
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(III)-catalyzed tandem annulative redox-neutral arylation/amidation of aromatic tethered alkenes†
Chao
Chen‡
ab,
Chen
Shi‡
ab,
Yaxi
Yang
abc and
Bing
Zhou
 *abc
*abc
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai 201203, China. E-mail: zhoubing@simm.ac.cn; zhoubing2012@hotmail.com
bUniversity of Chinese Academy of Sciences, 19 Yuquan Road, Beijing 100049, China
cSchool of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 16th October 2020
Abstract
Transition-metal-catalyzed directed C–H functionalization has emerged as a powerful and straightforward tool to construct C–C bonds and C–N bonds. Among these processes, the intramolecular annulative alkene hydroarylation reaction has received much attention because this intramolecular annulation can produce more complex and high value-added structural motifs found in numerous natural products and bioactive molecules. Despite remarkable progress, these annulative protocols developed to date remain limited to hydroarylation and functionalization of one side of alkenes, thus largely limiting the structural diversity and complexity. Herein, we developed a rhodium(III)-catalyzed tandem annulative arylation/amidation reaction of aromatic tethered alkenes to deliver a variety of 2,3-dihydro-3-benzofuranmethanamine derivatives bearing an all-carbon quaternary stereo center by employing 3-substituted 1,4,2-dioxazol-5-ones as an amidating reagent to capture the transient C(sp3)–Rh intermediate. Notably, by simply changing the directing group, a second, unsymmetrical ortho C–H amidation/annulation can be achieved to provide tricyclic dihydrofuro[3,2-f]quinazolinones in good yields.
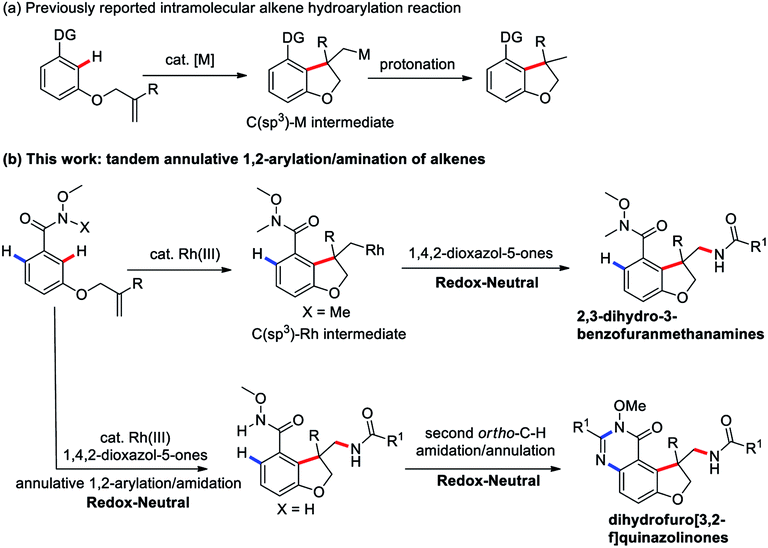
Transition-metal-catalyzed C–H functionalization for the direct conversion of C–H bonds to C–C bonds and C–N bonds has evolved into a widespread and effective strategy for fine chemical production.1 Among these processes, the hydroarylation of C–C double bonds via a C–H addition has been well-established and become an effective strategy to access synthetically useful structural motifs.1 Recently, this alkene hydroarylation reaction has received much attention in an intramolecular fashion2 (Scheme 1a) because this intramolecular annulation can produce more complex and high value-added structural motifs found in numerous natural products and bioactive molecules (Fig. 1).3 Despite remarkable progress in this area, most of the annulative protocols developed to date remain limited to one-component intramolecular alkene hydroarylation and functionalization of one side of alkenes. More challenging C–H arylation of intramolecular alkenes followed by a tandem coupling with a different coupling partner have unfortunately proven elusive thus far, thus largely limiting the structural diversity and complexity.
 | ||
| Scheme 1 Transition-metal-catalyzed C–H functionalization to construct the C–C and C–N bonds. | ||
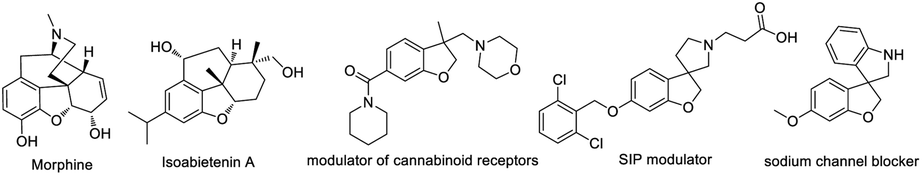 | ||
| Fig. 1 Representative bioactive 2,3-dihydrobenzofurans. | ||
On the other hand, nitrogen-containing molecules have gained great attention due to their widespread presence in natural products and widespread use in pharmaceutical science.4 During the last two decades, transition-metal-catalyzed direct C(sp2)–H amination/amidation assisted by chelating directing group is a well-established strategy.5 Recently, several examples of C(sp3)–H amination/amidation have also been reported for the efficient installation of C–N bonds.6,7 Mechanistically, the reaction is initiated by a chelation-assisted C–H metalation to form a C(sp3)–M species, which is then coupled with amination reagents to construct the C–N bonds.
