
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeparin reversal by an oligoethylene glycol functionalized guanidinocalixarene†
Qiaoxian
Huang‡
a,
Hong
Zhao‡
b,
Mingju
Shui
a,
Dong-Sheng
Guo
 *b and
Ruibing
Wang
*a
*b and
Ruibing
Wang
*a
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau SAR, China. E-mail: rwang@um.edu.mo
bCollege of Chemistry, Key Laboratory of Functional Polymer Materials (Ministry of Education), State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, China. E-mail: dshguo@nankai.edu.cn
First published on 28th August 2020
Abstract
Unfractionated heparin (UFH), a naturally occurring anionic polysaccharide, is widely used as an anticoagulant agent in clinical practice. When overdosed or used in sensitive patients, UFH may cause various risks and a UFH neutralizer needs to be administered immediately to reverse heparinization. However, the most common UFH neutralizer, protamine sulfate, often causes various adverse effects, some of which are life-threatening. Herein, we designed a highly biocompatible, oligoethylene glycol functionalized guanidinocalixarene (GC4AOEG) as an antidote against UFH. GC4AOEG and UFH exhibited a strong binding affinity, ensuring specific recognition and neutralization of UFH by GC4AOEG in vitro and in vivo. As a consequence, UFH-induced excessive bleeding was significantly alleviated by GC4AOEG in different mouse bleeding models. Additionally, no adverse effects were observed during these treatments in vivo. Taken together, GC4AOEG, as a strategically designed, biocompatible artificial receptor with strong recognition affinity towards UFH, may have significant clinical potential as an alternative UFH reversal agent.
Heparin sodium, often referred to as unfractionated heparin (UFH, also known as heparin), is a well-known anionic glycosaminoglycan consisting of long, helical, unbranched chains of repeating sulfonated disaccharide units (Fig. 1).1 It is currently a gold-standard life-saving drug to overcome blood coagulation by activating antithrombin-III to impede the coagulation process.2,3 Systemic heparinization is the most common anticoagulation procedure in surgical practice (e.g. open-heart surgery) and extracorporeal therapies such as kidney dialysis. At the end of surgery, excess heparin often needs to be deactivated by using a heparin neutralizer; otherwise patients have risks of low blood pressure and a slow heart rate, and may develop internal bleeding.4 Therefore, the neutralization of heparin has been a topic of significant research interest in the biomedical field.
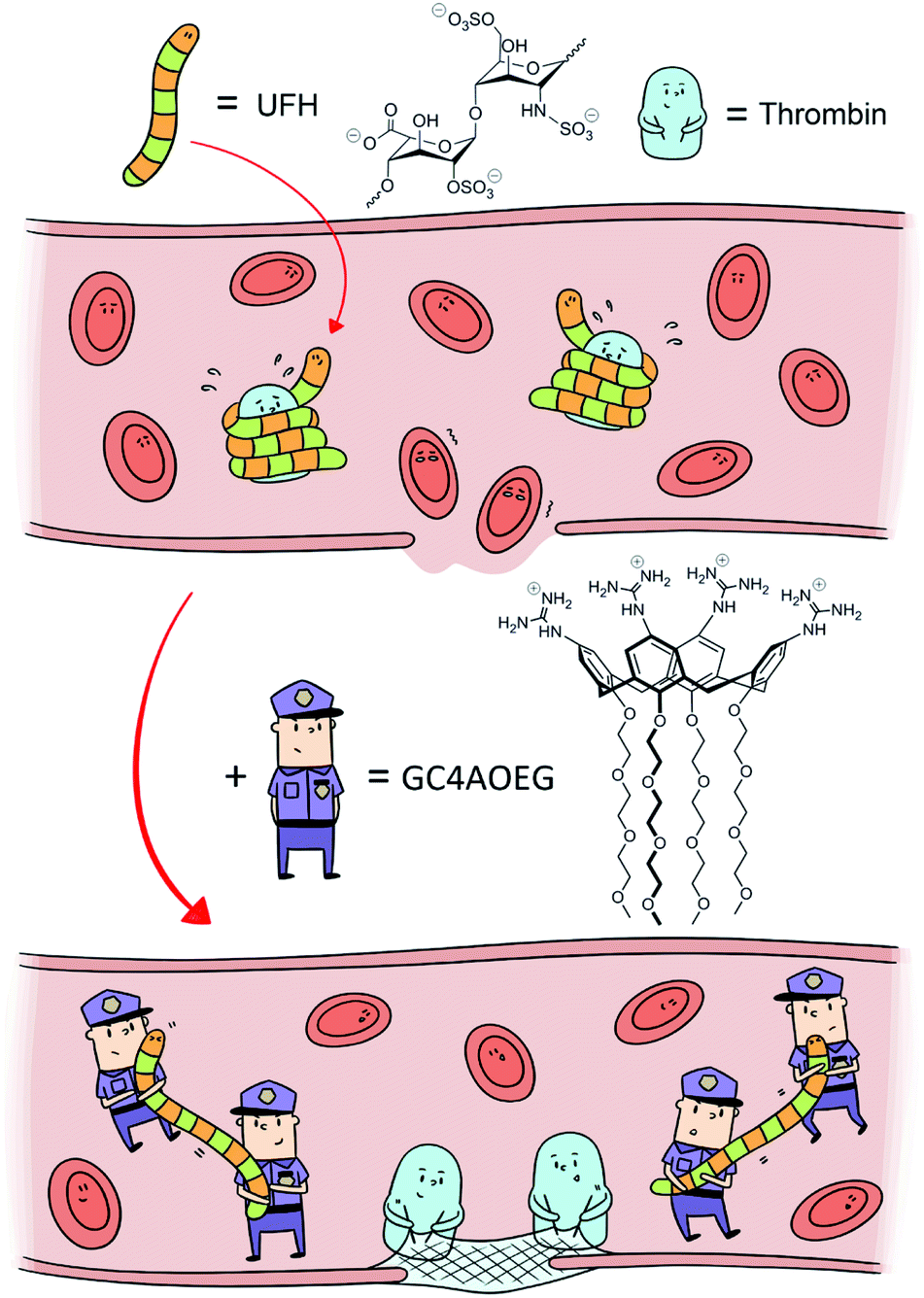 | ||
| Fig. 1 Scheme of heparin reversal by GC4AOEG in the circulatory system. | ||
Protamine sulfate, the only FDA-approved neutralizer of UFH, possesses a highly positive charge density due to its polymeric nature and rich arginine residues. Thus, electrostatic interactions are the major driving force in the formation of a UFH–protamine complex, leading to the neutralization and deactivation of UFH.1,5 However, due to its non-specific interactions, protamine sulfate often causes various adverse effects such as bradycardia, hypotension and pulmonary hypertension, as well as allergic reactions including life-threatening anaphylactic reactions in some patients.5 When overdosed, protamine may further impair the intricate balance in the blood and cause coagulopathy.5–7 Given these issues, there has been a medical need for alternative, safe UFH neutralizers that can specifically counteract UFH without causing serious adverse effects.8
