
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-controlled E/Z isomerization vs. [2 + 2] photocycloaddition mediated by supramolecular polymerization†
Torsten Dünnebacke,
Kalathil K. Kartha,
Johannes M. Wahl ,
Rodrigo Q. Albuquerque and
Gustavo Fernández*
,
Rodrigo Q. Albuquerque and
Gustavo Fernández*
Organisch-Chemisches Institut, Westfälische-Wilhelms Universität Münster, Corrensstraße, 40, 48149 Münster, Germany. E-mail: fernandg@uni-muenster.de
First published on 11th September 2020
Abstract
Control over the photochemical outcome of photochromic molecules in solution represents a major challenge, as photoexcitation often leads to multiple competing photochemical and/or supramolecular pathways resulting in complex product mixtures. Herein, we demonstrate precise and efficient control over the photochemical behaviour of cyanostilbenes in solution using a straightforward solvent-controlled approach based on supramolecular polymerization. To this end, we designed a π-extended cyanostilbene bolaamphiphile that exhibits tuneable solvent-dependent photochemical behaviour. Photoirradiation of the system in a monomeric state (in organic solvents) exclusively leads to a highly reversible and efficient E/Z photoisomerization, whereas a nearly quantitative [2 + 2] photocycloaddition into a single cyclobutane (anti head-to-tail) occurs in aqueous solutions. These results can be rationalized by a highly regular and preorganized antiparallel J-type arrangement of the cyanostilbene units that is driven by aqueous supramolecular polymerization. The presented concept demonstrates a novel approach towards solvent-selective and environmentally friendly photochemical transformations, which is expected to broaden the scope of supramolecular polymerization.
Introduction
Self-assembly of π-conjugated systems has become a key tool to create ordered and dynamic functional supramolecular nanomaterials.1–6 Particularly relevant in this regard are photoresponsive assemblies,5,7,8 as these systems can provide access to adaptive behaviour,9,10 one of key elements of intelligent matter.11 However, given that the energy landscape of photoresponsive supramolecular assemblies is extremely sensitive to minor changes in molecular organization as well as irradiation wavelength, intensity and time, controlling light-induced supramolecular phenomena remains a major challenge. For instance, photochromic building blocks that are molecularly dissolved or randomly organized within an assembly preferentially undergo a Z/E isomerization upon photoirradiation, leading in the latter case to a partial or complete disruption of the assembly.12 On the other hand, photochemical reactions such as cycloadditions may also occur if suitable photochromic molecules are located in close proximity and aligned in a preferred parallel fashion (<4.2 Å).13 Hence, control over the molecular order of photoresponsive systems in solution is key to predict the final outcome of the photochemical process. In a recent elegant example, Ajayaghosh and co-workers have exploited a gel as a confined medium in non-polar solvents to bias the photochemical behaviour of a 9-phenylethynylanthracene derivative, leading to a single product—the [4 + 2] photocycloadduct—in almost quantitative yield (>90%).14In addition to organic solvents, the use of water as medium for self-assembly is particularly relevant considering its environmentally friendly nature and its relevance in biological processes.15,16 In contrast to organic solvents, self-assembly in aqueous media is relatively less predictable given the difficulty in controlling the competition between hydrophobic and other non-covalent interactions.17,18 However, using rational design strategies, i.e. hydrophilic/hydrophobic ratio, and precisely modulating different experimental parameters, control over aqueous self-assembly processes can be achieved.16,19,20
In this context, programmed supramolecular polymerization represents a versatile tool to finely tune the spatial organization of molecules,1–4,10,21 which may be advantageous for controlling the outcome of photocycloadditions in solution. Expanding this strategy to aqueous media would be particularly relevant,16,17,20,22 as the poor solubility of most organic reactants undergoing photocycloadditions in water requires additional steps of encapsulation and release processes associated with the use of a nanocontainer.23 Given the need for green and sustainable chemical processes,15 supramolecular approaches in aqueous media would help to make photochemical transformations more simple, selective and environmentally benign.
Inspired by these principles, we selected cyanostilbenes as substrates due to their well-known ability to undergo [2 + 2] photocycloadditions and their strong bias towards predefined molecular organization via CN⋯H-bonds.24,25 In addition, cyanostilbenes exhibit excellent photophysical, electronic and stimuli-responsive properties,26,27 which have allowed their implementation in optoelectronics and as biomaterials.28 However, an inherent challenge of cyanostilbenes is the difficulty in controlling their photochemistry, as multiple pathways such as [2 + 2] cycloadditions, photocyclizations7,25,29,30 or E/Z photoisomerizations31 often occur concurrently. In recent elegant examples, microcrystallization methods29 and molecular confinement in nanocontainers32 have been used by Tang and co-workers to achieve regio- and stereoselective [2 + 2] photocycloadditions of cyanostilbenes. Notably, efficient and selective [2 + 2] cycloadditions were achieved for a cyanostilbene-pyridinium salt in aqueous suspensions using UV light.29 By modifying the experimental conditions, the authors could also control the formation of photocyclization and photoisomerization products. Additionally, the photocycloaddition product could be detected by exposure of crystals to room light for two weeks. This relatively long exposure time can be ascribed to the low penetration depth of light in solid matter. Despite these recent advances, “green” supramolecular approaches to control the photochemical outcome of cyanostilbenes in homogeneous aqueous solutions have not been accomplished to date.
Herein, we demonstrate precise and efficient control over E/Z isomerization and [2 + 2] photocycloaddition of cyanostilbenes in solution using visible light via a straightforward solvent-controlled approach based on supramolecular polymerization. This could be achieved by rational design of a linear cyanostilbene bolaamphiphile (Z)-CN-OPE, where the cyanostilbene core is conjugated with two oligophenyleneethynylene (OPE)-based aromatic wedges featuring peripheral glycol chains (Fig. 1a). While the extension of the π-surface is expected to simultaneously enhance the aggregation tendency of the cyanostilbene moiety and shift the irradiation wavelength to the visible region, the incorporation of glycol chains should ensure sufficient solubility in aqueous media. We anticipate that this appropriate balance of hydrophilic and hydrophobic structural elements would enable the required topochemical control for a [2 + 2] photocycloaddition to occur efficiently and selectively in aqueous media without the need of additional templating or confinement processes. On the other hand, the absence of close intermolecular contacts in organic “good” solvents is expected to hinder the photocycloaddition and preferentially favor a reversible E/Z photoisomerization pathway. In order to understand all these photochemical process in detail, both geometrical isomers of the target molecule (Z)- and (E)-CN-OPE were synthesized separately and fully characterized (see ESI for details†).
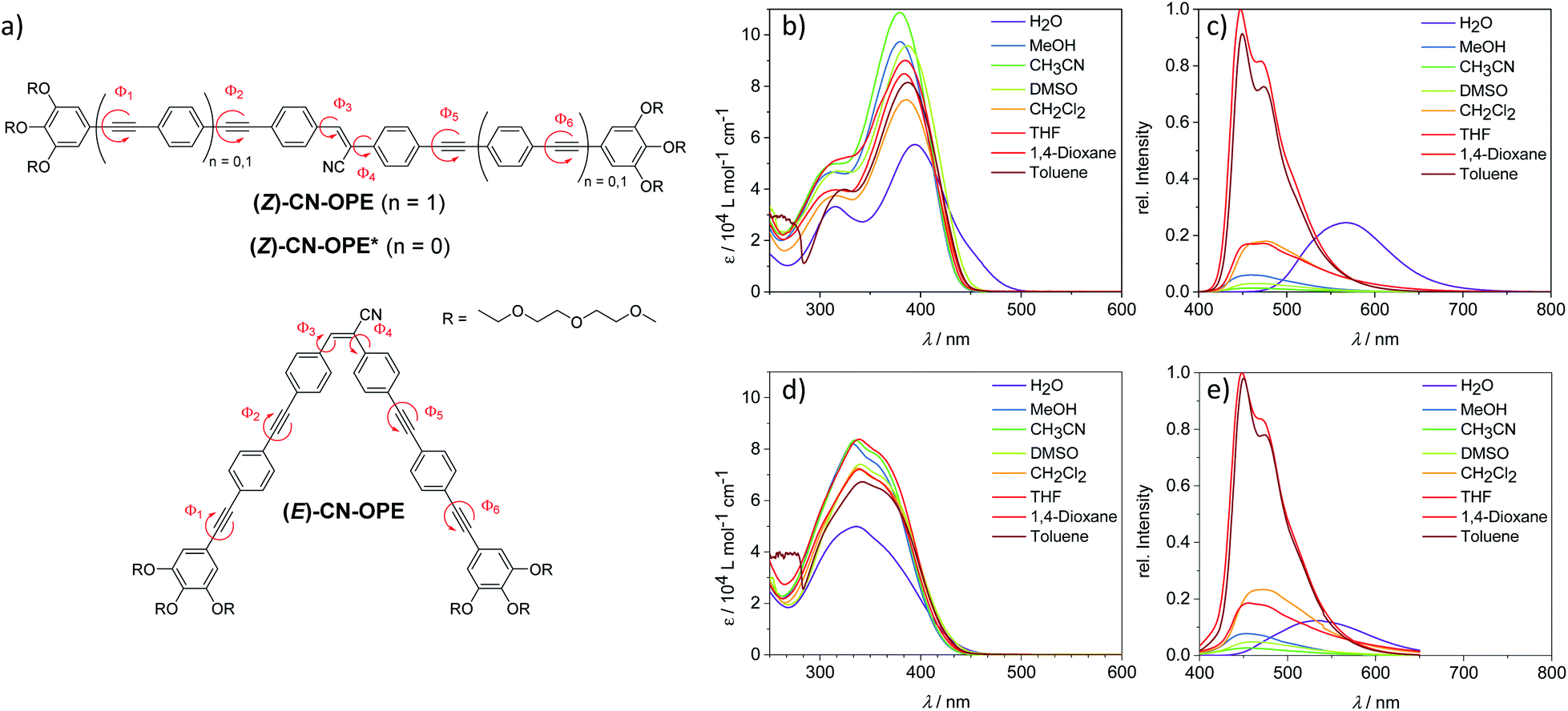 | ||
| Fig. 1 (a) Molecular structures of (Z)-CN-OPE, (E)-CN-OPE and reference compound (Z)-CN-OPE* with all designated internal rotations along the aromatic core (Φ1 to Φ6). (b) UV-Vis spectra of (Z)-CN-OPE in different selected solvents at c = 10−5 M. (c) Corresponding fluorescence spectra in different selected solvents at c = 10−5 M (excitation at λabs,max respectively). (d) UV-Vis spectra of (E)-CN-OPE in different selected solvents at c = 10−5 M. (e) Corresponding fluorescence spectra in different selected solvents at c = 10−5 M (excitation at λabs,max respectively). | ||
Results and discussion
Self-assembly behaviour in solution
We initially examined the self-assembly of the linear isomer (Z)-CN-OPE through UV-Vis studies in different solvents (298 K, 10−5 M). The system is molecularly dissolved in a wide range of organic solvents, whereas the use of alcohols and water promotes aggregation (Fig. S12 and S13†). The typical spectral pattern of molecularly dissolved (Z)-CN-OPE (i.e. in dichloromethane; DCM) exhibits a main absorption band with maximum (λmax) located around 385 nm along with a shoulder centered at 320 nm (Fig. 1b and Table S1†). In “poor” solvents such as water, significant spectral changes are observed: a bathochromic shift of λmax to 395 nm and the appearance of a shoulder band at around 450 nm that is accompanied by an overall broadening and decrease in the absorption (Fig. 1b, S12c, S13 and Table S1†). Nearly identical spectral features for the aggregate species are observed in protic solvents, i.e. alcohols, at a higher concentration (c ≥ 10−4 M, Fig. S13†). This behaviour suggests a J-type aggregation, which appears to be more pronounced in water due to a more significant red shift and lower extinction coefficients compared to the aggregates in alcohols. The observed behavior, however, deviates from a perfect J-type exciton coupling, as indicated by the broader absorbance and the lower overall extinction coefficient. In the herein presented system, a variety of different rotational angles in combination with bulky peripheral moieties may contribute to disturb a perfect J-type arrangement.On the other hand, the (E)-CN-OPE isomer showed no significant differences in the UV-Vis spectra (c = 10−5 M) (Fig. 1d and S14†) upon changing the solvents. Only in water, the occurrence of hypochromism is indicative of aggregation. However, the absence of additional spectral changes, i.e. shifts, suggests a weaker and less ordered aggregation than the linear (Z)-CN-OPE isomer. This assumption can be further supported by emission lifetime measurements (Fig. S15†). Despite that the 12-fold increase in the average emission lifetime of (E)-CN-OPE in aqueous solutions (0.141 ns in THF vs. 1.674 ns in H2O) implies an occurring aggregation process, the resulting average lifetime for aggregated (Z)-CN-OPE is significantly higher (4.471 ns in H2O), indicating a less ordered structure for (E)-CN-OPE in aqueous solutions. Furthermore, all attempts to obtain AFM images of these solutions showed no identifiable objects (Fig. S16†).
The photoluminescence (PL) spectra of both isomers are comparable in most of the tested solvents, with emission maxima between 455 and 472 nm (Fig. 1c, e and S17†), indicating that both isomers have the same emissive state.33,34 This is further validated by nearly identical average lifetimes of both isomers in THF (Fig. S15a and b†). The comparatively larger Stokes shifts (ΔλST) observed for (E)-CN-OPE (ΔλST = 109–128 nm) compared to (Z)-CN-OPE (ΔλST = 63–82 nm) (Fig. 1c, e and Table S1†) are also in agreement with different ground states, regardless of the same emissive state. Significantly different PL spectra were found for these isomers in water (Fig. 1c, e and S17†), which can be due to the effect of aggregation. For both isomers, the substantial bathochromic shift in the emission band (198 nm for (Z)-CN-OPE and 172 nm for (E)-CN-OPE) corroborates a J-type exciton coupling, which is more pronounced for the (Z)-CN-OPE isomer (Fig. 1c, e, S17 and Table S1.† For further details on the photophysical behaviour of both isomers, see ESI†).
Due to the strong aggregation propensity of linear (Z)-CN-OPE in water at room temperature even at concentrations as low as 3 × 10−7 M, the use of a co-solvent was necessary for mechanistic investigations.35 This approach also helps attenuate the marked lower critical solution temperature (LCST) behaviour of (Z)-CN-OPE in pure water (Fig. S18†), by which dehydration of the glycol chains upon heating (for the present system above 328 K at 10−5 M) results in turbid dispersions (see ESI for details†).36 Addition of a co-solvent (THF, volume fraction of water (fw) < 0.80, Fig. S18d†) or decreasing the concentration to 6 × 10−6 M in combination with heating enabled the derivation of the thermodynamic parameters of the self-assembly. Absorption and emission studies of (E)- and (Z)-CN-OPE (c = 10−5 M) in THF/water mixtures were initially carried out (Fig. 2a and S19†). Addition of up to 70 vol% water (fw ≤ 0.70) leads to an overall increase in absorption of (Z)-CN-OPE without any shift in λmax due to a possible restriction of internal rotation around the Φ1 to Φ6 bonds and associated planarization of the OPE scaffolds (see Fig. 1a).37 A comparison of the fluorescence excitation spectra in THF and water suggests an aggregation-induced planarization of the molecules (Fig. S20†). While the existence of two distinct excitation spectra in THF indicates different conformations of (Z)-CN-OPE in the molecularly dissolved state, a more planarized species is preferred in aqueous media. Upon further addition of water (fw > 0.75), a bathochromic shift (Δλmax = 18 nm), accompanied by a shoulder formation around 450 nm and a decrease in absorption are observed. This red-shifted absorption band further supports the formation of J-type aggregates.38 Several cooling rates (0.1 K min−1 to 5 K min−1) and THF/water ratios (fw = 0.70–0.80) were applied to demonstrate that (Z)-CN-OPE only forms a single, thermodynamically favored aggregate in solution (Fig. S21a–e†). The plots of αaggvs. temperature for (Z)-CN-OPE in water yielded non-sigmoidal curves (Fig. 2d) that were fitted to the cooperative nucleation–elongation model (Fig. S21a–e†),39 as also observed for other OPE-based bolaamphiphiles.40 In contrast to (Z)-CN-OPE, UV-Vis absorption studies of the V-shaped isomer (E)-CN-OPE at different THF/water ratios (fw = 0.10–1.00) showed no significant shifts in the λmax apart from a slight hypochromic behaviour (Fig. S19†).
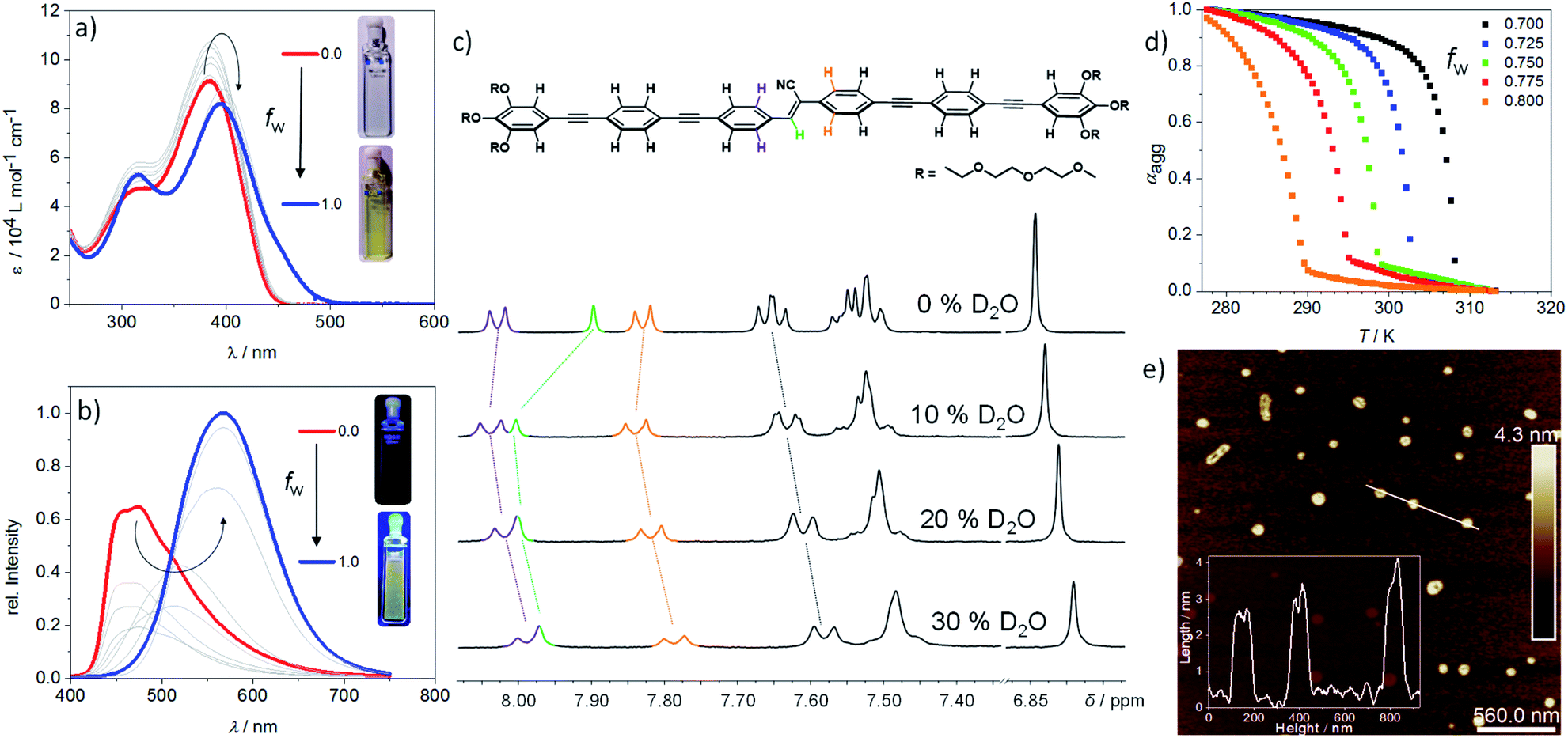 | ||
| Fig. 2 (a) UV-Vis spectra of (Z)-CN-OPE in different THF/H2O mixtures upon increasing volume fraction fw of water at c = 10−5 M and room temperature. (b) Corresponding fluorescence spectra under identical conditions. (c) Partial 1H NMR (THF-d8/D2O; 300 MHz; 298 K) showing the aromatic region of (Z)-CN-OPE under subsequent addition of D2O at a concentration of 3.96 × 10−3 M. (d) Cooling curves (plot of αaggvs. T) of (Z)-CN-OPE at c = 10−5 M using different THF/water ratios. (e) AFM image and height profile of aggregates in aqueous solution of (Z)-CN-OPE at c = 10−6 M drop-casted onto mica. | ||
Subsequently, fluorescence changes of (Z)-CN-OPE were monitored in different THF/water mixtures (fw = 0.10–1.00) as shown in Fig. 2b. Initial addition of 10% vol water (fw = 0.10) to a THF solution of (Z)-CN-OPE results in a drastic reduction of the fluorescence with no shifts in the emission maxima (λmax = 487 nm, Fig. 2b). Further addition of water induces a gradual red-shift in the emission band, which reaches a maximum of 570 nm at volume fractions of 80% (fw = 0.80). Simultaneously, the fluorescence is enhanced by 1.5 fold compared to that in pure THF (Fig. 2b). This behaviour can be attributed to the aggregation-induced emission enhancement (AIEE) commonly observed for linear (Z)-cyanostilbenes,41,42 which is caused by the restriction of internal rotation around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C axis upon photoirradiation.33,34 Increasing fw beyond 0.80 shows no significant changes in the fluorescence, indicating saturation of aggregation. On the other hand, the main emission band of (E)-CN-OPE undergoes a less significant red-shift in solvent-dependent experiments, even using large water contents (fw > 0.85; see Fig. S19d†). Thus, based on absorption and emission properties, only the more preorganized, undistorted linear isomer (Z)-CN-OPE is prone to efficient aggregation.
C axis upon photoirradiation.33,34 Increasing fw beyond 0.80 shows no significant changes in the fluorescence, indicating saturation of aggregation. On the other hand, the main emission band of (E)-CN-OPE undergoes a less significant red-shift in solvent-dependent experiments, even using large water contents (fw > 0.85; see Fig. S19d†). Thus, based on absorption and emission properties, only the more preorganized, undistorted linear isomer (Z)-CN-OPE is prone to efficient aggregation.
To elucidate the molecular packing, 1H NMR experiments were recorded by adding increasing volume fractions of D2O (fD = 0.0 to 0.3) to a monomer solution of (Z)-CN-OPE in THF-d8 (Fig. 2c, S22 and S23†). Addition of up to 10% vol water (fD = 0.0–0.10) causes a weak upfield shift of the aromatic signals (Δδ = 0.03 ppm) along with a more significant downfield shift for the signal of the vinylic proton (Δδ = 0.10 ppm) (Fig. 2c). Further addition of D2O (fD > 0.1) leads to an overall upfield shift for the signals corresponding to all aromatic rings as well as that of the vinylic proton. This trend, which has been recently observed for hydrophobic linear OPEs,43 points towards a plausible two-step supramolecular polymerization process: (i) an initial pre-nucleation event at fD < 0.10 driven by partial π–π stacking of the OPE units and H-bonding between the vinyl proton of one molecule and the cyano nitrogen of a neighbouring one (Cvinyl–H⋯N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, Fig. 3a, top); (ii) a subsequent elongation process mostly triggered by aromatic and hydrophobic interactions when fD exceeds 0.10. Such a two-step mechanism is also supported by UV-Vis experiments in THF/water mixtures as discussed above.
C, Fig. 3a, top); (ii) a subsequent elongation process mostly triggered by aromatic and hydrophobic interactions when fD exceeds 0.10. Such a two-step mechanism is also supported by UV-Vis experiments in THF/water mixtures as discussed above.
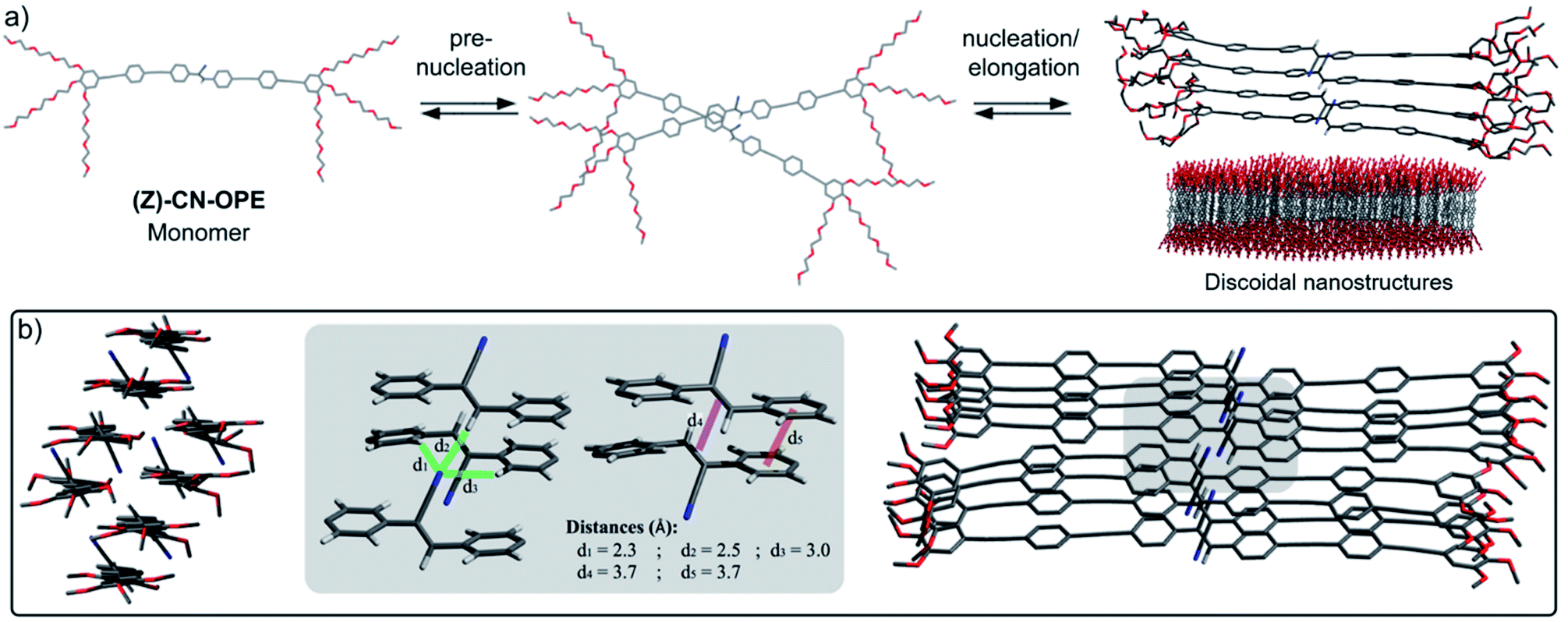 | ||
| Fig. 3 (a) Proposed stepwise aggregation process of (Z)-CN-OPE involving pre-nucleation, nucleation and elongation (the tetramer stack depicted in (a) has been geometry-optimized (PM6)). (b) Top and side view (left and right side of the image b, respectively) of two PM6 geometry-optimized tetramer stacks of (Z)-CN-OPE with TEG chains being replaced by OMe groups. Middle panel: magnification of the neighbouring cyanostilbene moieties with chosen intermolecular distances. | ||
2D ROESY NMR spectra were recorded at 299 K in THF-d8/D2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to shed more light on the packing of (Z)-CN-OPE. The appearance of a correlation signal between the vinylic proton and the adjacent aromatic protons, which is absent in COSY, corroborates a slight offset-stacking of the monomers of (Z)-CN-OPE (Fig. S24†).
:1) to shed more light on the packing of (Z)-CN-OPE. The appearance of a correlation signal between the vinylic proton and the adjacent aromatic protons, which is absent in COSY, corroborates a slight offset-stacking of the monomers of (Z)-CN-OPE (Fig. S24†).
FTIR measurements of (Z)-CN-OPE in THF (monomer) and thin film of an aggregate solution confirmed the existence of intermolecular CN⋯H interactions in the aggregated state, as the CN stretch shifts to lower wavenumbers (from 2216 to 2203 cm−1) upon aggregation (Fig. S25†).
Morphological and topological features of the (Z)-CN-OPE aggregates were obtained from Dynamic Light Scattering (DLS) experiments and Atomic Force Microscopy (AFM) (see Fig. S26 and S27†). Angular-dependent DLS of (Z)-CN-OPE in water (c = 10−5 M) revealed size distributions with average hydrodynamic radii RH of around 67 nm. Although the derived size distributions slightly vary (∼73 nm to ∼60 nm) within this set of measurements, the aggregates can be considered of spherical shape, as the RH values are stable over a wide range of angles (Fig. S26†).44 AFM imaging of the same aggregate solution drop-casted onto mica reveals the formation of disk-like aggregates with an average diameter of ∼80 nm and an average height of ∼3.5 nm (Fig. 2e, S27a and b†). The difference in the observed aggregate size when comparing the results from DLS and AFM can be explained by the hydration shell present in solution. Additional AFM studies increasing the amount of drop-casted material in combination with extended deposition time show the growth of several anisotropic nanostructures (Fig. S27c†). The height profile shows values of 3.5 ± 0.5 nm, which are in the same range as the isolated disks. The obtained height profiles are slightly lower than the length of the aromatic system of (Z)-CN-OPE (ca. 4.2 nm). This suggests that the monomer units within the assemblies are not arranged exactly perpendicular to the mica surface, but rather slightly tilted. Combining these results, we envisaged a stepwise self-assembly process involving a pre-nucleation, nucleation and elongation events that ultimately result in discoidal nanostructures, as illustrated in Fig. 3a.
Quantum mechanical calculations at the semiempirical dispersion-corrected PM6 level were carried out to provide insights on the structural features of self-assembled (Z)-CN-OPE. As shown in Fig. 3a and b, the cyano groups tend to arrange alternately in opposite directions within the optimized tetramer stacks, thereby cancelling the total electric dipole moment.45 The OPE units are planar and, consequently, involved in intermolecular aromatic interactions, whereas the CC double bond displays a distinct out of plane twist. While in isolated tetramer stacks intermolecular CN⋯H hydrogen bonds are not feasible (Fig. 3a, right and S28†), this can efficiently occur if two tetramers interact laterally to form an octamer, as revealed from the optimized stack shown in Fig. 3b. Hereby, the cyanostilbene strands formed by the respective tetramers align in a parallel fashion with a slight offset, allowing two corresponding, non-aromatic substituents attached to the CC double bond to form a head-to-tail like arrangement (see grey box in Fig. 3b).
The intermolecular distances from the cyano nitrogen to its neighbouring protons are d = 2.5 Å (CN⋯H–CC) and d = 2.3–3.0 Å (CN⋯Harom) (Fig. 3b, grey box). These theoretical results are in good agreement with intermolecular interactions determined by crystallographic data of previously reported cyanostilbenes.29,45,46 Furthermore, the calculated center-to-center distance between two adjacent stilbenic CC double bonds is 3.7 Å, which is very close to the values found for various stilbene13,47 and cyanostilbene29,48 assemblies undergoing [2 + 2] cycloaddition reactions. On this basis, we envisaged that the ideal dye arrangement of (Z)-CN-OPE can facilitate an efficient and selective light-induced cycloaddition reaction in the self-assembled state and a reversible geometrical photoisomerization in the isotropic state.17,18
Photoresponsive behaviour in the molecularly dissolved state
After detailed analysis of the experimental conditions under which (Z)-CN-OPE exists either in a monomeric or aggregated form, we next tested the photoresponsive behaviour. Upon exposing solutions of the (E)- (colorless) and (Z)-CN-OPE (yellow) isomers in CD2Cl2 (c = 8 × 10−3 M) to normal sunlight, a color change can be observed by the naked eye, suggesting a reversible photoswitching between both forms. These changes were further examined by time-dependent 1H NMR experiments upon irradiating separate solutions of the individual isomers at 298 K at different time intervals (Fig. 4a). Irrespective of the isomer (E or Z) that is irradiated, a photostationary state of 35:65 (Z:E) can be reached after 20 min of exposure to sunlight. This can be readily monitored by integration of the respective 1H signals of the corresponding isomers after each irradiation cycle (Fig. 4a).49
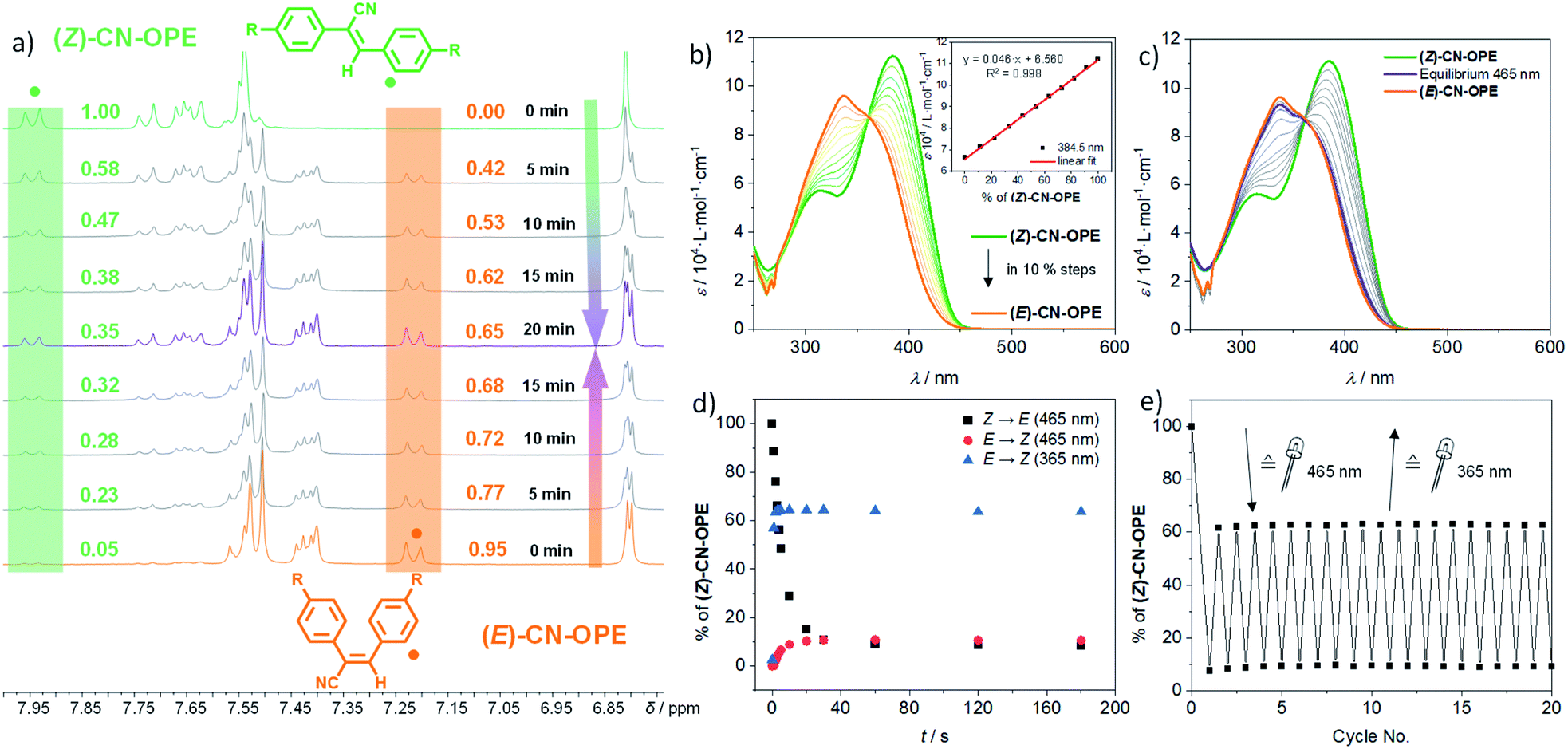 | ||
| Fig. 4 (a) 1H NMR spectra of (Z)-CN-OPE (top, green) and (E)-CN-OPE (bottom, orange) at c = 8 × 10−3 M in CD2Cl2 under exposure to sunlight, until photoequilibrium is reached (purple spectrum). (b) UV-Vis-spectra of E/Z-mixtures in DCM in 10% ratio steps at c = 10−5 M with equilibration function at λ = 384.5 nm. (c) UV-Vis-spectra of (Z)-CN-OPE (green) and (E)-CN-OPE (orange) at c = 10−5 M in DCM upon irradiation with λirr = 465 nm until the photoequilibrium is reached and determination of the E to Z ratio as a function of irradiation time. (d) Evolution of the E to Z ratio as a function of irradiation time while irradiating solutions of (Z)-CN-OPE and (E)-CN-OPE at different wavelengths. (e) Reversibility of the E/Z-photoisomerization upon irradiation at λirr = 465 nm and λirr = 365 nm starting from (Z)-CN-OPE in DCM at c = 10−5 M over 20 cycles. | ||
Further investigations of (Z)-CN-OPE under more dilute conditions (c = 10−5 M) using UV-Vis spectroscopy resulted in a photostationary state of ≥90% of (E)-CN-OPE. As this concentration range is too low for 1H NMR measurements, a spectroscopic method to determine the E:Z ratio was also employed (see ESI for details†).
Initially, UV-Vis absorption changes of (Z)-CN-OPE in DCM by adding increasing aliquot amounts of (E)-CN-OPE were monitored to identify the spectral features at different ratios (Fig. 4b). The plot of molar absorptivity ε vs. the percentage (%) of (Z)-CN-OPE yielded a linear regression (Fig. 4b inset). This calibration function was subsequently used to determine the Z:E equilibrium under light irradiation. Exposing solutions of either (Z)- or (E)-CN-OPE to visible light (λexc = 465 nm) for defined time intervals displays intermediate absorption spectra that cross via a defined isosbestic point.
This supports the presence of only two species (E- and Z-isomers) in equilibrium (Fig. 4c). Plotting the Z:E ratio as a function of time reveals a plateau after 20 s regardless of the initial pure isomeric ratio (Fig. 4d). The achieved photoequilibrium state mainly consists of the (E)-CN-OPE isomer (6:94 (Z:E), Fig. 4; for detailed information on the calculated equilibria ratios see ESI†). UV irradiation (λexc = 365 nm) triggers the back isomerization (E-to-Z) by re-establishing the photoequilibrium of 67:33% (Z:E) (Fig. 4d and Table 1). The time required to reach the equilibrium upon irradiation with UV light was around 2–3 s (Fig. 4d), which is considerably faster than in the presence of visible light. This interconversion between (E) and (Z) is highly reversible, no degradation being observed even after 20 consecutive cycles of alternated irradiation at 465 and 365 nm (Fig. 4e).
| Solvent in decreasing order of polarity45 | Equilibrium λirr = 465 nm, Z:E |
k Z→E,465 nm/10−2 s−1 | Equilibrium λirr = 365 nm, Z:E |
|---|---|---|---|
| MeOH | 21:79 |
2.49 | 66:34 |
| CH3CN | 57:43 |
1.03 | 87:13 |
| DMSO | 57:43 |
3.08 | 90:10 |
| DCM | 6:94 |
15.48 | 67:33 |
| THF | 28:72 |
7.30 | 85:15 |
| Dioxane | 3:97 |
18.91 | 47:53 |
| Toluene | 6:94 |
34.01 | 45:55 |
We also analysed the impact of the solvent on the kinetics and efficiency of the photoisomerization process (Fig. S29 and S30†). The results reveal that the equilibrium concentration of (E)-CN-OPE, as well as the first-order rate constant for the Z-to-E conversion, are dependent on the chosen solvent (Table 1). In rough approximation, the rate constant kZ→E is higher in non-polar solvents compared to polar solvents, while the equilibrium E/Z ratio exhibits only a weak trend in solvent polarity. Analysis of the ground and excited states using a Lippert–Mataga-plot50 revealed a more polar excited state (Fig. S31†), as previously reported for polar cyanostilbenes.26,42 We therefore assume that the influence of the solvent on the excited state may affect the reaction rate by possibly facilitating competing relaxation pathways such as twisted intramolecular charge transfer (TICT).51,52 Nevertheless, due to the polar, bulky TEG chains and the conjugated aromatic core, which induce different solvent–solute interactions, other solvent properties like viscosity/solvent friction should also be taken into account.52
Photochemical behaviour in the aggregated state
In sharp contrast to the behaviour in good solvents such as DCM, light irradiation (λexc = 465 nm) of an aqueous solution of (Z)-CN-OPE (c = 10−5 M, 298 K) leads to the formation of a new blue-shifted absorption band at 336 nm at the expense of the band at 400 nm (Fig. 5a, blue and brown solid lines). Significant changes are also observed in emission studies upon light irradiation. The emission band at around 570 nm that was previously assigned to J-type aggregates gradually decreases and blue shifts reaching an emission maximum of 490 nm (Fig. 5a, blue and brown dotted lines). These spectral changes suggest a shortening of the effective length of the conjugated π-system upon light irradiation, which might be the result of a [2 + 2] cycloaddition reaction31 of (Z)-CN-OPE in the aggregated state. Further analysis using Gel Permeation Chromatography (GPC) reveals that the species obtained upon photoirradiation has a larger molecular weight than the (Z)-CN-OPE and (E)-CN-OPE monomers (Fig. 5b). In order to identify the newly formed species, the resulting photoirradiation product was examined by High-Resolution Mass Spectrometry (HRMS) (Fig. 5c). These experiments shed m/z values of 1072.1886 and 1073.8404 that are in accordance with a [2 + 2] cycloaddition product of (Z)-CN-OPEvia a cyclobutane ring formation. Thus, the overall irradiation studies in different solvents clearly reveal that the [2 + 2] cycloaddition reaction of (Z)-CN-OPE only occurs in the aggregated state, where close interactions and optimal arrangement of the central CC double bonds is possible,29,53 as predicted by the PM6 calculations (Fig. 3). The newly formed species is obtained quantitatively (96% yield) as a single product, showing the high efficiency of cycloaddition of (Z)-CN-OPE in water/THF (99:1). Assuming that only the more stable (Z)-isomer is present in aqueous solution under standard conditions—the (E)-isomer is significantly less active for aggregation—, four different regioisomeric cyclobutanes can be formed with equal probability depending on the relative orientation of the reactants:29syn head-to-head, anti head-to-head, syn head-to-tail and anti head-to-tail (Fig. 5d). Chiral HPLC studies suggest that only one of these isomers is formed during the photocycloaddition of (Z)-CN-OPE (Fig. S32†). 1H NMR studies (c = 4 × 10−3 M, CDCl3) reveal only one clear singlet at δ = 5.23 ppm for the aliphatic protons of the cyclobutane ring (Fig. S33†). 13C NMR (CDCl3, 30 mM, 293 K) of this sample shows two carbon signals for the cyclobutane ring between δ = 55.9 and 47.8 ppm (Fig. S10†), revealing that the obtained isomer is highly symmetrical. The overall experimental results match well with those obtained for related cyanostilbenes undergoing selective [2 + 2] cycloadditions into the anti head-to-tail cylobutane (Fig. 5b, bottom right).29 On this basis and given the antiparallel arrangement of the cyanostilbene units in the aqueous assemblies (vide supra), this strongly suggests that the anti head-to-tail isomer is the most probable outcome of the photochemical reaction of (Z)-CN-OPE in water. To further demonstrate the advantage of our approach, the reaction was performed under comparable conditions (water, room temperature, λexc = 365 nm for 80 min) with the precursor (Z)-2,3-bis(4-((trimethylsilyl)ethynyl)phenyl)acrylonitrile (8) (see Scheme S1 and Fig. S34†). 1H NMR analysis of the reaction mixture revealed nearly no reaction. Only minor additional peaks, neither corresponding to the tested molecule nor the respective E-isomer, were detectable (Fig. S34a†). In the region of the cyclobutane protons, a set of different signals appears, indicating a mixture of the few synthesized photoproducts (Fig. S34b†).
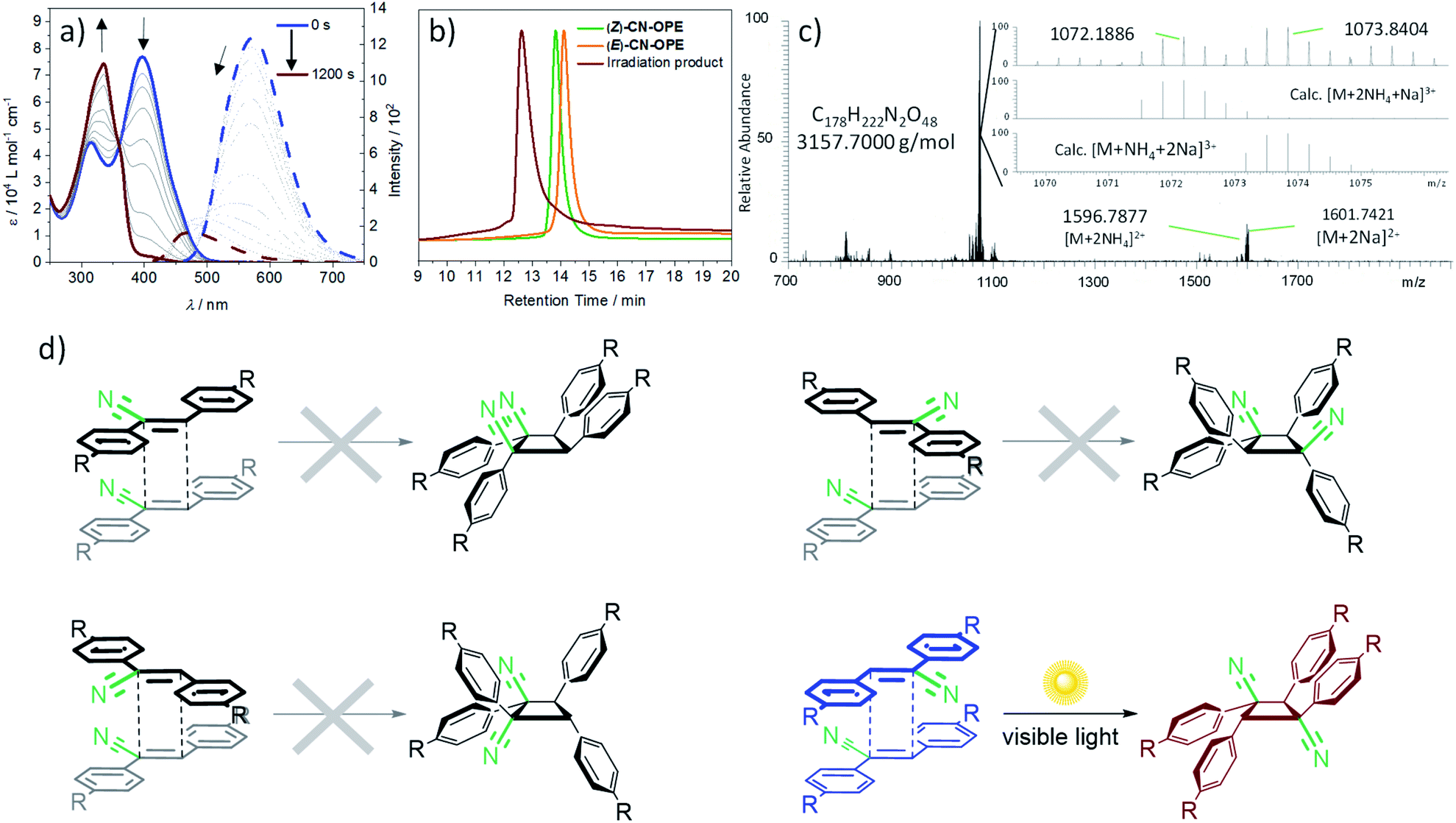 | ||
| Fig. 5 (a) Time-dependent UV-Vis (solid line) and fluorescence (dashed line) spectra of (Z)-CN-OPE in water at c = 10−5 M under exposure to visible light (λexc = 465 nm). The arrows depict the progression of the spectra over time. (b) GPC of both initial isomers compared to the photoproduct after irradiation. (c) HRMS spectra of the obtained photoproduct after irradiation. (d) Schematic representation of the possible [2 + 2] photocycloaddition reactions of (Z)-CN-OPE. Due to strong aggregation and intermolecular contacts in water, isomers arising from initial E/Z isomerization of CN-OPE are less likely and are therefore neglected. | ||
Once the specific isomer was identified, we further examined the efficiency of the [2 + 2] photoreaction in water by selecting additional light sources with different energies (λexc = 365 and 520 nm) (Fig. S35a–d†). Irradiation with UV light (λexc = 365 nm) causes an increase in the reaction rate, however at the expense of a slight decrease in the absorbance intensity, which might arise from other, less favourable side reactions (Fig. S35a†). Interestingly, the selective [2 + 2] photocycloaddition also occurs efficiently without side products when the aqueous solution of aggregated (Z)-CN-OPE is exposed to green light (λexc = 520 nm; Fig. S35c†). Plotting the absorption changes at 399 nm vs. time revealed a second order kinetics (Fig. S35a–c†). As the sun remains the most economical and sustainable light source, we finally exposed the aqueous assembly of (Z)-CN-OPE to normal sunlight and followed the photochemical reaction. Nearly complete conversion of (Z)-CN-OPE was observed in only 120 s, indicating the fast and efficient photochemical reaction under green conditions (normal daylight and in water; Fig. S35d†).
Finally, to further demonstrate the advantage of a supramolecular approach in aqueous media, we synthesized a structurally related (Z)-CN-OPE* (see Fig. 1), in which the aromatic surface is reduced at each side of the stilbene moiety (for synthesis and characterization, see ESI†). Even though the aggregation tendency is reduced by this structural modification (Fig. S36a†), we were able to reproduce the solvent-dependent photochemical behaviour observed by (Z)-CN-OPE. While irradiation of (Z)-CN-OPE* in the molecularly dissolved state with either visible or UV light (λ = 465 nm and 365 nm) triggers the reversible E/Z isomerization (Fig. S36c and d†), an efficient (90% yield) photocycloaddition into a single product (cyc-CN-OPE*) occurs in water (Fig. S36b and S11†), in good agreement with the behaviour of (Z)-CN-OPE.
Conclusions
We have reported a new linear bolaamphiphilic π-extended cyanostilbene derivative (Z)-CN-OPE that undergoes solvent-dependent E/Z photoisomerization or [2 + 2] photocycloadditions in a highly controlled manner. In solvents that favour a molecularly dissolved state (a wide range of organic solvents), a highly reversible and quantitative E/Z photoisomerization is observed. This could be monitored in detail by comparing the light-responsive behaviour of the target (Z)-CN-OPE and that of its geometrical isomer (E)-CN-OPE, which was synthesized separately. In contrast, the use of aqueous solutions induces the formation of emissive J-type supramolecular polymers that undergo a highly efficient and selective [2 + 2] photocycloaddition upon visible light irradiation. Extensive experimental studies supported by theory demonstrate that the combination or aromatic, hydrophobic and CN⋯H interactions enable the precise topochemical control for a specific cyclobutane (anti head-to-tail) to be formed efficiently. Our results have demonstrated that the photoresponsive behaviour of cyanostilbenes in solution can be precisely tuned by solvent-controlled supramolecular polymerization, which is expected to broaden the scope of stimuli-responsive materials.Experimental
See ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. F., K. K. K. and T. D. thank the European Commission for funding (ERC-StG-2016 SUPRACOP-715923). We also thank Prof. C. A. Strassert and Stefan Buss (WWU Münster) for emission lifetime measurements.References
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730 CAS.
- Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem.–Eur. J., 2019, 25, 5848 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef.
- C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS.
- (a) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS; (b) L. Maggini and D. Bonifazi, Chem. Soc. Rev., 2012, 41, 211 CAS; (c) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS.
- N. Bäumer, K. K. Kartha, N. K. Allampally, S. Yagai, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 15626 CrossRef.
- (a) S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325 CAS; (b) M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536 RSC; (c) E. Borré, J.-F. Stumbé, S. Bellemin-Laponnaz and M. Mauro, Chem. Commun., 2017, 53, 8344 RSC; (d) M. Yamauchi, T. Ohba, T. Karatsu and S. Yagai, Nat. Commun., 2015, 6, 8936 CrossRef CAS; (e) S. Yagai, Bull. Chem. Soc. Jpn., 2015, 88, 28 CrossRef; (f) R. D. Mukhopadhyay, V. K. Praveen and A. Ajayaghosh, Mater. Horiz., 2014, 1, 572 RSC; (g) C. García-Iriepa, M. Marazzi, L. M. Frutos and D. Sampedro, RSC Adv., 2013, 3, 6241 RSC.
- A. Walther, Adv. Mater., 2020, 32, 1905111 CrossRef CAS.
- G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757 RSC.
- (a) H. Zeng, P. Wasylczyk, D. S. Wiersma and A. Priimagi, Adv. Mater., 2018, 30, e1703554 CrossRef; (b) H. Zhou, X. Ding, Z. Zheng and Y. Peng, Soft Matter, 2013, 9, 4956 RSC.
- (a) K. Higashiguchi, G. Taira, J.-i. Kitai, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2015, 137, 2722 CrossRef CAS; (b) A. K. Mandal, M. Gangopadhyay and A. Das, Chem. Soc. Rev., 2015, 44, 663 RSC; (c) J. Li, H. Jiang, W. Hu, G. Zou and Q. Zhang, J. Photochem. Photobiol., A, 2012, 245, 28 CrossRef CAS; (d) S. Mahesh, A. Gopal, R. Thirumalai and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 7227 CrossRef CAS.
- R. Medishetty, I.-H. Park, S. S. Lee and J. J. Vittal, Chem. Commun., 2016, 52, 3989 RSC.
- S. Das, N. Okamura, S. Yagi and A. Ajayaghosh, J. Am. Chem. Soc., 2019, 141, 5635 CrossRef CAS.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747 CrossRef CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414 CrossRef CAS.
- A. Sikder and S. Ghosh, Mater. Chem. Front., 2019, 3, 2602 RSC.
- E. Krieg and B. Rybtchinski, Chem.–Eur. J., 2011, 17, 9016 CrossRef CAS.
- (a) Y. Wang and M. Lee, ChemPlusChem, 2020, 85, 711 CrossRef CAS; (b) H. Wang and M. Lee, Macromol. Rapid Commun., 2020, 41, e2000138 CrossRef; (c) I. Helmers, B. Shen, K. K. Kartha, R. Q. Albuquerque, M. Lee and G. Fernández, Angew. Chem., Int. Ed., 2020, 59, 5675 CrossRef CAS; (d) E. Cohen, Y. Soffer, H. Weissman, T. Bendikov, Y. Schilt, U. Raviv and B. Rybtchinski, Angew. Chem., Int. Ed., 2018, 57, 8871 CrossRef CAS; (e) W. Li, Y. Kim and M. Lee, Nanoscale, 2013, 5, 7711 RSC.
- E. Krieg, A. Niazov-Elkan, E. Cohen, Y. Tsarfati and B. Rybtchinski, Acc. Chem. Res., 2019, 52, 2634 CrossRef CAS.
- (a) M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38 CrossRef CAS; (b) G. Ghosh, T. Ghosh and G. Fernández, ChemPlusChem, 2020, 85, 1022 CrossRef CAS; (c) C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Acc. Chem. Res., 2017, 50, 1928 CrossRef CAS; (d) D. van der Zwaag, T. F. A. de Greef and E. W. Meijer, Angew. Chem., Int. Ed., 2015, 54, 8334 CrossRef CAS; (e) R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241 CrossRef CAS; (f) C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661 CrossRef CAS; (g) J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527 CrossRef CAS.
- (a) M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440 CrossRef CAS; (b) M. R. Molla and S. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 26672 RSC; (c) W. Li, Y. Kim, J. Li and M. Lee, Soft Matter, 2014, 10, 5231 RSC; (d) Y. Kim, W. Li, S. Shin and M. Lee, Acc. Chem. Res., 2013, 46, 2888 CrossRef CAS; (e) D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328 CrossRef.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914 CrossRef CAS.
- Y. Zhang, J. Sun, X. Lv, M. Ouyang, F. Cao, G. Pan, L. Pan, G. Fan, W. Yu, C. He, S. Zheng, F. Zhang, W. Wang and C. Zhang, CrystEngComm, 2013, 15, 8998 RSC.
- D. R. Turner, A. J. Edwards and R. O. Piltz, CrystEngComm, 2012, 14, 6447 CAS.
- X. Wang, Z. Ding, Y. Ma, Y. Zhang, H. Shang and S. Jiang, Soft Matter, 2019, 15, 1658 RSC.
- Y. Zhang, J. Hu, G. Yang, H. Zhang, Q. Zhang, F. Wang and G. Zou, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2458 CrossRef CAS.
- (a) M. Martínez-Abadía, R. Giménez and M. B. Ros, Adv. Mater., 2018, 30, 1704161 CrossRef; (b) M. Martínez-Abadía, S. Varghese, P. Romero, J. Gierschner, R. Giménez and M. B. Ros, Adv. Opt. Mater., 2017, 5, 1600860 CrossRef.
- P. Wei, J.-X. Zhang, Z. Zhao, Y. Chen, X. He, M. Chen, J. Gong, H. H.-Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 1966 CrossRef CAS.
- Y.-J. Gao, X.-P. Chang, X.-Y. Liu, Q.-S. Li, G. Cui and W. Thiel, J. Phys. Chem. A, 2017, 121, 2572 CrossRef CAS.
- K. Y. Kim, S. H. Jung, S. Lee, C. J. Moon, Y. Choi, M. Y. Choi and J. H. Jung, J. Phys. Chem. C, 2018, 122, 22143 CrossRef CAS.
- P. Wei, Z. Li, J.-X. Zhang, Z. Zhao, H. Xing, Y. Tu, J. Gong, T. S. Cheung, S. Hu, H. H.-Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Mater., 2019, 31, 1092 CrossRef CAS.
- N. Yamamoto, J. Phys. Chem. C, 2018, 122, 12434 CrossRef CAS.
- K. Kokado, T. Machida, T. Iwasa, T. Taketsugu and K. Sada, J. Phys. Chem. C, 2018, 122, 245 CrossRef CAS.
- D. Horn and J. Rieger, Angew. Chem., Int. Ed., 2001, 40, 4330 CrossRef CAS.
- (a) D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094 CrossRef; (b) D. Görl, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2016, 7, 6786 RSC.
- M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291 CrossRef CAS.
- (a) A. Rödle, M. Lambov, C. Mück-Lichtenfeld, V. Stepanenko and G. Fernández, Polymer, 2017, 128, 317 CrossRef; (b) T. Rudolph, N. Kumar Allampally, G. Fernández and F. H. Schacher, Chem.–Eur. J., 2014, 20, 13871 CrossRef CAS; (c) M. J. Mayoral, C. Rest, J. Schellheimer, V. Stepanenko and G. Fernández, Chem.–Eur. J., 2012, 18, 15607 CrossRef CAS.
- (a) Y. Ma, M. Cametti, Z. Džolić and S. Jiang, J. Mater. Chem. C, 2016, 4, 10786 RSC; (b) S. Shin, S. H. Gihm, C. R. Park, S. Kim and S. Y. Park, Chem. Mater., 2013, 25, 3288 CrossRef CAS; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (d) J. W. Chung, B.-K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750 CrossRef CAS.
- Y. Zhang, J. Sun, G. Bian, Y. Chen, M. Ouyang, B. Hu and C. Zhang, Photochem. Photobiol. Sci., 2012, 11, 1414 RSC.
- D. S. Philips, K. K. Kartha, A. Politi, T. Krüger, R. Q. Albuquerque, G. Fernandez, A. T. Politi and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 4732 CrossRef CAS.
- B.-S. Kim, D.-J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333 CrossRef CAS.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS.
- (a) W. Jia, P. Yang, J. Li, Z. Yin, L. Kong, H. Lu, Z. Ge, Y. Wu, X. Hao and J. Yang, Polym. Chem., 2014, 5, 2282 RSC; (b) Y. Li, F. Li, H. Zhang, Z. Xie, W. Xie, H. Xu, B. Li, F. Shen, L. Ye, M. Hanif, D. Ma and Y. Ma, Chem. Commun., 2007, 231 RSC.
- M. Nagarathinam, A. M. P. Peedikakkal and J. J. Vittal, Chem. Commun., 2008, 5277 RSC.
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163 CrossRef CAS.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef.
- (a) A. Kawski, Z. Naturforsch., 2002, 57a, 255 Search PubMed; (b) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690 CrossRef CAS.
- (a) S. Sasaki, G. P. C. Drummen and G.-i. Konishi, J. Mater. Chem. C, 2016, 4, 2731 RSC; (b) L. Zhu and Y. Zhao, J. Mater. Chem. C, 2013, 1, 1059 RSC; (c) J. Svoboda and B. König, Chem. Rev., 2006, 106, 5413 CrossRef CAS.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415 CrossRef CAS.
- X. Jin, D. Yang, Y. Jiang, P. Duan and M. Liu, Chem. Commun., 2018, 54, 4513 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03442h |
| This journal is © The Royal Society of Chemistry 2020 |