In this context, we wondered if a catalytic annulative C–H arylation of a O-bearing olefin-tethered arenes might be possible, thus leading to a C(sp3)–M intermediate, which upon capture with a amidation reagent to construct a new C–N bond and provide bioactive 2,3-dihydro-3-benzofuranmethanamine derivatives. Inherently, the tandem annulative 1,2-arylation/amidation of alkenes has several challenges. First, the resulting C(alkyl)–M intermediate is liable to undergo protonation to provide the alkene hydroarylation products.1,2 Moreover, a potential competing β-H elimination of the resulting C(alkyl)–M intermediate also required to be suppressed. In addition, compared with the C(sp2)–M species, the resulting C(alkyl)–M species is relatively unstable and also has a low reactivity.
To address these challenges and with our continuing interest in the Rh(III)-catalyzed C–H functionalization,8 we introduced a Weinreb amide as a directing group and 3-substituted 1,4,2-dioxazol-5-ones as the amide sources9 to trigger a new tandem annulative 1,2-arylation/amidation of alkenes via a Rh(III)-catalyzed C–H activation,10 providing a variety of synthetically challenging 2,3-dihydro-3-benzofuranmethanamine derivatives bearing an all-carbon quaternary stereo center (Scheme 1b). More importantly, through simply changing the directing group, a second, unsymmetrical ortho C–H amidation/annulation could be realized to provide tricyclic dihydrofuro[3,2-f]quinazolinone derivatives. This protocol provides a good complement to previously reported carboamination reactions.11
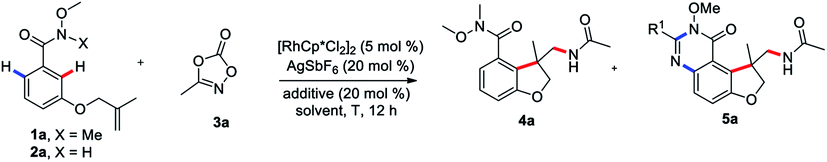
To begin our studies, Weinreb amide 1a was reacted with methyl dioxazolone 3a in the presence of various catalyst and AgSbF6 at 70 °C in DCE (Table 1, entries 1–4). The use of [Cp*RhCl2]2 as the catalyst was found to be crucial to give the desired tandem annulative product 4a, with other catalysts, such as [Ru(p-cymene)Cl2]2, [Cp*IrCl2]2, and Cp*Co(CO)I2, resulting in no desired product. Attempt to increase or lower the reaction temperature led to a slightly low yield (entries 5 and 6). Interestingly, when employing a NH–OMe amide 2a as the substrate and using 3 equivalent of 3a, a second, unsymmetrical ortho C–H amidation/annulation was achieved to provide the tricyclic dihydrofuro[3,2-f]quinazolinone 5a in 50% yield (entry 7). A screen of additives (entries 8–10) identified LiOAc as the optimal additive, affording the desired product 5a in 93% yield (entry 10). The Rh(III) catalyst was found to be crucial for this tandem annulative arylation/amidation reaction, with no reactivity in its absence (entries 11 and 12).