
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInter-regulated d-band centers of the Ni3B/Ni heterostructure for boosting hydrogen electrooxidation in alkaline media†
Fulin
Yang‡
,
Pengyu
Han‡
,
Na
Yao‡
,
Gongzhen
Cheng
 ,
Shengli
Chen
and
Wei
Luo
*
,
Shengli
Chen
and
Wei
Luo
*
College of Chemistry and Molecular Sciences, Wuhan University Wuhan, Hubei 430072, P. R. China. E-mail: wluo@whu.edu.cn
First published on 8th October 2020
Abstract
Ni3B/Ni heterostructures have been constructed, which exhibit exceptional catalytic performance toward the hydrogen oxidation reaction (HOR) under alkaline media, with the mass activity being about 10 times greater than that of Ni3B and Ni, respectively, ranking among the most active platinum-group-metal-free electrocatalysts. Experimental results and theoretical calculations confirm electron transfer from Ni3B to Ni at the Ni3B/Ni interface, resulting in inter-regulated d-band centers of these two components. This inter-regulation gives rise to optimized binding energies of intermediates, which together contribute to enhanced alkaline HOR activity.
Alkaline exchange membrane fuel cells (AEMFCs) as potential alternatives to proton exchange membrane fuel cells (PEMFCs) have attracted renewed attention.1–3 Compared to PEMFCs, it is possible to utilize platinum-group-metal-free (PGM-free) catalysts at both the anodes and cathodes in AEMFCs for the electrochemical hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR).4,5 Consequently, a large number of PGM-free catalysts have been developed for the alkaline ORR with Pt-like activity,6,7 however, the development of PGM-free HOR catalysts still remains a great challenge. More critically, even for those PGM-based electrocatalysts under alkaline electrolytes, the HOR kinetics are about 2 to 3 orders of magnitude lower than those under acidic conditions.8–10 Until now, only limited Ni-based materials have been reported with moderated catalytic activities, which are still far behind those of PGM-based electrocatalysts.11–13
Hydrogen binding energy (HBE) theory is widely recognized for hydrogen electrocatalysis, and the binding strength of the adsorbed hydrogen intermediate (H*) is considered as a sufficient descriptor to evaluate catalytic activity in general, following a classical volcano-type relationship.14,15 Due to the relatively strong hydrogen bonding ability of Ni, mainly two strategies including alloying with another metal16–18 or non-metal element19–21 and tuning the nickel–support interaction22–25 have been developed to boost the HOR performance through weakening the HBE of Ni. Besides, the bi-functional theory proposed by Markovic et al. suggests that the adsorbed OH (OH*) might also participate in the hydrogen electrode reaction directly.26,27 In this case, the properly strengthened oxophilicity of the catalyst can further accelerate the Volmer step, as well as the whole HOR process in alkaline media.28 Therefore, it is rational to design Ni-based catalysts with optimally balanced active sites for the adsorption of both H* and OH*. Generally, changing the d-band center of the catalyst could tune the binding strength of the adsorbed species on active sites.29 The upshifted or downshifted d-band center will lead to strengthened or weakened H*/OH* binding energy simultaneously. Thus, it is plausible to design catalysts with heterogeneous nanostructures that involve charge transfer at the interface to implement inter-regulated d-band centers of the multiple components to synergistically tailor the H*/OH* binding energy, as shown in Fig. 1.
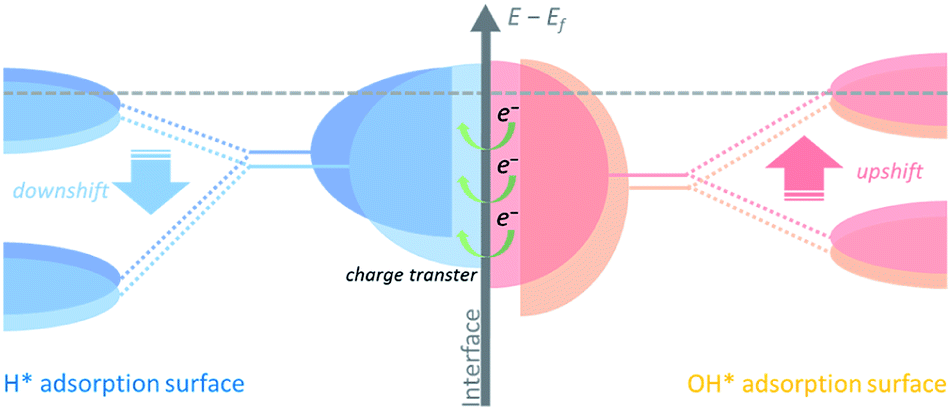 | ||
| Fig. 1 Schematic illustration describing the inter-regulated d-band centers at the interface. The downshifted d-band center of the H* adsorption surface and the upshifted d-band center of the OH* adsorption surface are induced by charge transfer between the two phases in the heterostructure. The gray dashed line represents the Fermi level. | ||
Herein we designed a heterostructured Ni-based nanocatalyst constructed from Ni nanocrystallines located on Ni3B, which could be in situ generated through annealing the amorphous Ni–B precursor. This in situ formed heterostructure provides strong contact between the two phases at the interface. Charge transfer from Ni3B to Ni with a relatively higher work function could increase the Fermi level of Ni,30 namely via downshifting the d-band center of Ni, and thus upshifting the d-band center of Ni3B. As a result, the inter-regulated d-band centers could optimize the binding energies of the intermediate species, e.g., weaken hydrogen adsorption and strengthen hydroxide adsorption, and thereby significantly boost the HOR performance under alkaline media.
A facile two-step approach was adopted to prepare the Ni3B/Ni heterostructure. Typically, a series of samples were obtained from annealing of the amorphous Ni–B precursor which was synthesized by reducing Ni2+ with NaBH4. Different annealing temperatures were preliminarily controlled to implement phase separation to in situ construct the well-defined Ni3B/Ni interface.31–33 These samples are denoted as NiB-T (T is the annealing temperature). Powder X-ray diffraction (PXRD) analysis was carried out to confirm the crystalline phases of the sample. As shown in Fig. 2a, the PXRD pattern of the amorphous Ni–B precursor remains unchanged until the annealing temperature rises to 300 °C. For NiB-300, the main peaks could be assigned to orthorhombic Ni3B (PDF#82-1699) and face-centered cubic (fcc) Ni (PDF#87-0712). When the annealing temperature was further elevated up to 400 °C, the characteristic diffraction pattern of Ni is retained and that of Ni3B almost disappears, indicating the transformation of Ni3B into Ni due to the excessive annealing temperature.31 Thus, it is suggested that appropriate annealing temperature (300 °C) triggers spontaneous crystallization and phase separation, which leads to the co-existence of both the metal and metal boride phases, resulting in the emergence of a large number of interfaces.
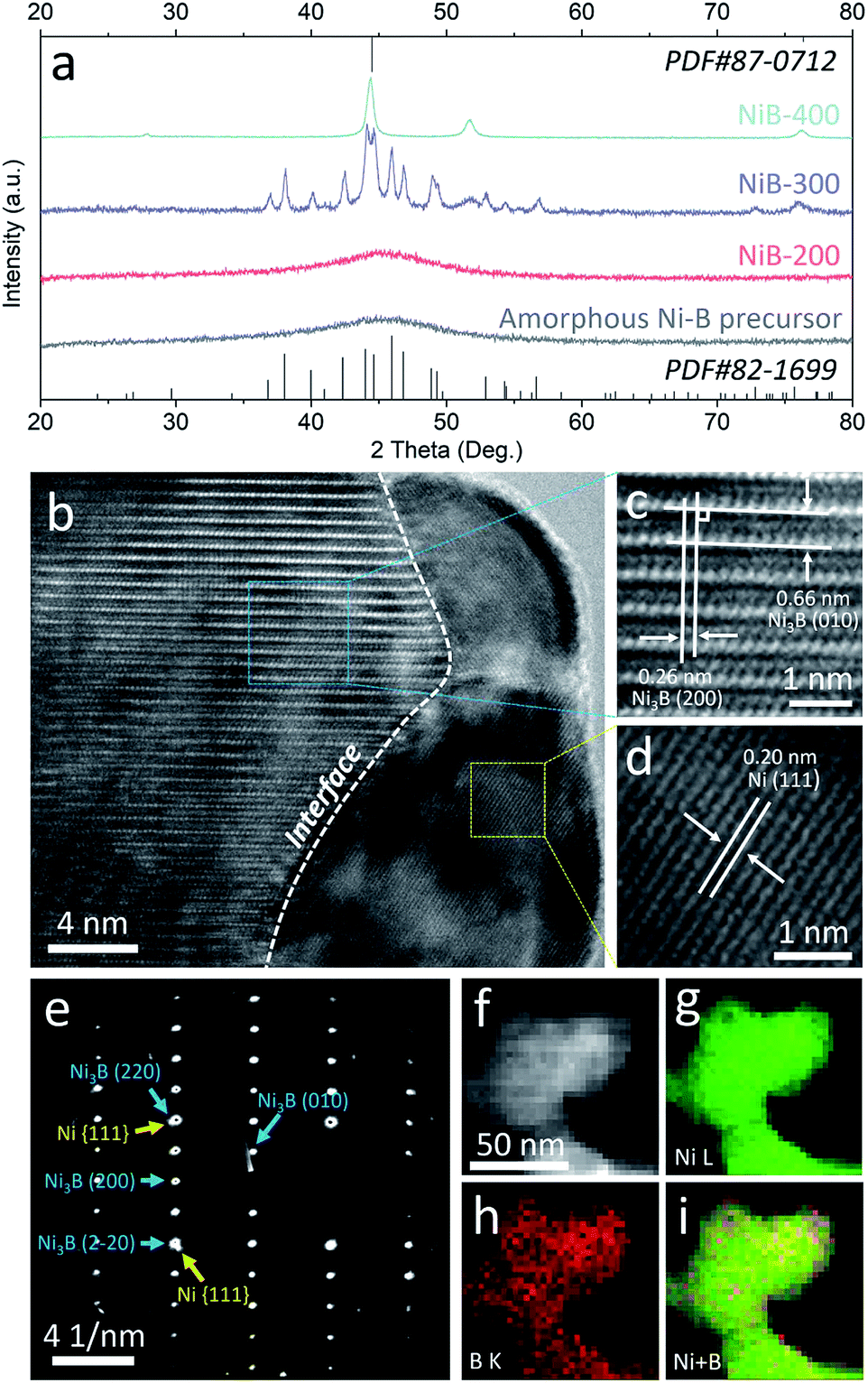 | ||
| Fig. 2 (a) XRD patterns of the amorphous Ni–B precursor and NiB-T; HRTEM images (b–d), SAED pattern (e), and STEM-EDX elemental mapping images (f–i) of NiB-300. | ||
Transmission electron microscopy (TEM) images reveal that the amorphous Ni–B precursor and three annealed samples possess a similar morphology that is composed of aggregated nanoparticles (Fig. S1–S4†). For NiB-300, the high-resolution TEM (HRTEM) images shown in Fig. 2b–d clearly display the presence of an interface underlined by the white dashed line between two different crystallized phases. The interplanar spacings on the left side are measured to be 0.26 and 0.66 nm, corresponding to the mutually perpendicular (200) and (010) crystal planes of orthorhombic Ni3B (Fig. 2c), respectively. The one on the right side depicts an interplanar spacing of about 0.20 nm, corresponding to the (111) plane of fcc Ni (Fig. 2d). Moreover, selected area electron diffraction (SAED) patterns also verify both the crystallized Ni3B and Ni as marked by blue and yellow arrows (Fig. 2e). Scanning TEM-energy dispersive X-ray (STEM-EDX) elemental mapping analysis shows the homogeneous distribution of Ni and B elements, suggesting the existence of a large number of interfaces (Fig. 2f). For comparison, Ni3B and Ni were also synthesized with similar morphology, and characterized by XRD and TEM (Fig. S5 and S6†).