Discovering and developing new heparin neutralizers has been a popular area of research.8,9 During the past two decades, a variety of different UFH neutralizers including small molecules,10 cationic polymers (e.g. polybrene),11–14 peptides,11,15 and nanoparticles16,17 have been designed and evaluated in vitro and/or in vivo. For instance, surfen, as a small-molecule antagonist of UFH, may electrostatically bind with UFH; however only modest neutralizing effects against UFH were observed in rats,10,18 likely attributed to the lack of strong, specific recognition. On the other hand, polycationic species, including polybrene19 and poly-DL-lysine,20 exhibited stronger binding with UFH and significant potential as UFH neutralization agents. However, toxicity was still a key concern of these species due to their intrinsic electrostatic interactions with red blood cells (RBC).21 Meanwhile, some UFH antagonists have achieved preliminary success in preclinical studies and even moved to clinical evaluations. For instance, ciraparantag (PER977), as a synthetic antidote against several anticoagulants, is currently being evaluated in phase II clinical trials.22 UHRA (Universal Heparin Reversal Agent), a synthetic multivalent dendrimer polymer in the form of nanoparticles with positively charged surfaces, can reverse the activity of all clinically available heparins and it is currently undergoing preclinical studies and will likely move to clinical investigations.23 However, the oligo- and poly-cationic nature of these species suggests their general tendency towards any negatively charged species, making them “universal” or function against several anticoagulants, implying their low specificity towards heparin.
More recently, the sequestration and reversal of toxic agents by supramolecular host molecules have attracted increasing attention, and a typical example of clinical and commercial success is sugammadex, a carboxylated derivative of gamma-cyclodextrin that may specifically reverse the activity of non-depolarizing neuromuscular blocking agents.24 Inspired by this clinical success, several macrocycles were designed and synthesized to selectively bind UFH. For instance, Liu et al. synthesized amphiphilic multi-charged cyclodextrins (AMCD), and AMCD-assembly was utilized for selective heparin binding.16 Nitz et al. derivatized a cyclodextrin with amide and guanidino groups as a polycationic receptor to recognize and detect UFH.25 Kostiainen and co-workers studied cationic, quaternary ammonium functionalized pillar[5]arene because of its potential complexation with UFH.26 Additionally, cationic calixarene derivatives were designed for UFH binding and guanidinocalixarenes exhibited stronger binding affinity with UFH than their quaternary amine-functionalized counterparts.27,28 In spite of decent binding affinities and selective recognition of UFH, these macrocycles still possess various limitations such as non-specific toxicity induced mostly by cationic charges, which may disrupt cell membranes and induce blood coagulation.29,30
An ideal UFH neutralizer should full-fill the following three requirements: (1) binding strongly towards UFH in a specific manner; (2) excellent biocompatibility and safety profile, and (3) a clearly defined molecular structure to facilitate batch-to-batch consistency. Thus far, none of the clinical UFH antagonists or previously reported candidates has fulfilled these conditions. Herein we designed an artificial receptor, an oligoethylene glycol functionalized guanidinocalixarene, GC4AOEG, by leveraging the asymmetrical structure of calixarene to strategically add guanidinium groups on one side and oligoethylene glycol (OEG) groups on the other side (Fig. 1). We anticipated that the guanidinium-enriched upper rim would bind strongly with UFH via salt bridges (charge-assisted hydrogen bonds).28,31 In addition, the biocompatible OEG-functionalized lower rim may help improve the water-solubility and biocompatibility of the host molecule.32,33
GC4AOEG was synthesized in 5 steps starting from the maternal calix[4]arene (Fig. 2). Briefly, p-tert-butylcalix[4]arene 1 was alkylated with tosylate 234 to obtain compound 3 with a well-defined cone conformation, and replacement of the tert-butyl with nitro groups via an ipso-nitration reaction afforded compound 4.35 Subsequently, compound 4 was hydrogenated in the presence of SnCl2·2H2O, affording the tetramine derivative 5. Subsequently, compound 6 was obtained via a reaction between compound 5 and di-Boc-protected thiourea units. The removal of the protecting groups was achieved using SnCl4 in ethyl acetate, to yield the target GC4AOEG (the characterization of intermediates (Fig. S1 and S2) and GC4AOEG (Fig. S3) are in the ESI†).
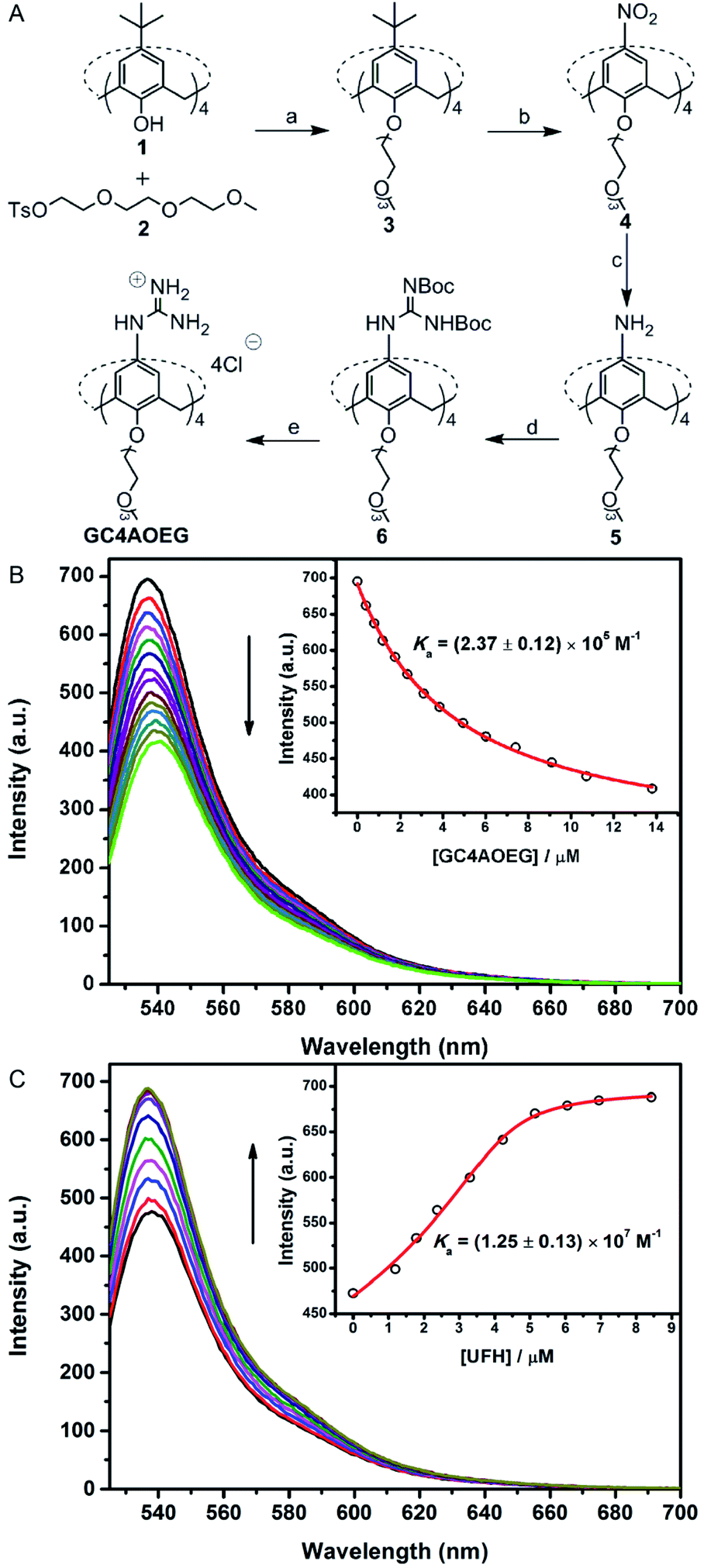 | ||
Fig. 2 Synthetic route of GC4AOEG and fluorescence titrations. (A(a)) NaH, dry DMF, and 75 °C; (b) HNO3, AcOH, dry CH2Cl2, and r.t.; (c) SnCl2·2H2O, C2H5OH/AcOEt (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), and reflux; (d) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, Et3N, AgNO3, dry CH2Cl2, and r.t.; (e) SnCl4, AcOEt, and r.t. (B) Direct fluorescence titration of 0.5 μM EY with different concentrations of GC4AOEG (up to 13.8 μM) in HEPES buffer (10 mM, pH = 7.4), and λex = 517 nm. (Inset) The associated titration curve at λem = 537 nm and best fit according to a 1:1 binding stoichiometry. (C) Competitive fluorescence titration of GC4AOEG·EY (4.0/0.5 μM) with UFH (up to 8.4 μM in the concentration of monomer units of UFH), and λex = 517 nm. (Inset) The associated titration curve at λem = 537 nm and best fit according to a n:1 competitive binding model, where n = 0.88. :1, v/v), and reflux; (d) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, Et3N, AgNO3, dry CH2Cl2, and r.t.; (e) SnCl4, AcOEt, and r.t. (B) Direct fluorescence titration of 0.5 μM EY with different concentrations of GC4AOEG (up to 13.8 μM) in HEPES buffer (10 mM, pH = 7.4), and λex = 517 nm. (Inset) The associated titration curve at λem = 537 nm and best fit according to a 1:1 binding stoichiometry. (C) Competitive fluorescence titration of GC4AOEG·EY (4.0/0.5 μM) with UFH (up to 8.4 μM in the concentration of monomer units of UFH), and λex = 517 nm. (Inset) The associated titration curve at λem = 537 nm and best fit according to a n:1 competitive binding model, where n = 0.88. | ||
The binding affinity between GC4AOEG and UFH was firstly investigated via a competitive titration approach. In this paper, we defined the repeated disaccharide unit as the UFH monomer unit, and the UFH concentration in this paper is the UFH monomer unit concentration. Eosin Y (EY) was selected as the reporter dye, owing to its strong complexation with GC4AOEG and the drastic fluorescence quenching after complexation. The equilibrium association constant (Ka), between GC4AOEG and EY, was determined by direct fluorescence titration and fitted as (2.37 ± 0.12) × 105 M−1 with 1:1 binding stoichiometry (Fig. 2B). The displacement of EY from GC4AOEG·EY by gradual addition of UFH resulted in the recovery of the intrinsic emission of EY. The best-fitting of the competitive titration model afforded ca. 1:1 binding stoichiometry between GC4AOEG and each monomer unit of UFH, as well as an ultrahigh binding affinity Ka of (1.25 ± 0.13) × 107 M−1 (Fig. 2C).
For in vitro analysis of the effectiveness of GC4AOEG against UFH, the activated partial thromboplastin time (aPTT) assay was conducted. The result (Fig. S8†) indicates that one equivalent of GC4AOEG (to UFH monomer) fully neutralized UFH, similar to protamine. Very importantly, it is obvious that protamine alone negatively influenced the aPTT time. In contrast, GC4AOEG alone did not affect the clotting time, suggesting that GC4AOEG can specifically bind with UFH directly with minimal side influences. The coagulation factor X levels in the plasma analyzed via the enzyme-linked immunosorbent assay (ELISA) further confirmed the safety and reversal effect of GC4AOEG towards UFH (Fig. S9†).
Next, the biocompatibility of GC4AOEG was investigated in vitro. As an alkyl derivative of guanidinocalixarene, GC4A-6C (Fig. S4 and S5†), which has a similar number of carbons (hexyl groups) at the lower rim to that of GC4AOEG, was also synthesized and examined in this study for comparative purposes. As shown in Fig. 3A and B, GC4AOEG (up to 200 μM) showed remarkably low cytotoxicity in several cell lines via MTT assays, in dramatic contrast to the relatively high cytotoxicity of GC4A-6C (Fig. 3C and D). The cellular toxicity of GC4A-6C was consistent with previous literature.36 In addition, alkyl derivatives of calixarene were generally more toxic than those without alkyl chains,37 likely attributed to their amphiphilic properties that may facilitate cell membrane disruption.38–40 The results suggested that the much-improved safety profile of GC4AOEG was attributed to oligoethylene glycol functionalization. Meanwhile, it is well known that cationic polymers or oligomers often show poor biocompatibility in the circulation system due to their non-selective binding to negatively charged RBC, resulting in RBC aggregation or hemolysis.41 Therefore, hemolysis and hemagglutination assays were conducted according to a method previously reported,42,43 with experimental details described in the method. The percent hemolysis of GC4A-6C (25, 50, 100 and 200 μM, respectively) was over 90%, which would limit its application in the circulatory system (Fig. S6†), as a hemolysis ratio below 5% is considered safe.44 Conversely, GC4AOEG exhibited nearly negligible (less than 3%) hemolytic activity at concentrations of up to 200 μM, and no agglutination was visualized during incubation with RBC (Fig. 3F), implying that OEG functionalization at the lower rim reduced non-specific interactions with the RBC membrane, resulting in less disturbance of the membrane structure and function or cellular aggregations.
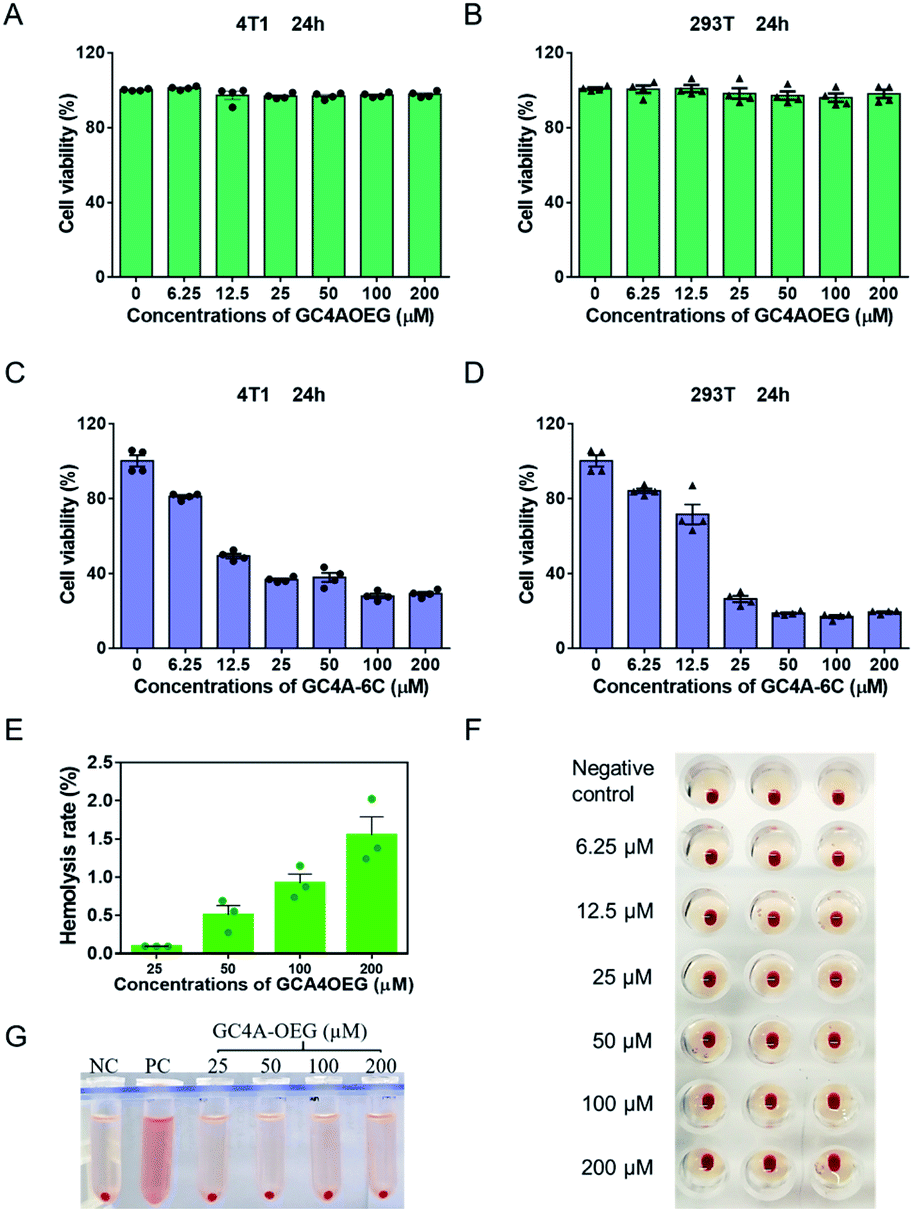 | ||
| Fig. 3 Biocompatibility study in cell lines and RBC. Cell viabilities of (A, C) 4T1 and (B, D) 293T, cells treated with different concentrations of GC4AOEG or GC4A-6C for 24 h. Each data point represents the mean ± S.E.M. from a set of experiments (n = 4). (E, G) Hemolysis test of GC4AOEG at different concentrations (NC = negative control; PC = positive control). Each data point represents the mean ± S.E.M. from a set of experiments (n = 3). (F) Agglutination test of RBC incubated with GC4AOEG at 2.0% hematocrit in normal saline. | ||
Inspired by the above findings, we further examined whether GC4AOEG may reverse bleeding in different mouse bleeding models under heparinization (with the experimental details described in the method, and the standard curve for the quantification of blood loss volume is showed in Fig. S7†),45 with both the total time of bleeding and total volume of lost blood evaluated for each model. As a proof of concept, 200 U kg−1 UFH and 2.245 mg kg−1 GC4AOEG (molar ratio of GC4AOEG and each monomer unit of UFH = 1:1) were respectively used, as representative doses in the study and the dose of UFH was based on a literature report.46 In a mouse tail transection model as an external bleeding model, as shown in Fig. 4A–C, after tail transection, the bleeding time and blood loss volume for mice treated with normal saline were 58.9 ± 10.7 min and 72.2 ± 15.8 μL, respectively. As expected, treatment with UFH increased the bleeding time and blood volume to 121.5 ± 20.2 min and 264.0 ± 43.6 μL, respectively. In contrast, the bleeding time was dramatically reduced down to the blank control level, when the mice were treated with GC4AOEG at the same time of, or 30 s after, i.v. administration of UFH (53.8 ± 11.4 min and 89.0 ± 13.3 min, respectively). Accordingly, the blood loss volume of mice successively treated with UFH and GC4AOEG (1:1 ratio) reached the control level (72.6 ± 14.3 μL), indicating that the strong binding affinity between GC4AOEG and UFH ensured their recognition in vivo. Of note, there was no significant difference between the GC4AOEG treated group (without heparinization) and the saline treated group, suggesting a decent safety profile of the artificial receptor.
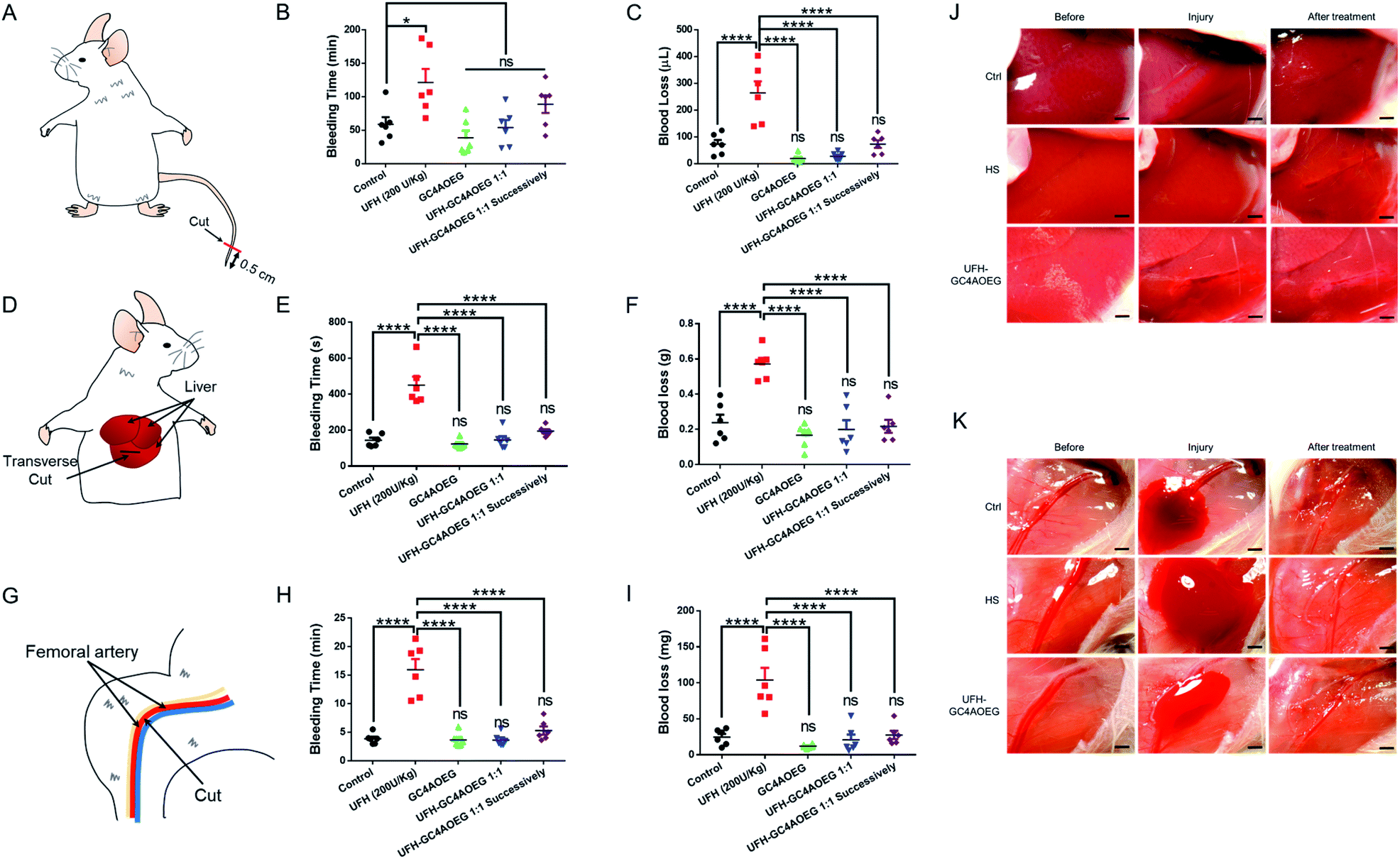 | ||
| Fig. 4 Reversal efficacy in in vivo mouse models. (A–C) Mouse tail transection model. (A) Scheme of the mouse tail transection model. (B) Total time of bleeding and (C) blood loss volume. (D–F & J) Mouse liver injury model. (D) Scheme of the mouse liver injury model. (E) Total time of bleeding and (F) blood loss weight. (J) Pictures exhibiting bleeding in liver injury before and after treatment. (G–I & K) Mouse femoral artery model. (G) Scheme of the mouse femoral artery model. (H) Total time of bleeding and (I) blood loss weight. (K) Pictures exhibiting bleeding in the femoral artery before and after treatment. All of those models were i.v. administration with normal saline (control), GC4AOEG (2.245 mg kg−1), or UFH (200 U kg−1) without and with GC4AOEG (2.245 mg kg−1, 1:1 molar stoichiometry of GC4AOEG and the monomer unit of UFH), and UFH–GC4AOEG 1:1 successively (GC4AOEG at a dose of 2.245 mg kg−1 30 s after UFH administration) respectively were quantified. Data presented are the mean ± S.E.M. (n = 6). *p < 0.05, ****p < 0.001, and ns represents “no significant difference” between the experimental group and the control group. | ||
In addition to external bleeding, internal bleeding such as liver injury model (Fig. 4D) was established in mice, and GC4AOEG's reversal of UFH was further evaluated in vivo. Mice were i.v. administered with normal saline (control), GC4AOEG (2.245 mg kg−1), or UFH (200 U kg−1) without and with GC4AOEG (2.245 mg kg−1), and successive UFH–GC4AOEG 1:1 (30 s in between), respectively. In 2 minutes, the abdomen was surgically opened to expose the liver. A wound of 0.5 cm length and 2 mm depth, in the left lobe of the liver, was created. Considerable bleeding was immediately observed in the UFH treatment group (Fig. 4J), with the total bleeding time lasting for 450.5 ± 46.8 s, and the total blood loss of 571.0 ± 35.0 mg, in contrast to 143.7 ± 14.7 s total bleeding time and 238.0 ± 45.0 mg total blood loss observed in the saline treated group. Interestingly, the UFH–GC4AOEG treated group showed no significant difference from the normal saline treated group. To simulate the clinical use scenario, GC4AOEG was injected after UFH's administration, and significantly reduced bleeding (from both time and volume perspectives) was observed, suggesting effective inhibition of the adverse effects of UFH, by GC4AOEG (Fig. 4E and F). GC4AOEG alone (without heparinization) did not exhibit any hematological toxicity in this model. To further evaluate the inhibitory effects of GC4AOEG against UFH in a preclinical model, a more serious internal bleeding model, femoral artery bleeding mouse model, was employed, and the treatment plan followed the previous two models described as above. Upon administration, the skin of the right leg and the overlying muscles were removed to expose the femoral artery and sciatic nerve. After an open injury at the middle segment of the femoral artery was created with a surgical scissor, blood gushed out immediately from the injured site (Fig. 4G and K). As shown in Fig. 4H and I, the longest average bleeding time (16.0 ± 1.9 min) and blood loss weight (103.8 ± 16.9 mg) were observed in the UFH treatment group of mice, in dramatic contrast to the bleeding time and blood loss of 3.9 ± 0.4 min and 24.7 ± 4.5 mg, respectively, in the normal saline treated group of mice. A bleeding time of 3.6 ± 0.4 min and blood loss of 20.8 ± 7.4 mg were recorded in the UFH–GC4AOEG treatment group. When UFH and GC4AOEG (at 1 molar equivalent) were successively injected, a bleeding time of 5.3 ± 0.7 min and blood loss of 27.7 ± 5.8 mg were noted, suggesting the significant reversal effects of GC4AOEG on UFH. Collectively, in all of the three bleeding models including internal and external bleeding models, i.v. administration of GC4AOEG significantly reversed UFH-induced excessive bleeding in external and internal injuries. More importantly, GC4AOEG alone exhibited negligible hematological activity, unlike other previously reported cationic small molecules, polymers, oligomers and macrocycles.
Furthermore, in order to further verify the safety profile of GC4AOEG at the effective dose in vivo, acute toxicity evaluation was performed in a mouse model. After the i.v. injection of GC4AOEG in mice at a dose of 2.245 mg kg−1 (i.v. injection of normal saline as the control group), the body weight, behaviors, and overall survival of the treated mice were monitored every day for 3 weeks. All the treated mice remained alive and showed normal behaviors, as well as normal body weight evolvement similar to that of the control group (Fig. 5A). On day 21 post administration, mice were euthanized for blood and organ samples were harvested (for details see the method). The organ indexes of representative major organs including the heart, liver, spleen, lungs, and kidneys isolated from the GC4AOEG treated mice were comparable to those of the mice administered with normal saline, with no significant differences observed (Fig. 5B). Hematological parameters such as the counts of whole blood cells (WBCs), red blood cells (RBCs), platelets (PLTs) and hemoglobin (HGB) (Fig. 5C), as well as the serum concentrations of liver and kidney function biomarkers including blood urea nitrogen (BUN), creatinine (crea), urea alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were all analyzed thoroughly (Fig. 5D and E). These results indicated that the hematological parameters, renal and hepatic functions of the mice treated with GC4AOEG were comparable with those of the mice in the normal saline treated group. Moreover, histopathological examinations of the major organs of the GC4AOEG treated mice showed normal microstructures comparable with those of the control group (Fig. 5F). Collectively, these results suggested that the i.v. administration of GC4AOEG at the therapeutic dose is safe.
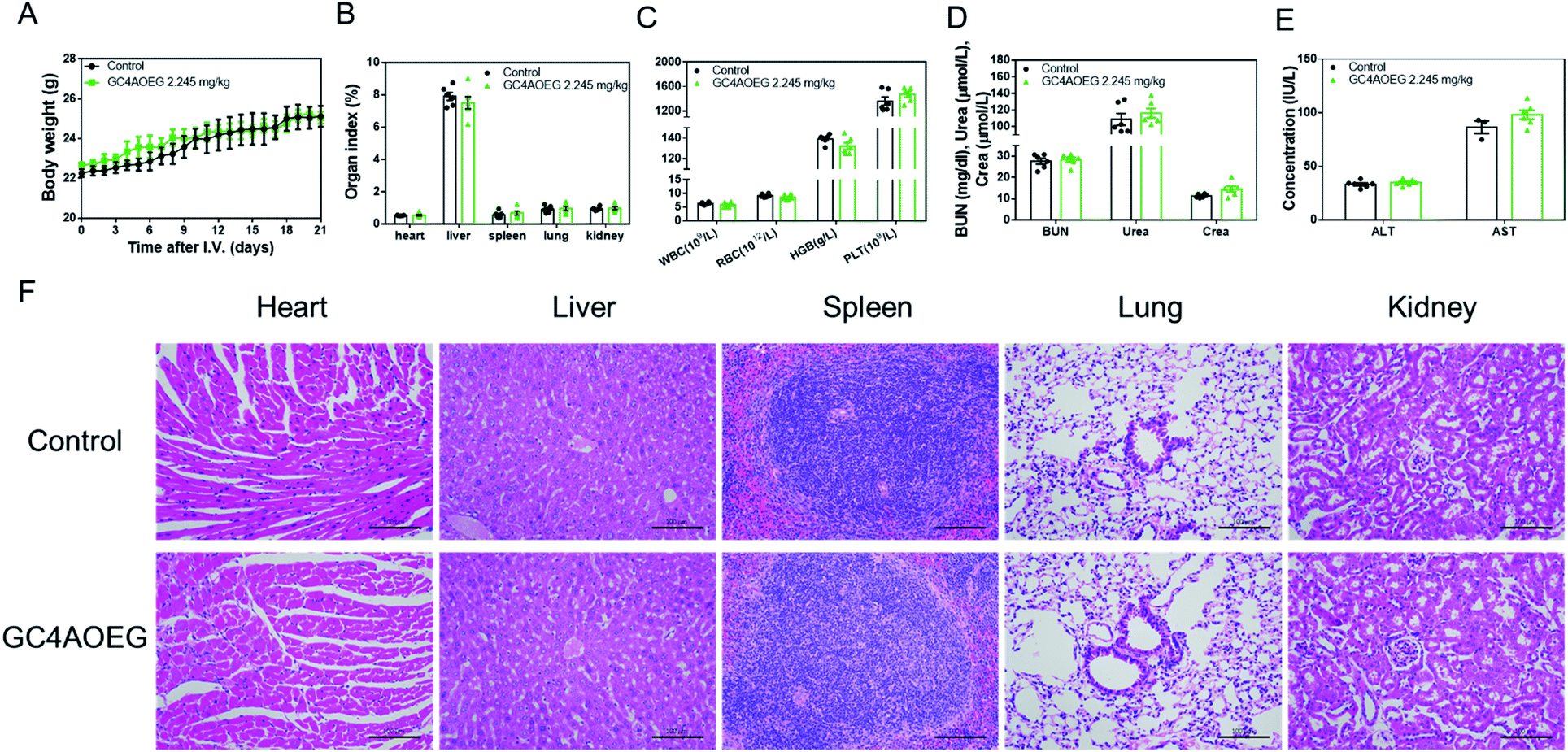 | ||
| Fig. 5 Preliminary acute toxicity evaluations on GC4AOEG. (A) Weight changes of mice after i.v. administration with a single dose of GC4AOEG. (B) Major organ indexes of the mice on day 21 post-administration with GC4AOEG. (C) Hematological parameters of the blood samples collected from the mice on day 21 after i.v. administration of GC4AOEG. (D) Renal and (E) hepatic functional biomarkers in the blood samples collected from the mice on day 21 after i.v. administration of GC4AOEG. Data are presented as mean ± S.E.M.; n = 6 for each group. (F) H&E histopathological analysis of the major organs from mice sacrificed 21 days after being injected with saline and GC4AOEG (2.245 mg kg−1). Scale bar = 100 μm. | ||
Conclusions
In conclusion, we designed GC4AOEG for safe, effective UFH neutralization. As an artificial macrocyclic receptor, GC4AOEG offers strong binding to UFH owing to the multiple guanidinium groups at the upper rim and meanwhile exhibits excellent biocompatibility in vitro and in vivo due to the presence of multiple OEG groups at the lower rim. Consequently, GC4AOEG significantly reversed UFH-induced excessive bleeding in different mouse bleeding models, including tail transection, liver and femoral artery injury bleeding models. Moreover, at the effective dose, no adverse effects were observed during treatments in vivo, and a systemic acute toxicity study further supported the safe use of the compound. Compared with neutralizers previously reported, the present macrocycle-based one demonstrates dual advantages of both small molecules and polymer species, that is, strong recognition (near-nanomolar affinity) of UFH and weak interactions with RBC. Moreover, GC4AOEG has a well-defined molecular structure with a precise size and molecular weight to ensure batch-to-batch consistency during preparation and manufacturing, which is an important parameter for regulatory approval and clinical translation. We reasonably envisage that GC4AOEG may become a new UFH antagonist with significant clinical potential. This study may also provide new insights into the design and development of reversal agents towards UFH and other toxic agents.Ethical statement
All animal experiments were conducted in accordance with the ethical guidelines of the Institute of Chinese Medical Sciences, University of Macau and the protocols were approved by the Animal Ethics Committee at the University of Macau.Conflicts of interest
R. W., D. G., Q. H. and H. Z. are currently filing a Chinese patent related to the content of this work. The other author(s) declare no competing interest.Acknowledgements
This work was supported by the Science and Technology Development Fund, Macao SAR (FDCT 020/2015/A1), NSFC (21871301, 51873090 and 21672112), University of Macau (MYRG2019-00059-ICMS) and Fundamental Research Funds for the Central Universities. Ms Qiaoxian Huang and Dr Dong-Sheng Guo are financially supported by the UM Macao Ph.D Scholarship and UM Macao Distinguished Visiting Scholar Award, respectively.Notes and references
- B. Casu, A. Naggi and G. Torri, Carbohydr. Res., 2015, 403, 60–68 CrossRef CAS PubMed.
- H. C. Hemker, J. Thromb. Haemostasis, 2016, 14, 2329–2338 CrossRef CAS PubMed.
- B. Dahlbäck, Lancet, 2000, 355, 1627–1632 CrossRef.
- G. M. Arepally, Blood, 2017, 129, 2864 CrossRef CAS PubMed.
- E. Sokolowska, B. Kalaska, J. Miklosz and A. Mogielnicki, Expert Opin. Drug Metab. Toxicol., 2016, 12, 897–909 CrossRef CAS PubMed.
- N. U. Khan, C. K. Wayne, J. Barker and T. Strang, Eur. J. Anaesthesiol., 2010, 27, 624–627 CrossRef CAS PubMed.
- C. Boer, M. I. Meesters, D. Veerhoek and A. B. A. Vonk, Br. J. Anaesth., 2018, 120, 914–927 CrossRef CAS PubMed.
- B. Ourri and L. Vial, ACS Chem. Biol., 2019, 14, 2512–2526 CrossRef CAS PubMed.
- M. T. Kalathottukaren, A. L. Creagh, S. Abbina, G. Lu, M. J. Karbarz, A. Pandey, P. B. Conley, J. N. Kizhakkedathu and C. Haynes, Blood Adv., 2018, 2, 2104–2114 CrossRef CAS PubMed.
- M. Schuksz, M. M. Fuster, J. R. Brown, B. E. Crawford, D. P. Ditto, R. Lawrence, C. A. Glass, L. Wang, Y. Tor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13075 CrossRef CAS PubMed.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9195 RSC.
- M. Corredor, D. Carbajo, C. Domingo, Y. Perez, J. Bujons, A. Messeguer and I. Alfonso, Angew. Chem., Int. Ed., 2018, 57, 11973–11977 CrossRef CAS PubMed.
- B. Ourri, J. P. Francoia, G. Monard, J. C. Gris, J. Leclaire and L. Vial, ACS Med. Chem. Lett., 2019, 10, 917–922 CrossRef CAS PubMed.
- B. Kalaska, J. Miklosz, K. Kaminski, B. Musielak, S. I. Yusa, D. Pawlak, M. Nowakowska, K. Szczubialka and A. Mogielnicki, RSC Adv., 2019, 9, 3020–3029 RSC.
- B. P. Schick, D. Maslow, A. Moshinski and J. D. San Antonio, Blood, 2004, 103, 1356–1363 CrossRef CAS PubMed.
- P. Y. Li, Y. Chen, C. H. Chen and Y. Liu, Chem. Commun., 2019, 55, 11790–11793 RSC.
- H. Y. Cheong, M. Groner, K. Hong, B. Lynch, W. R. Hollingsworth, Z. Polonskaya, J. K. Rhee, M. M. Baksh, M. G. Finn, A. J. Gale and A. K. Udit, Biomacromolecules, 2017, 18, 4113–4120 CrossRef CAS PubMed.
- D. T. Hunter Jr and J. M. Hill, Nature, 1961, 191, 1378–1379 CrossRef CAS PubMed.
- E. M. Jacobsen, E. J. Trettenes, F. Wisloff and U. Abildgaard, Thromb. J., 2006, 4, 3 CrossRef PubMed.
- R. J. Weiss, J. D. Esko and Y. Tor, Org. Biomol. Chem., 2017, 15, 5656–5668 RSC.
- W. H. Binder, M. J. Kunz and E. Ingolic, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 162–172 CrossRef CAS.
- J. E. Ansell, S. H. Bakhru, B. E. Laulicht, S. S. Steiner, M. Grosso, K. Brown, V. Dishy, R. J. Noveck and J. C. Costin, N. Engl. J. Med., 2014, 371, 2141–2142 CrossRef PubMed.
- M. T. Kalathottukaren, L. Abraham, P. R. Kapopara, B. F. L. Lai, R. A. Shenoi, F. I. Rosell, E. M. Conway, E. L. G. Pryzdial, J. H. Morrissey, C. A. Haynes and J. N. Kizhakkedathu, Blood, 2017, 129, 1368–1379 CrossRef CAS PubMed.
- A. Bom, M. Bradley, K. Cameron, J. K. Clark, J. Van Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M. Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 266–270 CrossRef PubMed.
- R. B. C. Jagt, R. F. Gómez-Biagi and M. Nitz, Angew. Chem., Int. Ed., 2009, 48, 1995–1997 CrossRef CAS PubMed.
- S. Valimaki, N. K. Beyeh, V. Linko, R. H. A. Ras and M. A. Kostiainen, Nanoscale, 2018, 10, 14022–14030 RSC.
- T. Mecca, G. M. Consoli, C. Geraci, R. La Spina and F. Cunsolo, Org. Biomol. Chem., 2006, 4, 3763–3768 RSC.
- Z. Zheng, W. C. Geng, J. Gao, Y. J. Mu and D. S. Guo, Org. Chem. Front., 2018, 5, 2685–2691 RSC.
- H. P. Fernandes, C. L. Cesar and L. Barjas-Castro Mde, Rev. Bras. Hematol. Hemoter., 2011, 33, 297–301 CrossRef PubMed.
- T. Mecca, G. M. L. Consoli, C. Geraci, R. La Spina and F. Cunsolo, Org. Biomol. Chem., 2006, 4, 3763–3768 RSC.
- E. Wexselblatt, J. D. Esko and Y. Tor, J. Org. Chem., 2014, 79, 6766–6774 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- S. Jevsevar, M. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef CAS PubMed.
- S. Kohmoto, E. Mori and K. Kishikawa, J. Am. Chem. Soc., 2007, 129, 13364–13365 CrossRef CAS PubMed.
- E. Galante, C. Geraci, S. Sciuto, V. L. Campo, I. Carvalho, R. Sesti-Costa, P. M. M. Guedes, J. S. Silva, L. Hill, S. A. Nepogodiev and R. A. Field, Tetrahedron, 2011, 67, 5902–5912 CrossRef CAS.
- F. Sansone, M. Dudic, G. Donofrio, C. Rivetti, L. Baldini, A. Casnati, S. Cellai and R. Ungaro, J. Am. Chem. Soc., 2006, 128, 14528–14536 CrossRef CAS PubMed.
- A. D. Martin, E. Houlihan, N. Morellini, P. K. Eggers, E. James, K. A. Stubbs, A. R. Harvey, M. Fitzgerald, C. L. Raston and S. A. Dunlop, ChemPlusChem, 2012, 77, 308–313 CrossRef CAS.
- J. Ranke, K. Molter, F. Stock, U. Bottin-Weber, J. Poczobutt, J. Hoffmann, B. Ondruschka, J. Filser and B. Jastorff, Ecotoxicol. Environ. Saf., 2004, 58, 396–404 CrossRef CAS PubMed.
- K. M. Docherty and C. F. Kulpa, Green Chem., 2005, 7, 185–189 RSC.
- Y. Chang, Z. Huang, Y. Jiao, J. F. Xu and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 21191–21197 CrossRef CAS PubMed.
- Z. H. Liu, Y. P. Jiao, T. Wang, Y. M. Zhang and W. Xue, J. Controlled Release, 2012, 160, 14–24 CrossRef CAS PubMed.
- Z. Zhen, X. L. Liu, T. Huang, T. F. Xi and Y. F. Zheng, Mater. Sci. Eng., C, 2015, 46, 202–206 CrossRef CAS PubMed.
- D. Pan, O. Vargas-Morales, B. Zern, A. C. Anselmo, V. Gupta, M. Zakrewsky, S. Mitragotri and V. Muzykantov, PLoS One, 2016, 11, e0152074 CrossRef PubMed.
- M. A. Dobrovolskaia and S. E. McNeil, J. Controlled Release, 2013, 172, 456–466 CrossRef CAS PubMed.
- J. Cheng, S. B. Feng, S. L. Han, X. J. Zhang, Y. D. Chen, X. Zhou, R. B. Wang, X. H. Li, H. Y. Hu and J. X. Zhang, ACS Nano, 2016, 10, 9957–9973 CrossRef CAS PubMed.
- K. Y. Lin, J. H. Lo, N. Consul, G. A. Kwong and S. N. Bhatia, ACS Nano, 2014, 8, 8776–8785 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials & methods and experimental details. See DOI: 10.1039/d0sc03922e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |