
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct electrochemical defluorinative carboxylation of α-CF3 alkenes with carbon dioxide†
Xiao-Tong
Gao
a,
Zheng
Zhang
a,
Xin
Wang
 b,
Jun-Song
Tian
a,
Shi-Liang
Xie
a,
Feng
Zhou
*a and
Jian
Zhou
ac
b,
Jun-Song
Tian
a,
Shi-Liang
Xie
a,
Feng
Zhou
*a and
Jian
Zhou
ac
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, P. R. China. E-mail: fzhou@chem.ecnu.edu.cn
bCollege of Chemistry, Sichuan University, Chengdu, Sichuan 610064, P. R. China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 11th September 2020
Abstract
An unprecedented γ-carboxylation of α-CF3 alkenes with CO2 is reported. This approach constitutes a rare example of using electrochemical methods to achieve regioselectivity complementary to conventional metal catalysis. Accordingly, using platinum plate as both a working cathode and a nonsacrificial anode in a user-friendly undivided cell under constant current conditions, the γ-carboxylation provides efficient access to vinylacetic acids bearing a gem-difluoroalkene moiety from a broad range of substrates. The synthetic utility is further demonstrated by gram-scale synthesis and elaboration to several value-added products. Cyclic voltammetry and density functional theory calculations were performed to provide mechanistic insights into the reaction.
Introduction
The carboxylation of organic halides using CO2 as an abundant and nontoxic C1 synthon1 is an important strategy to access carboxylic acids, which are widely distributed in natural products and pharmaceuticals.2 Despite good progress in the carboxylation of carbon–hetero bonds,3 the activation of C–F bonds for reaction development is largely undeveloped.4 This is possibly because the C–F bond is the strongest carbon–hetero bond with a high bond dissociation energy, the activation of which is kinetically unfavorable since fluorine is neither a good Lewis base nor a good leaving group.5 In this context, the selective C–F bond carboxylation of lightly fluorinated compounds could facilitate access to fluorinated carboxylic acids that are much sought-after substances for organic synthesis, materials science, and medicinal chemistry.6 However, to date, only a limited number of catalytic processes have been realized (Scheme 1A).7 Feng and co-workers combined photoredox/Pd dual catalysis to realize an sp2 C–F bond carboxylation of gem-difluoroalkenes with moderate Z/E selectivity.7a A Cu-catalyzed formal carboxylation was reported by Yu7b and by us7c respectively, in which carboxylation of the vinylboronate intermediate yielded α-fluoroacrylic acids with high Z-selectivity. Yu and co-workers also achieved a formal sp3 C–F bond carboxylation of α-CF3 styrenes, which was conducted at 80 °C with 1.5 equivalents of diboron reagent and 3.0 equivalents of base, giving α,α-difluorocarboxylates regioselectively.7b Because the C–F bond cleavage of α-CF3 alkenes might lead to both α- and γ-carboxylation, it is interesting to develop γ-carboxylation of α-CF3 alkenes using CO2 for the synthesis of carboxylic acids with a gem-difluoroalkene moiety. As a carbonyl bioisostere with less susceptibility to in vivo metabolism, gem-difluoroalkene is a prominent structural motif that is found widely in biologically active compounds (Scheme 1C).8 Moreover, they are versatile fluorinated building blocks in organic synthesis.9 Therefore, the efficient γ-carboxylation of α-CF3 alkenes using CO2 under mild conditions with broad substrate scope is highly desirable.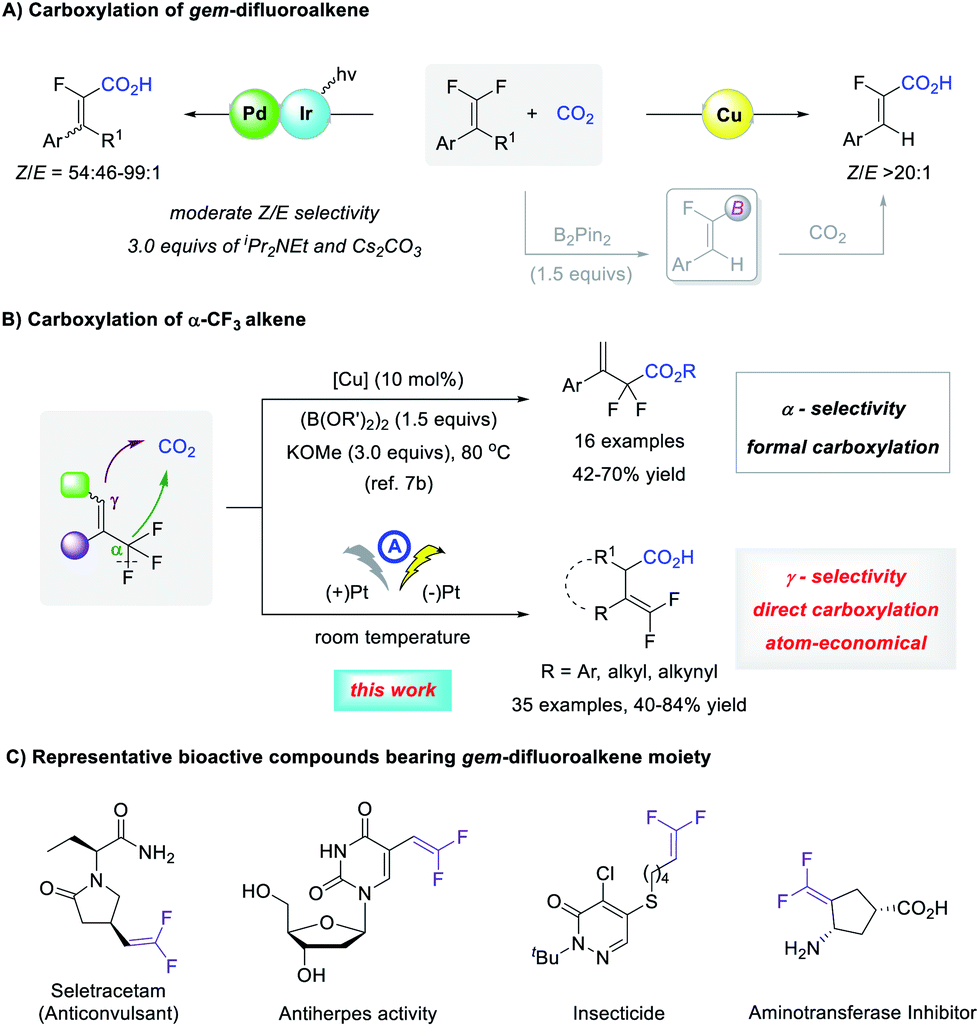 | ||
| Scheme 1 C–F bond carboxylation with CO2. | ||
Recently, increasing attention has been paid to synthetic organic electrochemistry.10 Using electricity as a driving force, the use of toxic or expensive reducing agents might be avoided, and room temperature is often sufficient to promote the reaction. Interestingly, electrochemical processes facilitated access to high-energy species or new mediators, thus affording opportunities that conventional chemistry may not have achieved.11 Intrigued by these attractive features, we postulated that electrochemistry might be a powerful strategy to develop direct and green defluorinative carboxylation of α-CF3 alkenes to secure a level of γ-carboxylation unattainable by copper catalysis and avoid the use of diboron reagent and bases. Here, we wish to report our results in electrochemical γ-selective carboxylation of α-CF3 alkenes with CO2 to structurally diverse γ,γ-difluoro vinylacetic acids with good functional group tolerance, using platinum plate as both cathode and nonsacrificial anode under mild conditions (Scheme 1B).
Currently, most electrocarboxylations are conducted with sacrificial anodes and/or require (quasi-)divided cell to forestall the undesired oxidation of starting material or carboxylic acid products.12 From the viewpoint of practicability and sustainability, the development of non-sacrificial metal system is more desirable.13 Just recently, Malkov and Buckley achieved highly regioselective electrosynthetic hydrocarboxylation of β,β-trisubstituted alkenes and conjugated dienes using non-sacrificial anode system.14 Encouraged by these elegant advances in nonsacrificial metal-based electrochemical carboxylation of alkenes,15 along with the seminal study on electrochemical C–F bond carboxylation of benzotrifluoride by Troupel,16 we tried to develop electrochemical carboxylation of α-CF3 alkenes using a nonsacrificial anode, with our interest in chemical fixation of CO2 to value-added chemicals.17 Accordingly, the reaction of α-CF3 substituted styrene 1a and bubbling CO2 was undertaken in DMF containing Et4NOTs to evaluate different nonsacrificial anodes, at a constant current of 8 mA in an undivided cell, with Pt-plate as cathode (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Anode | Cathode | Electrolyte | Solvent | Z (mA) | Yieldb (%) |
| a Reaction conditions: electrolyte (0.06 M), CO2 bubbling in solvent (6 mL). b Isolated yield. c With nBu4NClO4 (0.07 M), DMF (7 mL). d 7 hours. | ||||||
| 1 | C | Pt | Et4NOTs | DMF | 8 | 32 |
| 2 | RVC | Pt | Et4NOTs | DMF | 8 | 14 |
| 3 | Pt | Pt | Et4NOTs | DMF | 8 | 57 |
| 4 | Pt | C | Et4NOTs | DMF | 8 | 50 |
| 5 | Pt | RVC | Et4NOTs | DMF | 8 | 38 |
| 6 | Pt | Pt | Et4NOTs | DMF | 10 | 54 |
| 7 | Pt | Pt | Et4NOTs | DMF | 6 | 42 |
| 8 | Pt | Pt | n Bu4NOTs | DMF | 8 | 60 |
| 9 | Pt | Pt | n Bu4NClO4 | DMF | 8 | 72 |
| 10 | Pt | Pt | n Bu4NClO4 | DMA | 8 | 59 |
| 11 | Pt | Pt | n Bu4NClO4 | DCE | 8 | 20 |
| 12 | Pt | Pt | n Bu4NClO4 | THF | 8 | 70 |
| 13c | Pt | Pt | n Bu4NClO4 | DMF | 8 | 83 |
| 14c,d | Pt | Pt | n Bu4NClO4 | DMF | 8 | 82 |
Results and discussion
First, we evaluated graphite, RVC and Pt anodes, and found that the reaction indeed proceeded at room temperature for 4 hours, with a total charge of 6 Faraday/mol to yield the γ-carboxylation product 2a in 32, 14, and 57% yields, respectively, and no α-carboxylation adducts were detected (entries 1–3). This result unambiguously supported our working hypothesis and encouraged us to conduct further optimization studies using the Pt anode. Varying the cathode from Pt-plate to graphite or RVC gave no better results (entries 4 and 5). Increasing the current to 10 mA resulted in almost no change in yield but decreasing the current to 6 mA led to a reduction in the yield to 42% due to incomplete reaction (entries 6 and 7). Since the supporting electrolyte could affect the local environment near the electrode as part of the electrical double-layer,18 we next evaluated its influence. Using nBu4NClO4 as the electrolyte, the yield of 2a improved significantly to 72% (entry 9). The solvent effects were also investigated, but DMF still proved to be the solvent of choice (entries 10–12 vs. 9). Finally, by increasing the electrolyte concentration to 0.07 M and performing the reaction with an ElectraSyn 2.0 instrument (see ESI†), the desired carboxylic acid, 2a could be obtained in 83% yield (entry 13). To investigate the stability of the carboxylation product under constant current conditions in an undivided cell,19 we extended the reaction time to 7 hours and found that almost the same yield of 2a was obtained as that after 4 hours (entry 14 vs. 13). This indicated the carboxylation product was sufficiently stable and did not decompose during the reaction (for detail of optimization, see ESI†).Having established the optimal reaction conditions, we next evaluated the scope of the reaction with respect to α-CF3 styrene derivatives. With various substituents on the phenyl rings, including methyl, tert-butyl, phenyl, terminal alkene, halogen, ether, cyano, and ester groups, alkenes 1b–r readily afforded the corresponding γ,γ-difluoro vinylacetic acids, 2b–r in 45–84% yields. The electronic and steric effects of the phenyl substituents had little impact on the carboxylation. For instance, the reaction of isomeric substrates with a methyl, chloro, fluoro, or OCF3 group at the para- or meta-position proceeded smoothly to afford the corresponding products 2b, 2c, 2j–l and 2p–r in similarly good to high yields. The α-CF3 alkene derivatives bearing 2-naphthyl, 2-thiophenyl, or 2-benzofuranyl group also worked well to furnish 2s–u in 40–62% yields.
Notably, α-CF3 alkenes with α-alkyl substituents were also viable substrates. For example, alkenes bearing an α-phenethyl, phenylpropyl, or n-nonyl group produced the γ-carboxylation products 3a–c in 46–57% yields. Furthermore, alkenes with an α-alkynyl moiety worked well to give the desired acids, 3d and 3e in 48 and 65% yields, respectively. Trisubstituted alkenes were further investigated, furnishing corresponding acids 3f–i in 40–56% yields. Cyclic alkenes based on 1,2-dihydronaphthalene skeleton were amenable, giving 3j and 3k in 52 and 55% yields, respectively. To our delight, several complex substrates derived from β-D-glucose, estrone and fructose also reacted well, affording the desired adducts 4a–c in reasonable yields. These results clearly demonstrated the good functional group tolerance of our method. For some substrates indicated in Table 2, the addition of H2O was beneficial for the reaction yield, but the reason for this is not clear.20
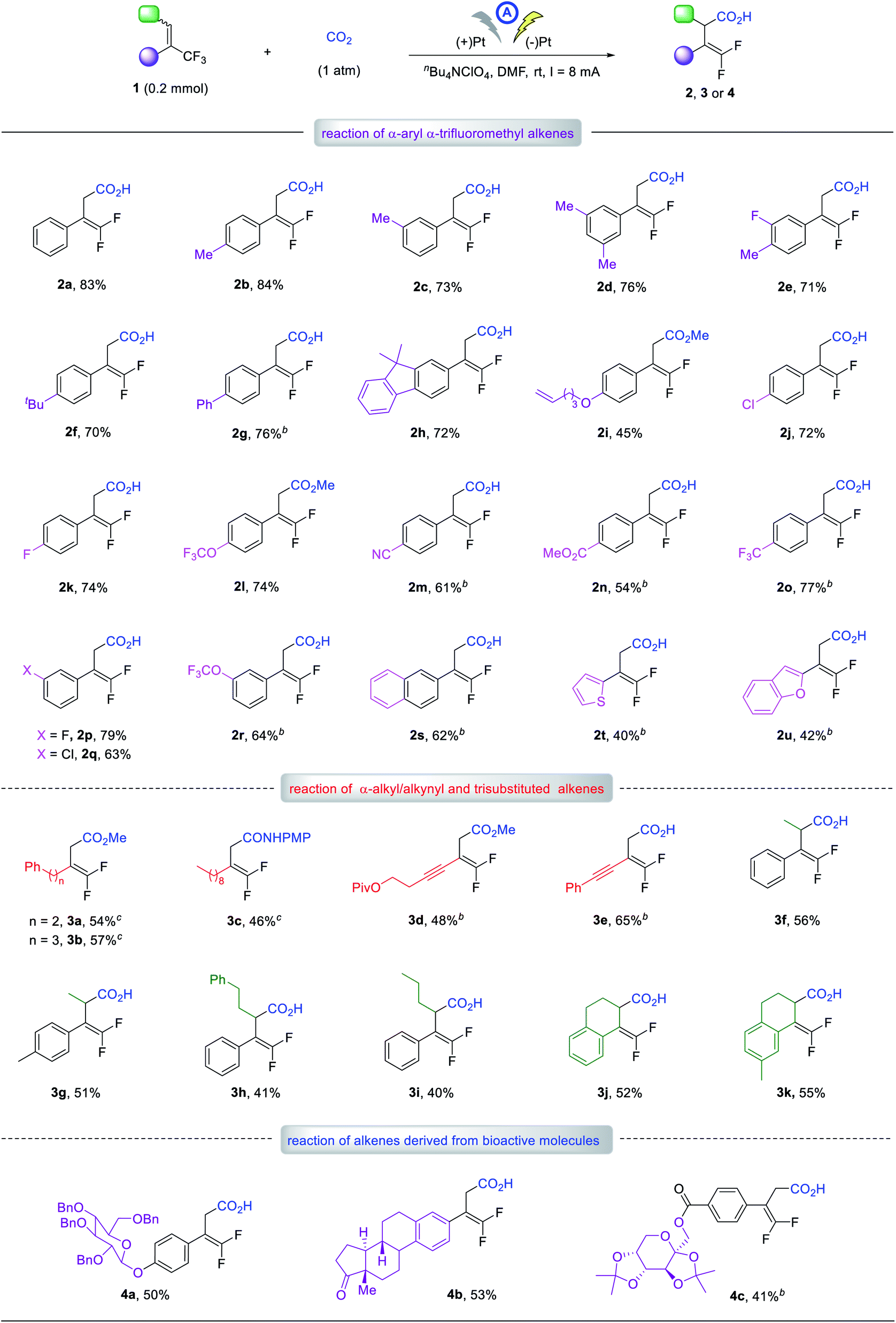
Given that a wide range of functional groups are tolerated, such as alkene, alkyne, and halogen groups, this methodology should be orthogonal to classical cross-coupling chemistry, which would further extend its synthetic utility. Also noteworthy was the perfect regioselectivity: only γ-carboxylation occurred for all the reactions discussed above, and no α-carboxylation products were detected. In addition, for substrates bearing a fluoro, CF3, or OCF3 group, possessing different sp2 or sp3 C–F bonds, the defluorinative carboxylation occurred at the α-CF3 alkenes moiety exclusively.
To further demonstrate the practicability of the developed electrochemical carboxylation, a gram-scale reaction of 1a was conducted on a 6.0 mmol scale, and the product 2a was isolated in 0.93 g with comparable yield (78%; Scheme 2). Moreover, the thus obtained carboxylic acid could be readily elaborated to valuable fluorine-containing molecules. Under Pd/C catalysis, the alkene moiety of 2a could be readily hydrogenated to give β-difluoromethyl carboxylic acid 5 in 90% yield. The methylation and subsequent TBAF promoted olefin isomerization delivered 6 in 78% yield. Condensation with amine afforded direct access to amide 7 in 92% yield, and reduction of the carboxylic acid moiety with LiAlH4 delivered alcohol 8 in 82% yield. The α-fluoro dihydrofuran 9 could be obtained in 40% yield via LiAlH4-mediated reduction and base-promoted defluorinative cyclization.
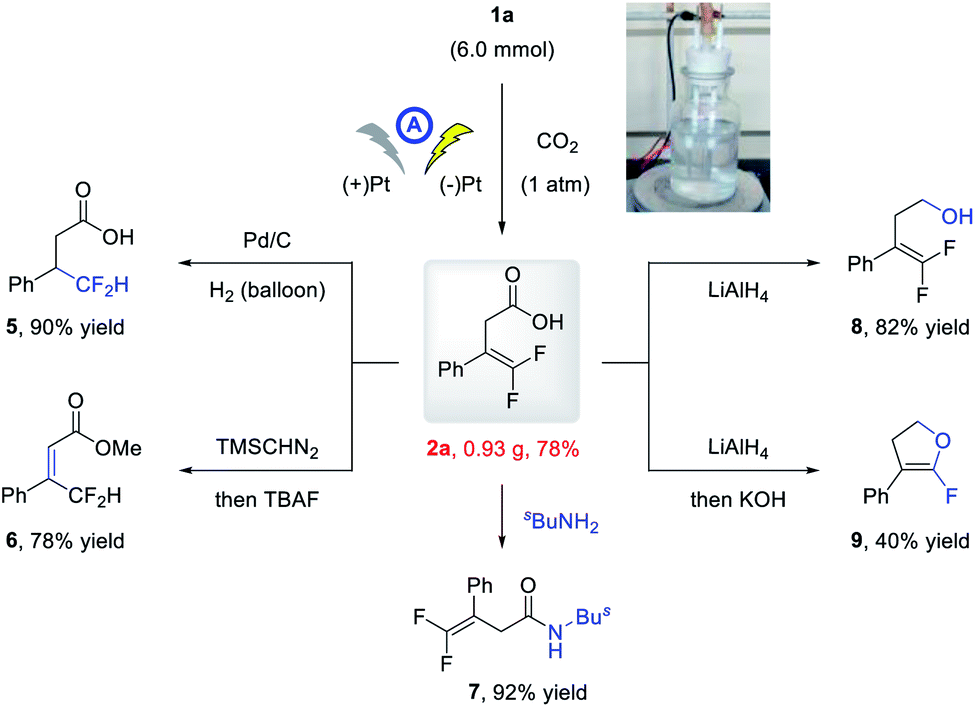 | ||
| Scheme 2 Synthetic elaboration of 2a. | ||
The reaction mechanism was then studied experimentally and computationally to shed light on the two attractive features of our protocol: the γ-carboxylation complementary to the α-carboxylation obtained by copper catalysis,7b and the obviation of a sacrificial anode. First, cyclic voltammetry (CV) analyses were conducted to investigate the electrochemical process on the cathode (Fig. 1).
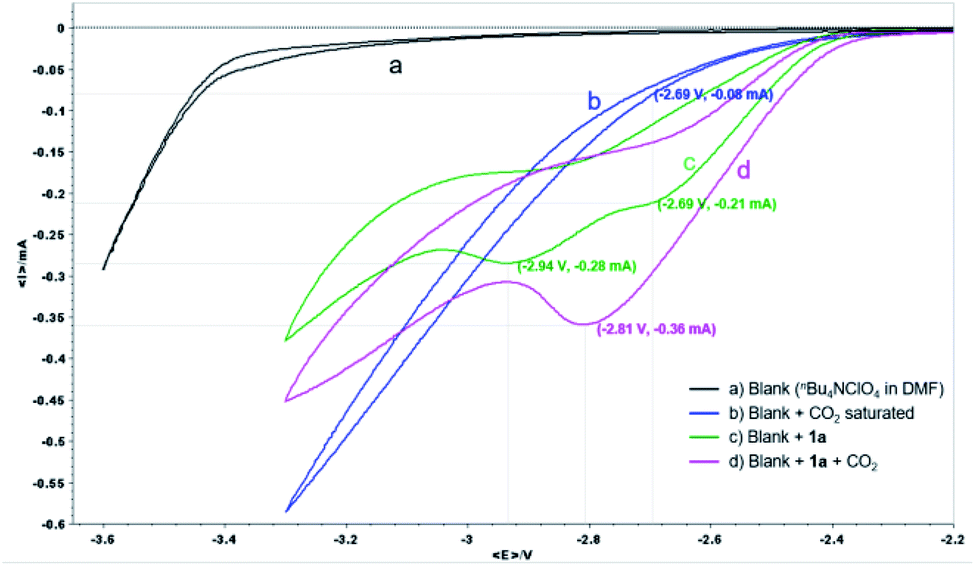 | ||
| Fig. 1 CV analysis of 1a. nBu4NClO4 (0.07 M) in DMF as electrolyte and a 100 mV s−1 scan rate using a Pt disk WE and Pt pillar CE along with Ag/AgNO3 as RE. | ||
For the CV of α-CF3 styrene 1a, a one-electron reduction peak in the potential at −2.69 V and a second at −2.94 V was observed (green line), whereas at a potential of −2.69 V, the reduction current of CO2 was less than 0.1 mA (blue line), indicating that 1a is easier to reduce than CO2. After the solution of 1a was saturated with CO2 (pink line), only one reduction peak was observed at −2.81 V with an associated peak current increase from 0.21 to 0.36 mA (ca. 1.7 times). The influence of potential on the reaction was further studied by constant potential electrolysis. When the reaction was conducted with cathode potential less than −2.70 V, the yield decreased gradually (Table S7 in ESI†). These results suggested that an ECEC process might be involved, in which a radical anion that could react immediately with CO2 might be generated after the first one-electron electroreduction, then the second electron transfer is facilitated at a less negative potential thus leading to a significant increase in current observed. Accordingly, since a different species is being reduced in the presence of CO2, the second peak at −2.94 V is not observed.21 Due to the higher stability of the tertiary alkyl radical, after the first one-electron electroreduction, carboxylation at the less substituted carbon of the alkene moiety should be favored.
To gain more evidence for the intermediacy of a radical anion, we subjected 1a to electrocarboxylation conditions in the presence of several known radical traps. Unfortunately, we were unable to trap the putative radical anion generated via the single-electron reduction or tertiary alkyl radical formed after the addition of CO2. This might be because the radical reduction and subsequent defluorination are favored under electroreduction conditions. However, the addition of 2.0 equivalents of TEMPO to the reaction in the absence of CO2 led to the formation of TEMPO adduct 10, in 40% yield. Furthermore, when the reaction was conducted in the absence of TEMPO and CO2, the allylic radical dimerization product 11, was obtained in 33% yield (Scheme 3). These results suggested that the radical anion was involved during the reaction and that its defluorination produced the allylic radical.
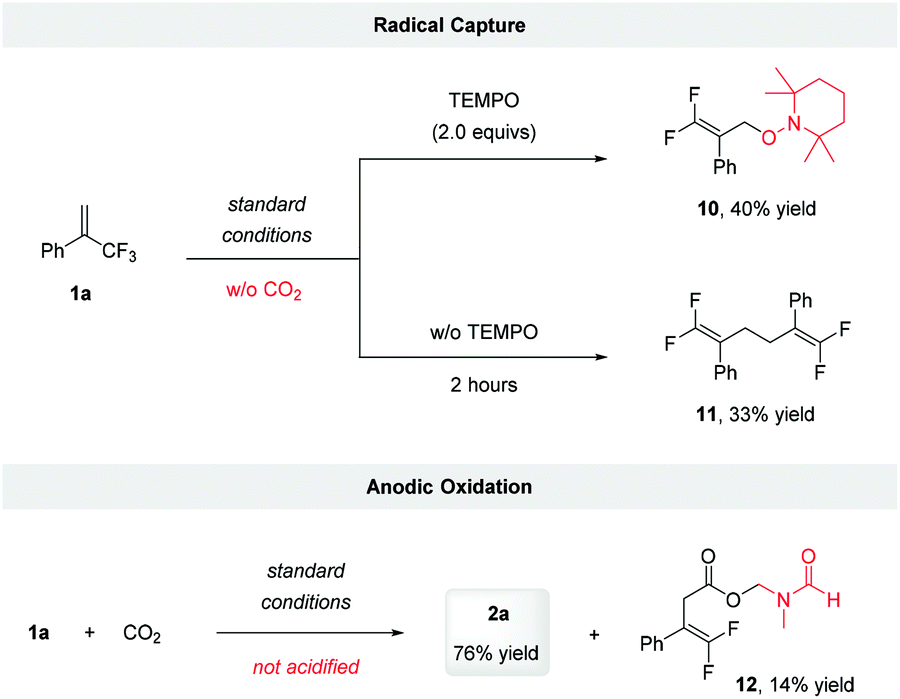 | ||
| Scheme 3 Control experiments. Standard conditions: constant current electrolysis under 8 mA using Pt-plate as cathode and anode with nBu4NClO4 (0.07 M) in DMF, 7 hours. | ||
Subsequently, density functional theory (DFT) calculations were performed as shown in Fig. 2. The results revealed that the reaction of CO2 with radical anion I, generated via one-electron reduction of α-CF3 alkene, was thermodynamically spontaneous with a low free-energy barrier of 8.4 kcal mol−1. Defluorination or protonation of radical anion I had higher free energy barriers of 13.3 and 19.3 kcal mol−1, respectively. These results are consistent with the experimental data and give a good explanation for the high regioselectivity and chemical selectivity of the reaction process.
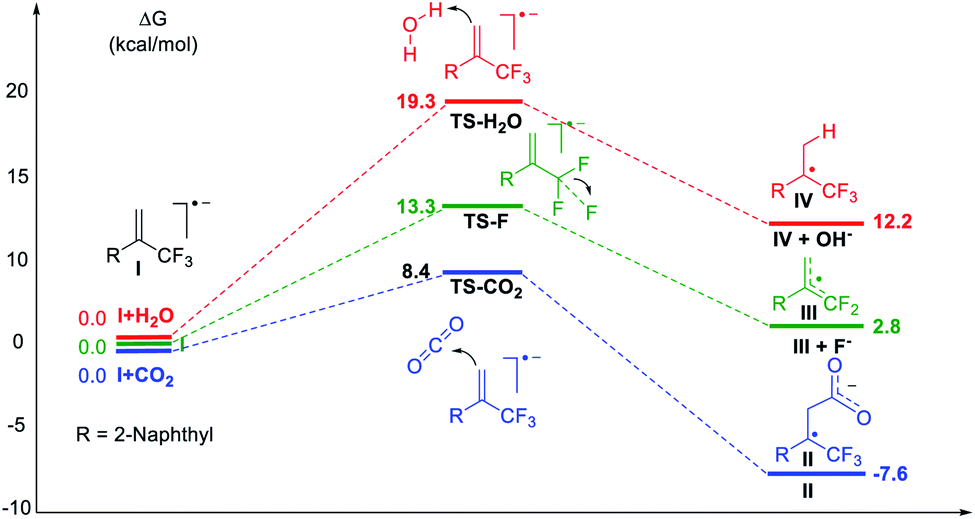 | ||
| Fig. 2 DFT calculations. Gibbs free-energy profile for possible reaction channels at the IEFPCM-M06-2X/6-311++G(d,p) level in solvent DMF. | ||
To identify the sacrificial reductant on the anode, we analyzed the reaction mixture of 1a with CO2 directly without acidification, and detected a DMF-protected carboxylate 12 (Scheme 3). This result suggested that a Shono oxidation of DMF might occur.22 Considering that water has a lower oxidation potential than DMF (1.23 and 1.9 V vs. SHE, respectively),23 it was more likely to act as sacrificial reductant.14b Inspired by Chen's work,24 the capture of oxygen generated via the potential anode oxidation of H2O was conducted using labeled H218O as additive. However, probably due to oxygen exchange of H218O with CO2via the formation of H2CO3, only the release of 16O2 was detected (for details, see ESI†).
Based on the above investigation of the mechanism, a putative reaction pathway was proposed, as shown in Fig. 3. Initially, a one-electron reduction of α-CF3 alkene generated the corresponding radical anion I, which reacted immediately with CO2 at the γ-position to give tertiary alkyl radical II. The secondary, one-electron reduction was then followed by a defluorination process to form carboxylate anion V. Meanwhile, the oxidation of DMF or H2O occurred at the anode, delivering the imine cation or hydrogen cation, both of which can interact with carboxylate anion V to yield the protected carboxylates or deliver the carboxylic acids directly.
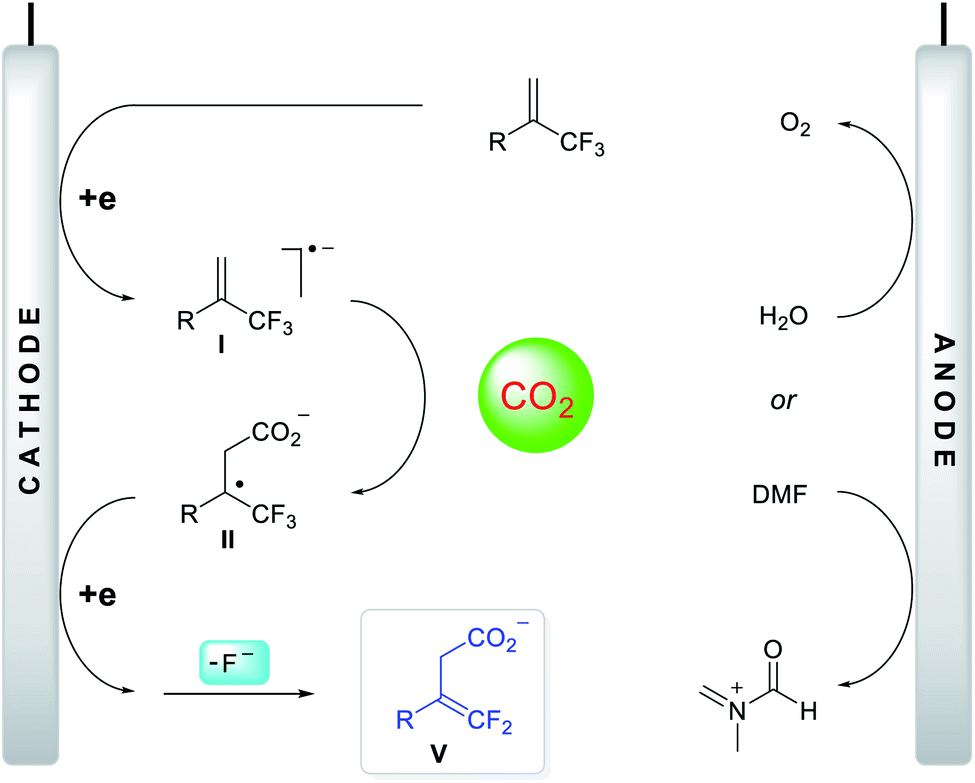 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed a regioselective electrochemical γ-carboxylation of α-CF3 alkenes using CO2 in a user-friendly undivided cell under constant current conditions, without the sacrifice of the anode. Both di- and trisubstituted α-CF3 alkenes work well to afford structurally diverse vinylacetic acids bearing a gem-difluoroalkene moiety in acceptable yields under mild conditions, with good tolerance of functional groups. Notably, this protocol constitutes a rare example of using an electrochemical process to secure regioselectivity that differs from that of the metal-catalyzed process, suggesting the potential of the electrochemistry approach for divergent synthesis. The application of this atom-economical electrochemical method for the synthesis of a diverse range of fluorine-containing carboxylic acids from well-known greenhouse gases CO2 and hydrofluorocarbons,25 is now in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from NSFC (21871090), the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities are highly appreciated. We thank Prof. Tian-Sheng Mei at Shanghai institute of Organic Chemistry for the guidance on electrochemical techniques and Prof. Xu Cheng in Nanjing University for the help in mechanism study.Notes and references
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (b) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 CrossRef; (c) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (d) R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259 CrossRef CAS; (e) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef; (f) X.-B. Lu, Carbon Dioxide and Organometallics, Springer, Switzerland, 2016 CrossRef; (g) Y. Yang and J.-W. Lee, Chem. Sci., 2019, 10, 3905 RSC; (h) M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937 CrossRef CAS; (i) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439 CrossRef CAS; (j) P. Xu, S.-Y. Wang, Y. Fang and S.-J. Ji, Chin. J. Org. Chem., 2018, 38, 1626 CrossRef CAS; (k) Z. Zhang, L. Gong, X.-Y. Zhou, S.-S. Yan, J. Li and D.-G. Yu, Acta Chim. Sinica, 2019, 77, 783 CrossRef.
- For reviews, see: (a) M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739 CrossRef; (b) S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666 RSC; (c) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS; (d) C. S. Yeung, Angew. Chem., Int. Ed., 2019, 58, 5492 CrossRef CAS; (e) W. Zhang, C. Guo and X. Lü, Chin. J. Catal., 2016, 37, 215 CrossRef CAS; (f) Y.-G. Chen, X.-T. Xu, K. Zhang, Y.-Q. Li, L.-P. Zhang, P. Fang and T.-S. Mei, Synthesis, 2018, 50, 35 CrossRef CAS; (g) Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, ACS Catal., 2020, 10, 10871 CrossRef.
- For selected examples, see: (a) A. Correa and R. Martin, J. Am. Chem. Soc., 2009, 131, 15974 CrossRef CAS; (b) T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106 CrossRef CAS; (c) T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221 CrossRef; (d) Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212 CrossRef CAS; (e) S. Zhang, W.-Q. Chen, A. Yu and L.-N. He, ChemCatChem, 2015, 7, 3972 CrossRef CAS; (f) M. Börjesson, T. Moragas and R. Martin, J. Am. Chem. Soc., 2016, 138, 7504 CrossRef; (g) Q.-Y. Meng, S. Wang and B. König, Angew. Chem., Int. Ed., 2017, 56, 13426 CrossRef CAS; (h) K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467 CrossRef CAS; (i) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef; (j) L.-L. Liao, G.-M. Cao, J.-H. Ye, G.-Q. Sun, W.-J. Zhou, Y.-Y. Gui, S.-S. Yan, G. Shen and D.-G. Yu, J. Am. Chem. Soc., 2018, 140, 17338 CrossRef CAS; (k) D. J. Charboneau, G. W. Brudvig, N. Hazari, H. M. C. Lant and A. K. Saydjari, ACS Catal., 2019, 9, 3228 CrossRef CAS; (l) H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122 CrossRef CAS; (m) Y.-G. Chen, B. Shuai, C. Ma, X.-J. Zhang, P. Fang and T.-S. Mei, Org. Lett., 2017, 19, 2969 CrossRef CAS; (n) W.-J. Zhou, Z.-H. Wang, L.-L. Liao, Y.-X. Jiang, K.-G. Cao, T. Ju, Y. Li, G.-M. Cao and D.-G. Yu, Nat. Commun., 2020, 11, 3263 CrossRef CAS.
- (a) J. L. Kiplinger, T. G. Richmond and C. E. Osterbeg, Chem. Rev., 1994, 94, 373 CrossRef CAS; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS; (c) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS; (d) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (b) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390 CrossRef CAS.
- (a) H.-J. Lehmler, M. O. Oyewumi, M. Jay and P. M. Bummer, J. Fluorine Chem., 2001, 107, 141 CrossRef CAS; (b) Q. Zhu, D. Ji, T. Liang, X. Wang and Y. Xu, Org. Lett., 2015, 17, 3798 CrossRef CAS; (c) J. I. Juncosa, K. Takaya, H. V. Le, M. J. Moschitto, P. M. Weerawarna, R. Mascarenhas, D. Liu, S. L. Dewey and R. B. Silverman, J. Am. Chem. Soc., 2018, 140, 2151 CrossRef CAS; (d) H. Pinfold, C. Greenland, G. Pattison and G. Costantini, Chem. Commun., 2020, 56, 125–128 RSC.
- (a) C. Zhu, Y.-F. Zhang, Z.-Y. Liu, L. Zhou, H. Liu and C. Feng, Chem. Sci., 2019, 10, 6721 RSC; (b) S.-S. Yan, D.-S. Wu, J.-H. Ye, L. Gong, X. Zeng, C.-K. Ran, Y.-Y. Gui, J. Li and D.-G. Yu, ACS Catal., 2019, 9, 6987 CrossRef CAS; (c) S.-L. Xie, X.-Y. Cui, X.-T. Gao, F. Zhou, H.-H. Wu and J. Zhou, Org. Chem. Front., 2019, 6, 3678 RSC.
- (a) Y. Pan, J. Qiu and R. B. Silverman, J. Med. Chem., 2003, 46, 5292 CrossRef CAS; (b) S. Messaoudi, B. Tréguier, A. Hamze, O. Provot, J.-F. Peyrat, J. R. De Losada, J.-M. Liu, J. Bignon, J. Wdzieczak-Bakala, S. Thoret, J. Dubois, J.-D. Brion and M. Alami, J. Med. Chem., 2009, 52, 4538 CrossRef CAS; (c) P. V. der Veken, K. Senten, I. Kertèsz, I. De Meester, A.-M. Lambeir, M.-B. Maes, S. Scharpé, A. Haemers and K. Augustyns, J. Med. Chem., 2005, 48, 1768 CrossRef.
- (a) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS; (b) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375 CrossRef CAS; (c) S. Koley and R. A. Altman, Isr. J. Chem., 2020, 60, 313 CrossRef CAS.
- (a) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (b) A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef CAS; (c) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS; (d) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC; (e) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS; (f) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS; (g) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS; (h) K. D. Moeller, Chem. Rev., 2018, 118, 4817 CrossRef CAS; (i) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309 CrossRef CAS; (j) F. Marken and J. D. Wadhawan, Acc. Chem. Res., 2019, 52, 3325 CrossRef CAS; (k) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS; (l) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300 CrossRef CAS; (m) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547 CrossRef CAS.
- (a) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS; (c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef; (d) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS; (e) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS; (f) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC ; also see ref. 10e.
- For reviews, see: (a) B. P. Sullivan, K. Krist and H. E. Guard, Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, Elsevier, Amsterdam, 1993 Search PubMed; (b) R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484 CrossRef; (c) L. Rossi, Curr. Green Chem., 2015, 2, 77 CrossRef CAS; (d) H. Senboku and A. Katayama, Current Opinion in Green and Sustainable Chemistry, 2017, 3, 50 CrossRef; (e) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644 CrossRef CAS.
- See ref. 11c: “a drawback of electrochemical carboxylations is that sacrificial electrodes are required”. Also see ref. 12e “the general utilization for a sacrificial anode in current electrochemical CO2 fixation should be avoided in order to prevent generation of stoichiometric metal waste”.
- (a) A. Alkayal, V. Tabas, S. Montanaro, I. A. Wright, A. V. Malkov and B. R. Buckley, J. Am. Chem. Soc., 2020, 142, 1780 CrossRef CAS; (b) A. M. Sheta, M. A. Mashaly, S. B. Said, S. S. Elmorsy, A. V. Malkov and B. R. Buckley, Chem. Sci., 2020, 11, 9109 RSC.
- For other elegant electrochemical carboxylation bearing non-sacrificial anode, see: (a) D.-T. Yang, M. Zhu, Z. J. Schiffer, K. Williams, X. Song, X. Liu and K. Manthiram, ACS Catal., 2019, 9, 4699 CrossRef CAS; (b) L. Muchez, D. E. De Vos and M. Kim, ACS Sustainable Chem. Eng., 2019, 7, 15860 CrossRef CAS; (c) R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, ChemElectroChem, 2015, 2, 73 CrossRef CAS; (d) H. Tateno, Y. Matsumura, K. Nakabayashi, H. Senboku and M. Atobe, RSC Adv., 2015, 5, 98721 RSC; (e) X. Gao, G. Yuan, H. Chen, H. Jiang, Y. Li and C. Qi, Electrochem. Commun., 2013, 34, 242 CrossRef CAS.
- C. Saboureau, M. Troupel, S. Sibille and J. Périchon, J. Chem. Soc., Chem. Commun., 1989, 1138 RSC.
- (a) X.-T. Gao, C.-C. Gan, S.-Y. Liu, F. Zhou, H.-H. Wu and J. Zhou, ACS Catal., 2017, 7, 8588 CrossRef CAS; (b) F. Zhou, S.-L. Xie, X.-T. Gao, R. Zhang, C.-H. Wang, G.-Q. Yin and J. Zhou, Green Chem., 2017, 19, 3908 RSC; (c) X.-T. Gao, S.-L. Xie, F. Zhou, H.-H. Wu and J. Zhou, Chem. Commun., 2019, 55, 14303 RSC; (d) S. Xie, X. Gao, F. Zhou, H. Wu and J. Zhou, Chin. Chem. Lett., 2020, 31, 324 CrossRef CAS.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72 CrossRef CAS.
- (a) D. Seebach, Helv. Chim. Acta, 2019, 102, e1900072 CrossRef; (b) M. C. Leech and K. Lam, Acc. Chem. Res., 2020, 53, 121 CrossRef CAS.
- Considering that the H2O possesses an oxidation potential of 1.23 V vs. SHE, which might act as sacrificial oxidant to avoid the unexpected oxidation. See: J. J. Medvedev, X. V. Medvedeva, F. Li, T. A. Zienchuk and A. Klinkova, ACS Sustainable Chem. Eng., 2019, 7, 19631 CrossRef CAS.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2001 Search PubMed; (b) N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197 CrossRef CAS; (c) C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404 RSC.
- (a) S. D. Ross, M. Finkelstein and R. C. Petersen, J. Am. Chem. Soc., 1964, 86, 2745 CrossRef CAS; (b) P. Alfonso-Súarez, A. V. Kolliopoulos, J. P. Smith, C. E. Banks and A. M. Jones, Tetrahedron Lett., 2015, 56, 6863 CrossRef; (c) H. Senboku, K. Nagakura, T. Fukuhara and S. Hara, Tetrahedron, 2015, 71, 3850 CrossRef CAS.
- T. S. Rodrigues, M. Zhao, T.-H. Yang, K. D. Gilroy, A. G. M. da Silva, P. H. C. Camargo and Y. Xia, Chem.–Eur. J., 2018, 24, 16944 CrossRef CAS.
- X. Liu, R. Liu, J. Qiu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2020, 59, 13962 CrossRef CAS.
- (a) J. Hansen, M. Sato, R. Ruedy, A. Lacis and V. Oinas, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9875 CrossRef CAS; (b) G. J. M. Velders, D. W. Fahey, J. S. Daniel, M. McFarland and S. O. Andersen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10949 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04091f |
| This journal is © The Royal Society of Chemistry 2020 |