Due to the different chemical environments and work functions between Ni3B and Ni, well-contacted interfaces might result in the reconstruction of electron structure. X-ray photoelectron spectroscopy (XPS) was performed to further prove this viewpoint. Fig. S7† shows the survey spectrum of NiB-300, confirming the existence of Ni and B elements. In the high-resolution Ni 2p3/2 XPS spectrum (Fig. S8a†), the peaks at about 852.1 eV and 856.6 eV can be attributed to metallic and oxidized Ni, respectively.34 Meanwhile, the high-resolution B 1s XPS spectrum shows two peaks at 187.6 and 192.8 eV, corresponding to boride and oxidized boron, respectively (Fig. S8b†).35 It is worth noting that NiB-300 exhibits positively shifted binding energies of both Ni0 and B0 compared to those in pure-phased Ni3B (about 851.9 eV and 187.2 eV, respectively), which demonstrates the reconstruction of the electron structure at the interface. Moreover, when compared to pure Ni (852.7 eV), the binding energy of Ni0 in NiB-300 is negatively shifted. Hence it is plausible to suggest that there is charge transfer from Ni3B to Ni in NiB-300. In addition, Ni 2p spectra also exhibit stepwise shifted binding energies when the temperature rises gradually during the annealing process (Fig. S9†), agreeing well with the XRD results shown in Fig. 2a that the phases change from amorphous Ni–B to Ni3B/Ni heterostructures and to Ni (with only traces of Ni3B) gradually.
The HOR performances of these obtained products were estimated using a rotating disk electrode (RDE) measurement system in a standard three-electrode system with 0.1 M KOH as the electrolyte. Before the electrochemical tests, all the Ni-based materials were loaded on carbon black with the loadings of about 50 wt%. The accurate loadings of Ni of these samples were further confirmed by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), as shown in Table S1.† Cyclic voltammetry (CV) curves of these catalysts were recorded in Ar-saturated 0.1 M KOH solution with a scan rate of 50 mV s−1, and the corresponding anodic polarization curves are shown in Fig. S10.† The peaks located at about 0.3 V (vs. RHE) could be attributed to the oxidation of Ni species.36,37 It should be noted that this anodic peak of NiB-300 exhibits about 5 mV negative shift compared to Ni but 27 mV positive shift to Ni3B, suggesting the changed electronic configuration of the catalyst surface, which agrees well with XPS results (Fig. S8†). This reconstructed electron structure might induce inter-regulated d-band centers of these two components that lead to the optimized catalytic performance. The polarization curves were obtained by linear sweep voltammetry (LSV) tests in H2-saturated 0.1 M KOH at a rotating speed of 2500 rpm with a scan rate of 5 mV s−1, as presented in Fig. 3a. Among them, NiB-300 unambiguously shows the highest HOR activity. HOR kinetic current densities (jk) could be extracted based on the Koutecky–Levich equation by eliminating the effect from mass transfer of H2.8 As shown in Fig. 3b, HOR polarization curves, typically for NiB-300, were recorded at different rotating speeds from 2500 rpm to 400 rpm. The Koutecky–Levich plot constructed at the overpotential of 0.05 V by fitting j−1vs. ω−1/2 could be obtained with a slope of 14.78 cm2 per mA rpm1/2, in good agreement with the theoretical value.38 Following this mass transport resistance correction, the mass activity denoted as jk at the overpotential of 0.05 V after normalization by the mass of the Ni element of NiB-300 is 24.71 mA mgNi−1, which is about an order of magnitude higher than that of Ni (3.91 mA mgNi−1) and Ni3B (2.21 mA mgNi−1), ranking among the most active Ni-based catalysts (Fig. 3d, Tables S2 and S3†).16–20,22,24,38–41
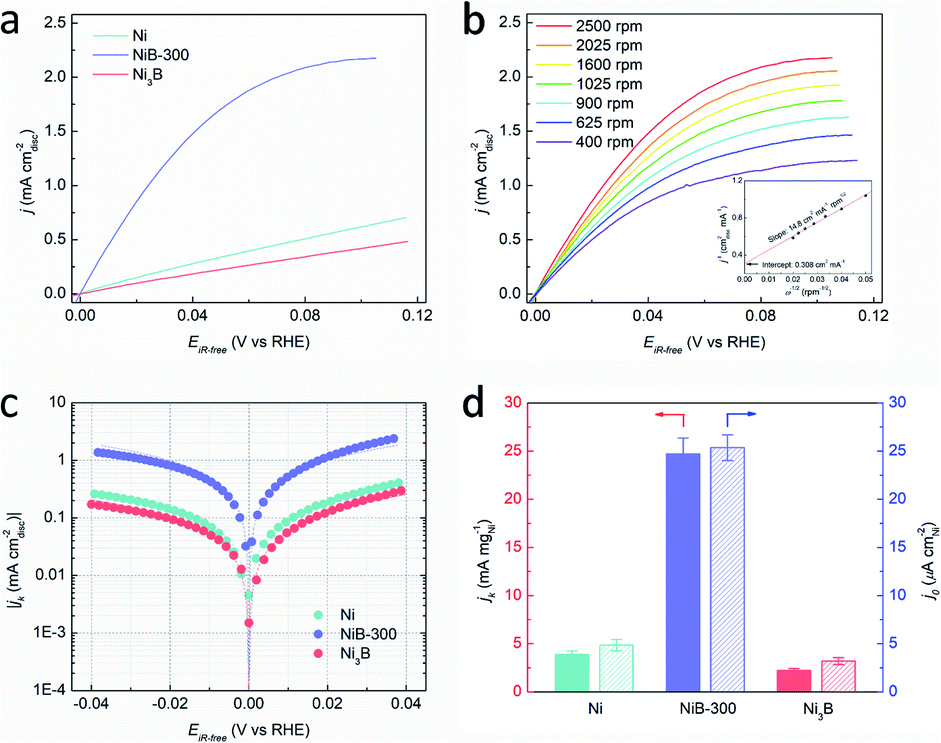 | ||
| Fig. 3 (a) HOR polarization curves of Ni, NiB-300, and Ni3B in H2-saturated 0.1 M KOH solution at a rotating speed of 2500 rpm with a scan rate of 5 mV s−1. (b) Polarization curves of NiB-300 in H2-saturated 0.1 M KOH solution with a scan rate of 5 mV s−1 at the rotating speeds varied from 2500 to 400 rpm. Inset shows the Koutecky–Levich plot at 50 mV (vs. RHE). (c) Tafel plots derived from (a) as well as the corresponding fits to the Butler–Volmer equation. (d) Comparison of jk at the overpotential of 50 mV (unpatterned) and j0 (patterned) of the three catalysts. | ||
The exchange current densities (j0) on these Ni-based catalysts could be further extracted by fitting the kinetic current densities according to the Butler–Volmer equation (Fig. 3c).8 The value of j0 of NiB-300 normalized by the electrochemically active surface area (ECSA) that was determined from the accumulated charge quantity of OH desorption22 from the surface of Ni via CV (Fig. S11†) is 25.37 μA cmNi−2, which is about 5 times and 7 times higher than that of Ni (4.85 μA cmNi−2) and Ni3B (3.19 μA cmNi−2), respectively, and is even comparable to those of some Pd-based catalysts (Tables S2 and S3†).10,16–20,24,38–43 In order to avoid the contribution from non-faradaic current, a steady-state polarization curve was also obtained for comparison to separate the hydrogen electrocatalytic response as shown in Fig. S12.† It could be observed that these two polarization curves are almost coincident, indicating that the use of a transient polarization curve obtained at the scanning rate of 5 mV s−1, as shown in Fig. 3a, to calculate the j0 will not result in obvious over/under-estimation. In addition, the value of j0 could also be estimated from the micro-polarization region within a small potential window from −10 to 10 mV according to the simplified Butler–Volmer equation22 (Fig. S13†), with almost the same results. Moreover, those Ni-based samples obtained at other annealing temperatures, such as the Ni–B precursor, NiB-200, and NiB-400, were also analyzed by electrochemical tests (Fig. S14 and S15†), and the values of jk and j0 are also calculated and presented in Table S3.† It can be seen that NiB-300 still shows the highest active performance.
An accelerated durability test (ADT) was conducted to explore the stability of NiB-300, with the potential range between −0.08 and 0.42 V, which includes the complete redox process of Ni/Ni2+. The schematic protocol of the potential scanning protocol is presented in Fig. S16a† with a sweep rate of 100 mV s−1. CV and LSV tests were performed in Ar/H2-saturated 0.1 M KOH solution respectively, as depicted in Fig. S16b–d,† which indicates obviously decreased ECSA and j0. This inferior stability could be attributed to the irreversible formation of β-Ni(OH)2.34,35,44 Nevertheless, the j0 of NiB-300 after ADT (0.015 mA cmNi−2) is still superior to those of the pure Ni (0.005 mA cmNi−2)and Ni3B (0.003 mA cmNi−2).
The Ni3B/Ni heterostructure could also be constructed by the annealing process from pure Ni3B with well-defined crystal structure, because the structure of Ni3B is not stable and can transform to Ni at relatively high temperature.31–33 The sample with the Ni/Ni3B interface synthesized by this method is denoted as Ni3B-300, and from XRD patterns (Fig. S17†) it was confirmed that both the characteristic diffraction patterns of orthorhombic Ni3B and fcc Ni are present. The nanoparticle sizes of both these heterostructures (NiB-300 and Ni3B-300) are similar (Fig. S18†). Fig. S19† depicts the HOR polarization curve of Ni3B-300, which shows the obviously higher apparent activity than Ni and Ni3B. As presented in Table S2,† the j0 of Ni3B-300 is measured to be 22.85 μA cmNi−2, comparable to that of NiB-300. Thus, it is suggested that the Ni3B/Ni interface might significantly enhance the intrinsic alkaline HOR activity resulting from the charge transfer induced regulation of the d-band centers.
DFT calculations were employed to further elucidate the origin of the enhanced intrinsic activity of the Ni3B/Ni heterostructure toward the HOR in alkaline electrolytes. We construct the Ni(111)/Ni3B(001) interface to represent the heterostructure of Ni3B/Ni due to the most matched interface in the light of the finite strain theory.45Fig. 4a–c exhibit the schematic model of the heterostructure of Ni3B/Ni, as well as those of pure-phased Ni and Ni3B for comparison. Firstly, the electronic structure of the heterostructure was calculated through Mulliken charge analysis. Five kinds of active sites are considered as marked by the red circles in Fig. 4b, in which four sites are located in the Ni3B phase while the other one belongs to the Ni phase. For comparison, the Mulliken charge on Ni3B and Ni was also analyzed. From Fig. 4d we can see that the Ni site in the Ni phase of the Ni3B/Ni heterostructure possesses lower Mulliken charge compared to that in pure-phased Ni, while the sites in the Ni3B phase of the Ni3B/Ni heterostructure possess larger Mulliken charge compared to that in pure-phased Ni3B. This result indicates that there is strong redistribution of the electronic structure in the interfacial area, which leads to charge transfer from Ni3B to Ni, consistent with the XPS analysis mentioned above.
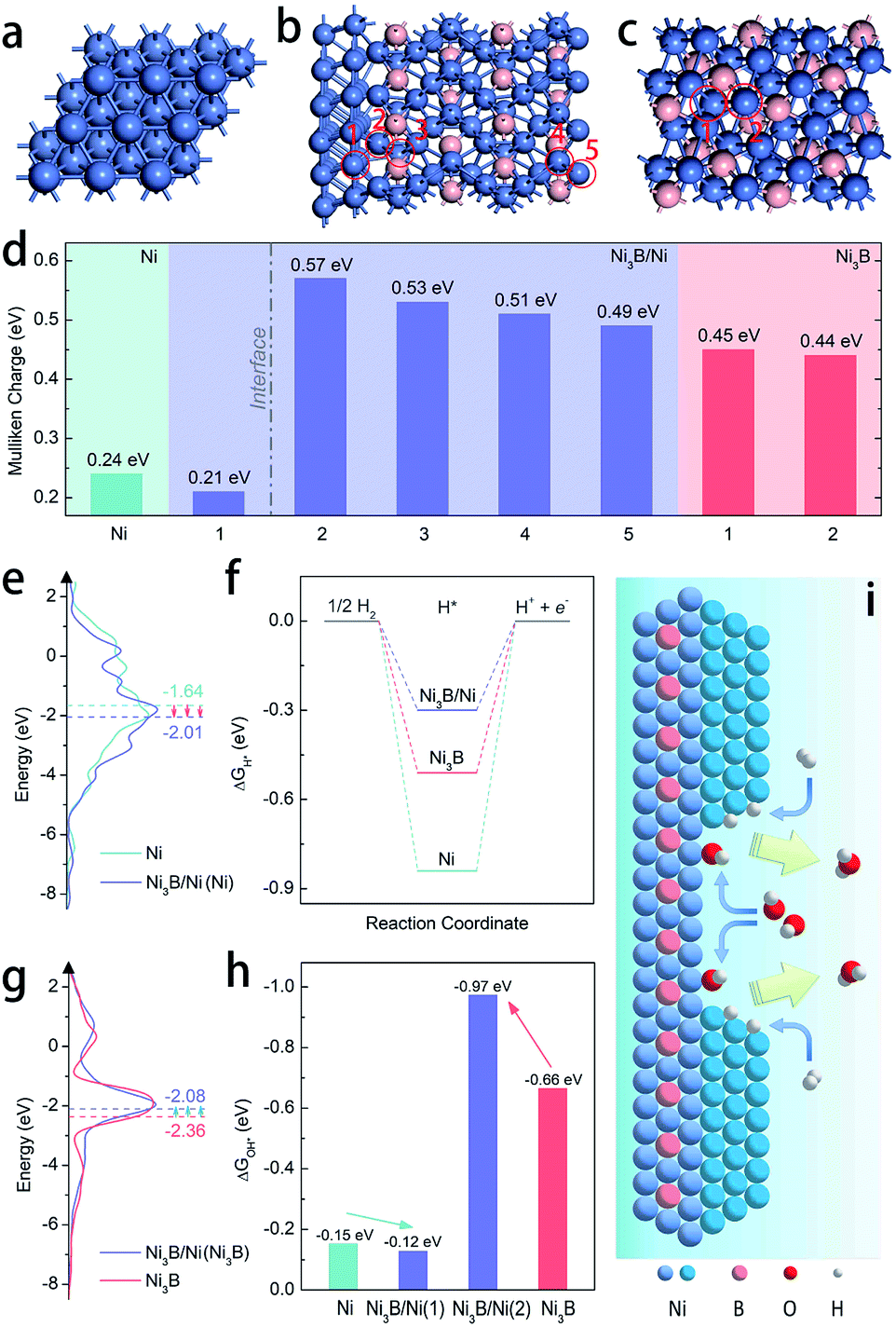 | ||
| Fig. 4 Atomic structure diagrams of the optimized models of (a) Ni, (b) the Ni3B/Ni heterostructure, and (c) Ni3B. (d) Mulliken charges for hollow sites in Ni (111), 1–5 sites in the Ni3B/Ni heterostructure, and 1-2 sites in Ni3B (001), as marked by red circles in (b) and (c). (e) DOS plots of Ni and the Ni3B/Ni heterostructure (each d-band center is highlighted by a dashed line). (f) Free-energy diagram for H* adsorption on Ni, the Ni3B/Ni heterostructure, and Ni3B. (g) DOS plots of the Ni3B/Ni heterostructure and Ni3B (each d-band center is highlighted by a dashed line). (h) Free-energy diagram for OH* adsorption on Ni, the Ni3B/Ni heterostructure, and Ni3B. (i) Schematic illustration of the accelerated HOR process at the interface between Ni and Ni3B in the Ni3B/Ni heterostructure. | ||
Moreover, the work functions were also calculated, showing the values of 3.594 eV and 3.028 eV for Ni and Ni3B, respectively. The work function (ϕ) result was also obtained experimentally from ultraviolet photoelectron spectroscopy (UPS) to further confirm the trend. As shown in Fig. S20,† the value of ϕ for Ni (4.12 eV) is larger than that of Ni3B (4.01), which shows the same trend as the calculated results. Therefore, the d-band center might also be changed correspondingly as suggested before.30 In addition, the d-band density of states (d-DOS) of Ni in these three models was calculated. As shown in Fig. 4e, the DOS near the Fermi level (Ef) shows that the d-band center of Ni in the Ni3B/Ni heterostructure is about −2.01 eV, which is more far away from Ef than that in pure-phased Ni (−1.64 eV). As a result, it is suggested that in this case a downshift of the d-band center indeed occurs when the Ni3B/Ni interface is constructed. In general, the lower d-band center could be corresponding to the weaker binding strength of adsorbates such as H* and OH*, due to the higher band filling of the antibonding states.46–48 Ni-based catalysts that are located on the left branch of the volcano plot possess overstrong HBE, and this weakening might boost the Volmer step that is usually recognized as the rate determining step and thereby facilitate alkaline HOR, which is confirmed in Fig. 4f which shows that the Ni3B/Ni heterostructure displays the optimal ΔGH* among the three models. Besides that, the binding strength of OH* on Ni sites is also weakened (Fig. 4h). In this case for the Ni3B/Ni heterostructure, while the d-band center of the Ni-phase downshifts due to the charge transfer through the interface, that of the Ni3B-phase would also be regulated. From Fig. 4g, we can observe that the d-band center of the Ni3B-phase of the Ni3B/Ni heterostructure (−2.08 eV) is upshifted closer to the Femi level compared to that of the pure-phased Ni3B (−2.36 eV), revealing the enhanced binding strength of OH* as illustrated in Fig. 4h. In consequence, both the weakened H binding energy and strengthened OH binding energy of the Ni3B/Ni heterostructure effectively promote the HOR in alkaline electrolytes24,25,36 as schematically illustrated in Fig. 4i, supporting our experimental results.
Considering the fact that more than 90% of the commercial H2 is produced from the industrial steam reforming from hydrocarbons, which makes CO inevitable in the available H2, and CO tolerance is an additional criterion in the pursuit of effective electrocatalysts. Due to the strong adsorption of CO on the surface of Pt, commercial Pt/C suffers a rapid decrease of the anodic current density during the chronoamperometry test in CO-containing (100 ppm) H2-saturated 0.1 M KOH at a constant potential of 0.05 V (Fig. 5a). Meanwhile, NiB-300 maintains a relatively stable anodic current density. After the chronoamperometry test, the LSV polarization curve of NiB-300 displayed a slight decrease compared with its initial state (Fig. 5b). In contrast, Pt/C shows an obviously lower activity even though under pure H2 conditions again since CO poisoning is irreversible under this HOR potential area. Hence, this NiB-300 electrocatalyst exhibits excellent CO tolerance compared to commercial Pt/C, and is comparable to some other Ni-based catalysts.19–21
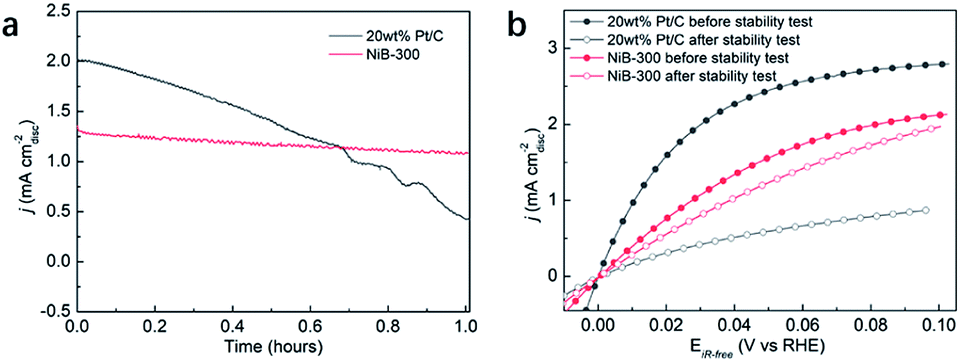 | ||
| Fig. 5 (a) Chronoamperometry curves of 20 wt% Pt/C and NiB-300 in 0.1 M KOH saturated with H2 containing 100 ppm CO measured at 0.05 V. (b) Comparison of the HOR polarization curves of 20 wt% Pt/C and NiB-300 in 0.1 M KOH saturated with H2 before and after the chronoamperometry test. | ||
Conclusions
In conclusion, the Ni3B/Ni heterostructure has been constructed through annealing amorphous Ni–B or crystallized Ni3B precursors. The obtained Ni3B/Ni heterostructure exhibits great CO-tolerance and the highest mass activity among PGM-free electrocatalysts toward alkaline HOR, which is about 10 times higher than that of Ni3B and Ni, respectively. Combining experimental analysis and theoretically calculated results, we find that the inter-regulated d-band center of the Ni3B/Ni heterostructure induced by charge transfer at the interface leads to weakened H* adsorption on Ni and strengthened OH* adsorption on Ni3B, which is regarded as the key factor in boosting the HOR performance. To the best of our knowledge, this is the first report on transition metal boride-based electrocatalysts toward the HOR. This work provides a promising new direction for developing low-cost and efficient PGM-free catalysts toward AEMFCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFB1502302), National Natural Science Foundation of China (21972107, 21832004, and 21633008), and National Natural Science Foundation of Hubei Province (2020CFA095). The DFT calculations have been done on the supercomputing system in the Supercomputing Center of Wuhan University.Notes and references
- B. P. Setzler, Z. Zhuang, J. A. Wittkopf and Y. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- Z. F. Pan, L. An, T. S. Zhao and Z. K. Tang, Prog. Energy Combust. Sci., 2018, 66, 141–175 CrossRef.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef CAS.
- G. A. Goenaga, A. L. Roy, N. M. Cantillo, S. Foister and T. A. Zawodzinski Jr, J. Power Sources, 2018, 395, 148–157 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasche, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- E. S. Davydova, S. Mukerjee, F. Jaouen and D. R. Dekel, ACS Catal., 2018, 8, 6665–6690 CrossRef CAS.
- A. G. Oshchepkov, G. Braesch, A. Bonnefont, E. R. Savinova and M. Chatenet, ACS Catal., 2020, 10, 7043–7068 CrossRef CAS.
- J. K. Norskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- W. Sheng, A. P. Bivens, M. Myint, Z. Zhuang, R. V. Forest, Q. Fang, J. G. Chen and Y. Yan, Energy Environ. Sci., 2014, 7, 1719–1724 RSC.
- O. V. Cherstiouk, P. A. Simonov, A. G. Oshchepkov, V. I. Zaikovskii, T. Y. Kardash, A. Bonnefont, V. N. Parmon and E. R. Savinova, J. Electroanal. Chem., 2016, 783, 146–151 CrossRef CAS.
- S. Kabir, K. Lemire, K. Artyushkova, A. Roy, M. Odgaard, D. Schlueter, A. Oshchepkov, A. Bonnefont, E. Savinova, D. C. Sabarirajan, P. Mandal, E. J. Crumlin, I. V. Zenyuk, P. Atanassov and A. Serov, J. Mater. Chem. A, 2017, 5, 24433–24443 RSC.
- W. Ni, A. Krammer, C. –S. Hsu, H. M. Chen, A. Schugler and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 7445–7449 CrossRef CAS.
- T. Wang, M. Wang, H. Yang, M. Xu, C. Zuo, K. Feng, M. Xie, J. Deng, J. Zhong, W. Zhou, T. Cheng and Y. Li, Energy Environ. Sci., 2019, 12, 3522–3529 RSC.
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Nat. Commun., 2018, 9, 4531 CrossRef.
- Z. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. Yan, Nat. Commun., 2016, 7, 10141 CrossRef CAS.
- S. A. Giles, Y. Yan and D. G. Vlachos, ACS Catal., 2019, 9, 1129–1139 CrossRef CAS.
- F. Yang, X. Bao, Y. Zhao, X. Wang, G. Cheng and W. Luo, J. Mater. Chem. A, 2019, 7, 10936–10941 RSC.
- S. Jiang, Q. Cheng, L. Zou, Z. Zou, Y. Li, Q. Zhang, Y. Gao and H. Yang, Chem. Phys. Lett., 2019, 728, 19–24 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS.
- E. Liu, J. Li, L. Jiao, H. T. T. Doan, Z. Liu, Z. Zhao, Y. Huang, K. M. Abraham, S. Mukerjee and Q. Jia, J. Am. Chem. Soc., 2019, 141, 3232–3239 CrossRef CAS.
- B. Hammer and J. K. Nørskov, in Adv. Catal., ed. H. K. Bruce and C. Gates, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- J. Zheng, D. A. Cullen, R. V. Forest, J. A. Wittkopf, Z. Zhuang, W. Sheng, J. G. Chen and Y. Yan, ACS Catal., 2015, 5, 1468–1474 CrossRef CAS.
- H. Li, H. Li and J. –F. Deng, Mater. Lett., 2001, 50, 41–46 CrossRef CAS.
- S. Wei, Z. Li, S. Yin, X. Zhang, W. Liu and X. Wang, J. Synchrotron Radiat., 2001, 8, 566–568 CrossRef CAS.
- J. Guo, Y. Hou, B. Li and Y. Wang, J. Appl. Electrochem., 2016, 46, 1177–1186 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, in Handbook of X-ray photoelectron spectroscopy, ed. G. E. Muilenburg, Perkin-Elmer Corporation, 1979, pp. 80–81 Search PubMed.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, in Handbook of X-ray photoelectron spectroscopy, ed. G. E. Muilenburg, Perkin-Elmer Corporation, 1979, pp. 36–37 Search PubMed.
- D. Floner, C. Lamy and J. M. Leger, Surf. Sci., 1990, 234, 87–97 CrossRef CAS.
- D. S. Hall, C. Bock and B. R. MacDougall, J. Electrochem. Soc., 2013, 160, F235–F243 CrossRef CAS.
- F. Yang, X. Bao, P. Li, X. Wang, G. Cheng, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2019, 58, 14179–14183 CrossRef CAS.
- Y. Yang, X. Sun, G. Han, X. Liu, X. Zhang, Y. Sun, M. Zhang, Z. Cao and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 10644–10649 CrossRef CAS.
- L. Gao, Y. Wang, H. Li, Q. Li, N. Ta, L. Zhuang, Q. Fu and X. Bao, Chem. Sci., 2017, 8, 5728–5734 RSC.
- W. Ni, T. Wang, P. A. Schouwink, Y. –C. Chuang, H. M. Chen and X. Hu, Angew. Chem., Int. Ed., 2020, 59, 10797–10801 CrossRef CAS.
- S. Henning, J. Herranz and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, F178–F189 CrossRef CAS.
- T. Bhowmik, M. K. Kundu and S. Barman, ACS Catal., 2016, 6, 1929–1941 CrossRef CAS.
- E. S. Davydova, F. D. Speck, M. T. Y. Paul, D. R. Dekel and S. Cherevk, ACS Catal., 2016, 6, 1929–1941 CrossRef.
- T. Liu, A. Li, C. Wang, W. Zhou, S. Liu and L. Guo, Adv. Mater., 2018, 30, 1803590 CrossRef.
- E. Nikolla, J. Schwank and S. Linic, J. Am. Chem. Soc., 2009, 131, 2747–2754 CrossRef CAS.
- T. Komatsu and S. Furukawa, Mater. Trans., 2015, 56, 460–467 CrossRef CAS.
- C. K. Poh, Z. Tian, J. Gao, Z. Liu, J. Lin, Y. P. Feng and F. Su, J. Mater. Chem., 2012, 22, 13643–13652 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03917a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |