracil (5hmU) and 5-formyluracil (5foU) in the human genome.18,19 However, there is still ongoing discussion about if these modifications are just aberrant DNA damage, or they are key epigenetic marks regulating gene transcription.20–22 This is partly due to the low abundance of 5mC derivatives3,4,23,24 and their quick removal by DNA glycosylases, which make the interrogation of their biological functions extremely challenging. Synthetic oligodeoxynucleotides (ODNs) containing 5mC derivatives serve as powerful surrogates to examine the chemical and enzymatic conversions of modified nucleobases, and to probe the mechanism of action and substrate specificity of DNA glycosylases. However, there are limited synthetic methods available for accessing these ODNs.25–29 And many of these synthetic strategies involve the use of toxic chemicals25–27,29 and often suffer from low yield and difficulty in purification. More importantly, there is no unified method to generate all the 5mC related phosphoramidites. Herein, we report an efficient and concise strategy to synthesize 5mC, 5hmC, 5foC, 5caC, thymine (T), 5hmU, 5foU and 5-carboxyluracil (5caU) phosphoramidites through one common intermediate, 5-iodo-2′-deoxyuridine. Subsequently, these modified nucleobases, along with C and U, were incorporated into synthetic ODNs in a site-specific fashion via standard solid-phase DNA synthesis. Ultimately, the ODNs were subjected to a panel of DNA glycosylases as well as mammalian cell extracts. These enzymes and cell extracts demonstrate diverse substrate preference and cleavage efficiency. This survey provides the first comprehensive inventory of base selection for various glycosylases. The novel divergent synthesis and chemical probes developed in the current study should facilitate the probing of epigenetic reprogramming pathways, the biological significance of modified nucleobases and the diversity in chemical lesion repair.
Experimental
Reagents and instruments
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA) and were of the highest purity commercially available. Commercially available phosphoramidites and sublimed 1H-tetrazole in anhydrous acetonitrile were purchased from Glen Research (Sterling, VA). Thin layer chromatography (TLC) was performed on precoated silica gel 60 F254, 5 cm × 20 cm, 250 μM thick plates purchased from EMD (Gibbstown, NJ). [γ-32P]-ATP was purchased from MP Biomedicals (Irvine, CA). E coli UDG and human SMUG1 and T4 polynucleotide kinase were purchased from New England Biolab (Ipswich, MA). Human TDG was purchased from Antagene (Santa Clara, CA). HeLa cell nuclear extract was purchased from Promega (Madison, WI).1H, 13C, and 31P NMR spectra were obtained using a Bruker Ultrashield 300 MHz spectrometer (Manning Park, MA). HPLC analysis was performed with a ThermoFinnigan (Waltham, MA) Surveyor HPLC system with a photodiode array and a single quad mass spectrometer detector. Oligonucleotides were synthesized with a Millipore Expedite nucleic acids synthesis system (Billerica, MA). Mass spectra were obtained with a Bruker Autoflex II MALDI-TOF mass spectrometer operated in positive ion reflectron mode. High-resolution mass spectra were obtained with either a Waters Micromass Q-TOF Ultima (Milford, MA) or a Thermo Scientific Q-Exactive Hybrid Quadrupole Orbitrap (Waltham, MA).
ODN synthesis and characterization
ODNs were prepared by solid-phase DNA synthesis as described previously.30–32 Following synthesis and deprotection, ODNs were purified by HPLC using Hamilton PRP-1 column and a gradient of 0 to 25% CH3CN in 10 mM of potassium phosphate buffer (pH ∼ 6.8). ODNs were then detritylated with 80% aqueous acetic acid at room temperature for 30 min. Following detritylation, they were purified by HPLC using Vydac C18 column and a gradient of 0 to 70% CH3CN in water. ODN purity was examined by MALDI-TOF,33 and the free base composition was verified by HPLC, following enzymatic digestion34 using a Aquasil C18 column and a gradient of 0 to 60% CH3CN in 20 mM of ammonium acetate (pH ∼ 5.5). The presence of modified bases was also verified by GC/MS following acid hydrolysis and conversion to the t-butyldimethylsilyl ethers.35Expression and purification of human MBD4
pET28-MBD4 was a gift from Cheryl Arrowsmith (Addgene plasmid # 25155). Recombinant, N-terminally His-tagged protein was expressed in E coli BL21 (DE3) Codon plus RIL cells (Agilent). The cells were culture in LB media supplemented with 50 μg mL−1 kanamycin at 37 °C until OD600 reached 0.6. Protein expression was induced with 100 μM of IPTG. The culture was grown for another 16 h at 15 °C before the cells were pelleted and lysed with 4 rounds of sonication. The protein was purified using Ni-NTA resin (Thermo Fisher) affinity column and eluted with increasing concentrations of imidazole. The purified protein was dialyzed against a solution of 50 mM Tris–HCl (pH 7.5), 1 mM EDTA, 15% glycerol and 2 mM DTT. The protein was aliquote, flash frozen and stored at −80 °C. Protein concentration was determined using Bradford assay. The protein was >95% pure as determined with SDS-PAGE gel electrophoresis.ODN labeling and annealing
5′-End radiolabeling was performed using [γ-32P]-ATP and T4 polynucleotide kinase under the conditions recommended by the enzyme supplier. Labeled mixture was subsequently centrifuged through G-50 Sephadex columns (Roche) to remove excess unincorporated nucleotide. Labeled single-stranded ODNs were annealed to a 2-fold molar excess of unlabeled complementary strand in 20 mM Tris–HCl (pH 8.0), 1 mM EDTA and 50 mM NaCl. The mixture was heated at 95 °C for 5 min and slowly cooled down to room temperature.Recombinant DNA glycosylase assay
The glycosylase assay was performed in a 10 μL reaction mixture containing the appropriate reaction buffer (20 mM Tris–HCl at pH 8, 1 mM DTT, 1 mM EDTA and 0.1 mg mL−1 BSA for eUDG, hTDG and hMBD4; 10 mM Bis–Tris–Propane–HCl at pH 7, 10 mM MgCl2, 1 mM DTT and 0.1 mg mL−1 BSA for hSMUG1). DNA substrate (50 nM) was incubated with the glycosylase (200 nM) at 37 °C in the reaction buffer. The reaction was terminated by the addition of 5 μL of 100 mM NaOH and an equal volume of Maxam–Gilbert loading buffer (98% formamide, 10 mM EDTA, 1 mg mL−1 xylene cyanol and 1 mg mL−1 bromophenol blue) and 50 pmol of an unlabeled ODN as a reannealing competitor. The backbone was cleaved at the abasic site with NaOH by heating at 95 °C for 30 min. For ODNs containing 5foC or 5foU, the reactions were stopped by heating to 75 °C for 5 min and then cooled to room temperature. Reaction mixtures were then resolved on a 15% denaturing polyacrylamide gel. The bands corresponding to the substrate and product were visualized and quantified using a Typhoon 9400 phosphorimager (GE Healthcare). Phosphorimager scans were quantified with ImageQuant TL (GE Healthcare) by generating rectangular boxes around the substrate or product. Lane-specific background was subtracted by using analogous rectangles on a portion of the gel lane that did not correspond to any signal. The percentage conversion was calculated by dividing the product band intensity by the sum of the remaining substrate intensity and product intensity.Nuclear extract glycosylase assay
The nuclear extracts from U87 and hES cells were generous gifts from Dr Kangling Zhang (University of Texas Medical Branch in Galveston). 5′-End labeled duplex (50 nM) was incubated with nuclear extract (0.5 mg mL−1) in a buffer containing 20 mM Tris–HCl at pH 8, 1 mM DTT, 1 mM EDTA and 0.1 mg mL−1 BSA to a total volume of 10 μL. The reactions were incubated at 37 °C before being quenched with the addition of 5 μL of 100 mM NaOH, an equal volume of Maxam–Gilbert loading buffer and 50 pmol of an unlabeled ODN as a reannealing competitor. The backbone was cleaved at the abasic site with NaOH by heating at 95 °C for 30 min. For ODNs containing 5foC or 5foU, the reactions were stopped by heating to 75 °C for 5 min and then cooled to room temperature. Reaction mixtures were then resolved on a 18% denaturing polyacrylamide gel. The bands corresponding to the substrate and product were visualized and quantified using a Typhoon 9400 phosphorimager as described above.Results and discussion
Synthesis of phosphoramidites containing modified C or U
Careful literature investigation suggests that commercially available 5-iodo-2′-deoxyuridine (5IU) serves as an ideal starting point for constructing 5-substituted 2′-deoxyuridine derivatives. TBDMS-protected 5IU, compound 1, can be further functionalized at the 5-position through a metal–halogen exchange reaction (Fig. 1).36 When BuLi was used alone, deiodinated product predominated, suggesting a competition between deprotonation and lithium–iodine exchange. Instead, NaH was introduced first to remove the imide proton. Subsequently, BuLi was added to effect the metal–halogen exchange and substitution. In previous studies, the 5-formyl group was installed either by oxidation of the methyl by K2S2O8 (ref. 25) or by Pd-catalyzed Stille coupling reaction with carbon monoxide.37 The former reaction resulted in low yield, while the latter one utilized a toxic organotin reagent as well as CO. In our study, compound 1 was incubated with NaH and nBuLi sequentially followed by treatment with DMF at −78 °C, leading to the formation of the protected 5foU in 80% yield after purification (Fig. 1, foU). When DMF was replaced by dimethyl carbonate, 5caU was obtained under similar conditions with good yield (Fig. 1, caU). The versatility of our synthetic strategy was further demonstrated in the preparation of 5hmU. 5-Hydroxymethyl group has routinely been generated by the reduction of 5-formyl group.27,38 The lithiation of compound 1, together with the subsequent treatment with paraformaldehyde, allowed 5hmU to be created directly in excellent yield (Fig. 1, hmU). Not surprisingly, the scope of this method can also be extended to methylation by methyl iodide to afford T (Fig. 1, T). So far, T, 5hmU, 5foU and 5caU were synthesized from one common ancestor under mild reaction conditions, by simply changing the electrophiles accordingly. Furthermore, our strategy potentially provides easy access to isotopically labeled 5mC derivatives when stable isotope or radioisotope labeled electrophiles or reagents are used.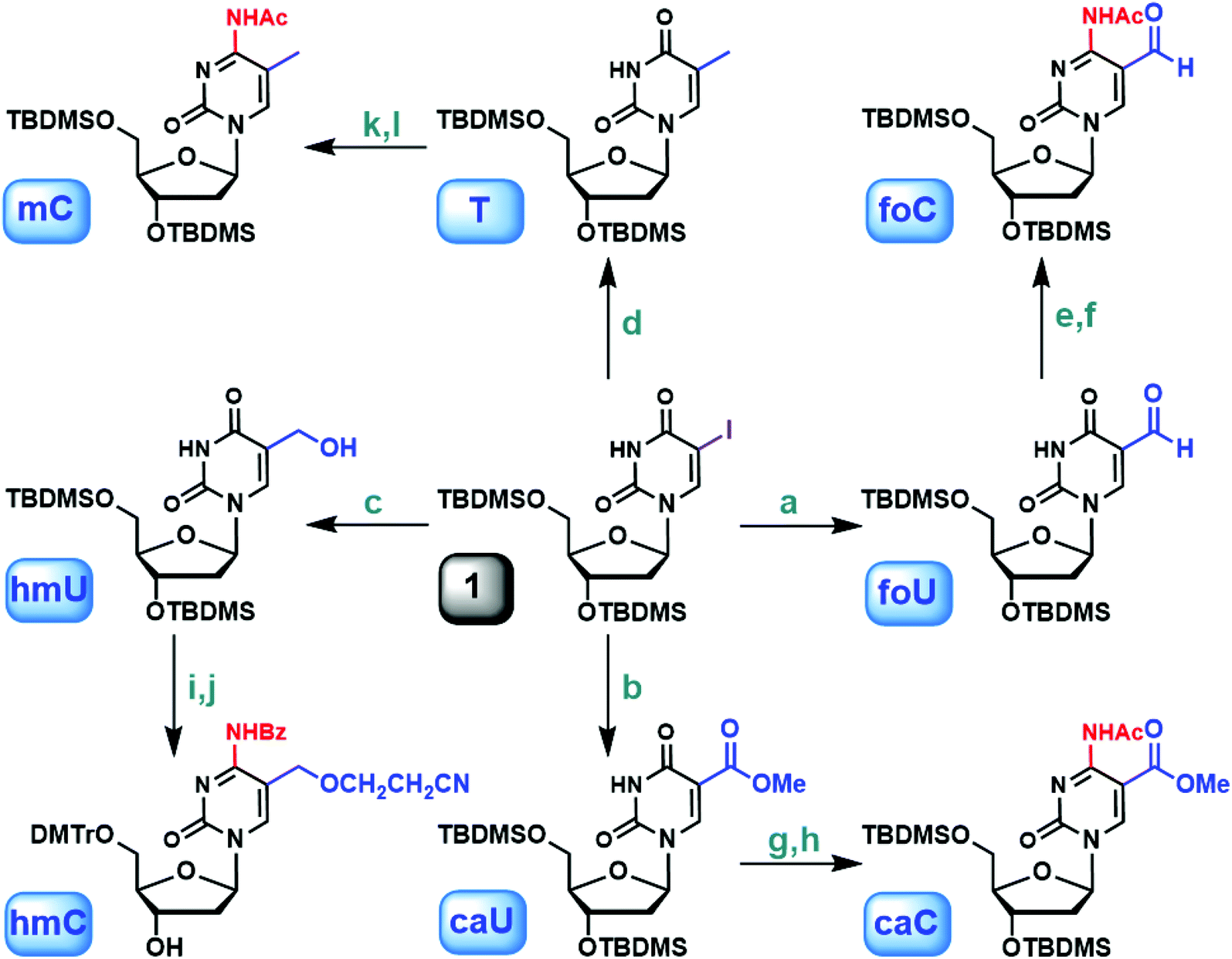 | ||
| Fig. 1 Synthesis of 5-methyl-2′-deoxycytidine and derivatives via 5-iodo-2′-deoxyuridine. (a) (1) NaH, THF; (2) nBuLi, then DMF, −78 °C (80%); (b) (1) NaH, THF; (2) nBuLi, then dimethyl carbonate, −78 °C (71%); (c) (1) NaH, THF; (2) nBuLi, then paraformaldehyde, −78 °C (86%); (d) (1) NaH, THF; (2) nBuLi, then MeI, −78 °C (93%); (e) (1) TEA, DMAP, TPSCl, CH3CN; (2) 25% NH4OH (90%); (f) Ac2O, DMF (92%); (g) (1) TEA, DMAP, TPSCl, CH3CN; (2) 25% NH4OH (72%); (h) Ac2O, DMF (88%); (i) TBAF, THF (85%); (j) ref. 29; (k) (1) TEA, DMAP, TPSCl, CH3CN; (2) 25% NH4OH (85%); (l) Ac2O, DMF (93%). | ||
The protected 2′-deoxyuridine analogs can be further elaborated to the corresponding phosphoramidites following the standard protocols (Fig. 2): TBDMS deprotection, tritylation of the 5′-hydroxyl group, and finally coupling with 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphorodiamidite (see ESI† for more details).39
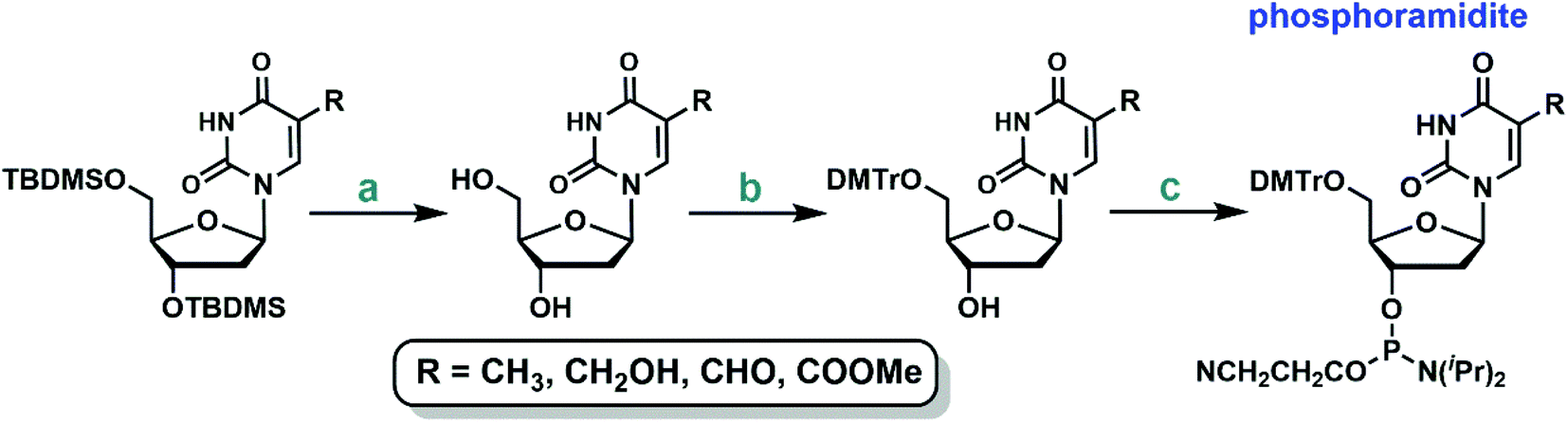 | ||
| Fig. 2 Standard protocols to synthesize the phosphoramidites of 5-substituted-2′-deoxyuridine analogs. (a) TBAF, THF (77–92%); (b) DMTrCl, pyridine (79–92%); (c) DIPEA, tetrazole in CH3CN, 2-cyanoethyl tetraisopropyl phosphoramidite (88–98%). | ||
With the substituted 2′-deoxyuridine derivatives readily available, easy access to 2′-deoxycytidine analogs became possible via in situ amination.40,41 For example, the uracil in protected 5foU was transformed into cytosine with 2,4,6-triisopropylbenzenesulfonyl chloride (TPSCl) in the presence of triethylamine (TEA) and N,N-dimethylaminopyridine (DMAP) followed by NH4OH treatment. Subsequently, the 4-amino group was acetylated to allow further modifications (Fig. 1, foC). Using the same method, 5caC and 5mC were obtained from 5caU and T, respectively (Fig. 1, caC and mC). For 5foC, 5caC and 5mC, the corresponding phosphoramidites were synthesized by the consecutive desilylation, tritylation and phosphitylation sequence. The formation of 5hmC was slightly different: the TBDMS-protected 5hmU was desilylated first, and the 5hmC phosphoramidite was then generated as described previously (Fig. 1, hmC).42 Several phosphoramidites mentioned above are also commercially available. We selected 5-carboxy-dC-CE phosphoramidite from Glen Research and 5-cadC-CE phosphoramidite from our study for comparison. The 0.25 g (0.276 mmol) package at Glen is priced at $1200. Our synthesis includes 7 steps starting from 5-iodo-2′-deoxyurdine. The overall cost to prepare 0.276 mmol 5cadC-CE phosphoramidite is less than $250 considering all the chemicals, solvents and disposables. Our strategy is straightforward, cost-friendly with synthetic easiness.
Compound 1 acted as a common precursor for eight different pyrimidine 2′-deoxynucleosides involved in the active demethylation pathway. In the first round, nucleophilic substitution at 5-position gave rise to four 2′-deoxyuridine analogs. In the second round, 4-position was modified to gain access to the 2′-deoxycytidine derivatives. Most of the steps were under mild conditions with good to excellent yields. The divergent synthesis described in this article greatly expanded our capacity in the production of epigenetically important nucleosides and related phosphoramidites, which paves the way for a better understanding of the epigenetic reprogramming and DNA repair pathways.
Synthesis of ODNs containing modified C or U
ODNs containing modified pyrimidine nucleobases at a specific location in a short sequence were prepared using standard solid-phase DNA synthesis.43,44 The sequence is shown in Fig. 3A. Special attention was given to the deprotection of ODNs containing labile functionalities. In 5caC and 5caU phosphoramidites, the carboxylate exists in the methyl ester form. The cleavage and deprotection were carried out in 0.4 M methanolic NaOH for 17 h at room temperature before being neutralized with acetic acid. The formyl group in both 5foC and 5foU phosphoramidites was not protected. It was compatible with the mild deprotection condition (0.1 M K2CO3 in methanol/water) and the standard deprotection treatment with NH4OH at room temperature for an extended period of time. All the synthetic ODNs were purified by HPLC, and their purities were examined by MALDI-TOF (Table S1†).33 The free base composition was verified by HPLC following enzymatic digestion,34 or by GC/MS following acid hydrolysis and conversion to the t-butyldimethylsilyl ethers.35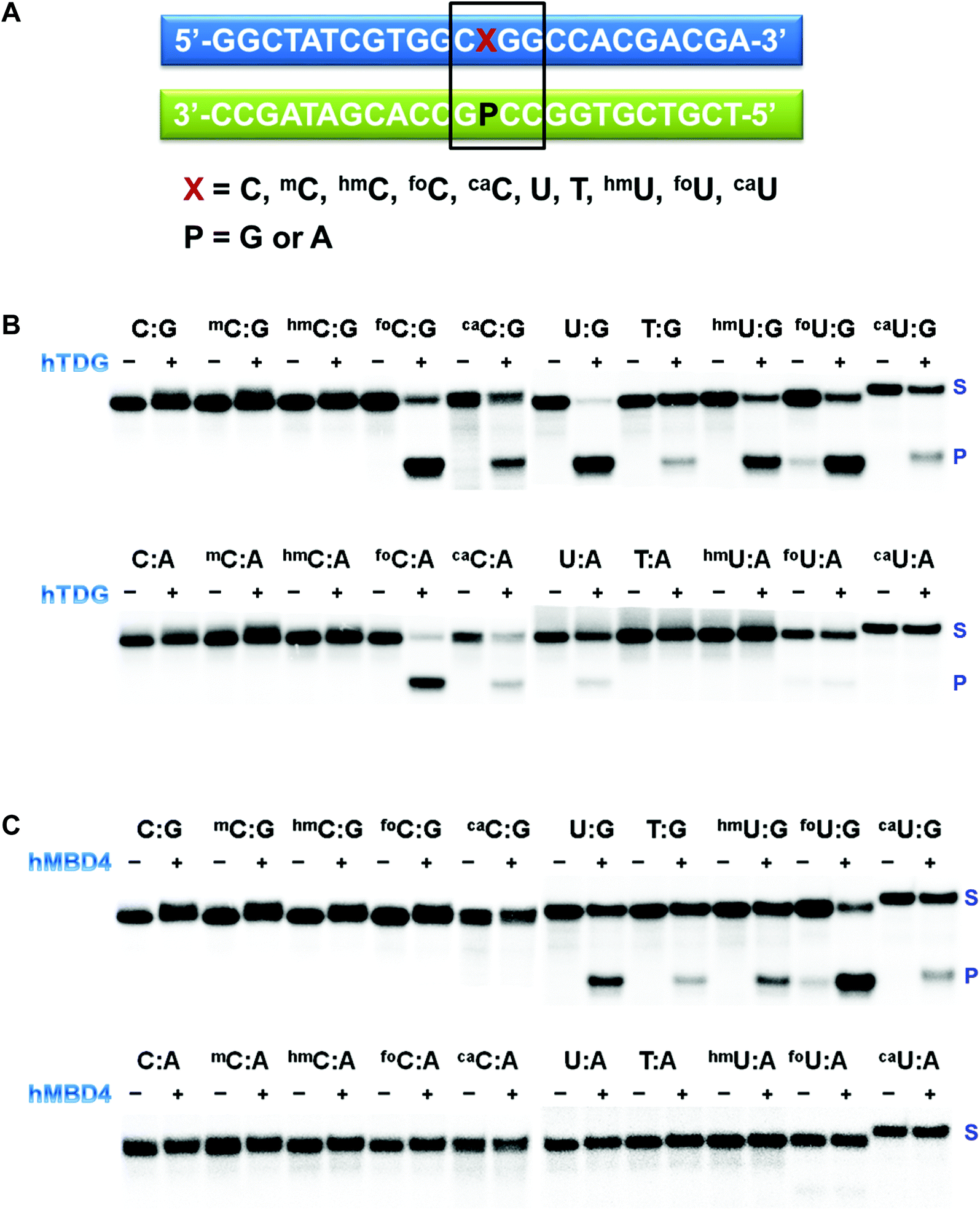 | ||
| Fig. 3 Substrate selectivity of recombinant UDGs. (A) Sequences of the synthetic ODNs used for the glycosylase assay; (B and C). Electrophoretic assays of hTDG (B) and hMBD4 (C) activity on the synthetic ODNs confirmed by autoradiography. The recombinant glycosylase was incubated with 32P end-labeled duplex DNA at 37 °C, followed by alkaline-induced quenching and cleavage of the DNA at glycosylase-generated abasic sites. S: substrate; P: product. | ||
Elucidation of substrate preference of recombinant DNA glycosylases using synthetic ODNs
The last decade has witnessed an ever-increasing interest in epigenetic editing, especially the reversible DNA modifications. Emerging evidence suggests that uracil DNA glycosylases (UDGs) play critical roles in the removal of 5mC and related pyrimidines and thus influence a broad spectrum of cellular events such as transcription silencing, cell differentiation and development. There have been sporadic reports on individual UDG's action on a limited number of cytosine modifications.10,11,45–50 However, a comprehensive analysis of the substrate selectivity and cleavage efficiency of the UDG superfamily is still not available. After securing the panel of synthetic ODNs bearing ten different cytosine derivatives, we set out to survey the activity of the UDG family members including E. coli uracil DNA glycosylase (eUDG), human thymine DNA glycosylase (hTDG), human single-stranded selective monofunctional uracil DNA glycosylase (hSMUG1) and human methyl-CpG binding domain protein 4 (hMBD4). In the first series of experiments, the C or U analogs were paired up with a guanine (G) in the complementary strand to form the duplex. The duplex was then 32P-labeled at the 5′-end followed by incubation with purified recombinant UDG. Finally, the alkaline strand cleavage would lead to the formation of a 32P-labeled short DNA fragment that can be visualized on a denaturing polyacrylamide DNA gel with a phosphorimager. As expected, U:G pair was well recognized by all four UDGs examined (Fig. 3B, C and S1†), and it was the only mismatch targeted by eUDG under our assay conditions. On the contrary, 5foC:G and 5caC:G mispairs were only acted upon by hTDG, consistent with the previous reports.11,46 Furthermore, hTDG was more active towards mismatched Us in DNA duplexes, removing U, T, 5hmU, 5foU and 5caU to different extents (Fig. 3B and Table 1). TDG has a strong preference for bases paired up with G and are followed by G,51–53 suggesting a pivotal role of TDG in the active demethylation pathway since 5mC is frequently formed in the CpG islands. Similar substrate scopes were observed for hSMUG1 and hMBD4: hSMUG1 targeted U and its analogs except for T (Fig. S1C†), while hMBD4 acted on all the U analogs with a preference for 5foU:G pair (Fig. 3C).
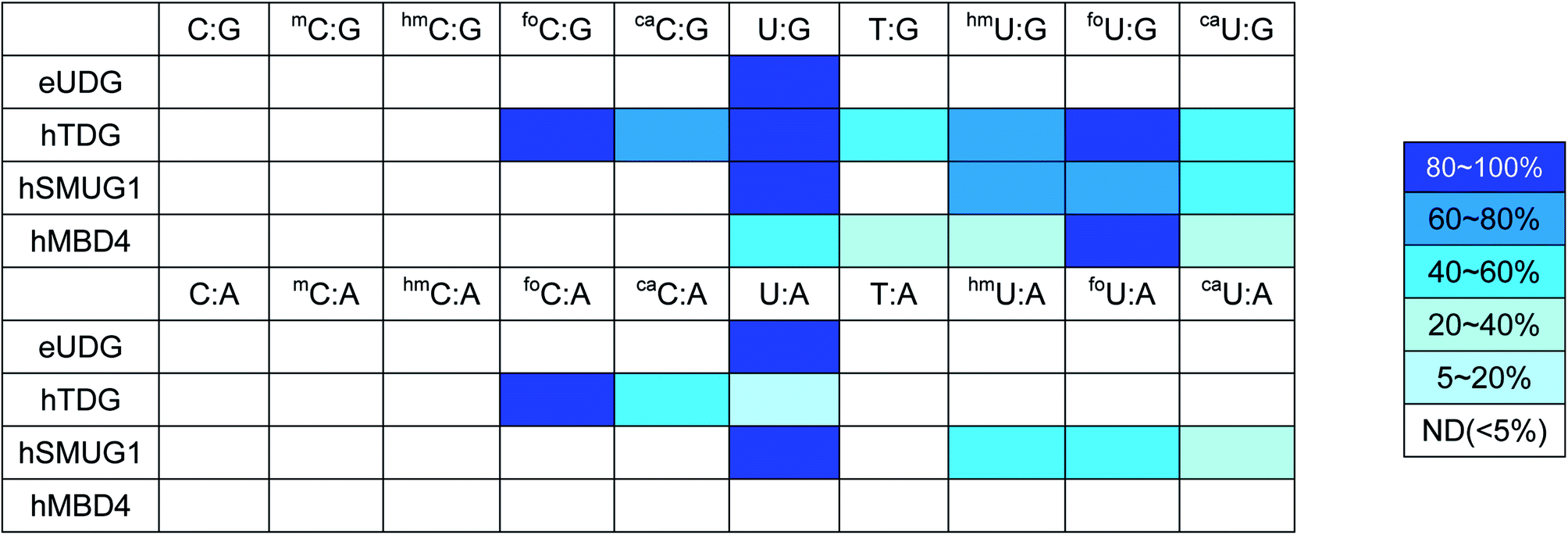
|
In the second set of experiments, the aforementioned 5mC analogs were paired opposite adenine (A). eUDG acted exclusively on U:A pair (Fig. S1A†). hTDG showed rapid excision of 5foC from 5foC:A mismatch, and also removed 5caC and U to lesser extents (Fig. 3B). hSMUG1, when first discovered, was thought to only have activity on single-stranded DNA (ssDNA).54 Recent studies suggest that the substrate scope of this glycosylase can be extended to double-stranded DNA (dsDNA).12,55–59 Indeed, hSMUG1 demonstrated functional group recognition in the U:A, 5hmU:A, 5foU:A and 5caU:A pairs, similar to the trend observed in the C/U:G series (Fig. S1C†). Despite the broad substrate tolerance in the first series, hMBD4 failed to remove any mismatched bases in the C/U:A series (Fig. 3C).
The UDGs use a base-flipping mechanism for substrate recognition: the target base in the duplex is flipped into the active site prior to the hydrolytic cleavage of the N-glycosidic bond.60,61 The substrate specificity is mainly governed by steric and electronic factors, as well as the stability of the DNA duplex. Among all the UDGs examined, eUDG exhibited the narrowest substrate range by removing only the U residue in the mispairs. Structural analysis suggests that the presence of Tyr66 in the binding pocket of eUDG excludes sterically bulky 5-substituted pyrimidines.62 hTDG, on the other hand, is considered a versatile DNA glycosylase by targeting a wide range of 5mC analogs. These modified bases can be accommodated in the hTDG binding pocket via π–π stacking interaction with Tyr152.46 Additionally, a network of hydrogen bond donors and acceptors provides greater flexibility in the binding site.46 Moreover, the substrate discrimination of hTDG can be explained by the electronic inductive property of the 5-substituents. Formyl group, an electron-withdrawing substituent, is expected to increase the acidity of the cleaved base to increase excision activity. Methyl and hydroxymethyl substituted bases are relatively poorer substrates due to their electron-donating effects. Unlike hTDG, hSMUG1 does not exploit the electronic effects of the substituents.12 Rather, it relies on the duplex stability for substrate discrimination.12 A previous study indicates that hMBD4 specifically recognizes T and 5hmU.49 Our study expanded its substrate repertoire to include U, 5foU and 5caU, but only when they are opposite G. The lack of detectable glycosylase activity against the C/U:A pairs requires further mechanistic and structural analysis.
Probing DNA glycosylase activity in cell extracts using synthetic ODNs
The processing of 5mC and its derivatives is believed to involve either enzymatic oxidation or deamination followed by glycosylase removal and DNA repair, indicating a potentially complicated intersection of DNA damage repair with epigenetic reprogramming. This pathway has taken on substantially greater significance because emerging data suggest that it is operational during cellular differentiation and in normal stem cells, but is uniformly defective in human cancer cells potentially due to defects in formation as well as processing.63,64 With the array of above-mentioned synthetic ODNs, it is possible to interrogate the DNA glycosylase activity in cell extracts from both cancer cells and normal stem cells. First, UDG activity in HeLa nuclear extract was probed using the panel of duplexes containing the C/U:G pairs (Fig. 4A and S2A†). Substantial activities against U:G, 5hmU:G and 5foU:G were detected. It appeared that HeLa nuclear extract lacks significant hTDG activity. The observed excision of 5hmU and 5foU was likely due to hSMUG1 activity, in agreement with a previous study.65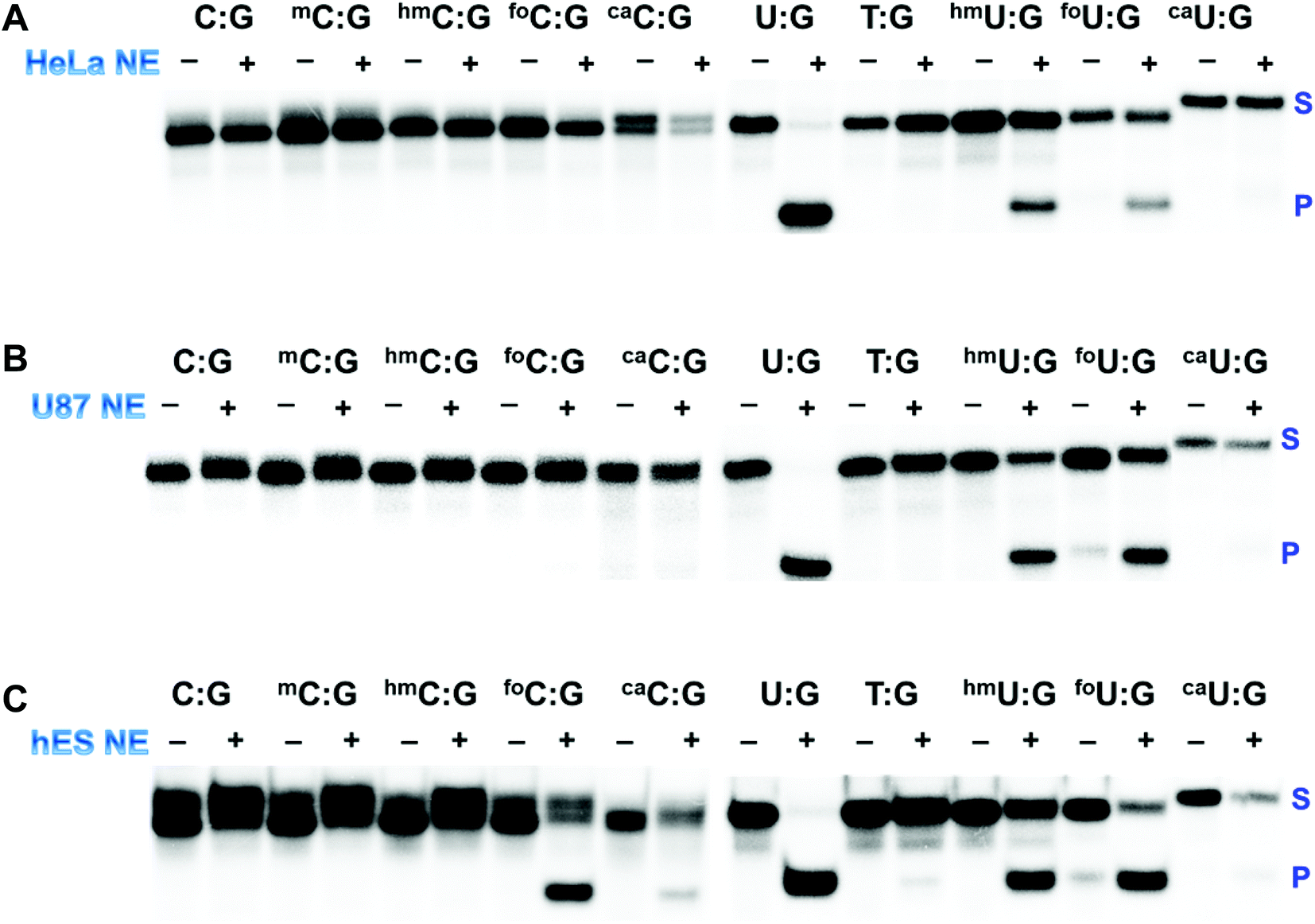 | ||
| Fig. 4 DNA glycosylase activity in cell extracts. The glycosylase activity in the nuclear extract (NE) from HeLa (A), U87 (B) and hES (C) cells to generate alkaline-sensitive sites was assayed using 32P end-labeled duplex DNAs. | ||
hTDG activity is known to be regulated transcriptionally by both promoter methylation and by tumor suppressor protein p53.66,67 The mRNA and protein levels of hTDG can be stimulated in a p53-dependent fashion. It has been reported that HeLa cells lack detectable levels of p53 protein.68 The absence of p53 results in the downregulation of hTDG. Furthermore, it was reported that high levels of hTDG causes cell cycle arrest in the S-phase in several human cell lines, including HeLa cells.69 SUMOylation-mediated degradation warrants an hTDG-free progression until the G2-phase. The downregulation of hTDG could contribute to the abundant transition mutations at methylated CpG sites frequently observed in human cancer cells. Our synthetic ODNs, together with the bioassay, were powerful tools in identifying a defective component of the demethylation pathway in HeLa cells.
This screening strategy was then expanded to human U87 glioblastoma cells and human embryonic stem (hES) cells (Fig. 4B, C, S2B and S2C†). U87 cells, similar to HeLa cells, demonstrated robust glycosylase activity against U, 5hmU and 5foU, suggesting a common mechanism underlying the epigenetic regulation of cell proliferation in these cell lines. hES cells, on the other hand, exhibited dramatically different excision patterns. In addition to several U analogs, hES nuclear extract was also able to remove 5foC efficiently and cleave 5caC to a much lesser extent (Fig. 4C), consistent with the substrate preference of hTDG. Recent studies indicate that mouse embryonic stem (mES) cells have high levels of 5hmC, significantly lower level of 5foC and almost undetectable levels of 5caC.23,70 Furthermore, the nuclear extract of mES cells is known to harbor 5caC-selective TDG activity.10 DNA methylation is considered an important epigenetic regulation of pluripotency and differentiation of stem cells. Previous results and our own data suggest that BER mediated by TDG plays critical roles in maintaining epigenetic stability during embryonic development.71
Conclusions
In summary, we report the divergent synthesis of phosphoramidites carrying epigenetically important 5mC analogs from one common precursor. Nucleophilic substitutions at 5-position of 5-iodo-2′-deoxyuridine furbished a set of U derivatives including T, 5hmU, 5foU and 5caU. In situ amination then converted the U series to C analogs. Our synthetic strategy provided unprecedented easiness and yields in generating these important DNA building blocks. The modified pyrimidines were incorporated into 24 mer ODNs in a site-specific manner using solid-support DNA synthesis. This battery of ODNs allowed the interrogation of substrate preference and excision efficiency of a family of recombinant UDGs. We further envisaged the evaluation of DNA glycosylase activity in human cell extracts using these synthetic ODNs, giving attention to the cancer cells as well as ES cells. Given the importance of epigenetic reprogramming in cell development, differentiation and disease progression, the synthetic methods and chemical probes we developed will be widely utilized for a better understanding of this intriguing biological event.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by start-up funding from VCU (to Y. C.) and from NSF (CHE-1846785 to Y. C.).References
- S. Kumar, V. Chinnusamy and T. Mohapatra, Front. Genet., 2018, 9, 640 CrossRef CAS.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS.
- T. Pfaffeneder, B. Hackner, M. Truss, M. Munzel, M. Muller, C. A. Deiml, C. Hagemeier and T. Carell, Angew. Chem., Int. Ed., 2011, 50, 7008–7012 CrossRef CAS.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS.
- Z. Liutkeviciute, E. Kriukiene, J. Licyte, M. Rudyte, G. Urbanaviciute and S. Klimasauskas, J. Am. Chem. Soc., 2014, 136, 5884–5887 CrossRef CAS.
- A. Schon, E. Kaminska, F. Schelter, E. Ponkkonen, E. Korytiakova, S. Schiffers and T. Carell, Angew. Chem., Int. Ed., 2020, 59, 5591–5594 CrossRef.
- A. R. Ramiro and V. M. Barreto, Trends Biochem. Sci., 2015, 40, 172–181 CrossRef CAS.
- E. L. Fritz and F. N. Papavasiliou, Genes Dev., 2010, 24, 2107–2114 CrossRef CAS.
- S. Cortellino, J. Xu, M. Sannai, R. Moore, E. Caretti, A. Cigliano, M. Le Coz, K. Devarajan, A. Wessels, D. Soprano, L. K. Abramowitz, M. S. Bartolomei, F. Rambow, M. R. Bassi, T. Bruno, M. Fanciulli, C. Renner, A. J. Klein-Szanto, Y. Matsumoto, D. Kobi, I. Davidson, C. Alberti, L. Larue and A. Bellacosa, Cell, 2011, 146, 67–79 CrossRef CAS.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS.
- A. Maiti and A. C. Drohat, J. Biol. Chem., 2011, 286, 35334–35338 CrossRef CAS.
- A. Darwanto, J. A. Theruvathu, J. L. Sowers, D. K. Rogstad, T. Pascal, W. Goddard 3rd and L. C. Sowers, J. Biol. Chem., 2009, 284, 15835–15846 CrossRef CAS.
- H. Wu and Y. Zhang, Cell, 2014, 156, 45–68 CrossRef CAS.
- H. Ide and M. Kotera, Biol. Pharm. Bull., 2004, 27, 480–485 CrossRef CAS.
- E. Li and Y. Zhang, Cold Spring Harbor Perspect. Biol., 2014, 6, a019133 CrossRef.
- D. P. Barlow and M. S. Bartolomei, Cold Spring Harbor Perspect. Biol., 2014, 6, a018382 CrossRef.
- C. X. Song and C. He, Trends Biochem. Sci., 2013, 38, 480–484 CrossRef CAS.
- F. Kawasaki, S. Martinez Cuesta, D. Beraldi, A. Mahtey, R. E. Hardisty, M. Carrington and S. Balasubramanian, Angew. Chem., Int. Ed., 2018, 57, 9694–9696 CrossRef CAS.
- E. A. Raiber, R. Hardisty, P. van Delft and S. Balasubramanian, Nat. Rev. Chem., 2017, 1, 0069 CrossRef CAS.
- R. Olinski, M. Starczak and D. Gackowski, Mutat. Res., Rev. Mutat. Res., 2016, 767, 59–66 CrossRef CAS.
- M. Janouskova, Z. Vanikova, F. Nici, S. Bohacova, D. Vitovska, H. Sanderova, M. Hocek and L. Krasny, Chem. Commun., 2017, 53, 13253–13255 RSC.
- Z. Vanikova, M. Janouskova, M. Kambova, L. Krasny and M. Hocek, Chem. Sci., 2019, 10, 3937–3942 RSC.
- D. Globisch, M. Munzel, M. Muller, S. Michalakis, M. Wagner, S. Koch, T. Bruckl, M. Biel and T. Carell, PLoS One, 2010, 5, e15367 CrossRef CAS.
- C. X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C. H. Chen, W. Zhang, X. Jian, J. Wang, L. Zhang, T. J. Looney, B. Zhang, L. A. Godley, L. M. Hicks, B. T. Lahn, P. Jin and C. He, Nat. Biotechnol., 2011, 29, 68–72 CrossRef CAS.
- K. Sato, W. Hirose and A. Matsuda, Curr. Protoc. Nucleic Acid Chem., 2008, 1.21.1–1.21.19 Search PubMed.
- Q. Dai and C. He, Org. Lett., 2011, 13, 3446–3449 CrossRef CAS.
- M. Munzel, D. Globisch, C. Trindler and T. Carell, Org. Lett., 2010, 12, 5671–5673 CrossRef CAS.
- A. S. Schroder, J. Steinbacher, B. Steigenberger, F. A. Gnerlich, S. Schiesser, T. Pfaffeneder and T. Carell, Angew. Chem., Int. Ed., 2014, 53, 315–318 CrossRef.
- A. Ono, N. Haginoya, M. Kiyokawa, N. Minakawa and A. Matsuda, Bioorg. Med. Chem. Lett., 1994, 4, 361–366 CrossRef CAS.
- J. Fujimoto, L. Tran and L. C. Sowers, Chem. Res. Toxicol., 1997, 10, 1254–1258 Search PubMed.
- V. Guerniou, D. Gasparutto, S. Sauvaigo, A. Favier and J. Cadet, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1073–1075 CrossRef CAS.
- R. A. Paselk, Biochem. Educ., 1987, 15, 52 CrossRef.
- Z. Cui, J. A. Theruvathu, A. Farrel, A. Burdzy and L. C. Sowers, Anal. Biochem., 2008, 379, 196–207 CrossRef CAS.
- B. A. Connolly and P. C. Newman, Nucleic Acids Res., 1989, 17, 4957–4974 CrossRef CAS.
- J. I. Kang Jr and L. C. Sowers, Chem. Res. Toxicol., 2008, 21, 1211–1218 Search PubMed.
- M. Aso, T. Kaneko, M. Nakamura, N. Koga and H. Suemune, Chem. Commun., 2003, 1094–1095, 10.1039/b301425h.
- G. J. Crouch and B. E. Eaton, Nucleosides Nucleotides, 1994, 13, 939–944 CrossRef CAS.
- Q. Dai and C. He, Curr. Protoc. Nucleic Acid Chem., 2011, 4.47.1–4.47.18 Search PubMed.
- J. Nielsen, M. Taagaard, J. E. Marugg, J. H. van Boom and O. Dahl, Nucleic Acids Res., 1986, 14, 7391–7403 CrossRef CAS.
- Y. El Safadi, J. C. Paillart, G. Laumond, A. M. Aubertin, A. Burger, R. Marquet and V. Vivet-Boudou, J. Med. Chem., 2010, 53, 1534–1545 CrossRef CAS.
- N. S. Li and J. A. Piccirilli, J. Org. Chem., 2004, 69, 4751–4759 CrossRef CAS.
- S. Tardy-Planechaud, J. Fujimoto, S. S. Lin and L. C. Sowers, Nucleic Acids Res., 1997, 25, 553–559 CrossRef CAS.
- N. D. Sinha, J. Biernat, J. McManus and H. Koster, Nucleic Acids Res., 1984, 12, 4539–4557 CrossRef CAS.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- P. Liu, A. Burdzy and L. C. Sowers, DNA Repair, 2003, 2, 199–210 CrossRef CAS.
- H. Hashimoto, S. Hong, A. S. Bhagwat, X. Zhang and X. Cheng, Nucleic Acids Res., 2012, 40, 10203–10214 CrossRef CAS.
- H. Hashimoto, X. Zhang and X. Cheng, Nucleic Acids Res., 2012, 40, 8276–8284 CrossRef CAS.
- J. Otani, K. Arita, T. Kato, M. Kinoshita, H. Kimura, I. Suetake, S. Tajima, M. Ariyoshi and M. Shirakawa, J. Biol. Chem., 2013, 288, 6351–6362 CrossRef CAS.
- S. Morera, I. Grin, A. Vigouroux, S. Couve, V. Henriot, M. Saparbaev and A. A. Ishchenko, Nucleic Acids Res., 2012, 40, 9917–9926 CrossRef CAS.
- Q. M. Zhang, S. Yonekura, M. Takao, A. Yasui, H. Sugiyama and S. Yonei, DNA Repair, 2005, 4, 71–79 CrossRef CAS.
- M. T. Morgan, M. T. Bennett and A. C. Drohat, J. Biol. Chem., 2007, 282, 27578–27586 CrossRef CAS.
- T. R. Waters and P. F. Swann, J. Biol. Chem., 1998, 273, 20007–20014 CrossRef CAS.
- M. Abu and T. R. Waters, J. Biol. Chem., 2003, 278, 8739–8744 CrossRef CAS.
- K. A. Haushalter, M. W. Todd Stukenberg, M. W. Kirschner and G. L. Verdine, Curr. Biol., 1999, 9, 174–185 CrossRef CAS.
- J. E. Wibley, T. R. Waters, K. Haushalter, G. L. Verdine and L. H. Pearl, Mol. Cell, 2003, 11, 1647–1659 CrossRef CAS.
- M. Matsubara, T. Tanaka, H. Terato, E. Ohmae, S. Izumi, K. Katayanagi and H. Ide, Nucleic Acids Res., 2004, 32, 5291–5302 CrossRef CAS.
- R. J. Boorstein, A. Cummings Jr, D. R. Marenstein, M. K. Chan, Y. Ma, T. A. Neubert, S. M. Brown and G. W. Teebor, J. Biol. Chem., 2001, 276, 41991–41997 CrossRef CAS.
- A. Masaoka, M. Matsubara, R. Hasegawa, T. Tanaka, S. Kurisu, H. Terato, Y. Ohyama, N. Karino, A. Matsuda and H. Ide, Biochemistry, 2003, 42, 5003–5012 CrossRef CAS.
- K. Kemmerich, F. A. Dingler, C. Rada and M. S. Neuberger, Nucleic Acids Res., 2012, 40, 6016–6025 CrossRef CAS.
- L. H. Pearl, Mutat. Res., DNA Repair, 2000, 460, 165–181 CrossRef CAS.
- N. Schormann, R. Ricciardi and D. Chattopadhyay, Protein Sci., 2014, 23, 1667–1685 CrossRef CAS.
- G. Xiao, M. Tordova, J. Jagadeesh, A. C. Drohat, J. T. Stivers and G. L. Gilliland, Proteins, 1999, 35, 13–24 CrossRef CAS.
- G. Ficz and J. G. Gribben, Genomics, 2014, 104, 352–357 CrossRef CAS.
- C. X. Song and C. He, Genome Biol., 2012, 13, 173 CrossRef CAS.
- V. Rusmintratip and L. C. Sowers, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14183–14187 CrossRef CAS.
- H. Kim, J. Park, Y. Jung, S. H. Song, S. W. Han, D. Y. Oh, S. A. Im, Y. J. Bang and T. Y. Kim, Int. J. Oncol., 2010, 36, 1563–1572 CAS.
- N. M. da Costa, A. Hautefeuille, M. P. Cros, M. E. Melendez, T. Waters, P. Swann, P. Hainaut and L. F. Pinto, Cell Cycle, 2012, 11, 4570–4578 CrossRef.
- G. Matlashewski, L. Banks, D. Pim and L. Crawford, Eur. J. Biochem., 1986, 154, 665–672 CrossRef CAS.
- U. Hardeland, C. Kunz, F. Focke, M. Szadkowski and P. Schar, Nucleic Acids Res., 2007, 35, 3859–3867 CrossRef CAS.
- T. Pfaffeneder, F. Spada, M. Wagner, C. Brandmayr, S. K. Laube, D. Eisen, M. Truss, J. Steinbacher, B. Hackner, O. Kotljarova, D. Schuermann, S. Michalakis, O. Kosmatchev, S. Schiesser, B. Steigenberger, N. Raddaoui, G. Kashiwazaki, U. Muller, C. G. Spruijt, M. Vermeulen, H. Leonhardt, P. Schar, M. Muller and T. Carell, Nat. Chem. Biol., 2014, 10, 574–581 CrossRef CAS.
- D. Cortazar, C. Kunz, J. Selfridge, T. Lettieri, Y. Saito, E. MacDougall, A. Wirz, D. Schuermann, A. L. Jacobs, F. Siegrist, R. Steinacher, J. Jiricny, A. Bird and P. Schar, Nature, 2011, 470, 419–423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04161k |
| This journal is © The Royal Society of Chemistry 2020 |