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Catalyst (5 mol%) | Additive (20 mol%) | Solvent | T (°C) | Yield of 4a (%) | Yield of 5a (%) |
| a Conditions: 1a (0.1 mmol), 3a (0.12 mmol), catalyst (5 mol%), AgSbF6 (20 mol%) and additive (20 mol%) in DCE (1 mL) for 12 h. Yield isolated by column chromatography. b Conditions: 2a (0.1 mmol), 3a (0.3 mmol), catalyst (5 mol%), AgSbF6 (20 mol%), additive (20 mol%) in DCE (1 mL) for 12 h. Yield isolated by column chromatography. | |||||||
| 1 | Me | [Cp*RhCl2]2 | — | DCE | 70 | 89 | 0 |
| 2 | Me | [Ru(p-cymene)Cl2]2 | — | DCE | 70 | 0 | 0 |
| 3 | Me | [Cp*IrCl2]2 | — | DCE | 70 | 0 | 0 |
| 4 | Me | Cp*Co(CO)I2 | — | DCE | 70 | 0 | 0 |
| 5 | Me | [Cp*RhCl2]2 | — | DCE | 90 | 69 | 0 |
| 6 | Me | [Cp*RhCl2]2 | — | DCE | 50 | 70 | 0 |
| 7b | H | [Cp*RhCl2]2 | — | DCE | 70 | 0 | 50 |
| 8b | H | [Cp*RhCl2]2 | Cu(OAc)2 | DCE | 70 | 0 | 87 |
| 9b | H | [Cp*RhCl2]2 | KOAc | DCE | 70 | 0 | 86 |
| 10b | H | [Cp*RhCl2]2 | LiOAc | DCE | 70 | 0 | 93 |
| 11b | H | — | LiOAc | DCE | 70 | 0 | 0 |
| 12 | Me | — | — | DCE | 70 | 0 | 0 |
Having determined the optimal reaction conditions, we sought to evaluate the substrate scope (Scheme 2). First, the amidation reagents were explored and 1,4,2-dioxazol-5-ones substituted with primary alkyl (4a and 4e), secondary alky (4b, 4c and 4f), tertiary alkyl (4d) and aryl group (4g–j) all coupled smoothly with 1a, providing the 2,3-dihydro-3-benzofuranmethanamines 4a–4j in good yields. The structure of 4f was unambiguously confirmed by an X-ray crystallographic analysis (CCDC 2015893). The scope with regards to the arene moiety was then examined. The substrates 1 containing either electron-donating or electron-withdrawing substituents at different positions on the arene ring were well tolerated and provide the desired products 4k–t in good yields. We were pleased that 2-naphthalenecarboxamide effectively underwent this tandem annulative 1,2-arylation/amidation reaction, affording the desired product 4r in good yield. Notably, the various substituted allyl groups such as ethyl, cyclopentyl, phenyl, and phenoxymethyl groups were found to be compatible with the reaction conditions (4u–x). In addition, 3-N-tethered and 3-S-tethered substrates failed to give the desired tandem annulative products 4z and 4za.
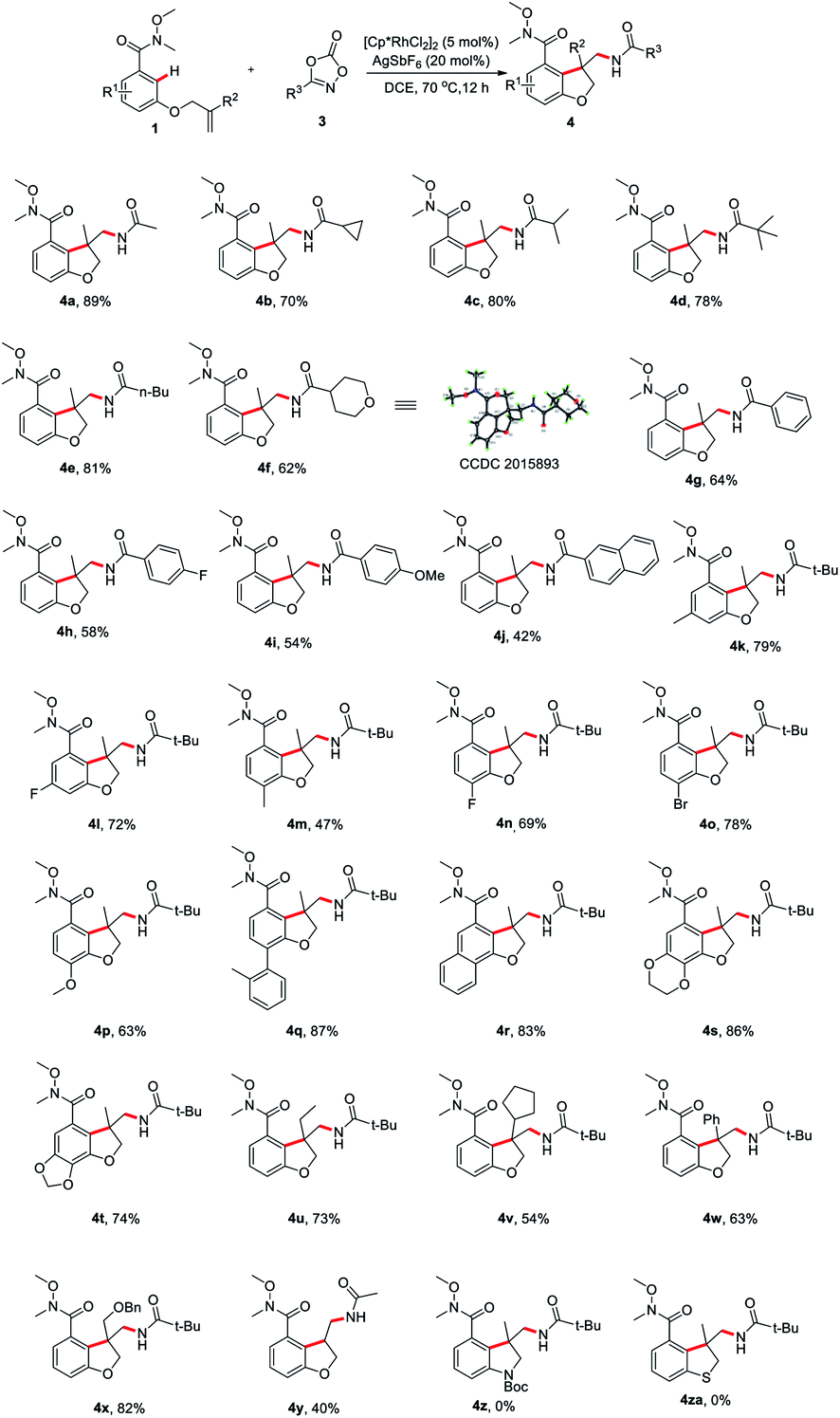 | ||
| Scheme 2 Substrate scope of tandem annulative arylation/amidation reaction of aromatic tethered alkenes. Conditions: 1 (0.1 mmol), 3 (0.12 mmol), [Cp*RhCl2]2 (5 mol%), AgSbF6 (20 mol%) in DCE (1 mL) at 70 °C for 12 h. Yield isolated by column chromatography. | ||
Next, we proceeded to explore the scope of this unsymmetrical twofold C–H functionalization reaction (Scheme 3). Under the optimal reaction conditions, amidating reagents bearing alkyl or aryl groups are fully tolerated, affording the tricyclic dihydrofuro[3,2-f]quinazolinones 5a–5h in good yields. The structure of 5e was unambiguously confirmed by an X-ray crystallographic analysis (CCDC 2014245). Electronic and steric modification of the aryl group was also tolerated. Both electron-deficient (5j, 5m–o) and electron-rich (5i, 5k, 5l, 5q and 5r) substrates gave the corresponding tricyclic systems in good yields. Meta and para substitutions of a methyl group were also tolerated and delivered the products 5i and 5k, indicating a high tolerance for steric hindrance. Interestingly, when 2-naphthalenecarboxamide was used, a third C–H amidation of naphthalene ring took place, affording the product 5s in 65% yield. Notably, the current method effectively resulted in the ethyl-, cyclopentyl, phenyl, and phenoxymethyl-substituted products 5t–w bearing an all-carbon quaternary stereo center in good yield, respectively.
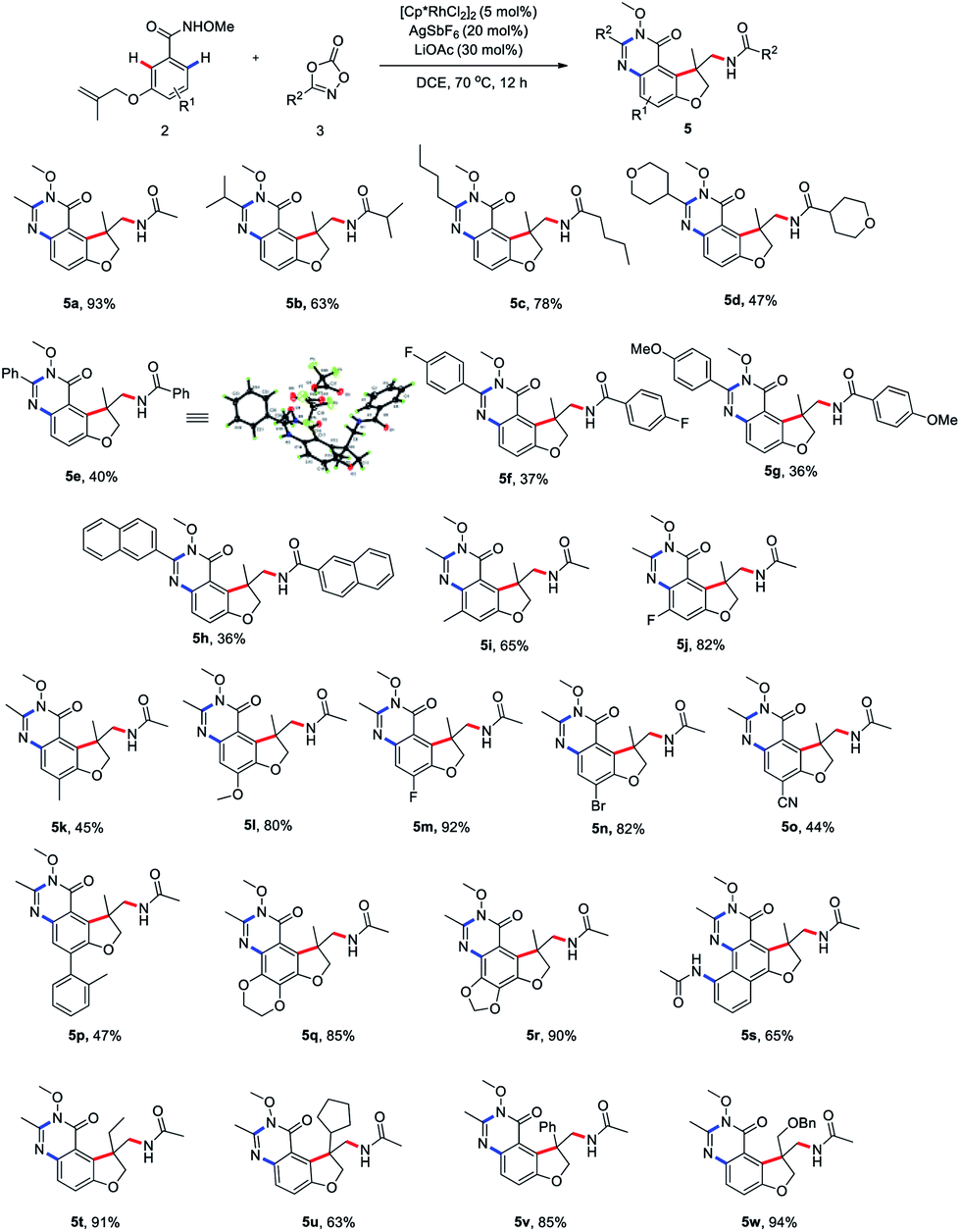 | ||
| Scheme 3 Substrate scope of unsymmetrical twofold C–H functionalization reaction. Conditions: 2 (0.1 mmol), 3 (0.30 mmol), [Cp*RhCl2]2 (5 mol%), AgSbF6 (20 mol%), LiOAc (20 mol%) and DCE (1 mL) at 70 °C for 12 h. Yield isolated by column chromatography. | ||
To check the practicability of this protocol, this two procedures could be readily scaled up with comparable efficiency in the presence of 2.5 mol% of Rh(III) catalyst on a 2.0 mmol scale (eqn (1) and (2)). The product 5a could be readily converted into potential useful intermediates, such as amines 6a and free amino quinazolinone analog 6b, respectively (eqn (3)).
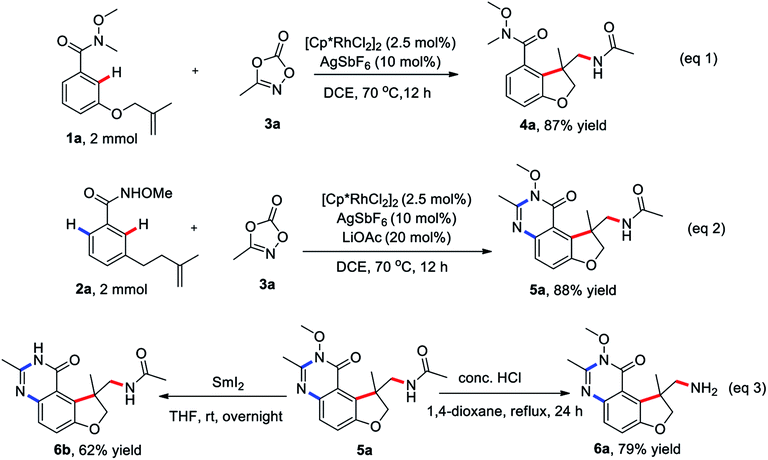
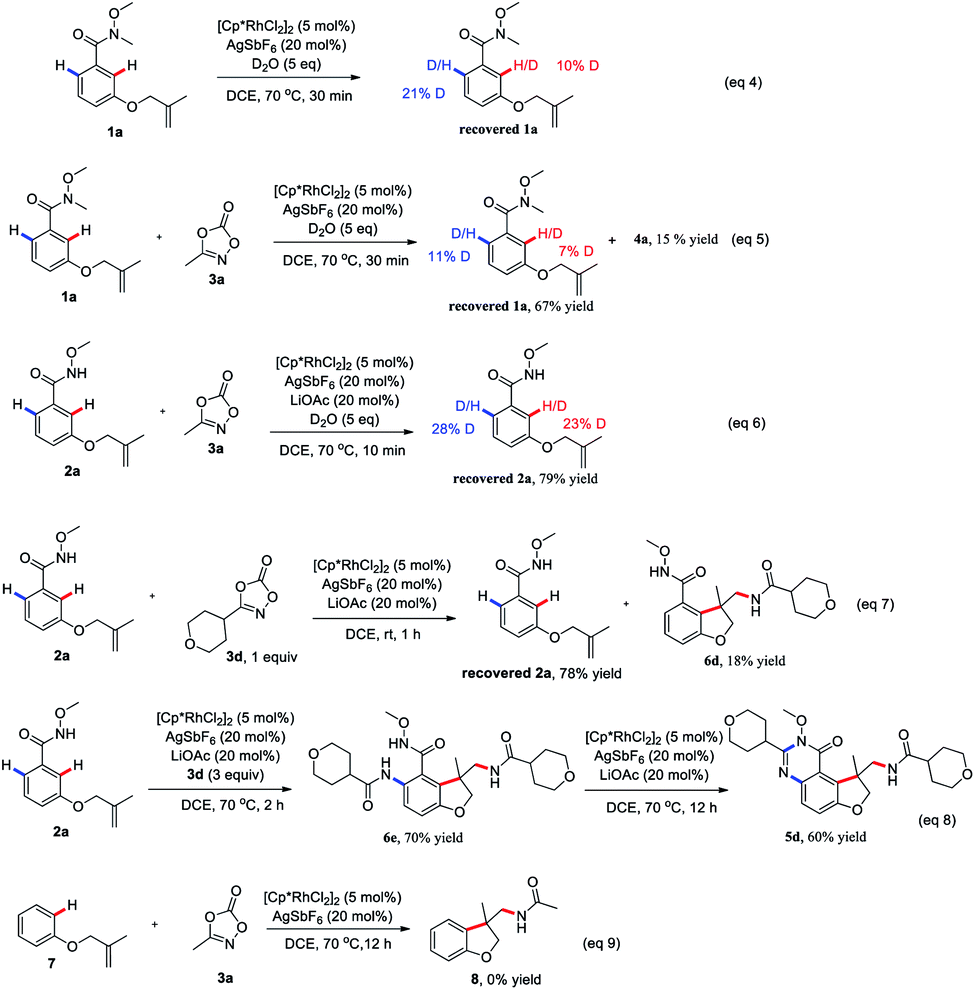
To gain insight into the reaction mechanism, hydrogen/deuterium (H/D) exchange were carried out. A H/D exchange at the ortho-position of the amide group in the re-isolated 1a and 2a was observed in the absence or presence of 3a, indicative of the reversibility of the ortho C–H activation (eqn (4)–(6)). Treatment of 2a with 1 equivalent of 3d at room temperature for 1 h delivered the 6d as the sole product, indicating that the intramolecular tandem annulative 1,2-arylation/amidation of alkenes is faster than ortho C–H amidation/annulation (eqn (7)). In addition, the use of 3 equivalent of 3d at 70 °C for 2 h provided 6e as the main product and subsequent treatment of 6e under the standard conditions gave 5d in 60% yield (eqn (8)), indicating that the second ortho C–H amidation occurs first, followed by an intramolecular dehydration to give the desired quinazolinone product. Finally, treatment of substrate 7 with 3a under the standard reaction conditions did not give any product 8, ruling out the possibility of the insertion of a nitrene to double bond (eqn (9)).
Based on above-mentioned experimental results, a plausible reaction pathway is proposed in Scheme 4. [Cp*RhCl2]2 precursor reacts with AgSbF6 to form an active cationic Rh(III) species, which undergoes a C–H bond activation to form cyclometalated complex Int-A. Coordination of the tethered olefin and a subsequent migratory insertion affords the intermediate Int-B, which undergoes an oxidative addition into the N–O bond of 3a, followed by a CO2 extrusion, to provide the Rh(V) nitrenoid species Int-C. Reductive elimination occurs to deliver the intermediate Int-D which then is protonated to release product 4a or Int-E and regenerate the catalyst. Int-E can undergo a second ortho C–H activation to give Int-F, which can be oxidized by 3a again to afford the Rh(V) nitrenoid species Int-G, with a CO2 extrusion. Subsequent reductive elimination and protonation give the Int-I which undergoes an intramolecular dehydration to deliver the product 5a.
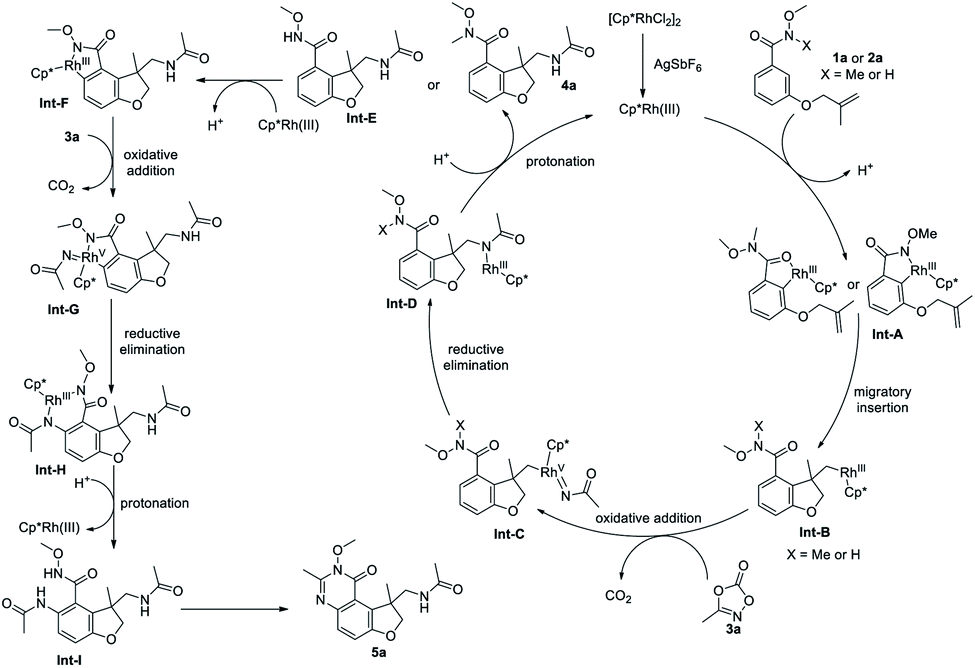 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed an unprecedented rhodium-catalyzed tandem annulative arylation/amidation reaction of aromatic tethered alkenes by using 3-substituted 1,4,2-dioxazol-5-ones as an amidating reagent. This robust transformation proceeds with a broad functional group tolerance under relatively mild and redox-neutral reaction conditions, releasing CO2 as the single byproduct. A wide variety of 2,3-dihydro-3-benzofuranmethanamine derivatives bearing an all-carbon quaternary stereo center can be accessed with high yields. Notably, by simply changing the directing group, a second, unsymmetrical ortho C–H amidation/annulation can be achieved to provide tricyclic dihydrofuro[3,2-f]quinazolinone derivatives in good yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by National Natural Science Foundation of China (No. 21702218, 81973166, 91753207, 81773568), the National major science and technology project “major new drug creation” (2018ZX09711002-006-001, 2018ZX09711002-008-005), Youth Innovation Promotion Association (2017333), Science and Technology Commission of Shanghai Municipality (18431907100) and K. C. Wong Education Foundation.References
- Selected reviews on transition metal-catalyzed C–H bond funtionalization: (a) P. Gandeepan, T. Mgller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS; (b) R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS; (d) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS; (e) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS; (f) D.-S. Kim, W.-J. Park and C.-Ho. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS; (g) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (h) L. C. M. Castro and N. Chatani, Chem. Lett., 2015, 44, 410–421 CrossRef CAS; (i) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS; (j) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS; (k) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- Selected examples: (a) R. K. Thalji, K. A. Ahrendt, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 9692–9693 CrossRef CAS; (b) T. A. Davis, T. K. Hyster and T. Rovis, Angew. Chem., Int. Ed., 2013, 52, 14181–14185 CrossRef CAS; (c) T. A. Davis, C. Wang and T. Rovis, Synlett, 2015, 26, 1520–1524 CrossRef CAS; (d) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507–511 CrossRef CAS; (e) Z. Shi, M. Boultadakis-Arapinis, D. C. Koester and F. Glorius, Chem. Commun., 2014, 50, 2650–2652 RSC; (f) K. Ghosh, R. K. Rit, E. Ramesh and A. K. Sahoo, Angew. Chem., Int. Ed., 2016, 55, 7821–7825 CrossRef CAS; (g) Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 8574–8578 CrossRef CAS; (h) D. F. Fernández, M. Gulías, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2017, 56, 9541–9545 CrossRef; (i) Z. Guan, S. Chen, Y. Huang and H. Yao, Org. Lett., 2019, 21, 3959–3962 CrossRef CAS; (j) K. Ghosh, M. Shankar, R. K. Rit, G. Dubey, P. V. Bharatam and A. K. Sahoo, J. Org. Chem., 2018, 83, 9667–9681 CrossRef CAS; (k) K. Mukherjee, E. Ramesh, K. Ghosh and A. K. Sahoo, Asian J. Org. Chem., 2018, 7, 1380–1384 CrossRef CAS; (l) K. Ghosh, A. Ghosh, K. Mukherjee, R. K. Rit and A. K. Sahoo, J. Org. Chem., 2020, 85, 8618–8626 CrossRef CAS.
- (a) M. N. Attala and P. Diaz, PCT, WO /012221 A1, 2009; (b) A. Stoit, W. I. Iwema Barker, H. K. A. C. Coolen, M. J. P. Van Dongen and N. J.-L. D. Leflemme, PCT, WO /004378 A1, 2012; (c) S. Chowdhury, J. Fu, R. Kamboj, S. Liu, Q. Jia, V. Raina and J. Sun, PCT, WO/046087 A2, 2008.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS; (b) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS; (c) B. M. Dancy and P. A. Cole, Chem. Rev., 2015, 115, 2419–2452 CrossRef CAS.
- Selected examples: (a) J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110–9113 CrossRef CAS; (b) J. Ryu, K. Shin, S. H. Park, J. Y. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 9904–9908 CrossRef CAS; (c) S. Yu, B. Wan and X. Li, Org. Lett., 2013, 15, 3706–3709 CrossRef CAS; (d) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2013, 15, 3014–3017 CrossRef CAS; (e) D.-G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS; (f) H. Zhao, Y. Shang and W. Su, Org. Lett., 2013, 15, 5106–5109 CrossRef CAS; (g) K.-H. Ng, Z. Zhou and W.-Y. Yu, Chem. Commun., 2013, 49, 7031–7033 RSC; (h) B. Zhou, J. Du, Y. Yang, H. Feng and Y. Li, Org. Lett., 2013, 15, 6302–6305 CrossRef CAS; (i) J. Du, Y. Yang, H. Feng, Y. Li and B. Zhou, Chem.–Eur. J., 2014, 20, 5727–5731 CrossRef CAS; (j) B. Zhou, J. Du, Y. Yang, H. Feng and Y. Li, Org. Lett., 2014, 16, 592–595 CrossRef CAS; (k) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492–2502 CrossRef CAS; (l) H. Kim, K. Shin and S. Chang, J. Am. Chem. Soc., 2014, 136, 5904–5907 CrossRef CAS; (m) C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 1597–1600 CrossRef CAS; (n) G. B. Boursalian, M.-Y. Ngai, K. N. Hojczyk and T. Ritter, J. Am. Chem. Soc., 2013, 135, 13278–13281 CrossRef CAS; (o) D. Zhu, G. Yang, J. He, L. Chu, G. Chen, W. Gong, K. Chen, M. D. Eastgate and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 2497–2500 CrossRef CAS.
- For selected reviews: (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS; (b) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS; (c) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC.
- For examples: (a) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS; (b) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7–10 CrossRef CAS; (c) T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141–4144 CrossRef CAS; (d) X. Huang, Y. Wang, J. Lan and J. You, Angew. Chem., Int. Ed., 2015, 54, 9404–9408 CrossRef CAS; (e) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707–4713 CrossRef CAS; (f) Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588–13592 CrossRef CAS; (g) Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496–3499 CrossRef CAS; (h) X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706–3710 CrossRef CAS; (i) S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. White, Nat. Chem., 2015, 7, 987–994 CrossRef CAS; (j) J. He, T. Shigenari and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 6545–6549 CrossRef CAS.
- (a) Y. Wu, Z. Chen, Y. Yang, W. Zhu and B. Zhou, J. Am. Chem. Soc., 2018, 140, 42–45 CrossRef CAS; (b) B. Zhou, Z. Chen, Y. Yang, W. Ai, H. Tang, Y. Wu, W. Zhu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 12121–12126 CrossRef CAS; (c) Y. Yang, X. Wang, Y. Li and B. Zhou, Angew. Chem., Int. Ed., 2015, 54, 15400–15404 CrossRef CAS.
- (a) Y. Park, K. T. Park, J. G. Kim and S. Chang, J. Am. Chem. Soc., 2015, 137, 4534–4542 CrossRef CAS; (b) H. Wang, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 13049–13052 CrossRef CAS; (c) S. Y. Hong, Y. Park, Y. Hwang, Y. B. Kim, M.-H. Baik and S. Chang, Science, 2018, 359, 1016–1021 CrossRef CAS; (d) H. Lei and T. Rovis, J. Am. Chem. Soc., 2019, 141, 2268–2273 CrossRef CAS; (e) T. Knecht, S. Mondal, J.-H. Ye, M. Das and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 7117–7121 CrossRef CAS; (f) J. S. Burman, R. J. Harris, C. M. B. Farr, J. Bacsa and S. B. Blakey, ACS Catal., 2019, 9, 5474–5479 CrossRef CAS.
- For reviews on Rh(III)-catalyzed C–H activations, see: (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS; (b) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212–11222 CrossRef CAS; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (d) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS; (e) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31–41 CAS; (f) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS; (g) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS; (h) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS; (i) S. Vasquez-Cespedes, X. Wang and F. Glorius, ACS Catal., 2018, 8, 242–257 CrossRef CAS; (j) T. Piou and T. Rovis, Acc. Chem. Res., 2018, 51, 170–180 CrossRef CAS.
- For selected examples of non-annulative carboamination reactions, see: (a) T. Piou and T. Rovis, Nature, 2015, 527, 86–90 CrossRef CAS; (b) A. Lerchen, T. Knecht, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 15166–15170 CrossRef CAS; (c) J. E. Ney and J. P. Wolfe, Angew. Chem., Int. Ed., 2004, 43, 3605–3608 CrossRef; (d) R. Lira and J. P. Wolfe, J. Am. Chem. Soc., 2004, 126, 13906–13907 CrossRef CAS; (e) D. N. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157–12159 CrossRef CAS; (f) D. R. White, J. T. Hutt and J. P. Wolfe, J. Am. Chem. Soc., 2015, 137, 11246–11249 CrossRef CAS; (g) W. Zeng and S. R. Chemler, J. Am. Chem. Soc., 2007, 129, 12948–12949 CrossRef CAS; (h) C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 9488–9489 CrossRef CAS; (i) G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474–1475 CrossRef CAS; (j) T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020–2023 CrossRef CAS; (k) A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224–7230 CrossRef CAS; (l) T. Pinkert, T. Wegner, S. Mondal and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 15041–15045 CrossRef CAS; (m) S. Maity, T. J. Potter and J. A. Ellman, Nat. Catal., 2019, 2, 756–762 CrossRef CAS; (n) D. Zhao, S. Vasquez-Cespedes and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 1657–1661 CrossRef CAS; (o) T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2015893 and 2014245. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04007j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |