
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyclic diaminocarbene-based Thiele, Chichibabin, and Müller hydrocarbons†
Avijit
Maiti‡
 a,
Shubhadeep
Chandra‡
b,
Biprajit
Sarkar
*b and
Anukul
Jana
*a
a,
Shubhadeep
Chandra‡
b,
Biprajit
Sarkar
*b and
Anukul
Jana
*a
aTata Institute of Fundamental Research Hyderabad, Gopanpally, Hyderabad-500046, Telangana, India. E-mail: ajana@tifrh.res.in
bUniversität Stuttgart, Fakultät Chemie, Lehrstuhl für Anorganische Koordinationschemie, Institut für Anorganische Chemie, Pfaffenwaldring 55, D-70569, Stuttgart, Germany. E-mail: biprajit.sarkar@iac.uni-stuttgart.de
First published on 2nd October 2020
Abstract
Thiele, Chichibabin and Müller hydrocarbons are considered as classical Kekulé diradicaloids. Herein we report the synthesis and characterization of acyclic diaminocarbene (ADC)-based Thiele, Chichibabin, and Müller hydrocarbons. The calculated singlet–triplet energy gaps are ΔES–T = −27.96, −3.70, −0.37 kcal mol−1, respectively, and gradually decrease with the increasing length of the π-conjugated spacer (p-phenylene vs. p,p′-biphenylene vs. p,p′′-terphenylene) between the two ADC-scaffolds. In agreement with the calculations, we also experimentally observed the enhancement of paramagnetic diradical character as a function of the length of the π-conjugated spacer. ADC-based Thiele's hydrocarbon is EPR silent and exhibits very well resolved NMR spectra, whereas ADC-based Müller's hydrocarbon displays EPR signals and featureless NMR spectra at room temperature. The spacer also has a strong influence on the UV-Vis-NIR spectra of these compounds. Considering that our methodology is modular, these results provide a convenient platform for the synthesis of an electronically modified new class of carbon-centered Kekulé diradicaloids.
Introduction
In recent years the chemistry of stable Kekulé diradicaloids has attracted special attention due to their unique properties and their significant importance in modern chemical physics.1 Kekulé diradicaloids possess a characteristic resonance structure between a closed-shell quinonoid and an open-shell diradical form. In this regard, Thiele's hydrocarbon Ia,2 Chichibabin's hydrocarbon Ib,3 and Müller's hydrocarbon Ic4 represent classical examples of Kekulé diradicaloids (Scheme 1). Kekulé diradicaloid analogues of Ia–c have been reported, and these range from polycyclic hydrocarbons II5 to a replacement of the diphenylcarbene-scaffold (Ph2C) by isoelectronic motifs such as aminium (Ar2N+) III,6 N-heterocyclic carbene (NHC) IV,7 and cyclic(alkyl)(amino)carbene (CAAC) V (Scheme 1).8 The properties arising from these Kekulé diradicaloids are strikingly different from each other. Therefore, the changing of the diphenylcarbene-scaffold of Ia–c with isoelectronic motifs should lead to electronically tunable novel diradicaloids.9 | ||
| Scheme 1 Chemical structures of I–VI (R = monoanionic organic substituents). | ||
In the case of carbene chemistry it has been documented that changing of substituents at the carbenic carbon-centre leads to the electronic state alteration from triplet to singlet and vice versa.10 For instance diphenylcarbene is triplet in its ground state and has been studied in matrix isolation experiments.11 Changing the phenyl-substituent with an amino-substituent leads to an isolable singlet carbene namely acyclic diaminocarbene (ADC)12 which has been used as a ligand in transition metal coordination chemistry13 and in catalysis.14 Due to the open-framework of acyclic diaminocarbenes (ADCs), the N–C(carbenic carbon)–N bond angle is wider in comparison to that of cyclic diaminocarbenes. Moreover, the conformational flexibility at the N-centres is much more than in cyclic diaminocarbenes. As a result the steric- as well as electronic-properties of acyclic diaminocarbene (ADC) are very much different from those of cyclic diaminocarbenes.15 For instance ADCs are stronger σ-donors and π-acceptors compared to both cyclic imidazolidin-2-ylidenes, which are non-aromatic 5-membered NHCs with a saturated backbone, and cyclic imidazole-2-ylidenes, which are aromatic 5-membered NHCs with an unsaturated backbone.16 Moreover, the synthetic routes for ADCs are relatively simpler than those of cyclic diaminocarbenes with a huge substrate scope.17 Furthermore, recent results show that carbene-derived diradicaloids18 can function as a new class of singlet fission materials.19 We were thus interested in considering the replacement of diphenylcarbene-scaffold (Ph2C) of Ia–c by an acyclic diaminocarbene (ADC)-motif. The envisaged compounds will represent a new class of diradicaloids with tunable spin states and electronic properties. Herein we present the synthesis and characterization of isolable acyclic diaminocarbene (ADC)-based Thiele, Chichibabin, and Müller hydrocarbons VI as Kekulé diradicaloids (Scheme 1).
Results and discussion
Keeping in mind that acyclic diaminocarbene (ADC)-based Thiele, Chichibabin, and Müller hydrocarbons possess a resonance structure of open-shell diradical form, we have considered the corresponding dications as a surrogate to gain synthetic access to these compounds.20 Accordingly, we have chosen 1,4-dibromobenezene 1a, 4,4′-dibromobiphenyl 1b, and 4,4′′-dibromo-p-terphenyl 1c as readily available precursors, respectively to synthesize the targeted dications in a modular approach (Scheme 2).21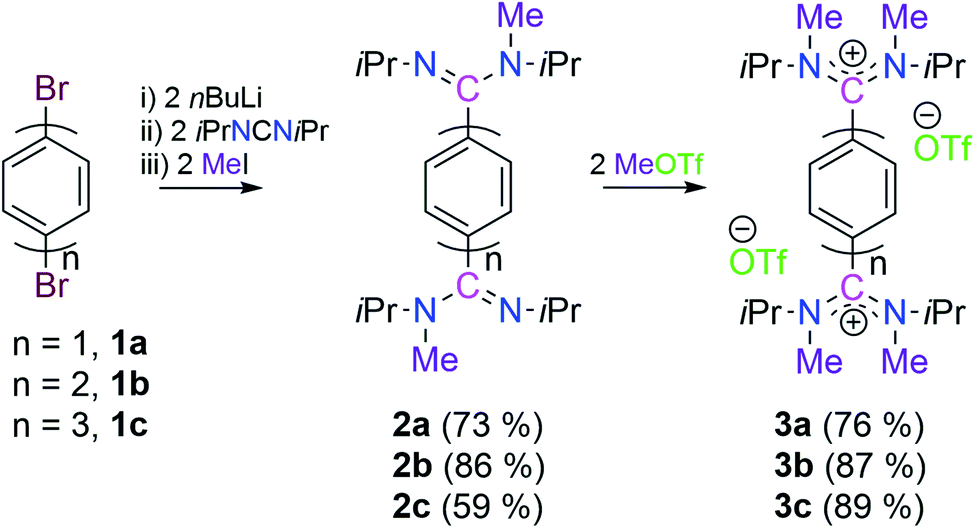 | ||
| Scheme 2 Synthesis of 3a, 3b and 3c. | ||
Three in situ sequential reactions of 1a, 1b, and 1c with two equivalents of nBuLi, N,N′-diisopropylcarbodiimide, and methyl iodide lead to 2a, 2b, and 2c, respectively in a good yield (Scheme 2).21 Formation of 2a, 2b, and 2c has been confirmed by 1H and 13C{1H} NMR spectroscopy. Further the formation of 2b and 2c has been confirmed by the solid-state molecular structure determination (see Fig. S57 in the ESI†).21 The subsequent reactions of 2a, 2b, and 2c with two equivalents of MeOTf lead to the bis-amidinium cations 3a (76%), 3b (87%), and 3c (89%), respectively in a very good yield (Scheme 2).21 Compounds 3a, 3b and 3c have been characterized by solid-state molecular structure analysis (see Fig. S58 in the ESI†)21 along with 1H, 13C{1H} and 19F{1H} NMR spectroscopy, elemental analysis, and HRMS. Among the possible conformations, bis-amidinium dications 3a–3c adopt a pseudo-cis orientation in the solid-state in which four methyl groups are in an anti-arrangement with respect to the π-conjugated spacer.22 The solid state molecular structures of 3a, 3b and 3c reveal the presence of C2-symmetry which also has been reflected in their solution state 1H and 13C{1H} NMR spectra. In the 13C{1H} NMR spectra the most downfield shifted signals are at δ = 169.5, 170.9, and 171.2 ppm, respectively for 3a, 3b and 3c which are for the 13C-nuclei that are flanked between the two nitrogen centres (see Fig. S4, S14 and S23 in the ESI†).
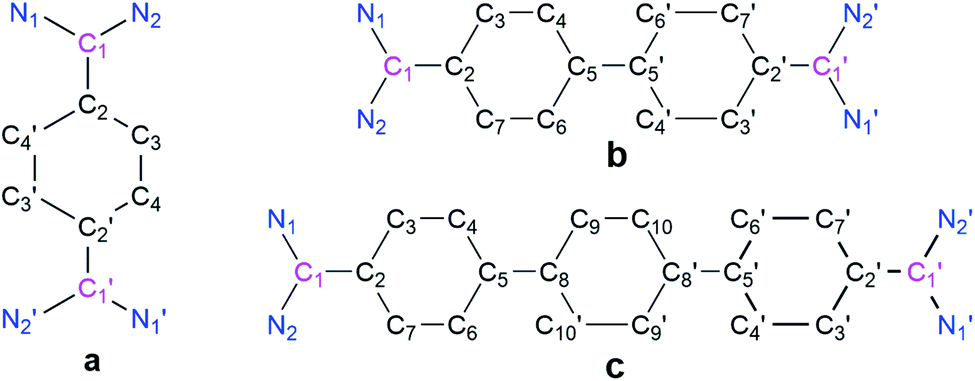
The cyclic voltammograms of 3a–3c were measured in CH3CN/0.1 M Bu4NPF6 using a glassy carbon (GC) working electrode (Fig. 1).21 A first reduction wave for 3a, 3b and 3c is observed at −1.42, −1.61 and −1.82 V vs. FcH/FcH+, respectively. For all three cases the peak-to-peak separation between the forward and the reverse wave is rather small (40–50 mV), and this fact already points to the two-electron nature of the reduction waves. UV-Vis-NIR spectroelectrochemical measurements deliver spectra (see Fig. S44–S46 in the ESI†) after the first reduction waves that match perfectly with the spectra of the isolated two-electron reduced species (vide infra), thus providing further evidence for the two-electron nature of the first reduction waves.21 This observation is similar to what we had recently reported for the dication of compound V (−1.36 V vs. FcH/FcH+), and shows the stability of the two-electron reduced form possibly enabled through a quinoidal structure.8 However, the dications for NHC-based Thiele and Chichibabin hydrocarbons (IVa–b) exhibit two one-electron redox waves.7a,b The first reduction occurs at relatively lower reduction potentials (−0.80 to −1.29 V vs. FcH/FcH+). This could be due to the relatively larger structural reorganization in the case of ADC-systems than that of NHC-systems after reduction which has been evidenced by the change of angle between the planes involving the carbene-scaffold (N1–C1(carbenic carbon)–N2) and adjacent aryl ring (C3–C2–C4′/C7) of starting dications (3a–c) to the corresponding two-electron reduced compounds (4a–c) (see Scheme 3 and Table 1). Compounds 3b and 3c display further irreversible reduction steps (see Fig. S42 and S43 in the ESI†), which we tentatively assign to the reduction of the extended spacers.21
 | ||
| Fig. 1 Cyclic voltammograms of 3a (black), 3b (blue) and 3c (red) displaying the 1st reduction in CH3CN/0.1 M Bu4NPF6 measured at a GC working electrode with 100 mV s−1. | ||
 | ||
| Scheme 3 Synthesis of 4a, 4b and 4c. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | N1–C1 | N2–C1 | C1–C2 | C2–C3 | C3–C4 | C5–C5'/C8 | ∠N1–C1–N2 | ∑N [°] | BLA [Å] | Angle between N1–C1–N2 and C3–C2–C4′/C7 planes |
| a IPr for imidazole-2-ylidene based NHC. b SIPr for imidazolin-2-ylidene based NHC. | ||||||||||
| 3a | 1.324 | 1.321 | 1.489 | 1.389 | 1.376 | — | 123.279 | 359.9 | 0.02 | 64.818 |
| 4a | 1.386 | 1.396 | 1.383 | 1.435 | 1.344 | — | 113.89 | 358.7 | 0.09 | 24.372 |
| Ia | — | — | 1.381 | 1.449 | 1.346 | — | — | — | 0.10 | — |
| IVaIPr | 1.415 | 1.417 | 1.376 | 1.452 | 1.355 | — | 104.577 | 353.9 | 0.09 | 9.171 |
| IVaSIPr | 1.407 | 1.405 | 1.365 | 1.448 | 1.347 | — | 107.878 | 350.9 | 0.10 | 13.263 |
| V | 1.396 | — | 1.381 | 1.452 | 1.352 | — | — | 358.1 | 0.10 | — |
| 3b | 1.334 | 1.320 | 1.489 | 1.390 | 1.377 | 1.480 | 123.448 | 359.9 | 0.02 | 66.674 |
| 4b | 1.382 | 1.396 | 1.387 | 1.437 | 1.347 | 1.407 | 114.11 | 358.5 | 0.09 | 25.079 |
| Ib | — | — | 1.415 | 1.424 | 1.372 | 1.448 | — | — | 0.05 | — |
| IVbIPr | 1.393 | 1.400 | 1.388 | 1.443 | 1.358 | 1.413 | 104.643 | 356.9 | 0.08 | 19.804 |
| IVbSIPr | 1.383 | 1.397 | 1.386 | 1.442 | 1.360 | 1.409 | 107.886 | 351.7 | 0.08 | 15.332 |
| 3c | 1.335 | 1.323 | 1.494 | 1.384 | 1.380 | 1.491 | 123.126 | 359.9 | 0.00 | 62.550 |
| 4c | 1.379 | 1.379 | 1.401 | 1.437 | 1.360 | 1.435 | 114.86 | 358.3 | 0.07 | 28.553 |
| IVcIPr | 1.399 | 1.400 | 1.401 | 1.439 | 1.364 | 1.431 | 104.529 | 358.65 | 0.07 | 5.433 |
| IVcSIPr | 1.382 | 1.391 | 1.398 | 1.436 | 1.361 | 1.433 | 107.985 | 349.9 | 0.07 | 19.031 |
Subsequently, the reduction of 3a, 3b, and 3c with two equivalents of KC8 led to 4a (89%, pale yellow), 4b (53%, dark blue), and 4c (64%, dark green), respectively as crystalline solids (Scheme 3). The 21 1H NMR spectrum of 4a shows a singlet at δ = 6.32 ppm for the central phenyl protons which is very much upfield shifted in comparison to 3a (δ = 7.81 ppm). In the case of 4b two doublets appeared for the central biphenyl protons at δ = 7.30 and 6.53 ppm with a coupling constant of 3J(1H,1H) = 9.6 Hz.
The 13C{1H} NMR spectra of 4a and 4b display downfielded resonances at δ = 147.4 and 150.0 ppm (see Fig. S7 and S17 in the ESI†), respectively for the 13C-nuclei which are flanked between the two nitrogen atoms. In the case of 4c the 1H NMR spectrum at room temperature displays a featureless broad signal only for the alkyl region, and there are no significant signals in the aromatic region (Fig. 2). The absence of the 1H NMR signals of the aromatic hydrogens at room temperature indicates a diradical contribution of 4c even at room temperature.23 On lowering the temperature to 233 K it shows proton signals at δ = 7.15, 6.96, and 6.31 ppm with equal intensity. The intensities of these signals also further increase with lowering the temperatures (Fig. 2). There was no 13C{1H} NMR signal for 4c even at 218 K (see Fig. S27 in the ESI†). In the case of 4b at higher temperatures the signal intensity gets reduced and peaks get broad due to a higher population of the paramagnetic triplet diradical state (see Fig. S18 in the ESI†).21
 | ||
| Fig. 2 Selected region of the temperature-dependent 1H NMR spectra of 4c in THF-d8. | ||
Compounds 4a and 4b are well soluble in benzene, toluene, and THF. This is in contrast to 4c which is soluble in THF only. Compounds 4a, 4b, and 4c are stable both in solution and in the solid state under an inert atmosphere for at least a month. 4a and 4b are stable in solution and in the solid state even after six months under an inert atmosphere. 4a and 4b are also stable after melting at 136 °C and 118 °C, respectively which was confirmed by measuring 1H NMR spectra (see Fig. S8 and S19 in the ESI†). Compound 4c starts to slowly decompose at room temperature after 5–6 days, whereas it is quite stable at −30 °C. It is stable for a month in the solid state but decomposes after melting at 118 °C. However, compounds 4a, 4b, and 4c are air-sensitive and decomposed within a minute in the solution state (see Fig. S39 and S40 in the ESI†).
The solid-state structures of 4a, 4b, and 4c (Fig. 3) suggest that the C–N bond lengths (1.38 to 1.39 Å) are longer than those in 3a, 3b, and 3c (1.32 to 1.33 Å) but shorter than those of the corresponding NHC-analogues IVa–c (Table 1).7 The N–C–N bond angles of 4a, 4b, and 4c are 113.89 (2), 114.11(1), and 114.86(1)°, respectively. These angles are smaller than that in bis(diisopropylamino)carbene (∠N–C–N = 121.0°)24 but close to that in N,N′-ditertiarybutyl-N,N′-diphenyl acyclic diaminocarbene-coordinated AuCl (∠N–C–N = 115.0°).25 Among the possible conformations, 4a–4c adopt a pseudo-cis orientation in which all four methyl groups are in a syn-arrangement with respect to the π-conjugated spacer, which is in contrast to that of 3a–3c. In 4a the dihedral angle between the acyclic diaminocarbene-scaffold and the central phenyl ring is 24.36° and the two acyclic diaminocarbene-motifs are parallel to each other with a distance of 0.261 Å. In 4b the two phenyl rings of the biphenyl motif are coplanar and the two acyclic diaminocarbene-scaffolds are parallel to each other with a distance of 0.661 Å. The dihedral angle between the acyclic diaminocarbene-scaffold and the central biphenyl ring is 25.25°. The C5–C5′ bond length is 1.407(19) Å which is shorter in comparison with that of Ib (1.445 Å) (Table 1).26
 | ||
| Fig. 3 Solid-state structures of 4a (left), 4b (top-right), and 4c (bottom right). Hydrogen atoms are omitted for clarity. | ||
In 4c the two terminal benzene rings of the p-terphenyl motif are parallel to each other with a distance of 0.597 Å, while the middle benzene ring is twisted with respect to the terminal benzene rings. The dihedral angle between the middle benzene ring and the terminal benzene rings is 6.21°. The dihedral angle between the acyclic diaminocarbene-scaffold and the connected phenyl ring is 28.89°. The C5–C8 bond length is 1.435(23) Å which is longer than that of C5–C5′ in 4b (1.407(19) Å) but very similar to that of Ib (1.445 Å) (Table 1). The sum of the bond angles around C1 is 360°, and N1/N2 are 358.6° (4a), 358.8° (4b), and 358.1° (4c) indicating the planarity of the respective centres. The bond length alternation (BLA) of the aromatic π-conjugated spacer is different for 4a (0.10 Å), 4b (0.08 Å), and 4c (0.06, and 0.04 for the middle phenyl ring). The BLA for 4a is same as that for Ia and for 4b is larger than that of Ib (0.05 Å) (Table 1).26
DFT calculations at the PBE0/def2-TZVP level of theory suggest a closed-shell singlet ground state for 4a–c. The singlet triplet energy gaps for 4a, 4b, and 4c are ΔES–T = −27.96, −3.70, −0.37 kcal mol−1, respectively.21 This indicates that the singlet–triplet energy gap gradually decreases with the increasing length of the π-conjugated spacer (p-phenylene vs. p,p′-biphenylene vs. p,p′′-terphenylene) between the two ADC-scaffolds. To further investigate the electronic structures we have also estimated the biradical character for compounds 4a–c. Theoretical calculations at the PBE0/def2-TZVP level of theory suggest that 4a has negligible biradical character while compounds 4b and 4c have a biradical character of 43% and 65% respectively.21 Accordingly, compound 4a is EPR silent just like V8 whereas 4b and 4c exhibit EPR signals both in the solid and solution states at 298 K with a g-value of 2.004 (Fig. 4 and see Fig. S47 in the ESI†).
 | ||
| Fig. 4 Solid state EPR spectra measured at room temperature and the calculated spin density plot (with an isovalue of 0.004) at the PBE0/def2-TZVP level of theory for 4b (a) and 4c (b). | ||
This fact indicates that even though the singlet state is the ground state for 4b (ΔES–T = −3.70 kcal mol−1) and 4c (ΔES–T = −0.37 kcal mol−1), due to the small singlet–triplet gap, the triplet state is populated to a certain extent at room temperature. Similar observations were made in related systems that were investigated recently.7c,9a,27 This is not the case for 4a which has a relatively larger singlet triplet gap (ΔES–T = −27.96 kcal mol−1).21 Gratifyingly, the results from the EPR experiments fit nicely with the observations from the NMR experiments (vide supra). Additionally, fitting of the variable temperature EPR data with the Bleaney–Bower equation furnished further information about the singlet–triplet gaps of ΔES–T = −2.3 kcal mol−1 for 4b and ΔES–T = −0.65 kcal mol−1 for 4c which also indicate significant population of the triplet state at room temperature (see Fig. S48 and S49 in the ESI†).21 The obtained singlet–triplet energy gaps from the solid-state variable temperature EPR study for 4b and 4c are very close to those of theoretically calculated values.21
The above observations indicate that the enhancement of paramagnetic triplet diradical character occurs with increasing the length of the π-conjugated spacer. Further support for this hypothesis comes from the bond lengths between the carbenic carbon-C1 and the attached carbon-C2 of the π-conjugated spacer (for 4a: 1.3827(38) Å, 4b: 1.3871(19), and 4c: 1.4009(22) Å) (Table 1). Spin density calculations of 4a–4c suggest that considerable unpaired electron density is located at the carbon-centre which is flanked between two N-centres (Fig. 4 and see Fig. S50 in the ESI†).21
The UV/Vis spectra of compounds 4a, 4b, and 4c exhibit their main absorption bands at λmax = 436 (ε = 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 185 L mol−1 cm−1), 591 (ε = 46656 L mol−1 cm−1), and 688 (ε = 44680 L mol−1 cm−1) nm, respectively (Fig. 5). TD-DFT calculations suggest these to be HOMO–LUMO transitions (see Fig. S51, S53 and S55 and Tables S3, S6, and S9 in the ESI†).21 This clearly shows that upon increasing the length of the aromatic π-conjugated spacer between the two acyclic diaminocarbene-scaffolds the HOMO–LUMO gap decreases.
185 L mol−1 cm−1), 591 (ε = 46656 L mol−1 cm−1), and 688 (ε = 44680 L mol−1 cm−1) nm, respectively (Fig. 5). TD-DFT calculations suggest these to be HOMO–LUMO transitions (see Fig. S51, S53 and S55 and Tables S3, S6, and S9 in the ESI†).21 This clearly shows that upon increasing the length of the aromatic π-conjugated spacer between the two acyclic diaminocarbene-scaffolds the HOMO–LUMO gap decreases.
 | ||
| Fig. 5 Comparison of the UV/Vis spectra of 4a, 4b, and 4c in THF at room temperature. | ||
Remarkably, compound 4c displays additional absorption at λmax = 879 nm (ε = 10572 L mol−1 cm−1) (Fig. 5). TD-DFT calculations on the triplet state of all three molecules suggest long wavelength bands. We hypothesize that the observance of a long wavelength band for 4c which also absorbs in the NIR region is related to the population of the triplet state for this molecule. These UV-Vis-NIR spectra match well with those obtained from the corresponding UV-Vis-NIR spectroelectrochemical measurements (see Fig. S44–S46 in the ESI†).21 Thus, the absorption maxima of these species can be tuned over a range of greater than 250 nm. Intriguingly, 4c, which displays the highest diradicaloid character displays absorptions that go deep into the NIR region.
Conclusions
In conclusion, we have designed and synthesized the first stable acyclic diaminocarbene (ADC)-based Thiele, Chichibabin, and Müller hydrocarbons in a modular approach. The ADC-analogues of the Chichibabin, and Müller hydrocarbons display population of the triplet state at room temperature, whereas the ADC-analogue of the Thiele hydrocarbon exhibits diamagnetic character. The straightforward synthetic methodology revealed in this study, and the resulting tuning of the spin states and the color of these compounds will be instrumental for the generation of new classes of carbon centre based Kekulé diradicaloids and polyradicaloids.28Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by intramural funds at the Tata Institute of Fundamental Research (TIFR) Hyderabad, Gopanpally, Hyderabad-500046, Telangana, India from the Department of Atomic Energy (DAE), Government of India, India and SERB (CRG/2019/003415), India. The National Facility for High-Field NMR, TIFR-Hyderabad, is highly acknowledged for the very convenient access to NMR spectrometers. Prof. Dr Andreas Köhn from the Institute of Theoretical Chemistry, University of Stuttgart is kindly acknowledged for valuable suggestions regarding the calculations of the biradical character. We are grateful to the reviewers for their critical insights to improve the quality of the manuscript.Notes and references
- Selected references are: (a) T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. D. Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291–11351 CrossRef CAS; (b) M. Abe, Diradicals, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS; (c) K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. R. Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, J. Am. Chem. Soc., 2020, 142, 4329–4340 CrossRef CAS; (d) G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS; (e) K. Kamada, S.-i. Fuku-en, S. Minamide, K. Ohta, R. Kishi, M. Nakano, H. Matsuzaki, H. Okamota, H. Higashikawa, K. Inoue, S. Kojima and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 232–241 CrossRef CAS; (f) C. Zhang, S. M. Rivero, W. Liu, D. Casanova, X. Zhu and J. Casado, Angew. Chem., Int. Ed., 2019, 58, 11291–11295 CrossRef CAS; (g) J. L. Zafra, L. Qiu, N. Yanai, T. Mori, M. Nakano, M. P. Alvarez, J. T. L. Navarrete, C. J. Gómez-García, M. Kertesz, K. Takimiya and J. Casado, Angew. Chem., Int. Ed., 2016, 55, 14563–14568 CrossRef CAS; (h) S. Dong, T. Y. Gopalakrishna, Y. Han, H. Phan, T. Tao, Y. Ni, G. Liu and C. Chi, J. Am. Chem. Soc., 2019, 141, 62–66 CrossRef CAS; (i) G. E. Rudebusch, G. L. Espejo, J. L. Zafra, M. P. Alvarez, S. N. Spisak, K. Fukuda, Z. Wei, M. Nakano, M. A. Petrukhina, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 12648–12654 CrossRef CAS; (j) X. Zhu, H. Tsuji, K. Nakabayashi, S. Ohkoshi and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 16342–16345 CrossRef CAS; (k) X. Hu, W. Wang, D. Wang and Y. Zheng, J. Mater. Chem. C, 2018, 6, 11232–11242 RSC.
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS.
- E. Müller and H. Pfanz, Ber. Dtsch. Chem. Ges. B, 1941, 74, 1051–1074 CrossRef.
- (a) W. Zeng, T. Y. Gopalakrishna, H. Phan, T. Tanaka, T. S. Herng, J. Ding, A. Osuka and J. Wu, J. Am. Chem. Soc., 2018, 140, 14054–14058 CrossRef CAS; (b) T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
- (a) Y. Su, X. Wang, Y. Li, Y. Song, Y. Sui and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 1634–1637 CrossRef CAS; (b) G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS.
- (a) D. Rottschäfer, N. K. T. Ho, B. Neumann, H.-G. Stammler, M. von. Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef; (b) D. Rottschafer, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem. Sci., 2018, 9, 4970–4976 RSC; (c) D. Rottschäfer, J. Busch, B. Neumann, H.-G. Stammler, M. von. Gastel, R. Kishi, M. Nakano and R. S. Ghadwal, Chem.–Eur. J., 2018, 24, 16537–16542 CrossRef.
- A. Maiti, J. Stubbe, N. I. Neuman, P. Kalita, P. Duari, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Angew. Chem., Int. Ed., 2020, 59, 6729–6734 CrossRef CAS.
- (a) J. Wang, X. Xu, H. Phan, T. S. Herng, T. Y. Gopalakrishna, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 14154–14158 CrossRef CAS; (b) M. A. Majewski, P. J. Chmielewski, A. Chien, Y. Hong, T. Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stępień, Chem. Sci., 2019, 10, 3413–3420 RSC.
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS; (b) K. Hirai, T. Itoh and H. Tomioka, Chem. Rev., 2009, 109, 3275–3332 CrossRef CAS.
- M. S. Platz, V. P. Senthilnathan, B. B. Wright and C. W. McCurdy Jr, J. Am. Chem. Soc., 1982, 104, 6494–6501 CrossRef CAS.
- (a) R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schütz, Angew. Chem., Int. Ed., 2004, 43, 5896–5911 CrossRef CAS; (b) J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS; (c) T. Schulz, D. Weismann, L. Wallbaum, R. Guthardt, C. Thie, M. Leibold, C. Bruhn and U. Siemeling, Chem.–Eur. J., 2015, 21, 14107–14121 CrossRef CAS.
- (a) V. P. Boyarskiya, K. V. Luzyanina and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef; (b) M. A. Kinzhalov and K. V. Luzyanin, Coord. Chem. Rev., 2019, 399, 213014 CrossRef CAS.
- (a) B. Dhudshia and A. N. Thadani, Chem. Commun., 2006, 668–670 RSC; (b) L. M. Slaughter, ACS Catal., 2012, 2, 1802–1816 CrossRef CAS.
- D. Martin, Y. Canac, V. Lavallo and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 5023–5030 CrossRef CAS.
- K. Denk, P. Sirsch and W. A. Herrmann, J. Organomet. Chem., 2002, 649, 219–224 CrossRef CAS.
- R. W. Alder, M. E. Blake, S. Bufali, C. P. Butts, A. G. Orpen, J. Schütz and S. J. Williams, J. Chem. Soc., Perkin Trans. 1, 2001, 1586–1593 RSC.
- (a) B. M. Barry, R. G. Soper, J. Hurmalainen, A. Mansikkamäki, K. N. Robertson, W. L. McClennan, A. J. Veinot, T. L. Roemmele, U. Werner-Zwanziger, R. T. Boeré, H. M. Tuononen, J. A. C. Clyburne and J. D. Masuda, Angew. Chem., Int. Ed., 2018, 57, 749–754 CrossRef CAS; (b) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS.
- (a) J. Messelberger, A. Grünwald, P. Pinter, M. M. Hansmann and D. Munz, Chem. Sci., 2018, 9, 6107–6117 RSC; (b) A. Japahuge, S. Lee, C. H. Choi and T. Zeng, J. Chem. Phys., 2019, 150, 234306 CrossRef; (c) T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020, 59, 7906–7914 CrossRef CAS.
- (a) D. Mandal, S. Sobottka, R. Dolai, A. Maiti, D. Dhara, P. Kalita, R. S. Narayanan, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Sci., 2019, 10, 4077–4081 RSC; (b) M. K. Nayak, J. Stubbe, N. I. Neuman, R. S. Narayanan, S. Maji, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Chem.–Eur. J., 2020, 26, 4425–4431 CrossRef CAS.
- See the ESI† for the experimental details, analytical data, NMR spectra, UV/Vis spectra, X-ray crystallographic details and details of theoretical calculations.
- (a) E. L. Rosen, M. D. Sanderson, S. Saravanakumar and C. W. Bielawski, Organometallics, 2007, 26, 5774–5777 CrossRef CAS; (b) M. S. Collins, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Organometallics, 2010, 29, 3047–3053 CrossRef CAS.
- (a) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS; (b) K. Kamada, S. Fuku-en, S. Minamide, K. Ohta, R. Kishi, M. Nakano, H. Matsuzaki, H. Okamoto, H. Higashikawa, K. Inoue, S. Kojima and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 232–241 CrossRef CAS; (c) Y. Su, X. Wang, X. Zheng, Z. Zhang, Y. Song, Y. Sui, Y. Li and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 2857–2861 CrossRef CAS; (d) G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123 CrossRef CAS.
- H. Seo, B. P. Roberts, K. A. Abboud, K. M. Merz Jr and S. Hong, Org. Lett., 2010, 12, 4860–4863 CrossRef CAS.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004–6011 CrossRef CAS.
- (a) C. Saalfrank, F. Fantuzzi, T. Kupfer, B. Ritschel, K. Hammond, I. Krummenacher, R. Bertermann, R. Wirthensohn, M. Finze, P. Schmid, V. Engel, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., DOI:10.1002/anie.202008206; (b) C. H. E. Chow, Y. Han, H. Phan and J. Wu, Chem. Commun., 2019, 55, 9100–9103 RSC.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data, NMR spectra, UV/Vis spectra, X-ray crystallographic details and details of theoretical calculations. CCDC 2010337 (2b), 2010338 (2c), 1983573 (3a), 1983580 (3b), 1983581 (3c), 1983587 (4a), 1983586 (4b) and 1983588 (4c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03622f |
| ‡ A. M. and S. C. contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |