
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalyzed remote functionalization of inert C(sp3)–H bonds of alkenes via 1,n-hydrogen-atom-transfer by C-centered radical relay†
Kang-Jie
Bian‡
,
Yan
Li‡
,
Kai-Fan
Zhang
,
Yan
He
,
Tian-Rui
Wu
,
Cheng-Yu
Wang
and
Xi-Sheng
Wang
 *
*
Hefei National Laboratory for Physical Sciences at the Microscale, Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China. E-mail: xswang77@ustc.edu.cn
First published on 9th September 2020
Abstract
As an alternative approach to traditional C–H activation that often involved harsh conditions, and vicinal or primary C–H functionalization, radical relay offers a solution to these long-held problems. Enabled by 1,n (n = 5, 6)-hydrogen atom transfer (HAT), we use a most prevalent moiety, alkene, as the precursor to an sp3 C-centered radical to promote selective cleavage of inert C(sp3)–H bonds for the generation of azidotrifluoromethylated molecules. Mild conditions, broad scope and excellent regioselective control (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) are observed in the reactions. Deuterium labelling studies disclose the kinetic characteristics of the transformations and verify a direct 1,n-HAT pathway. The key to this C-centered radical relay is that iron plays a dual role as a radical initiator and terminator to incorporate the azide functionality through radical oxidation via azido–ligand-transfer. The methods and the later derivatization promise expeditious synthesis of CF3-containing organic azides, γ-lactam and triazoles that are widely used in designing new fluorescent tags and functional materials.
:1) are observed in the reactions. Deuterium labelling studies disclose the kinetic characteristics of the transformations and verify a direct 1,n-HAT pathway. The key to this C-centered radical relay is that iron plays a dual role as a radical initiator and terminator to incorporate the azide functionality through radical oxidation via azido–ligand-transfer. The methods and the later derivatization promise expeditious synthesis of CF3-containing organic azides, γ-lactam and triazoles that are widely used in designing new fluorescent tags and functional materials.
Introduction
The facile construction of complex functional molecules in a straightforward, efficient and practical manner from easily available materials is a major target and challenge in organic synthesis. Transition-metal-catalyzed C–H functionalization is a highly step- and atom-economic tactic to synthesise target molecules with structural complexity and diversity and it has been developed as one of the most ideal methods meeting the requirements of synthetic chemistry.1 Indeed, the direct functionalization of C–H bonds is emerging as a powerful strategy for retrosynthetic disconnection in the total synthesis of complex molecules, especially for the omnipresent inert C(sp3)–H bonds in basic chemical raw materials.2 As an alternative approach to traditional C(sp3)–H activation, where metallacycles are usually formed in five-, six-membered rings and restricted mostly for vicinal functionalization of primary C(sp3)–H bonds, hydrogen-atom-transfer (HAT) through a radical pathway has been used as an efficient tool to activate the remote C(sp3)–H bond selectively.3 Of note is that, as the target inert C(sp3)–H bond is almost impossible to distinguish from ubiquitous C–H bonds on the alkyl chain, 1,n (normally 1,5)-hydrogen-atom-transfer (HAT) offers a reliable solution for selective cleavage of remote C(sp3)–H bonds in a highly chemo- and site-selective fashion. In addition, the radical relay strategy offers a complementary path to direct functionalization of steric-hindered tertiary C(sp3)–H bonds, which still remains a significant challenge in traditional transition-metal-catalyzed C–H activation reactions.Very recently, a ‘directing-group strategy’3g has been introduced through a 1,5-HAT process for direct functionalization of remote C(sp3)–H bonds on alkyl chains. Actually, in retrospect, this type of ‘directed-functionalization’ can be traced back to 1883. The pioneering work, the Hofmann–Löffler–Freytag (HLF)4 reaction, has been well documented as a highly efficient and site-selective strategy for remote C(sp3)–H amination, where a pre-decorated N-haloamine was required to serve as the precursor to a N-centered radical.
Furthermore, heteroatom-containing groups, such as amides, amines and alcohols, have been widely used as ‘directing-groups’ to generate the heteroatom-centered radical to initiate the radical relay process due to their intrinsic high reactivity, in which the directing group could be ‘recovered’ by abstraction of the hydrogen atom from C(sp3)–H bonds. Reasonably, it could be inferred that the carbon radical, likewise, should offer an alternative solution to initiate the 1,n-HAT process and promote in situ generation of more stable carbon radicals on the alkyl chain for diverse functionalization. Compared with the highly reactive heteroatom-centered radical that can easily undergo hydrogen abstraction, 1,n-HAT initiated by the C-centered radical was more difficult with regard to bond dissociation energies (BDEs)5 and the HAT rate constant,6 which led to a weaker driving force for remote functionalization. Actually, to efficiently produce a carbon radical for promotion of following radical relay,7 one of the most facile and direct methods is through the radical addition to unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, which is also a major method that is known for enhancing molecular complexity in a single step.8 Using this strategy, several pioneering examples were reported on copper-catalyzed9 or visible-light-mediated10 1,6-difunctionalizations of alkenes recently; however, among these studies, remote functionalization can only be achieved on relatively active C(sp3)–H bonds, requiring an adjacent heteroatom functionality such as a nitrogen or oxygen atom,9b,d and carbonyl group,9c or on benzylic C–H bonds abutting electron-donating, neutral or weakly-withdrawing arenes.10 It was proposed that the carbocation was likely to be involved in the catalytic cycle,9c,10a where the in situ generated carbon radical underwent oxidation through single electron transfer (SET) leading to the corresponding carbocation and then was quenched by nucleophiles (Fig. 1a). In contrast, we envisioned that by using metal-catalyzed radical oxidation,11 the rapid capture of the carbon radical via a ligand-transfer process could offer a solution to the long-held unresolved issue of remote functionalization of inert/unactivated12 C(sp3)–H bonds (Fig. 1b).
C bonds, which is also a major method that is known for enhancing molecular complexity in a single step.8 Using this strategy, several pioneering examples were reported on copper-catalyzed9 or visible-light-mediated10 1,6-difunctionalizations of alkenes recently; however, among these studies, remote functionalization can only be achieved on relatively active C(sp3)–H bonds, requiring an adjacent heteroatom functionality such as a nitrogen or oxygen atom,9b,d and carbonyl group,9c or on benzylic C–H bonds abutting electron-donating, neutral or weakly-withdrawing arenes.10 It was proposed that the carbocation was likely to be involved in the catalytic cycle,9c,10a where the in situ generated carbon radical underwent oxidation through single electron transfer (SET) leading to the corresponding carbocation and then was quenched by nucleophiles (Fig. 1a). In contrast, we envisioned that by using metal-catalyzed radical oxidation,11 the rapid capture of the carbon radical via a ligand-transfer process could offer a solution to the long-held unresolved issue of remote functionalization of inert/unactivated12 C(sp3)–H bonds (Fig. 1b).
 | ||
| Fig. 1 Remote difunctionalization of alkenes enabled by the 1,n-HAT strategy. | ||
Herein, we described the first example of iron-catalyzed remote azidation of an inert C–H bond through radical relay using an sp3 C-centered radical, in which a selective cleavage of the remote C(sp3)–H bond took place, enabled by a direct 1,n-HAT process, furnishing 1,6- and 1,7-azidotrifluoromethylated molecules. The key to success is the radical oxidation of the in situ generated carbon radical by ironIII–azide species.13 This azidotrifluoromethylation was achieved in an iron-catalyzed system, demonstrating high catalytic reactivity, mild conditions, and excellent regioselective control (mostly >20:1). The deuterium labeling reactions, for the first time indicate that an entropically less favored 1,6-HAT3i,14 follows a direct C(sp3)–H bond cleavage pathway.
Results and discussion
Iron is widely known as an efficient catalyst for diverse organic transformations due to its low price, non-toxicity and environmentally benign character. Our study commenced with diethyl 2-allyl-2-isobutylmalonate S1 as the pilot substrate, Togni-II agent as the trifluoroalkylating reagent and TMSN3 as the azide source in the presence of a catalytic amount of iron salts (entries 1–21, Table 1). To our delight, the desired remote difunctionalized product 1 was obtained in 75% of yield when Fe(acac)3 was used as the catalyst and L1 as the ligand (entry 1, Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Ligand | Solvent | Yield (%) |
|
a Reaction condition: S1 (0.1 mmol), TMSN3 (0.3 mmol), Togni-II (0.125 mmol), catalyst (10 mol%), solvent (1.0 mL), 60 °C, N2, 12 h. 1H NMR yield using CH2Br2 as the internal standard. Isolated yield in parentheses.
b 40 °C.
c 60 °C.
d Liu's condition (ref. 9c). The ratio of 1,6- and 1,2-azidotrifluoromethylation was 4.5:1.
e 1,6:1,2 = 5:1.
|
||||
| 1 | Fe(acac)3 | L1 | DCM | 75 |
| 2 | Fe(acac)3 | L2 | DCM | 68 |
| 3 | Fe(acac)3 | L3 | DCM | 79 |
| 4 | Fe(acac)3 | L4 | DCM | 71 |
| 5 | Fe(acac)3 | L5 | DCM | 72 |
| 6 | Fe(acac)3 | L6 | DCM | 70 |
| 7 | Fe(acac)3 | — | DCM | 84 (82) |
| 8b | Fe(acac)3 | — | DCM | 73 |
| 9c | Fe(acac)3 | — | DCM | 71 |
| 10 | Fe(acac)3 | — | DCE | 82 |
| 11 | Fe(acac)3 | — | MeCN | 73 |
| 12 | Fe(acac)3 | — | Toluene | 55 |
| 13 | Fe(acac)3 | — | MeOH | Trace |
| 14 | Fe(acac)3 | — | DMA | 57 |
| 15 | Fe(acac)3 | — | 1,4-Dioxane | 59 |
| 16 | Fe(OTf)2 | — | DCM | 18 |
| 17 | Fe2(SO4)3 | — | DCM | 57 |
| 18 | Fe(acac)2 | — | DCM | 55 |
| 19 | Fe(OAc)2 | — | DCM | 50 |
| 20 | FeCl2 | — | DCM | 42 |
| 21 | FeF2 | — | DCM | 72 |
| 22 | — | — | DCM | 0 |
| 23d | CuI | — | DCE | 45 |
| 24e | CuTc | DCM/1,4-dioxane | 30 | |
|
||||

A number of phenanthroline and bipyridine ligands were next examined carefully (entries 1–6, Table 1). Even though the reaction yields were slightly improved, the regioselective control (1,6- vs. 1,2-) under these ligand-addition conditions was lacking. Surprisingly, the removal of the ligand from the reaction system could affect the reactivity of the metal center furnishing the desired product 2a in 82% of isolated yield (in parentheses) with regioselectivity up to 139:1 (determined by 19F NMR, entry 7, Table 1). While increasing or decreasing the reaction temperature would reduce almost 10% of the yield (entries 8 and 9, Table 1), the investigation of the solvent effect indicated that the halogenated solvent (entry 10, Table 1) could offer comparable yield, but protonic solvent was not suitable in this reaction (entry 13, Table 1). Next, a careful screening of iron salts was then carried out. Except for the most lewis-acidic Fe(OTf)2 (entry 16), other Fe(II) or Fe(III) catalysts could also furnish 1 in moderate to good yield (entries 17–21, Table 1), but the best result was still for Fe(acac)3. Finally, control experiments indicated that the presence of the iron catalyst was indispensable (entry 22).
It should be noted that not only was the photocatalytic system not capable of activating remote inert C–H bonds,10a but also only moderate yield of the difunctionalized product (with poor site-selectivity) could be obtained by the replacement of the iron catalyst with a copper catalyst (entries 23 and 24, Table 1) (for more details, see the ESI†).
With the optimized conditions in hand, we next explored the substrate scope of this protocol for azidotrifluoromethylation. As shown in Table 2, the remote azidation of the inert C(sp3)–H site on the aliphatic chains was achieved smoothly, giving corresponding products in moderate to excellent yields (1–12). Of note is that, we also listed the yield (in parentheses) of the representative products using the optimal copper-catalyzed system to show the distinct contrast in efficiency between the two systems. Firstly, functionalization on the acyclic chain including the gem-dimethyl (1) and the elongated chains (2–4) was successful with high regioselective control (>20:1). Cyclic substrates were tested next and the products 5–11 were afforded in moderate to good yields. Interestingly, when using Fe(acac)3 as the catalyst, it was found that a significant amount of 1,2-difunctionlizational byproducts was generated (indicated by 19F NMR) and thus circumscribed the site-selectivity of this transformation; while using FeF2 as the catalyst, the remote azidated products were obtained in almost one isomer (regioselectivity > 20:1). In these tests, the functionalization of the bulkier tertiary C(sp3)–H such as tetrahydropyran (5), and N-tethered substrates such as N-Ts (6) and N-Boc (7) was also compatible with the iron-catalyzed system. Moreover, we then synthesized a series of cycloalkyl substrates and to our excitement, using FeF2, the functionalization at this remote C(sp3)–H site on four-membered rings to seven-membered rings (8–10) proceeded very well (69–80%) to give the corresponding 1,6-azidotrifluoromethylated products. Also, we examined the feasibility of this transformation at a more sterically congested site, where the remote functionalization could be achieved on the tricyclic ring and the tertiary azides (11) were furnished in moderate yield. Remarkably, we turned our attention to the more challenging aspect of this reaction-the hydrogen abstraction at the less reactive secondary C(sp3)–H sites. Of note is that there was almost no energy difference between the initiating carbon radical and the in situ generated secondary carbon radical and this interesting case (12) gave the corresponding product in 58% of yield. Another type of substrate that is equally difficult to apply, in comparison to the remote functionalization of C(sp3)–H adjacent to the hetero-atom containing functionality,9b–d is the remote functionalization of the benzylic C(sp3)–H site. Even though there were pioneer reports on the 1,6-difunctionalization on this type of unactivated alkenes, the substrate scope was largely confined to aryl rings bearing electron-donating (OMe and Me) and weak electron-withdrawing groups (halides) in previous systems.10 Satisfyingly, when using this iron-catalyzed system, a number of alkenes bearing a range of para-, meta-, and ortho-substituted aryl rings were smoothly azidotrifluoromethylized and gave the corresponding products in moderate to excellent yields (52–87%). Meanwhile, both electron-donating groups such as methyl (14, 25, and 28), phenyl (16), and methoxy (17, 26, and 29), and weak electron-withdrawing groups such as fluoro (18, and 30), chloro (19), and bromo (20, 27, and 31) can all be tolerated which enabled further synthetic elaboration of the products via transition-metal-catalyzed cross-coupling reactions. Of note is that, the previously problematic alkenes bearing strong electron-withdrawing groups such as trifluoromethyl (21), esters (22), cyano (23), and nitro (24) could also be compatible with our standard conditions. The protocol was also effective for alkenes bearing di-substituted aryl rings including di-trifluoromethylated (33) and di-cholorinated (34) alkenes. Furthermore, alkenes bearing fused-/hetero-arenes such as 2-naphthalene (35), 2H-benzofuran (32), thiophene (36), and pyridine (37) were also well difunctionalized under the standard conditions. Substrates with different substituents on the aliphatic chain (38–41) were also studied and accommodated in the reaction including the previously reported problematic N-Ts tethered alkenes,10a albeit in relatively lower yields (40). Another challenging substrate 1-allyl-2-ethylbenzene was tested and obtained in synthetically useful yield (41). Besides C–H bonds on aliphatic chains and at the benzylic site, the protocol also worked well for azidation of C(sp3)–H abutting a heteroatom (N or O). 42 and 43 could be afforded with high regioselective control in 68% and 54% of yield, respectively.
| Reaction conditions: substrate (0.2 mmol), TMSN3 (3 equiv.), Togni-II (1.25 equiv.), Fe(acac)3 (10 mol%), DCM (2.0 mL), N2, 50 °C, 12 h. Isolated yields.a With CuTc (10 mol%), DCM/1,4-dioxane (1:1) (2.0 mL), N2, 30 °C, 12 h.b 10 mol% of FeF2 was used.c 1,6-:1,2- = 1.1:1.d 1,4-Dioxane was used as solvent. |
|---|

|
Compared with 1,5-HAT, 1,6-HAT is less reported due to multiple factors including the rigid structure of intermediates that precludes the formation of six-membered rings.3i,14 To our great delight, not only remote 1,7-difunctionalization could be achieved on alkenes bearing aryl rings (45–47), remote functionalization at the unactivated C–H site on the aliphatic alkene (44) can also be smoothly realized with high regioselective control. Even though 1,6-HAT was often considered to be entropy less-favored compared with other competitive side reactions, the facile transformation under this system can furnish the products in synthetically useful yields.
To demonstrate the synthetic potential of these transformations, scale-up reactions and further derivatization studies were then carried out. As depicted in Scheme 1, the scale-up reaction was performed affording the desired product in 73% of yield. Accordingly, the reduction of the azide group was complete under 1 atm H2 and the reduced product swiftly underwent an intramolecular cyclization, giving the γ-lactam 48 in 78% of yield. Furthermore, Huisgen cyclization was next tested. With the addition of phenylacetylene, 71% yield of cyclo-addition product 49 was obtained from the azidotrifluoromethylated product.
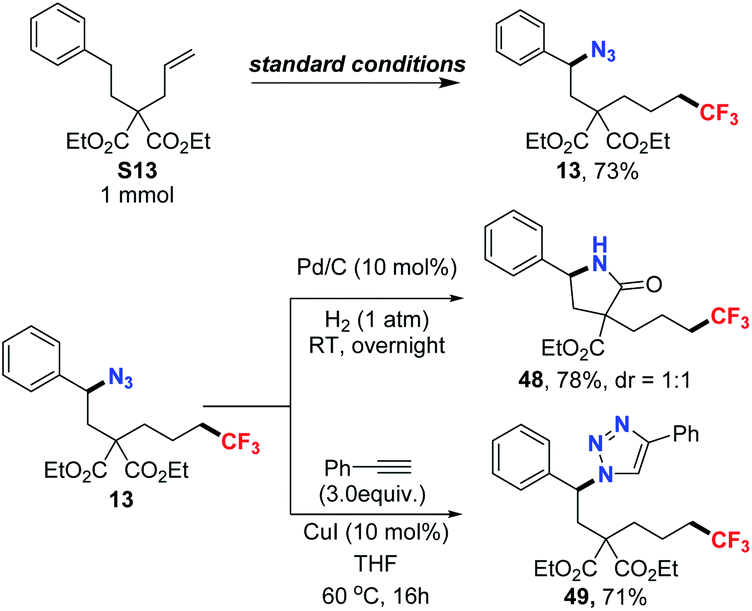 | ||
| Scheme 1 Scale-up reaction and derivative studies. | ||
To gain some insights into the mechanism of the transformations, elementary mechanistic studies were then carried out. Firstly, the subjection of some known radical scavengers, such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), and butylated hydroxytoluene (BHT), to the standard conditions afforded no desired product 50 where alkene could be fully recovered. The corresponding coupling (TEMPO-CF3) could be detected by 19F NMR analysis with 82% yield (eqn (1), Scheme 2). Moreover, a radical clock experiment using alkenyl cyclopropane was conducted, affording the cyclization product 51 in 52% yield (eqn (2), Scheme 2). These radical-trapping experiments confirmed that a free CF3 radical was generated to initiate the radical relay in the catalytic cycle.
 | ||
| Scheme 2 Elementary mechanistic studies. aUsing PhCF3 as the internal standard. | ||
Next, a series of deuterium labeling studies15 were performed to reveal the kinetic characteristics of the reaction and whether 1,n-HAT undergoes a direct or two-step pathway.
First of all, deuterium-transferred product 13-d2 and 1,2-difunctionalized product 13′-d2 were obtained in the yields of 47% and 26%, respectively (1.8:1) (Scheme 3a), which correlated with the direct 1,5-HAT process. For the generation of 13′-d2, more difficulty was observed in C–D bond cleavage than than in C–H bond cleavage, which resulted in the generation of the competitive 1,2-difunctionalized product. Secondly, the intramolecular and intermolecular kinetic isotope effect (KIE) was then investigated. In the intramolecular competition reaction, S13-d yielded a KIE value of 2.6 (Scheme 3b). The observation of the kinetic isotope effect doesn't indicate that C–H cleavage occurs during RDS but demonstrates irreversibility of C–H cleavage via the 1,5-HAT process. In the intermolecular competition reactions, one-pot experiments and parallel experiments gave KIE values of 1.3 and 1.27 respectively (for more details, see the ESI†). Collectively, these results demonstrated that the C–H cleavage through HAT is not reversible (primary KIE in intramolecular experiments) and this step is not the rate determining step (no primary KIE in intermolecular experiments, Scheme 3c).10a
 | ||
| Scheme 3 Deuterium labelling experiments. | ||
To understand the process of 1,7-difunctionalization, in which two possible pathways including direct 1,6-HAT or 1,5-HAT followed by subsequent 1,2-HAT were both likely to be involved, two more deuterium-labeled substrates were designed and studied under standard conditions. Interestingly, benzylic dideuterium-substituted alkene S46-d2 afforded 1,2-difunctionalized product 46-d2 alone, possibly due to the united effects of more difficult C–D cleavage and less favored 1,6-HAT. Alternatively, we designed another β-dideuterium-labeled (in regard to arene) analogue S46-d2′. Supposedly, if the reaction underwent a prior 1,5-HAT followed by 1,2-HAT, the deuterium would shift to the benzylic position; in contrast, no deuterium shifting would be observed through a direct 1,6-HAT process. To our great satisfaction, the desired product 46-d2 was obtained in 50% yield with complete deuterium retention (Scheme 3d). These results suggested that the direct 1,6-HAT process is more feasible for 1,7-difunctionalization.
Based upon the collective mechanistic evidence and previous reports,11,13,16 a possible mechanistic cycle was suggested, as shown in Fig. 2. The CF3 radical, generated from the Togni reagent through single electron transfer (SET) between Togni-II and an active Fe catalyst, is captured by alkene to generate a secondary carbon radical B, followed by the direct 1,n-HAT process affording the tertiary/secondary radical C in situ. Meanwhile, the higher-valent iron–azide (FeIII–N3) species will swiftly sequester the intermediate C to give the 1,6- (or 1,7-) difunctionalized product 1 via azido–ligand-transfer, with concurrent regeneration of the low-valent iron catalyst (path a). Alternatively, we speculated whether the tertiary/secondary carbocation is involved in the reaction, which would disclose a totally different reaction pathway (nucleophilic substitution instead of radical oxidation) (path b).
 | ||
| Fig. 2 Possible reaction pathway. | ||
First of all, the suspected coupling between acid anion A and the tertiary cation D was not detected.9a Meanwhile, as has been shown in Table 2, 12 can be afforded without the rearrangement byproduct, which otherwise would be often obtained if the carbocation is generated adjacent to the tert-butyl group. These collective results made us inclined towards path a.
Conclusions
In summary, we have developed the first example of iron-catalyzed remote azidation of inert C(sp3)–H bonds by sp3 C-centered radical relay; remote 1,6- and 1,7-azidotrifluoromethylation of alkenes were successfully achieved. This method demonstrates high catalytic reactivity, mild conditions, excellent functional group tolerance and great regioselectivity. The key to the transformation is that iron plays the dual role of a radical initiator to promote the generation of and a terminator where ironIII–azide can oxidize the in situ generated remote carbon radical and incorporate the azide functionality via azido–ligand-transfer. Deuterium studies revealed the direct 1,n- (n = 5, 6) HAT process. Further investigation of functionalization on fully linear chains and utilization of this strategy to construct potentially bioactive molecules is still underway in our laboratory.
and a terminator where ironIII–azide can oxidize the in situ generated remote carbon radical and incorporate the azide functionality via azido–ligand-transfer. Deuterium studies revealed the direct 1,n- (n = 5, 6) HAT process. Further investigation of functionalization on fully linear chains and utilization of this strategy to construct potentially bioactive molecules is still underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000) and the National Science Foundation of China (21971228 and 21602213) for financial support.Notes and references
- (a) R. Giri, B. F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (d) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS; (e) B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 186 Search PubMed; (f) Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640 CrossRef CAS; (g) T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296 CrossRef CAS.
- (a) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (b) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS; (c) X. Huang and J. T. Groves, ACS Catal., 2016, 6, 751 CrossRef CAS; (d) H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936 CrossRef CAS; (e) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS.
- For reviews on remote C–H functionalization: (a) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94 RSC; (b) Ž. Čeković, Tetrahedron, 2003, 59, 8073 CrossRef; (c) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603 RSC; (d) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC; (e) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (f) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC; (g) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS; (h) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569 CrossRef CAS; (i) M. A. Short, J. M. Blackburn and J. L. Roizen, Synlett, 2019, 31, 102 Search PubMed; (j) X. Wu and C. Zhu, Chinese Chemical Society Chemistry, 2020, 2, 813 Search PubMed; Recent examples of remote C–H functionalization via the HAT strategy: (k) N. Yoshikai, A. Mieczkowski, A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2010, 132, 5568 CrossRef CAS; (l) A.-F. Voica, A. Mendoza, W. R. Gutekunst, J. O. Fraga and P. S. Baran, Nat. Chem., 2012, 4, 629 CrossRef CAS; (m) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS; (n) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771 CrossRef CAS; (o) H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934 RSC; (p) E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204 CrossRef CAS; (q) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940 CrossRef CAS; (r) Y. Zhu, K. Huang, J. Pan, X. Qiu, X. Luo, Q. Qin, J. Wei, X. Wen, L. Zhang and N. Jiao, Nat. Commun., 2018, 9, 2625 CrossRef; (s) X. Wu, H. Zhang, N. Tang, Z. Wu, D. Wang, M. Ji, Y. Xu, M. Wang and C. Zhu, Nat. Commun., 2018, 9, 3343 CrossRef; (t) C. G. Na and E. J. Alexanian, Angew. Chem., Int. Ed., 2018, 57, 13106 CrossRef CAS; (u) D. Bafaluy, J. M. Muñoz-Molina, I. Funes-Ardroiz, S. Herold, A. J. de Aguirre, H. Zhang, F. Maseras, T. R. Belderrain, P. J. Pérez and K. Muñiz, Angew. Chem., Int. Ed., 2019, 58, 8912 CrossRef CAS; (v) Z. Liu, H. Xiao, B. Zhang, H. Shen, L. Zhu and C. Li, Angew. Chem., Int. Ed., 2019, 58, 2510 CrossRef CAS; (w) R. O. Torres-Ochoa, A. Leclair, Q. Wang and J. Zhu, Chem.–Eur. J., 2019, 25, 9477 CrossRef CAS; (x) Z. Li, R. O. Torres-Ochoa, Q. Wang and J. Zhu, Nat. Commun., 2020, 11, 403 CrossRef CAS.
- (a) A. W. Hofman, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef; (b) K. Löffler and C. Freytag, Ber. Dtsch. Chem. Ges., 1909, 42, 3427 CrossRef.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36 CrossRef CAS.
- For selective examples on sp2 C-radical initiated remote functionalization: (a) D. P. Curran, D. Kim, H. T. Liu and W. Shen, J. Am. Chem. Soc., 1988, 110, 5900 CrossRef CAS; (b) M. Murakami, M. Hayashi and Y. Ito, J. Org. Chem., 1992, 57, 793 CrossRef CAS; (c) S. E. Booth, T. Benneche and K. Undheim, Tetrahedron, 1995, 51, 3665 CrossRef CAS; (d) G. Han, M. G. LaPorte, M. C. McIntosh, S. M. Weinreb and M. Parvez, J. Org. Chem., 1996, 61, 9483 CrossRef CAS; (e) G. W. Gribble, H. L. Fraser and J. C. Badenock, Chem. Commun., 2001, 805 RSC; (f) A. L. J. Beckwith, V. W. Bowry, W. R. Bowman, E. Mann, J. Parr and J. M. D. Storey, Angew. Chem., Int. Ed., 2004, 43, 95 CrossRef; (g) F. Dénès, F. Beaufils and P. Renaud, Synlett, 2008, 2389 CrossRef; (h) N. Yoshikai, A. Mieczkowski, A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2010, 132, 5568 CrossRef CAS; (i) J. K. Vellucci and C. M. Beaudry, Org. Lett., 2015, 17, 4558 CrossRef CAS; (j) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340 CrossRef CAS; (k) J.-Q. Chen, Y.-L. Wei, G.-Q. Xu, Y.-M. Liang and P.-F. Xu, Chem. Commun., 2016, 52, 6455 RSC; (l) F.-Q. Huang, X. Dong, L.-W. Qi and B. Zhang, Tetrahedron Lett., 2016, 57, 1600 CrossRef CAS; (m) S. Wu, X. Wu and C. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1499 CrossRef CAS; (n) S. Wu, X. Wu and C. Zhu, Sci. China: Chem., 2019, 62, 1507 CrossRef CAS; (o) S. Yang, X. Wu, S. Wu and C. Zhu, Org. Lett., 2019, 21, 4837 CrossRef CAS; (p) T. Shang, J. Zhang, Y. Zhang, F. Zhang, X.-S. Li and G. Zhu, Org. Lett., 2020, 22, 3667 CrossRef CAS. For selective examples on sp3 C-radical initiated remote functionalization: (q) D. Crich, S. Sun and J. Brunckova, J. Org. Chem., 1996, 61, 605 CrossRef CAS; (r) Y. Ueno, K. Chino, M. Watanabe, O. Moriya and M. Okawara, J. Am. Chem. Soc., 1982, 104, 5564 CrossRef CAS; (s) G. Stork, R. Mook, S. A. Biller and S. D. Rychnovsky, J. Am. Chem. Soc., 1983, 105, 3741 CrossRef CAS; (t) H. Nishiyama, T. Kitajima, M. Matsumoto and K. Itoh, J. Org. Chem., 1984, 49, 2298 CrossRef CAS; (u) M. Masnyk, Tetrahedron Lett., 1997, 38, 879 CrossRef CAS; (v) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857 CrossRef CAS.
- For reviews on alkene difunctionalization: (a) R. M. Romero, T. H. Wöste and K. Muñiz, Chem.–Asian J., 2014, 9, 972 CrossRef CAS; (b) T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 CrossRef CAS; (c) T. Koike and M. Akita, Org. Chem. Front., 2016, 3, 1345 RSC; (d) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS; (e) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS.
- (a) H. Egami, R. Shimizu, S. Kawamura and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 4000 CrossRef CAS; (b) P. Yu, J.-S. Lin, S.-C. Zheng, Y.-P. Xiong, L.-J. Zhao, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 11890 CrossRef CAS; (c) L. Huang, J.-S. Lin, B. Tan and X.-Y. Liu, ACS Catal., 2015, 5, 2826 CrossRef CAS; (d) T. Li, P. Yu, Y.-M. Du, J.-S. Lin, Y. Zhi and X.-Y. Liu, J. Fluorine Chem., 2017, 203, 210 CrossRef CAS.
- (a) W. Shu, E. Merino and C. Nevado, ACS Catal., 2018, 8, 6401 CrossRef CAS; (b) L. Li, H. Luo, Z. Zhao, Y. Li, Q. Zhou, J. Xu, J. Li and Y.-N. Ma, Org. Lett., 2019, 21, 9228 CrossRef CAS.
- (a) M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS; (b) J. K. Kochi, Science, 1967, 155, 415 CrossRef CAS.
- The definition of inert C(sp3)–H bonds was introduced in ref. 3g, 7r and D. M. Peacock, C. B. Roos and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 647 CrossRef CAS.
- C.-L. Zhu, C. Wang, Q.-X. Qin, S. Yruegas, C. D. Martin and H. Xu, ACS Catal., 2018, 8, 5032 CrossRef CAS.
- M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS.
- P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03987j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |