
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost translational modification-assisted cancer immunotherapy for effective breast cancer treatment†
Shevanuja
Theivendran
a,
Jie
Tang
a,
Chang
Lei
a,
Yannan
Yang
a,
Hao
Song
 a,
Zhengying
Gu
ac,
Yue
Wang
a,
Yang
Yang
a,
Lei
Jin
b and
Chengzhong
Yu
*ac
a,
Zhengying
Gu
ac,
Yue
Wang
a,
Yang
Yang
a,
Lei
Jin
b and
Chengzhong
Yu
*ac
aAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, QLD 4072, Australia. E-mail: c.yu@uq.edu.au
bSchool of Medicine and Public Health, University of Newcastle, NSW 2308, Australia
cSchool of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China
First published on 10th September 2020
Abstract
Post translational modifications (PTM) such as phosphorylation are often correlated with tumorigenesis and malignancy in breast cancer. Herein, we report a PTM-assisted strategy as a simplified version of a personalized cancer vaccine for enhanced cancer immunotherapy. Titanium modified dendritic mesoporous silica nanoparticles (TiDMSN) are applied to assist the specific enrichment of phosphorylated tumor antigens released upon immunogenic cell death. This strategy significantly improved the tumor inhibition efficacy in a bilateral breast cancer model and the expansion of both CD8+ and CD4+ T cells in the distant tumor site. The nanotechnology based PTM-assisted strategy provides a simple and generalizable methodology for effective personalized cancer immunotherapy.
Introduction
Immunotherapy has revolutionized cancer treatment, utilizing the body's natural defense system to eradicate tumors with minimal side effects.1 One example of cancer immunotherapy is the discovery of immune checkpoint blockades (ICBs) that help the immune system to recognize and attack tumor cells.2 Nevertheless, checkpoint blockade immunotherapy exhibits a low response rate in poorly immunogenic tumor types such as breast cancer.2,3 Personalized cancer immunotherapy holds great promise in enhancing the immunogenicity by identifying immunogenic antigens from individual patients and engineering a custom-made vaccine.4,5 Despite its advantage of tumor specificity, this strategy is time consuming and expensive.6 Another strategy to improve the immunogenicity is introducing immunogenic cell death (ICD) of cancer cells.7 The ICD strategy uses chemo-drugs or radiation to trigger the in situ generated tumor antigens (TAs) and danger associated molecular patterns (DAMPs) which can attract more immune cells to the tumor site. TAs are subsequently processed by antigen presenting cells (APCs) to provoke systemic antitumor immunity. However, TAs are “hidden” in a large pool of self-proteins released during cell death and difficult to process effectively, posing a limitation for ICD translation.7To improve the effectiveness of the ICD strategy, nanoparticles have been employed to either amplify the ICD signals,8 or to enrich TAs for effective delivery to immune cells.7,9,10 Amplification of ICD using nanoparticles resulted in enhanced exposure of DAMPs.8 However, the effective delivery of antigens released by ICD would further improve the efficacy of immunotherapy. Previously, poly(lactic-co-glycolic acid),10 iron oxide,9 and upconversion nanoparticles,11 have been used for capturing TAs including DAMPs (collectively called TA hereafter) released from the ICD treatment to induce effective antitumor immunity. Nevertheless, the aforementioned nonporous nanoparticles with a low surface area exhibited limited enrichment performance and selectivity towards TAs.12 Importantly, post translational modifications (PTM) such as phosphorylation are closely related to cancer progression.13 Of note is that breast cancer is reported to have elevated levels of phosphorylation which contribute to malignant phenotypes and tumorigenesis.14,15 Besides, the phosphorylation of eukaryotic initiation factor 2 (eIF2) alpha is accompanied by the endoplasmic reticulum stress response during the ICD induction process.16,17 Moreover, DAMPs also can go through post translational alternations including phosphorylation18 (e.g. heat shock proteins HSP 90 and HSP 70).19,20 However, less attention has been paid in using these unique molecular features of cancer in the choice of nanoparticles for TA enrichment for cancer immunotherapy. Numerous nanomaterials especially nanoporous materials with tunable surface chemistry have been reported for selective phosphoprotein enrichment in proteomic studies,21,22 such as titanium modified dendritic mesoporous silica nanoparticles (TiDMSN) with high specificity for the enrichment of phosphoproteins.21 Nevertheless, such nanomaterials are rarely applied in cancer immunotherapy for the enrichment of phosphorylated tumor antigens (p-TAs) to elicit a robust antigen specific immune response.
Herein, we report the application of TiDMSN for in situ phosphoprotein enrichment in combination with ICD and ICB (PD-L1 antibody) immunotherapy for breast cancer treatment (Fig. 1). TiDMSN with large mesopores, a high surface area and phosphoprotein enriching capability21 are used to assist selective enrichment of p-TAs released upon doxorubicin (Dox) induced ICD. This strategy leads to dendritic cell (DC) maturation and T cell activation for realizing systemic antitumor immunity. In combination with ICB, this strategy has shown excellent anticancer performance in the inhibition of both treated primary and untreated distant tumors due to enhanced cytotoxic T lymphocyte (CTL) activation and infiltration into distant tumor sites. Our PTM-assisted design provides a simplified yet powerful strategy for enhanced cancer immunotherapy with cost-effectiveness and timely availability.
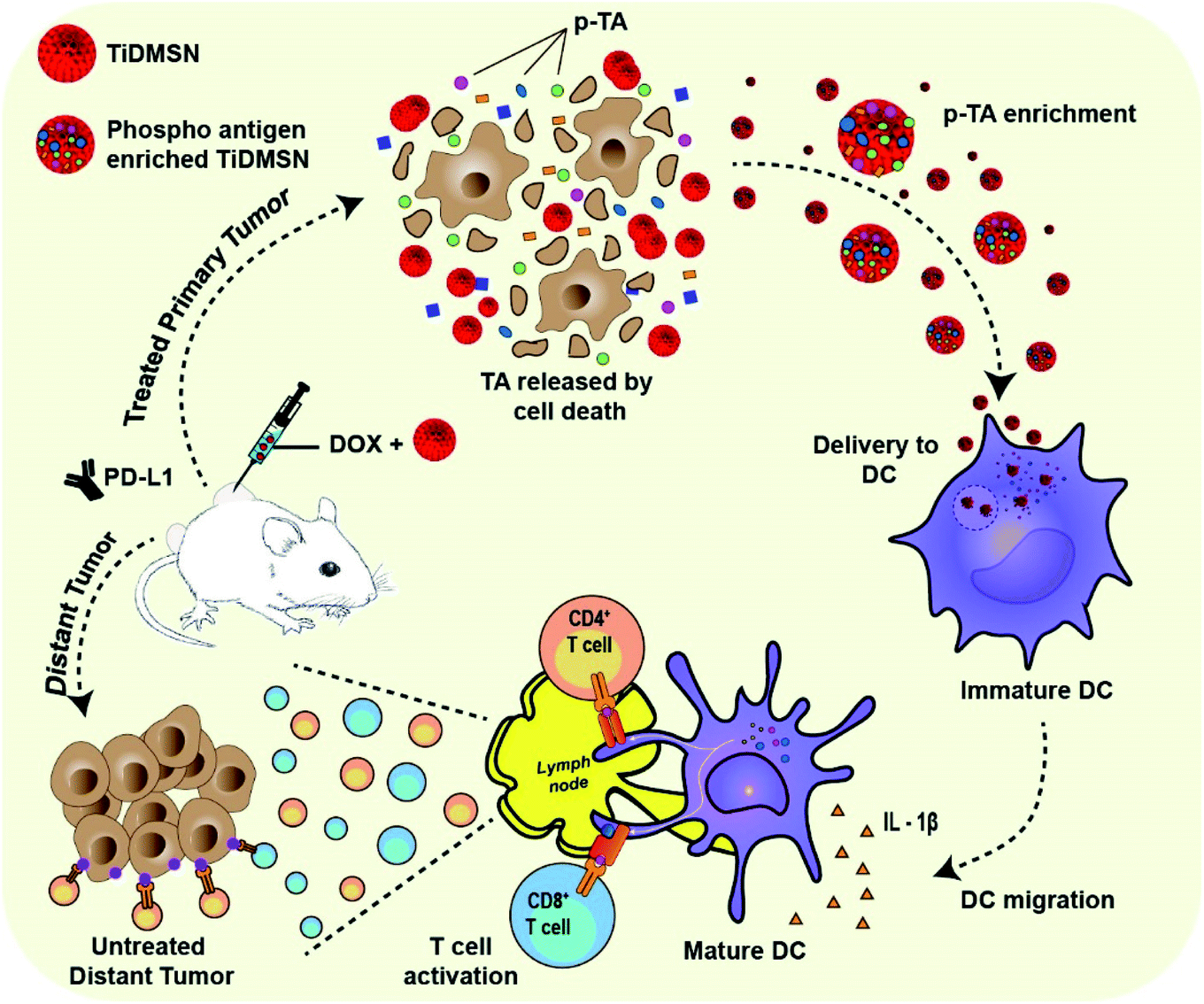 | ||
| Fig. 1 Schematic illustration of the phosphorylated TA enrichment for effective cancer immunotherapy. PTM-assisted cancer immunotherapy using TiDMSN to enrich p-TAs released after ICD to improve antigen specific anti-tumor immunity. | ||
Results and discussion
TiDMSN were prepared using a reported method with some modifications.21 Briefly, DMSN were prepared first23 and then a thin polydopamine (PDA) coating (named PDA–DMSN) was applied. PDA24 was chosen for its biocompatibility and chelating ability with metal ions. Finally, titanium modification was performed by treating PDA–DMSN with titanium tetrachloride to obtain the final TiDMSN product. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of TiDMSN are shown in Fig. 2. Monodispersed nanoparticles with an average diameter of ∼280 nm can be observed from Fig. 2a and b, where the existence of central-radial dendritic large pores can also be seen clearly. Compared to the TEM image of DMSN (Fig. S1†), the structure of TiDMSN is similar, indicating that the large-pore dendritic nanostructure shows little change during the synthesis process.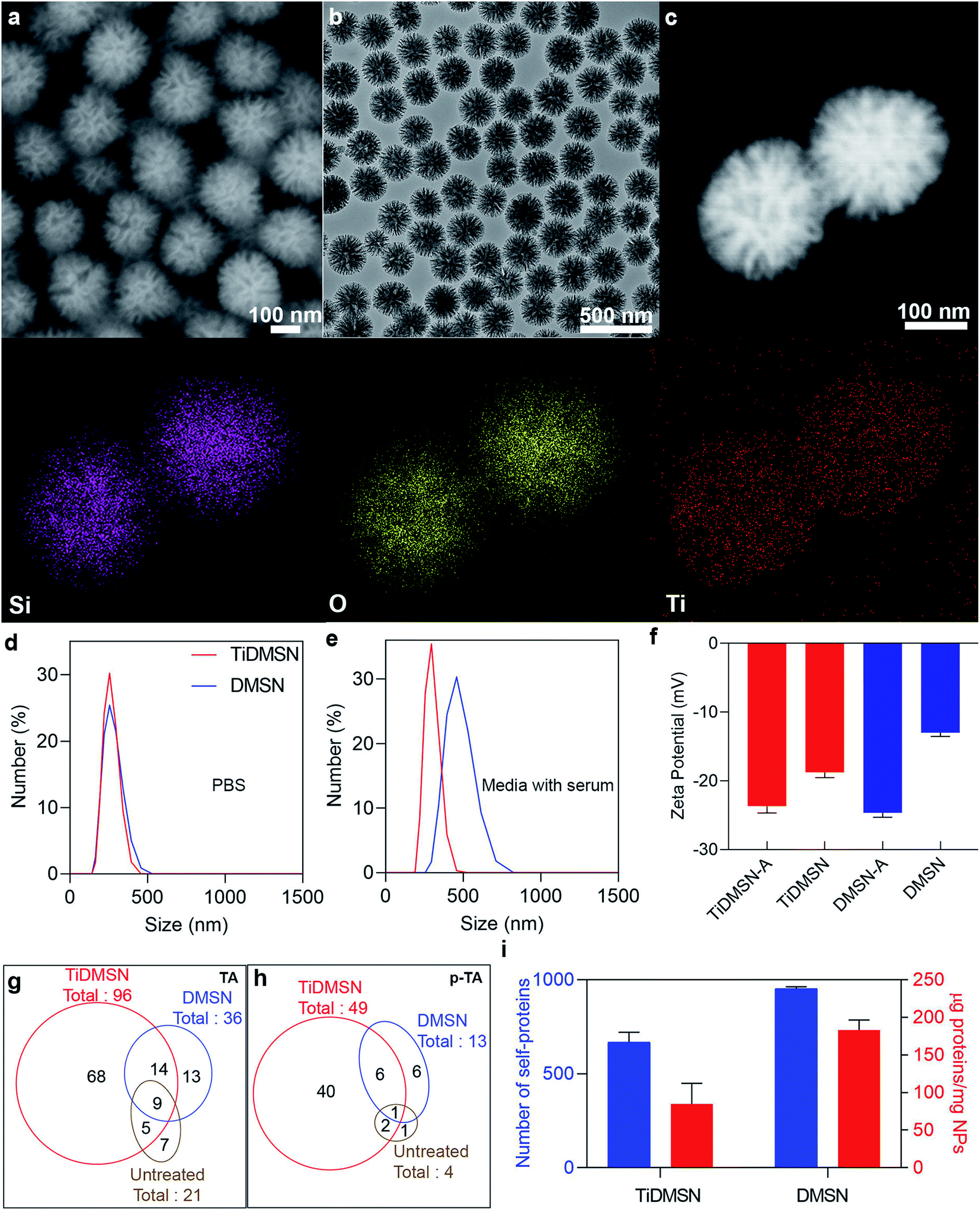 | ||
| Fig. 2 Characterization and antigen enrichment efficiency of nanoparticles. SEM image (a), TEM image (b), dark-field scanning TEM, EDS elemental mapping images of TiDMSN (c). Size distribution of TiDMSN and DMSN in PBS (d) and media with serum (e), zeta potential of TiDMSN and DMSN before and after antigen enrichment (f). Number of TAs and p-TAs enriched by different nanoparticle formulations (g and h). Quantification of proteins enriched by different nanoparticle formulations (i). | ||
Energy-dispersive X-ray spectroscopy (EDS) mapping results are shown in Fig. 2c, where the homogeneous distribution of silica, oxygen and titanium can be observed. The weight percentage of the PDA coating and Ti modification was calculated from thermogravimetric analysis (TGA), which was measured to be 18% and 4%, respectively (Fig. S2†). Furthermore, the thickness of the PDA coating was characterized by TEM (Fig. S3†) and found to be 1.9 nm in thickness. The titanium content of TiDMSN was measured to be 10.6 μg mg−1 by inductively coupled plasma optical emission spectrometry (ICP-OES) as shown in Table S1.† The successful PDA and Ti modifications on the nanoparticle surface were confirmed by attenuated total reflection-Fourier-transform infrared spectroscopy (ATR-FTIR, Fig. S4†) and X-ray photoelectron spectroscopy (XPS, Fig. S5†). As shown in Fig. S4,† characteristic peaks of the surface hydroxyl groups (–OH) and amine groups (–NH) can be observed in the spectra of PDA–DMSN and TiDMSN at 975 and 1636 cm−1, respectively. From XPS analysis of PDA–DMSN and TiDMSN, Ti 2p, N 1s, C 1s, Si 1s and Si 2p peaks at 458, 400, 286.2, 154.1, and 102.7 eV can be seen, further confirming the successful modification of titanium and polydopamine on the nanoparticles.
The N2 adsorption–desorption isotherms and pore size distribution curves of TiDMSN and DMSN are shown in Fig. S6.† The detailed information obtained from the gas sorption analysis is given in Table S1.† The Brunauer–Emmett–Teller (BET) surface area and the total pore volume of TiDMSN are 249.7 m2 g−1 and 1.0 cm3 g−1, respectively, both lower than those of DMSN (400.2 m2 g−1 and 1.37 cm3 g−1) due to the two-step modification process. However, the pore sizes are almost the same around 34 nm for TiDMSN and DMSN. The zeta potential of different nanoparticle formulations was measured (Fig. S7†), all showing negatively charged surfaces at pH 7. The isoelectric points of silica and PDA are ∼2 and 4,25,26 respectively, hence DMSN and PDA–DMSN exhibited negative zeta potential. After Ti modification, the zeta potential of TiDMSN slightly increased, mainly due to the positively charged Ti4+ chelation. However, the abundant catechol groups on PDA resulted in an overall negative charge at neutral pH.
Dynamic light scattering analysis (DLS) was used to determine the size distributions of nanoparticles in PBS and Dulbecco's modified Eagle medium supplemented with fetal bovine serum (DMEM + FBS). As shown in Fig. 2d, TiDMSN and DMSN show relatively narrow particle size distributions around 290 nm in PBS, slightly larger than the size determined from SEM/TEM observations. In DMEM + FBS (Fig. 2e), the particle size distributions of TiDMSN and DMSN are ∼300 nm and ∼460 nm with PDI values of 0.3 and 0.45 respectively. DMSN exhibited a relatively larger and broader size distribution compared to that of TiDMSN in DMEM + FBS, possibly due to a higher level of non-specific binding of negatively charged serum proteins on DMSN than TiDMSN which resulted in subsequent aggregation of nanoparticles.27
The above results are supported by the TGA and surface charge characterization (Fig. S7 and S8†). TiDMSN and DMSN incubated in DMEM + FBS showed an increased weight loss by 9% and 22%, respectively. Despite the overall negative charge of the serum proteins, random adsorption of serum proteins on negatively charged nanoparticles due to weak electrostatic pull initiated by the multiple positive sites28 of serum proteins has been reported previously.29,30 The zeta potential of DMSN is less negative than that of TiDMSN due to the extensive availability of hydroxyl groups on both silica and polydopamine,31,32 leading to less repulsion with negatively charged serum proteins, more aggregation of nanoparticles, and possibly more non-specific protein adsorption in comparison with TiDMSN.
To evaluate the TA and p-TA enrichment performance of TiDMSN after the induction of ICD, 4T1 breast cancer cell lines were treated with Dox, then the tumor lysates were collected for subsequent enrichment studies. The ICD of the cancer cells was confirmed by measuring the expression of calreticulin (CRT) after treating the cancer cells with Dox.8 As shown in Fig. S9,† the mean fluorescence intensity (MFI) of CRT in the sample treated with Dox was significantly higher compared to the PBS control group, indicating the successful induction of ICD. The change in zeta potential before and after antigen enrichment was compared as shown in Fig. 2f to gain information on protein enrichment on the nanoparticle surface. The use of antigen enriched nanoparticles was denoted by “-A” at the end of the nanoparticle name. For example, antigen enriched TiDMSN are denoted as TiDMSN-A. The zeta potential of both TiDMSN and DMSN increased after antigen enrichment; however, the difference in zeta potential between DMSN and DMSN-A is large compared to that of TiDMSN, indicating a higher level of non-specific adsorption of proteins in DMSN with a larger surface area and higher pore volume than those of TiDMSN.
The number of TAs and p-TAs enriched by TiDMSN and DMSN is summarized in Fig. 2g and h, Tables S2 and S3.† 96 TAs were detected in TiDMSN, of which 49 were phosphorylated. In comparison, only 36 TAs and 13 p-TAs were identified to be enriched by DMSN, while 21 TAs and 4 p-TAs were detected in the sample without any enrichment. Interestingly, out of the 96 TAs and 49 p-TAs enriched by TiDMSN, 68 and 40 were not identified in other control groups. The pTAs enriched by TiDMSN include both DAMPs and tumor associated antigens, various RAS33,34 and RAB35 oncogene proteins, heat shock proteins (HSP 90, HSP 70, and HSP 105),19,20 the HMG box transcription factor36 and the Rho family of proteins, to name a few.37 These proteins are reported to be over expressed in breast cancer, which can act as immunogens for the activation of the immune system. Overall, TiDMSN showed high efficiency in enrichment of p-TAs released upon ICD.
The relative abundance of various TAs and p-TAs was characterized to gain an insight into the relative amounts of antigens enriched by TiDMSN and DMSN. As illustrated in Fig. S10,† the relative abundance of p-TAs enriched by TiDMSN is also high compared to DMSN. Moreover, the relative abundance of p-TAs enriched by TiDMSN is higher than that of TAs. Hence, TiDMSN can specifically enrich more number as well as amount of p-TAs than DMSN. The most abundant TA and p-TA in the TiDMSN sample were histone 2A and anoctamin, respectively. Histone 2A is an overexpressed tumor associated antigen in breast cancer and responsible for the cell proliferation of breast cancer cells.38 These antigens can promote an inflammatory immune response as well as affect the immunogenicity of cancer cells.39 Anoctamin is another type of tumor antigen over expressed in breast cancer and related to poor prognosis of patients.40,41
To highlight the advantage of surface modification in specific binding, the total amount of proteins and the number of self-proteins enriched by each nanoparticle formulation were also quantified (Fig. 2i and Table S4†). As expected, the total amount of proteins enriched by DMSN (∼182.5 μg mg−1) is higher than that of TiDMSN (∼84 μg mg−1) due to the non-specific adsorption of proteins owing to the high surface area and pore volume in DMSN. The influence of nanoparticle concentration on the overall protein adsorption capacity was also tested (Fig. S11†). DMSN exhibited a higher protein adsorption capacity at all three nanoparticle concentrations compared to TiDMSN. With increased nanoparticle concentration, the protein adsorption capacity decreased, consistent with the literature reports.42 Hence, a low nanoparticle concentration is beneficial for enhanced antigen enrichment. Furthermore, the number of self-proteins enriched by DMSN is also higher than that of TiDMSN due to the high adsorption capacity of DMSN. The number of self-proteins enriched by TiDMSN and DMSN is ∼632 and ∼944, respectively. The above results demonstrate the importance of using TiDMSN to specifically enrich phosphorylated proteins and to reduce non-specific binding.
We further analyzed the possible inflammatory pathways activated by the TAs and p-TAs enriched by TiDMSN and DMSN using a bioinformatics tool, Ingenuity Pathway Analysis (IPA). The most possible pathways in TiDMSN compared to the DMSN group included dendritic cell maturation, IL-8 signaling, IL-22 signaling, PD-1, PD-L1 cancer immunotherapy pathway, and B cell development (Fig. S12a and b†). The results revealed that in comparison with DMSN, TiDMSN enriched with more TAs and pTAs can regulate the pathways associated with the immune system and immune responses. Moreover, IPA analysis of proteins enriched by TiDMSN and DMSN also indicated that the top related disease was cancer (Fig. S12c and d†), probably because the antigens are enriched by the nanoparticles after inducing ICD in cancer cells. These results based on IPA analysis further indicate that TiDMSN are more efficient than DMSN in recruiting TAs, especially p-TAs, that are highly related to cancer for activating the immune system.
The ICD induction process follows complicated intracellular events which results in the synthesis of different danger signals and tumor antigens. In addition, the cellular stress process leads to the synthesis of proteins with structural alterations by PTM which helps in improving immunogenicity.19,43 To highlight the importance of inducing ICD for the release of TAs, we performed the nanoparticle enrichment analysis on tumor lysates prepared by the freeze–thaw method (Fig. S13†) and compared the results to those obtained from the Dox treatment protocol (Fig. 2g and h). The TA number identified from freeze–thaw versus Dox treatment protocols was 22 vs. 21 in the untreated group, 30 vs. 36 in the DMSN group, and 41 vs. 96 in the TiDMSN group. The p-TA number in two protocols was 6 vs. 4, 3 vs. 13 and 14 vs. 49 in untreated, DMSN and TiDMSN groups, respectively. These results indicate the advantage of ICD induction for releasing more TAs, especially p-TAs, for subsequent enrichment by nanoparticles,44 however, only in the presence of the TiDMSN group that can efficiently enrich the p-TAs released upon the induction of ICD.
To determine a safe nanoparticle concentration for the rest of the experiments, the cell viability of nanoparticles towards kidney cells (HEK cells) and immunocytes (RAW 264.7 cells) at different nanoparticle concentrations was analyzed. As shown in Fig. 3a and S14,† the cell viability percentage was above 60% for both TiDMSN and DMSN at a maximum concentration of 50 μg ml−1, hence 25 μg ml−1 was chosen in the following experiments. To study the ex vivo immune-stimulatory performance of nanoparticles before and after antigen enrichment, splenocytes were incubated with nanoparticles for 20 h to determine the expression level of CD 86, MHC class I and MHC class II in both dendritic cells and macrophages45 (marked by anti-CD11C and anti-F4/80, respectively). As shown in Fig. S15,† when splenocytes were treated with nanoparticles prior to antigen enrichment, no significant difference in the expression of CD86, MHC class I and MHC class II was observed, indicating that bare TiDMSN and DMSN are biocompatible and can hardly induce antigen presenting cell (APC) maturation. However, TiDMSN-A induced the highest expression of CD86 in both dendritic cells and macrophages. The maturation of APCs induced by nanoparticles carrying immunogenic antigens further caused the expression of co-stimulatory proteins such as CD86 for the subsequent activation of T cells,46,47 also beneficial for the activation of antigen specific immune responses.
In addition to the maturation efficacy of antigen enriched nanoparticles, the antigen presentation efficiency was also tested. Antigen presentation through classic MHC class I and II is crucial for the subsequent T cell repertoire activation.48,49 Moreover, it is well known that the antigen cross presentation through MHC class I is essential for the successful activation of cytotoxic CD8 T cells, which drives the effective antitumor immune response,48 while the antigen presentation through MHC class II corresponds to the activation of CD4 helper T cells.48 It can be seen from Fig. 3b that macrophages and dendritic cells showed enhanced expression of both MHC class I and II in the TiDMSN-A treated group, indicating that the immunogenic antigens processed in the APC are successfully presented on MHC class I and II receptors47 which resulted in increased expression of the respective receptors on the APC surface. Next, macrophages were treated with different antigen enriched nanoparticles to determine the level of cytokine release. As shown in Fig. 3c and d, the secretion levels of pro-inflammatory cytokines IL-1β and IL-6 were also significantly higher for the group treated with TiDMSN-A due to the effective enrichment of immunogenic tumor antigens. Overall, these results confirm the significant improvement in antigen delivery and activation of the immune system using the PTM-targeting antigen enriched TiDMSN.
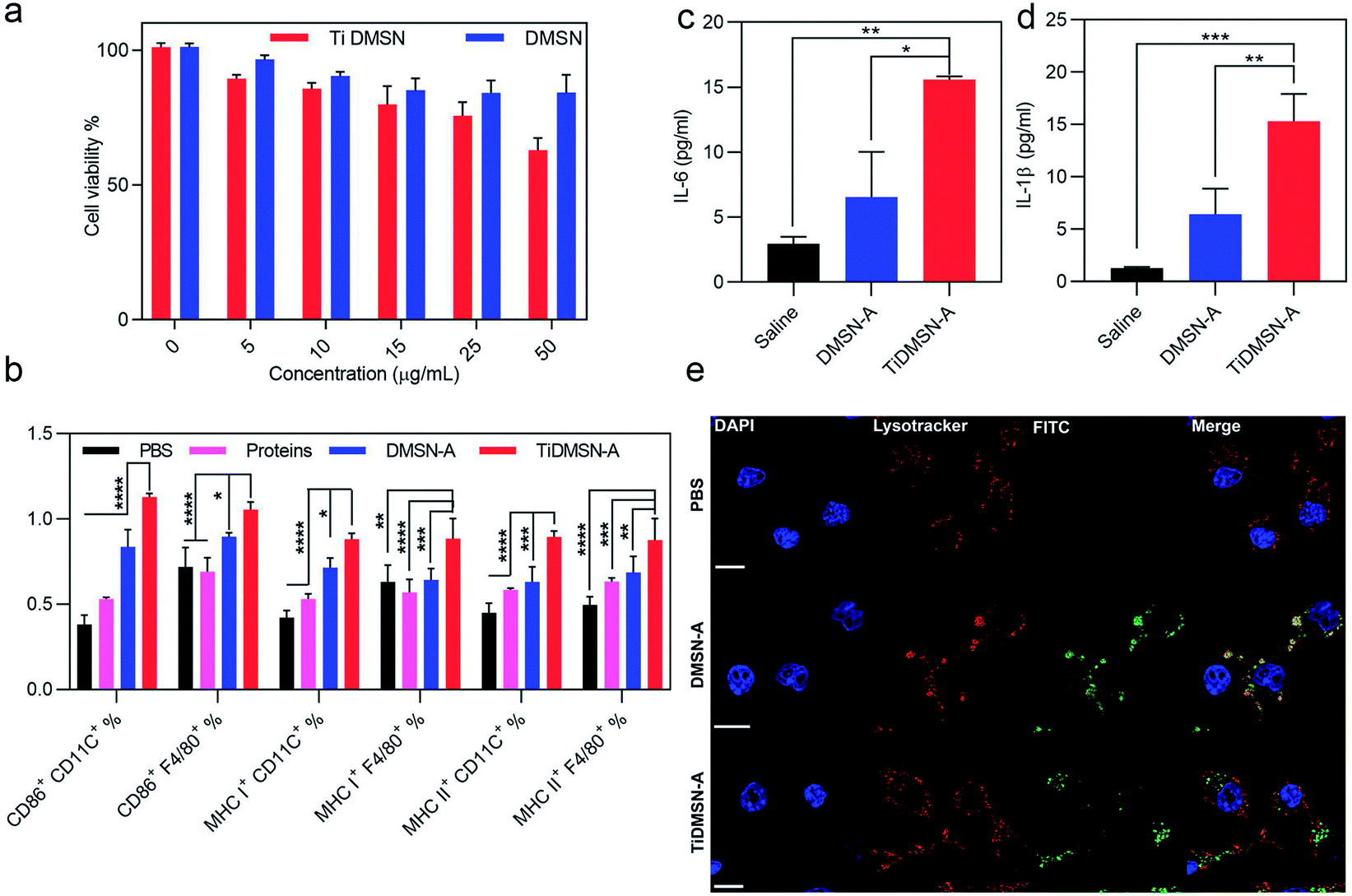 | ||
| Fig. 3 Cytotoxicity profile of nanoparticles, in vitro immune cell maturation and cytokine release efficiency of antigen enriched nanoparticles. HEK cell viability after 24 h treatment of TiDMSN and DMSN at various concentrations (a). Flow cytometry analysis of CD 86, MHC class I and MHC class II expression on splenocytes treated with PBS or different antigen enriched nanoparticle formulations (b). Macrophages were marked by the F4/80 antibody and the dendritic cells were marked by the CD 11C antibody. Cytokine secretion levels after treating macrophages with different antigen enriched nanoparticle formulations (c and d). CLSM images for analyzing the endosomal escape of the antigen enriched nanoparticles (e). The “proteins” group refers to the tumor lysates without enrichment. Data represent mean ± standard deviation (s.d.) from three independent experiments (n = 3) with significance levels *P < 0.05, **P < 0.01, ***P < 0.01, and ****P < 0.01. | ||
It is well known that the presentation of the internalized antigens on MHC class I molecules follows a cytosolic pathway, which requires the release of antigens from the endosomes. Hence, the endosomal escape efficacy of antigen enriched TiDMSN and DMSN was tested. It can be seen from Fig. 3e and S16† that DMSN-A labeled with FITC co-localizes with the Lysotracker Red labelled endosomes in the merged figure, which illustrates the inability of those nanoparticles to perform a successful endosomal escape.50 However, distinct bright green fluorescence can be seen for TiDMSN-A in the merged figure, indicating successful endosomal escape. Fig. S17† further demonstrates that the PDA–DMSN-A can undergo partial endosome escape due to the presence of amine groups in PDA.51,52 This might be the reason for the endosomal escape of TiDMSN, which is important for the cross presentation of antigens from TiDMSN-A for enhanced expression of MHC class I as shown in Fig. 3b.
In order to confirm that the antigen enriched by the nanoparticles can be cross presented on MHC class I for successful T cell activation, we further tested the binding efficiency of the antigens enriched by TiDMSN towards the MHC class I receptors using the immunoaffinity separation method. From the list of tumor antigens enriched by TiDMSN given in Table S2,† DNA repair and recombination protein RAD54B was found bound to the MHC class I receptors isolated from the RAW macrophages treated with TiDMSN-A. This is a protein produced during the process of DNA damage,53 possibly due to the induction of ICD by Dox in this study. In addition, RAD54B stands for a somatic mutation which is overexpressed in different cancer types including breast cancer.54,55 In previous reports, the analysis of TA presentation enriched by nanoparticles for the activation of the immune system is rarely reported.9–11 Our finding based on the immunoaffinity separation test supports our hypothesis that tumor antigens enriched by the nanoparticles can be successfully cross presented in the immune cells, which is crucial for the activation of CTLs to induce the antigen specific anti-tumor immunity.
To test whether the PTM-assisted antigen enrichment can improve immunotherapy in vivo, a bilateral 4T1 breast cancer mouse model was designed. Mice were randomly divided into 4 groups (saline, Dox, DMSN + Dox and TiDMSN + Dox) with 5 mice in each group. For DMSN + Dox and TiDMSN + Dox groups, Dox was mixed with nanoparticles to induce ICD. Mice were injected with four formulations to the right flank (primary tumor) on day 0 and subsequently subjected to PD-L1 treatment the next day, repeating three times as illustrated in Fig. 4a. The secondary tumor was left untreated. The immunotherapeutic efficacy and the stimulation of the abscopal effect was assessed by measuring the size of the primary and the secondary tumors at a regular interval of every 2 days. The mice in all the groups were sacrificed on day 12 as the tumor sizes of the mice in the saline group exceeded 1000 mm3. As shown in Fig. 4b and c, mice treated with TiDMSN + Dox exhibited enhanced tumor regression in both primary and secondary tumors compared to the mice treated with Dox or DMSN + Dox. In addition, the IFN-γ secretion (Fig. 4d) in the blood of the animal group treated with TiDMSN was also significantly higher compared to the control. It is concluded that TiDMSN can significantly improve the therapeutic efficacy and abscopal effect.
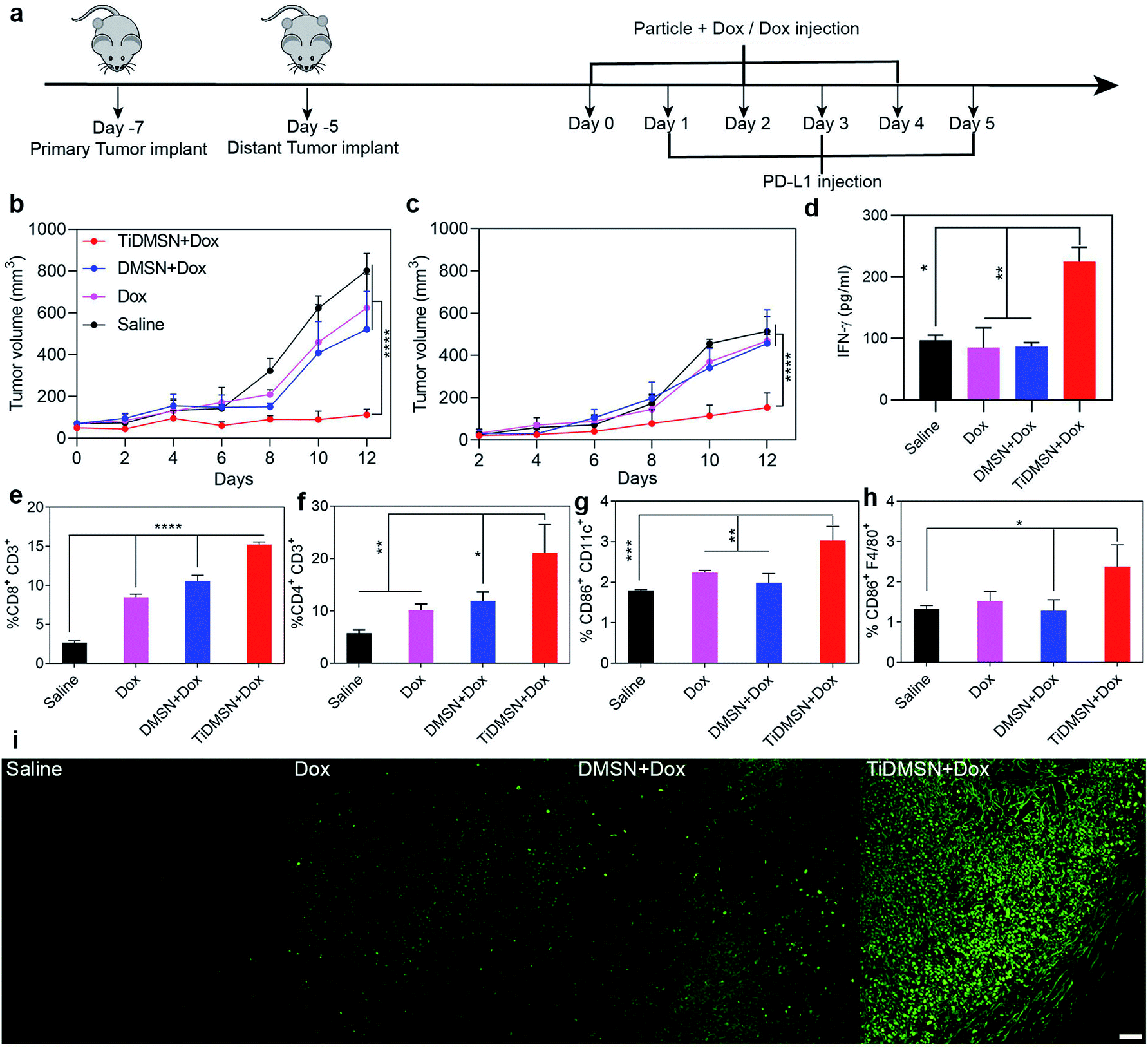 | ||
| Fig. 4 Summary of the results obtained from the in vivo experiments. (a) In vivo experimental design and (b and c) tumor-growth curves of mice treated with different nanoparticle formulations. Primary tumors (b) were treated with different formulations while the distant tumors (c) received no treatment. The IFN-γ level in the serum collected from mice after treatment (d). Infiltration of CD8+CD3+ T cells (e) and CD4+CD3+ T cells (f) in the distant tumor site. Maturation level (CD86+) of dendritic cells marked by the CD11C antibody (g) and macrophages marked by the F4/80 antibody (h) (n = 5, mean ± SD with significance levels *P < 0.05, **P < 0.01, ***P < 0.01, and ****P < 0.01). TUNEL staining of the distant tumors obtained from mice treated with different nanoparticle formulations (i); scale bar: 50 μm; the saline group received no treatment. | ||
In order to evaluate the antigen cross presentation and the subsequent T cell activation efficiency for an effective anti-tumor immune response, the CTL infiltration at the distant tumor site was evaluated. Fig. 4e and f show the CD8+ and CD4+ T cell infiltration at the distant tumor site induced by different nanoparticle formulations. Stronger CD8+ and CD4+ infiltration was observed for the animal groups treated with TiDMSN + Dox compared to Dox and DMSN + Dox treated groups. The significant improvement in the infiltration of CTLs in the distant tumor site of the animals treated with TiDMSN exemplifies the enhanced antigen cross presentation of the tumor antigens on the MHC class I molecules for the activation of CTLs to induce a strong immune response.56 In addition, the CD4 T cell's help is essential for the priming of the CD8 T cell.57,58 Hence, the enhanced expansion of CD4+ T cells in the group treated with TiDMSN together with the antigen specific CD8 T cells improved the effectiveness of the immunotherapy in combination with chemotherapy. Expression of CD8, CD4 and TNF-α in the secondary tumor site was further characterized with immunohistochemistry. As shown in Fig. S18,† enhanced expression of CD8, CD4 and TNF-α was observed for the TiDMSN + Dox group compared to Dox and DMSN + Dox groups. Furthermore, as shown in Fig. 4g and h, the groups treated with TiDMSN + Dox showed higher levels of CD 86 expression on dendritic cells and macrophages from the spleens collected at the end of the treatment. In addition, the secondary tumor samples were collected at the end of the treatment process (12 days) and the cell apoptosis of tumor tissues was analyzed using the TUNEL immunohistochemical staining method. As shown in Fig. 4i, brighter green fluorescence indicating enhanced cell apoptosis can be observed for the group treated with TiDMSN + Dox compared to Dox and DMSN + Dox treated groups, further confirming the therapeutic efficacy and abscopal effect of the PTM-assisted antigen enriched nanoparticles.
To study the ICD induction in vivo and the contribution of nanoparticles towards ICD, the CRT exposure in vivo in different treatment groups at the Dox dosage of 2.5 mg kg−1 was measured. This dosage was the same as that used in the above animal experiments, but lower than the dosage used in the literature for inducing potent tumor inhibition.8,59 As shown in Fig. S19,† a significant difference in CRT exposure between all the treatment groups and the untreated saline group was observed, indicating successful ICD induction by Dox. However, no significant difference was observed between Dox and Dox + nanoparticle treated groups, suggesting that the nanoparticles used in this study contributed little to ICD induction. This observation also indicates that the enhanced therapeutic efficacy of TiDMSN should be attributed to the efficient delivery of more immunogenic TAs to the immune cells for enhanced maturation and activation of the immune system compared to control groups.
The distribution and migration of nanoparticles during the in vivo conditions were further characterized. We first tested the time dependent imaging of intratumorally injected fluorescent labelled nanoparticles mixed with Dox. IR-780 was chosen as the fluorescent dye with excitation and emission wavelengths at 710 and 800 nm, respectively, which are different from those of Dox (Fig. S20†). As shown in Fig. S21a,† the nanoparticle accumulation inside the tumor was observed for more than 5 days, which is expected to provide a sufficiently long-time window to enrich the released antigens post ICD induction. The percentage of nanoparticles taken up by tumor infiltrating immune cells was also analyzed by flow cytometry (Fig. S21b†). Dendritic cells and macrophages in the tumor site were marked by CD11C and F4/80 markers. The uptake percentages of fluorescent labelled TiDMSN by dendritic cells and macrophages were significantly higher than those of DMSN. The percentage of TiDMSN in dendritic cells was higher than that in macrophages, possibly because dendritic cells are responsible for taking immunogenic tumor antigens enriched by TiDMSN whereas macrophages tend to take up nanoparticles by phagocytosis.60 Flow cytometry analysis of dendritic cells (marked by CD11C) isolated from lymph nodes (Fig. S21c†) post injection further indicates that TiDMSN were more effectively taken up by dendritic cells compared to DMSN, suggesting that dendritic cell mediated antigen presentation is prompted by TiDMSN for immune activation. Accumulation sites of fluorescent labelled nanoparticles in the tumor were also characterized by slicing the tumor (Fig. S22†). CLSM analysis showed that the nanoparticles were accumulated mainly in the exterior regions compared to core regions with limited perfusion, favorable for antigen delivery to immune cells also located in tumor margins.61,62
Finally, the biosafety of different nanoparticle formulations was analyzed. Heart, liver, kidney, lung, spleen, primary and secondary tumors were collected at the end of treatment for H & E staining (Fig. S23†). No significant damage in major organs was observed compared to the group that received no treatment. However, significant damage was observed in primary and secondary tumors treated with TiDMSN + Dox compared to Dox and DMSN + Dox treated groups. In addition, metastasis of cancer cells into the lung tissue also has been significantly reduced in the group treated with TiDMSN + Dox. No significant change in the body weight of mice was observed over the treatment period (Fig. S24†).
Bio-distribution of nanoparticles in the major organs and tumor tissues collected after 4 days of intratumor injection of nanoparticles was analyzed by measuring the silica content using ICP-OES. As shown in Fig. S25,† TiDMSN were mostly accumulated in the primary tumor site compared to other major organs and no significant migration of nanoparticles was observed to the distant tumor site. These results indicate the minimal side effects caused by the nanoparticles used in the treatment groups; and that the regression of the distant tumor was prompted by the abscopal effect induced by the immune response rather than nanoparticle migration.
Conclusions
In summary, we have designed a PTM-assisted cancer immunotherapy strategy to realize effective anti-tumor immunity for breast cancer treatment. Efficient inhibition of both primary and distant tumors has been demonstrated using our design with high p-TA enrichment efficiency. Further investigation on the establishment of a reliable protocol is required for assessing the in vivo phosphoprotein enrichment capability of nanoparticles. It is expected that the PTM assisted enriching strategy has the potential to facilitate personalized cancer immunotherapy in other cancer types.63 For example, colon cancer is reported to have aberrantly glycosylated cancer associated antigens,64 which can be enriched through the glycoprotein enriching strategies reported previously.65 Therefore, nanoparticles with specific enriching capability towards a PTM type can be designed for targeting tumor antigens in various cancer types according to their PTM signatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from the Australian Research Council and Queensland Government, the Queensland node of the Australian National Fabrication Facility, Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, UQ Flow Cytometry Facility, the University of Queensland, and Science and Technology Commission of Shanghai Municipality (19JC1412100), China. The authors also acknowledge Dr Amanda Nouwans for the assistance with mass spectrum data collection, Mr Barry Wood for the assistance with XPS, and animal house staffs for their assistance with the in vivo experiments.References
- M. Dougan, G. Dranoff and S. K. Dougan, Annu. Rev. Cancer Biol., 2019, 3, 55–75 CrossRef.
- Y.-J. Park, D.-S. Kuen and Y. Chung, Exp. Mol. Med., 2018, 50, 109 CrossRef.
- R. H. Vonderheide, S. M. Domchek and A. S. Clark, Clin. Cancer Res., 2017, 23, 2640–2646 CrossRef.
- A. Terbuch and J. Lopez, Vaccines, 2018, 6, 52 CrossRef CAS.
- U. Sahin and Ö. Türeci, Science, 2018, 359, 1355 CrossRef CAS.
- Ö. Türeci, M. Löwer, B. Schrörs, M. Lang, A. Tadmor and U. Sahin, Nat. Biomed. Eng., 2018, 2, 566–569 CrossRef.
- G. Kroemer, L. Galluzzi, O. Kepp and L. Zitvogel, Annu. Rev. Immunol., 2013, 31, 51–72 CrossRef CAS.
- Y. Yang, J. Tang, P. L. Abbaraju, M. Jambhrunkar, H. Song, M. Zhang, C. Lei, J. Fu, Z. Gu, Y. Liu and C. Yu, Angew. Chem., Int. Ed., 2018, 57, 11764–11769 CrossRef CAS.
- B. Wang, J. An, H. Zhang, S. Zhang, H. Zhang, L. Wang, H. Zhang and Z. Zhang, Small, 2018, 14, 1801372 CrossRef.
- Y. Min, K. C. Roche, S. Tian, M. J. Eblan, K. P. McKinnon, J. M. Caster, S. Chai, L. E. Herring, L. Zhang, T. Zhang, J. M. DeSimone, J. E. Tepper, B. G. Vincent, J. S. Serody and A. Z. Wang, Nat. Nanotechnol., 2017, 12, 877 CrossRef CAS.
- M. Wang, J. Song, F. Zhou, A. R. Hoover, C. Murray, B. Zhou, L. Wang, J. Qu and W. R. Chen, Adv. Sci., 2019, 6, 1802157 CrossRef.
- Q. Sun, M. Barz, B. G. De Geest, M. Diken, W. E. Hennink, F. Kiessling, T. Lammers and Y. Shi, Chem. Soc. Rev., 2019, 48, 351–381 RSC.
- C. Dai and W. Gu, Trends. Mol. Med., 2010, 16, 528–536 CrossRef CAS.
- H. J. Lin, F. C. Hsieh, H. Song and J. Lin, Br. J. Cancer, 2005, 93, 1372–1381 CrossRef CAS.
- Y. Cho, H. G. Kang, S.-J. Kim, S. Lee, S. Jee, S. G. Ahn, M. J. Kang, J. S. Song, J.-Y. Chung, E. C. Yi and K.-H. Chun, Cell Death Differ., 2018, 25, 1781–1795 CrossRef CAS.
- J. Fucikova, E. Becht, K. Iribarren, J. Goc, R. Remark, D. Damotte, M. Alifano, P. Devi, J. Biton, C. Germain, A. Lupo, W. H. Fridman, M.-C. Dieu-Nosjean, G. Kroemer, C. Sautès-Fridman and I. Cremer, Cancer Res., 2016, 76, 1746 CrossRef CAS.
- L. Bezu, A. Sauvat, J. Humeau, M. Leduc, O. Kepp and G. Kroemer, OncoImmunology, 2018, 7, e1431089 CrossRef.
- A. D. Garg, L. Galluzzi, L. Apetoh, T. Baert, R. B. Birge, J. M. Bravo-San Pedro, K. Breckpot, D. Brough, R. Chaurio, M. Cirone, A. Coosemans, P. G. Coulie, D. De Ruysscher, L. Dini, P. de Witte, A. M. Dudek-Peric, A. Faggioni, J. Fucikova, U. S. Gaipl, J. Golab, M.-L. Gougeon, M. R. Hamblin, A. Hemminki, M. Herrmann, J. W. Hodge, O. Kepp, G. Kroemer, D. V. Krysko, W. G. Land, F. Madeo, A. A. Manfredi, S. R. Mattarollo, C. Maueroder, N. Merendino, G. Multhoff, T. Pabst, J.-E. Ricci, C. Riganti, E. Romano, N. Rufo, M. J. Smyth, J. Sonnemann, R. Spisek, J. Stagg, E. Vacchelli, P. Vandenabeele, L. Vandenberk, B. J. Van den Eynde, S. Van Gool, F. Velotti, L. Zitvogel and P. Agostinis, Front. Immunol., 2015, 6, 588 Search PubMed.
- A. Serrano-del Valle, A. Anel, J. Naval and I. Marzo, Front. Cell Dev. Biol., 2019, 7, 50 CrossRef.
- T. Zininga, L. Ramatsui and A. Shonhai, Molecules, 2018, 23, 2846 CrossRef.
- Y. Hong, Y. Yao, H. Zhao, Q. Sheng, M. Ye, C. Yu and M. Lan, Anal. Chem., 2018, 90, 7617–7625 CrossRef CAS.
- Z.-G. Wang, N. Lv, W.-Z. Bi, J.-L. Zhang and J.-Z. Ni, ACS Appl. Mater. Interfaces, 2015, 7, 8377–8392 CrossRef CAS.
- Y. Yang, S. Bernardi, H. Song, J. Zhang, M. Yu, J. C. Reid, E. Strounina, D. J. Searles and C. Yu, Chem. Mater., 2016, 28, 704–707 CrossRef CAS.
- H. Zhang and F.-Q. Yang, J. Sep. Sci., 2019, 42, 342–359 CrossRef CAS.
- J. Liebscher, Eur. J. Org. Chem., 2019, 2019, 4976–4994 CrossRef CAS.
- J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healthc. Mater., 2018, 7, 1700831 CrossRef.
- M. P. Calatayud, B. Sanz, V. Raffa, C. Riggio, M. R. Ibarra and G. F. Goya, Biomaterials, 2014, 35, 6389–6399 CrossRef CAS.
- K. Kubiak-Ossowska, B. Jachimska and P. A. Mulheran, J. Phys. Chem. B, 2016, 120, 10463–10468 CrossRef CAS.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS.
- Z. J. Deng, M. Liang, I. Toth, M. Monteiro and R. F. Minchin, Nanotoxicology, 2013, 7, 314–322 CrossRef CAS.
- Z. Wang, Y. Xie, Y. Li, Y. Huang, L. R. Parent, T. Ditri, N. Zang, J. D. Rinehart and N. C. Gianneschi, Chem. Mater., 2017, 29, 8195–8201 CrossRef CAS.
- L. Klosterman and C. J. Bettinger, Int. J. Mol. Sci., 2016, 18, 14 CrossRef.
- L. B. Eckert, G. A. Repasky, A. S. Ülkü, A. McFall, H. Zhou, C. I. Sartor and C. J. Der, Cancer Res., 2004, 64, 4585 CrossRef CAS.
- S. Malaney and R. J. Daly, J. Mammary Gland Biol., 2001, 6, 101–113 CrossRef CAS.
- P. D. Gopal Krishnan, E. Golden, E. A. Woodward, N. J. Pavlos and P. Blancafort, Cancers, 2020, 12, 259 CrossRef.
- S. Martinotti, M. Patrone and E. Ranzato, ImmunoTargets Ther., 2015, 4, 101–109 CAS.
- R. B. Haga and A. J. Ridley, Small GTPases, 2016, 7, 207–221 CrossRef CAS.
- A. Svotelis, N. Gévry, G. Grondin and L. Gaudreau, Cell Cycle, 2010, 9, 364–370 CrossRef CAS.
- W. Sun, S. Lv, H. Li, W. Cui and L. Wang, Genes, 2018, 9, 633 CrossRef.
- A. Britschgi, A. Bill, H. Brinkhaus, C. Rothwell, I. Clay, S. Duss, M. Rebhan, P. Raman, C. T. Guy, K. Wetzel, E. George, M. O. Popa, S. Lilley, H. Choudhury, M. Gosling, L. Wang, S. Fitzgerald, J. Borawski, J. Baffoe, M. Labow, L. A. Gaither and M. Bentires-Alj, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, E1026–E1034 CrossRef CAS.
- H. Wu, H. Wang, S. Guan, J. Zhang, Q. Chen, X. Wang, K. Ma, P. Zhao, H. Zhao, W. Yao, F. Jin, Q. Xiao and M. Wei, Oncotarget, 2017, 8, 84996–85013 CrossRef.
- T.-X. Zhang, G.-Y. Zhu, B.-Y. Lu, C.-L. Zhang and Q. Peng, Nanomedicine, 2017, 12, 2757–2769 CrossRef CAS.
- A. Tesniere, T. Panaretakis, O. Kepp, L. Apetoh, F. Ghiringhelli, L. Zitvogel and G. Kroemer, Cell Death Differ., 2008, 15, 3–12 CrossRef CAS.
- R. Kuai, W. Yuan, S. Son, J. Nam, Y. Xu, Y. Fan, A. Schwendeman and J. J. Moon, Sci. Adv., 2018, 4, eaao1736 CrossRef.
- A. Alloatti, F. Kotsias, J. G. Magalhaes and S. Amigorena, Immunol. Rev., 2016, 272, 97–108 CrossRef CAS.
- J. C. Mbongue, H. A. Nieves, T. W. Torrez and W. H. R. Langridge, Front. Immunol., 2017, 8, 327 Search PubMed.
- L. Fong and E. G. Engleman, Annu. Rev. Immunol., 2000, 18, 245–273 CrossRef CAS.
- J. Neefjes, M. L. M. Jongsma, P. Paul and O. Bakke, Nat. Rev. Immunol., 2011, 11, 823 CrossRef CAS.
- P. A. Roche and K. Furuta, Nat. Rev. Immunol., 2015, 15, 203 CrossRef CAS.
- M. Jambhrunkar, M. Yu, H. Zhang, P. Abbaraju, A. K. Meka, A. Cavallaro, Y. Lu, N. Mitter and C. Yu, Nano Res., 2018, 11, 370–382 CrossRef CAS.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS.
- Y. Xing, J. Zhang, F. Chen, J. Liu and K. Cai, Nanoscale, 2017, 9, 8781–8790 RSC.
- U. S. Srinivas, B. W. Q. Tan, B. A. Vellayappan and A. D. Jeyasekharan, Redox Biol., 2018, 101084, DOI:10.1016/j.redox.2018.101084.
- Z. Zhang, X. Li, Y. Han, T. Ji, X. Huang, Q. Gao and D. Ma, Biomed. Pharmacother., 2019, 118, 109341 CrossRef CAS.
- T. Hiramoto, T. Nakanishi, T. Sumiyoshi, T. Fukuda, S. Matsuura, H. Tauchi, K. Komatsu, Y. Shibasaki, H. Inui, M. Watatani, M. Yasutomi, K. Sumii, G. Kajiyama, N. Kamada, K. Miyagawa and K. Kamiya, Oncogene, 1999, 18, 3422–3426 CrossRef CAS.
- E. Reeves and E. James, Immunology, 2017, 150, 16–24 CrossRef CAS.
- Y. Kumamoto, L. M. Mattei, S. Sellers, G. W. Payne and A. Iwasaki, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 8749 CrossRef CAS.
- T. Ahrends, A. Spanjaard, B. Pilzecker, N. Bąbała, A. Bovens, Y. Xiao, H. Jacobs and J. Borst, Immunity, 2017, 47, 848–861 CrossRef CAS.
- D. Iliopoulos, H. A. Hirsch and K. Struhl, Cancer Res., 2011, 71, 3196 CrossRef CAS.
- H. L. Herd, K. T. Bartlett, J. A. Gustafson, L. D. McGill and H. Ghandehari, Biomaterials, 2015, 53, 574–582 CrossRef CAS.
- C. Roma-Rodrigues, I. Pombo, L. Raposo, P. Pedrosa, A. R. Fernandes and P. V. Baptista, Front. Bioeng. Biotech., 2019, 7, 197 CrossRef.
- M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, L. M. Coussens, D. I. Gabrilovich, S. Ostrand-Rosenberg, C. C. Hedrick, R. H. Vonderheide, M. J. Pittet, R. K. Jain, W. Zou, T. K. Howcroft, E. C. Woodhouse, R. A. Weinberg and M. F. Krummel, Nat. Med., 2018, 24, 541–550 CrossRef CAS.
- B. S. Sharma, V. Prabhakaran, A. Desai, J. Bajpai, R. Verma and P. Swain, Oncogen, 2019, 2, 12 CrossRef.
- M. K. Sethi and S. Fanayan, Int. J. Mol. Sci., 2015, 16, 29278–29304 CrossRef CAS.
- C.-C. Chen, W.-C. Su, B.-Y. Huang, Y.-J. Chen, H.-C. Tai and R. P. Obena, Analyst, 2014, 139, 688–704 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02803g |
| This journal is © The Royal Society of Chemistry 2020 |