
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhenylene-bridged bis(benzimidazolium) (BBIm2+): a dicationic organic photoredox catalyst†
Takuya
Kodama
 a,
Maiko
Kubo‡
a,
Wataru
Shinji‡
a,
Kei
Ohkubo
*bc and
Mamoru
Tobisu
*a
a,
Maiko
Kubo‡
a,
Wataru
Shinji‡
a,
Kei
Ohkubo
*bc and
Mamoru
Tobisu
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: tobisu@chem.eng.osaka-u.ac.jp
bInstitute for Advanced Co-Creation Studies, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: ohkubo@irdd.osaka-u.ac.jp
cInstitute for Open and Transdisciplinary Research Initiatives, Osaka University, Suita, Osaka 565-0871, Japan
First published on 7th October 2020
Abstract
A dicationic photoredox catalyst composed of phenylene-bridged bis(benzimidazolium) (BBIm2+) was designed, synthesised and demonstrated to promote the photochemical decarboxylative hydroxylation and dimerisation of carboxylic acids. The catalytic activity of BBIm2+ was higher than that for a monocation analogue, suggesting that the dicationic nature of BBIm2+ plays a key role in these decarboxylative reactions. The rate constant for the decay of the triplet–triplet absorption of the excited BBIm2+ increased with increasing concentration of the carboxylate anion with a saturated dependence, suggesting that photoinduced electron transfer occurs within the ion pair complex composed of the triplet excited state of BBIm2+ and a carboxylate anion.
Introduction
Given the widespread availability of carboxylic acids from nature and commercial sources, the decarboxylative transformation of carboxylic acid derivatives represents an attractive synthetic organic method.1 However, the direct decarboxylation of unactivated carboxylic acids remains a challenge, and the use of stoichiometric amounts of metal oxidants,2 or pre-activation to acid chlorides or activated esters, such as Barton esters3 and N-(acyloxy)phthalimides (NHPI esters),4 are still required. Although organic electron acceptors such as acridinium,5 iminium,6 phthalimide,7 and cyanoarene8 have been reported to promote the decarboxylation of carboxylic acids under photoirradiation conditions, a stoichiometric amount of these acceptors is needed for the reaction to reach completion. The use of transition metal catalysts and photoredox catalysts for the catalytic decarboxylative transformation of carboxylic acids has extensively been studied (Scheme 1). In transition metal catalysed reactions, the metal carboxylates are generated in situ and then undergo decarboxylation to produce an aryl-metal species (Scheme 1a).1a,c–e However, the substrates are limited to the highly activated aromatic carboxylic acids bearing electron-withdrawing groups, such as nitro or fluorine group(s), at the ortho position, except for the Ni/PPh3-catalysed decarboxylative olefination of alkyl carboxylic acids.9 The other catalytic method involves the use of photoredox catalysis,1b,e,10 in which the photoinduced single electron oxidation of an alkyl carboxylate anion with a photoexcited catalyst occurs, with the formation of an alkyl carboxyl radical (Scheme 1b). The alkyl carboxyl radical then spontaneously undergoes decarboxylation to produce an alkyl radical, which can participate in subsequent reactions. However, the application of this protocol to simple carboxylic acids is still difficult due to their highly positive oxidation potentials (e.g., CH3COO−: E1/2(CH3COO−/CH3COO˙) = +1.47 V vs. SCE),11 when commonly used ruthenium polypyridyl complexes are employed as a photoredox catalyst (e.g., Ru(bpy)32+; bpy = 2,2′-bipyridyl: E1/2 (*RuII/RuI) = +0.77 V vs. SCE).12 The use of iridium complexes containing pyridine-based ligands bearing electron-withdrawing groups has recently emerged as viable photocatalysts for promoting the oxidative decarboxylation of simple aliphatic carboxylic acids.13,14 However, these transition-metal complexes are expensive and potentially toxic.15 In the light of the above situation, the development of a simple and robust catalyst system that enables oxidation of unactivated carboxylic acids without the use of harmful reagents would be highly desirable. Organic photoredox catalysts have the potential to address these issues.16–18 An organic acridinium dye could be used in photocatalytic decarboxylative reactions of alkyl carboxylic acids including hydrodecarboxylation and decarboxylative trifluoromethylthiolation.16,17 Yoshimi and co-workers reported on decarboxylative transformations of a wide variety of alkyl carboxylic acids using an arene/electron acceptor system by photoinduced electron transfer.18,19 Although these examples demonstrate the potential utility of organic photoredox catalysts for use in oxidative decarboxylative transformation of unactivated organic acids, new classes of organic catalysts20 continue to be developed to further expand the scope of these reactions. | ||
| Scheme 1 Direct catalytic transformation of carboxylic acids. | ||
We envisioned that dicationic organic salts could function as photoredox catalysts with high oxidation ability because their electron deficient nature allows the HOMO energy level to be lowered, thereby making the reduction potential in the excited state to be sufficiently high to allow carboxylates to be oxidised. To date, organic dicationic molecules have not been applied to photoredox reactions, except for 2,7-diazapyrenium,21 trisaminocyclopropenium22 and bisphosphonium.23 Herein, we report on the design, synthesis and properties of a new dicationic organic photocatalyst based on a bis(benzimidazolium) (BBIm2+) framework, and its application to photocatalytic decarboxylative reactions is also demonstrated.
Results and discussion
As cationic moieties, we focused on imidazolium scaffolds, which have found various applications in modern synthetic and material chemistry, for example, as precursors of N-heterocyclic carbenes24 and components of ionic liquids.25 Thermal stability,26 redox properties,27 and ease of structural modification would be desirable properties of such a catalyst. We designed a new phenylene-bridged bis(benzimidazolium) (BBIm2+), with the expectation that two cationic imidazolium moieties should result in sufficiently low HOMO energies that would permit unactivated carboxylic acids to be oxidised upon photoexcitation (Scheme 2a). Density functional theory calculations at the ωB97XD/6-31G(d,p) level of theory were performed to investigate the structural and electronic properties of the BBIm2+. Structural optimisation revealed that the benzimidazolium rings of BBIm2+ are twisted relative to the central phenyl ring by 47° with a C2h symmetry. The electrostatic potential indicated that the majority of the positive charges in BBIm2+ are distributed over the imidazolium rings (Scheme 2c). BBIm2+ possesses a lower HOMO (ψ161 = −13.48 eV) than that of monocation analogue 1+ (ψ91 = −11.60 eV, Scheme 2d, cf. Fig. S5 in the ESI† for details), possibly because of the more positive nature of the molecule. These results indicate that BBIm2+ would be reduced more easily than 1+, thereby serving as a stronger oxidant than 1+ as we initially envisioned. Indeed, the calculated HOMO energy of BBIm2+ was lower than that of 9-mesityl-10-methylacridinium (Acr-Mes+, ψ83 = −11.24 eV, cf. Fig. S5 in the ESI†), which can oxidize alkyl carboxylic acids in the photo excited state.16,17 The HOMO of BBIm2+ is distributed across the benzimidazolium and central phenylene rings, whereas the LUMO is largely located at the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N moiety of the benzimidazolium ring and the phenylene moiety.
C–N moiety of the benzimidazolium ring and the phenylene moiety.
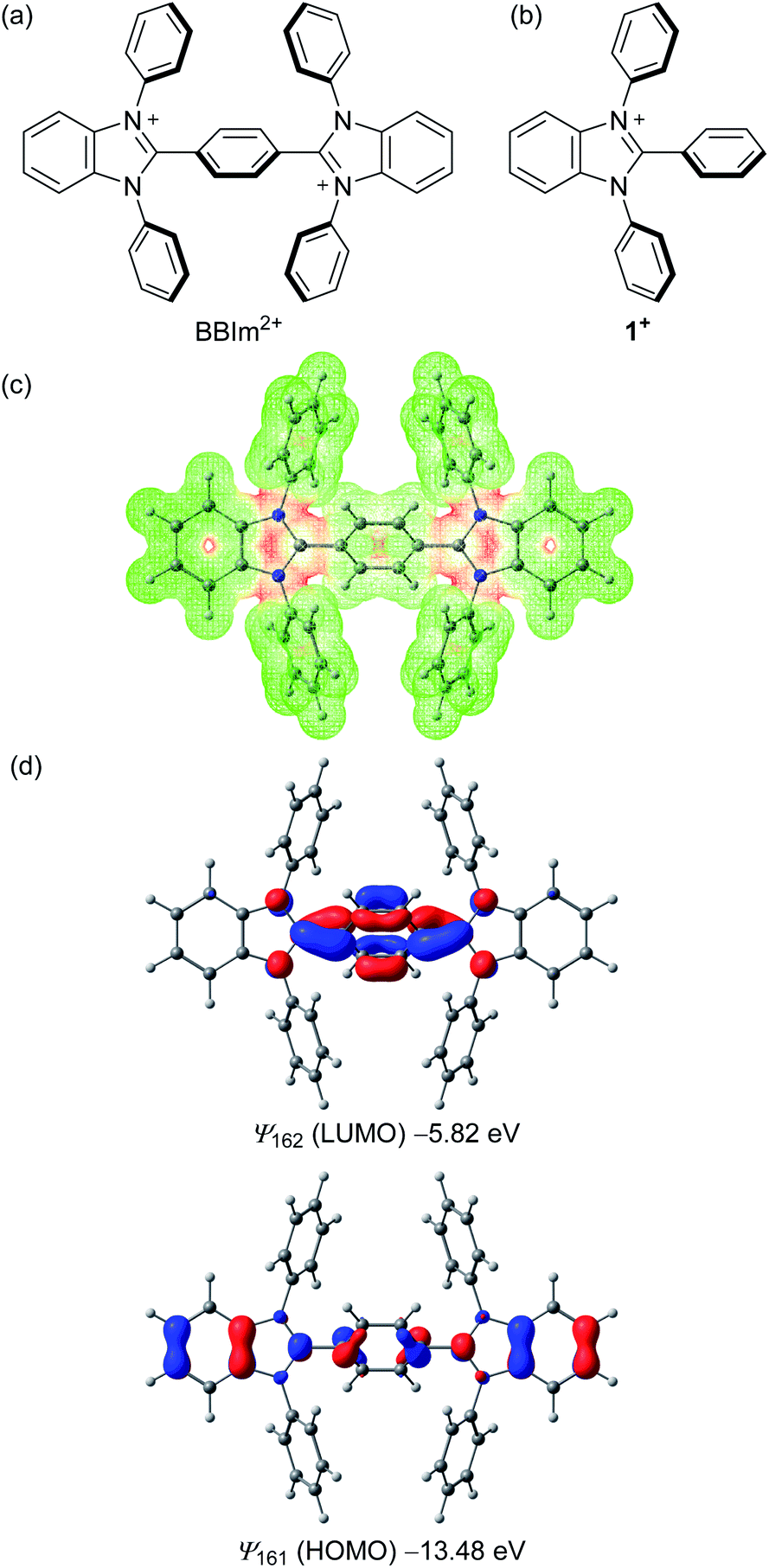 | ||
| Scheme 2 Molecular structures of (a) BBIm2+ and (b) monocation 1+; (c) SCF potential (red positive and green negative) and (d) frontier orbitals (HOMO, LUMO) of BBIm2+ generated by DFT calculations at the ωB97XD/6-31G(d,p) level of theory along with their energy levels. | ||
Having estimated theoretically that BBIm2+ has a sufficient oxidation ability to oxidise carboxylates by photoinduced electron transfer, we commenced our synthetic studies of BBIm2+. BBIm2+ was synthesised by a commonly used condensation reaction between a diamine and an aldehyde (Scheme 3).28 Thus, the treatment of diamine 2 with terephthalaldehyde and a catalytic amount of p-toluenesulfonic acid afforded the double cyclised product 3 in 83% yield. Subsequent oxidation of 3 with N-bromoacetamide provided BBIm2+·2[Br−]. Because of the poor solubility of BBIm2+·2[Br−] in common organic solvents, the counter anion was changed to PF6− by treatment with AgPF6 in acetonitrile to give BBIm2+·2[PF6−] in 89% yield (2 steps from 3), which is soluble in acetonitrile, DMF, and hexafluoroisopropanol.
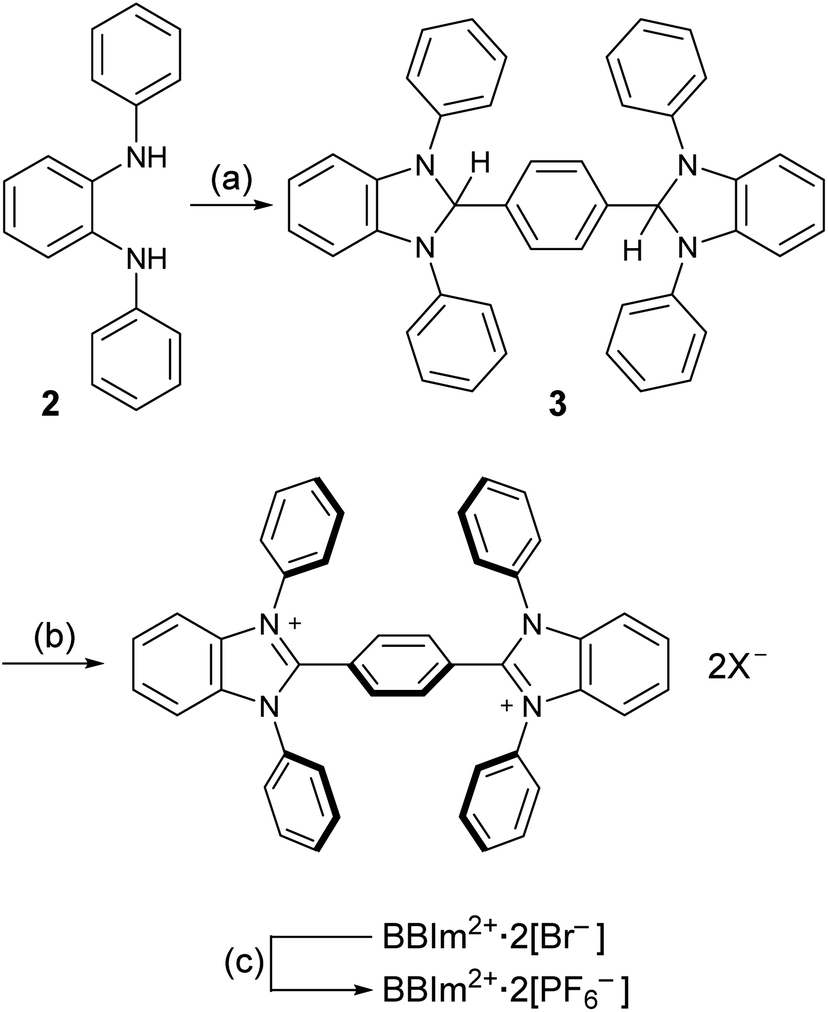 | ||
| Scheme 3 Synthesis of BBIm2+·2[PF6−]. aReaction conditions: (a) terephthalaldehyde, p-TSA, MgSO4, toluene, RT, 2 h, 83%. (b) N-Bromoacetamide, DMF, toluene, RT, 1 h. (c) AgPF6, MeCN, RT, 1 h, 89% (2 steps). p-TSA = p-toluenesulfonic acid, DMF = N,N-dimethylformamide, RT = room temperature. | ||
Recrystallization from DMSO/methanol gave colorless crystals of BBIm2+·2[PF6−], which were suitable for a single X-ray crystallographic analysis (Fig. 1a, see Fig. S1 in ESI†). The dihedral angle for the central phenyl ring and benzimidazolium moieties was found to be 59° (Fig. 1b), which was larger than the calculated value. This difference can be attributed to the existence of counter anions and/or a packing force. The counter anions were located close to the imidazolium moieties in the crystal, in good agreement with the sites predicted based on the calculated electrostatic potential (Scheme 1b).
 | ||
| Fig. 1 (a) ORTEP drawing of BBIm2+·2[PF6−] at the 50% probability level and (b) side view of BBIm2+·2[PF6−]. | ||
Absorption and emission spectra of BBIm2+·2[PF6−] (Fig. 2a) displayed an intense absorption band at around 290 nm with a weak broad tail in the longer wavelength region (∼500 nm), while a single emission peak with λmax at 428 nm was observed. A large Stokes shift (∼140 nm) of BBIm2+·2[PF6−] suggested that BBIm2+·2[PF6−] undergoes a significant structural change in the excited state. According to the time-dependent (TD) ωB97XD/6-31G(d,p) calculation of BBIm2+, the absorption band at around 290 nm of BBIm2+ is assigned to HOMO (ψ161) → LUMO (ψ162) transitions (λ = 288 nm and f = 0.99).
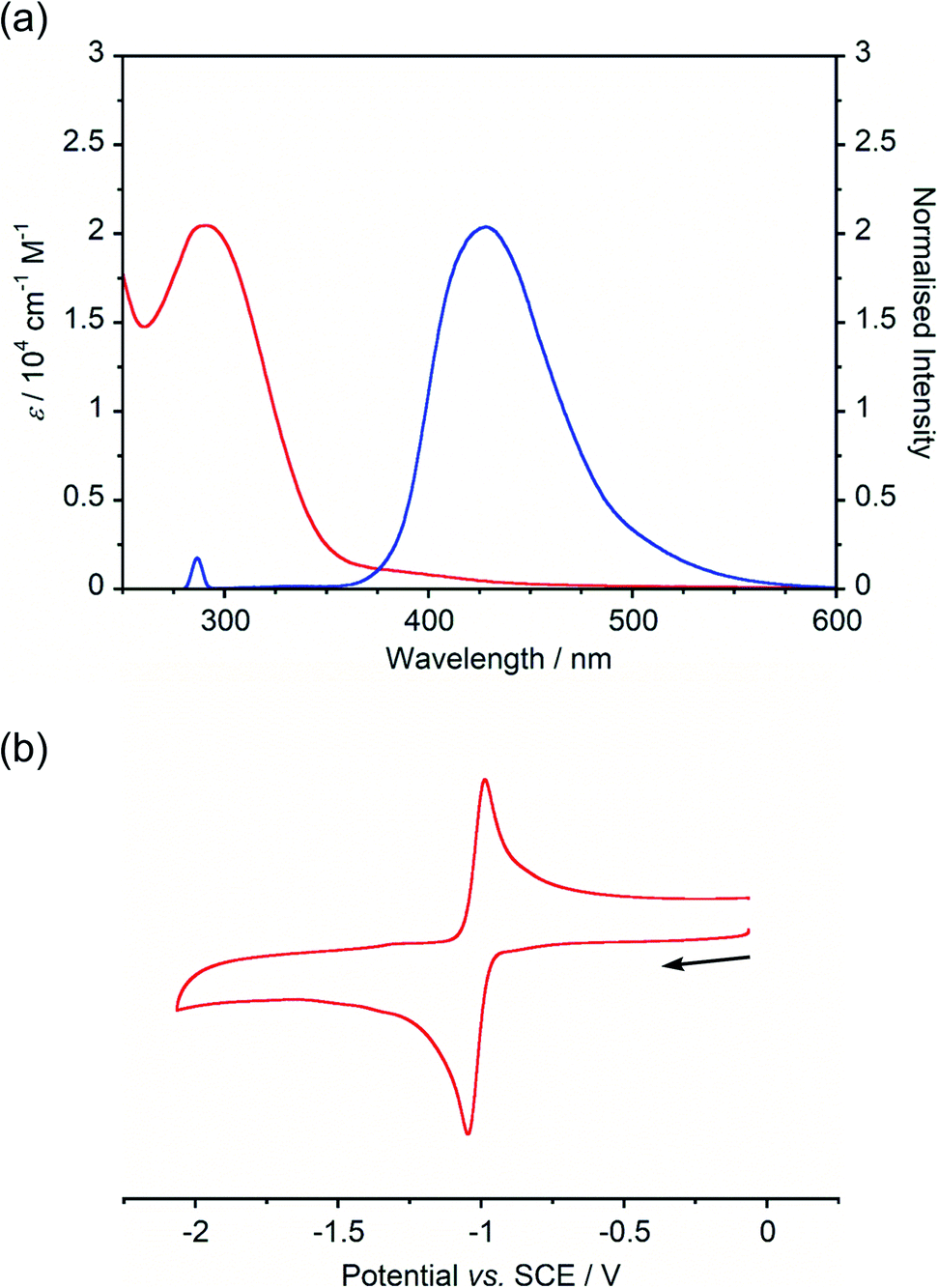 | ||
| Fig. 2 (a) UV-vis absorption (red) and emission (blue) (λex = 290 nm) spectra of BBIm2+·2[PF6−] (1.0 × 10−5 M in acetonitrile, room temperature); (b) cyclic voltammogram of BBIm2+·2[PF6−] (5.0 × 10−4 M in acetonitrile containing nBu4NPF6, room temperature, scan rate = 0.5 V s−1). | ||
The cyclic voltammogram (CV) of BBIm2+·2[PF6−] in acetonitrile exhibited a reversible redox wave (E1/2 = −1.02 V vs. SCE) (Fig. 2b).29 The corresponding monocation 1+·[Br−] displayed an irreversible wave (Ep/2 = −1.47 V vs. SCE, Fig. S6 in ESI†). Therefore, the advantage of using BBIm2+·2[PF6−] over the corresponding monocation 1+·[Br−] can be attributed to not only its higher reduction potential as we initially envisioned, but also to the stability of the reduced species. Optical and electrochemical measurements led us to estimate the excited singlet state reduction potential of BBIm2+·2[PF6−] to be 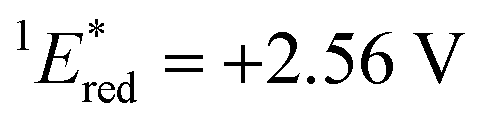 vs. SCE,30 the value of which is sufficiently high to permit a wide variety of organic compounds such as benzene31 or carboxylic acids to be oxidised.11 The triplet state reduction potential of BBIm2+·2[PF6−] was also estimated to be
vs. SCE,30 the value of which is sufficiently high to permit a wide variety of organic compounds such as benzene31 or carboxylic acids to be oxidised.11 The triplet state reduction potential of BBIm2+·2[PF6−] was also estimated to be  vs. SCE.30
vs. SCE.30
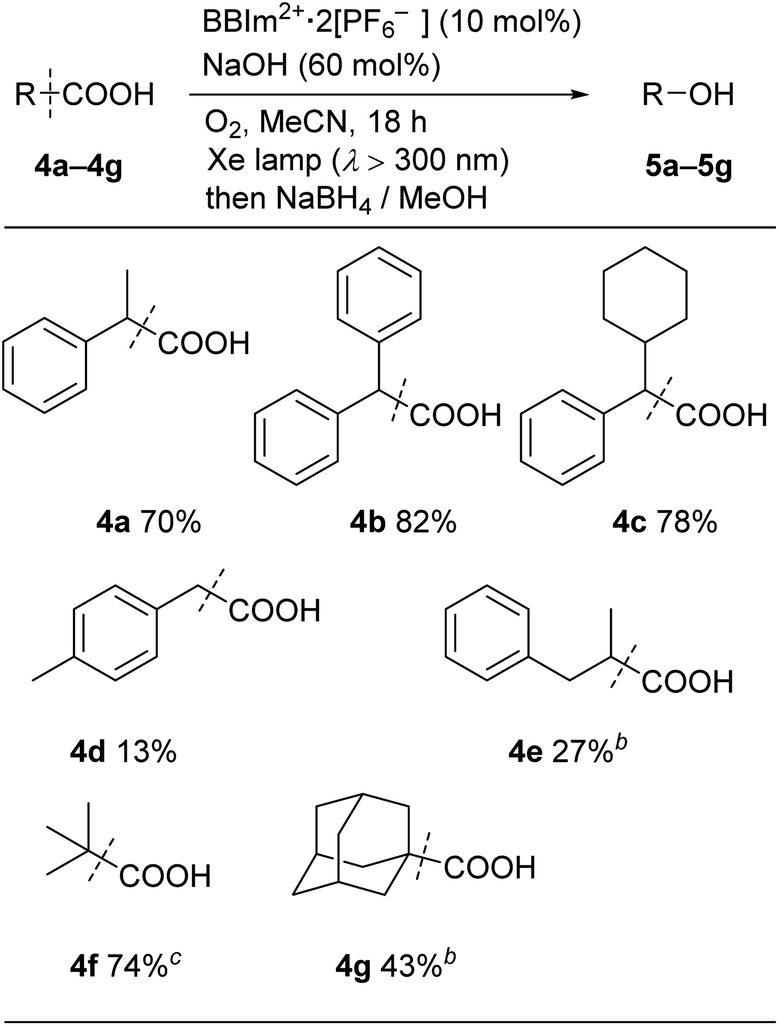
We next examined the catalytic activity of BBIm2+·2[PF6−] in the photocatalytic decarboxylative hydroxylation of carboxylic acids.16 Irradiation of an acetonitrile solution of carboxylic acid 4a with a xenon lamp (>300 nm) in the presence of BBIm2+·2[PF6−] (10 mol%) and NaOH (60 mol%) under an atmosphere of oxygen for 5 h afforded the desired alcohol 5a in 68% yield, after reductive work up with NaBH4 (Table 1, entry 1).16b Control experiments revealed that both BBIm2+·2[PF6−] and irradiation are essential for the reaction to efficiently proceed (entries 2 and 3). The reaction proceeded more efficiently under an atmosphere of oxygen compared with reactions performed under an atmosphere of nitrogen (entry 4). In the absence of NaOH, the yield was decreased to 32% (entry 5). The use of the corresponding monocation 1+·[Br−] (entry 6) or Acr-Mes+·[ClO4−]16a,32 (entry 7) as a photocatalyst also gave 5a, but in lower yields of 40% and 46%, respectively. In addition, the quantum yield for the photocatalytic hydroxylation reaction using BBIm2+·2[PF6−] was determined to be 0.60, which is significantly higher than that for 1+·[Br−] (0.004) or Acr-Mes+·[ClO4−] (0.23) catalysed reactions.33 The reaction also proceeded by using 365 nm or 405 nm LED lights instead of a xenon lamp (entries 8 and 9). It was possible to oxidise an array of carboxylic acids using BBIm2+ under the optimised conditions (Table 2). Benzylic carboxylic acids bearing methyl (4a), phenyl (4b) and cyclohexyl (4c) groups at the benzylic position were successfully decarboxylated to give the corresponding benzyl alcohols (5a–5c) in good yields. Whereas primary benzylic (4d) and secondary non-benzylic (4e) carboxylic acids were not good substrates in this decarboxylative hydroxylation, tertiary alkyl carboxylic acids 4f and 4g underwent the reaction to give products in moderate to good yield.
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | GC yields (%) |
| a Reaction conditions: (a) 4a (0.15 mmol), BBIm2+·2[PF6−] (0.015 mmol), NaOH (0.090 mmol), O2, in acetonitrile (6.0 mL), xenon lamp (500 W) irradiation for 5 h, RT; (b) NaBH4, MeOH. | ||
| 1 | None | 68 |
| 2 | Without BBIm2+·2[PF6−] | 0 |
| 3 | In the absence of light source | 0 |
| 4 | Under N2 | 8 |
| 5 | Without base | 32 |
| 6 | 1 +·[Br−] instead of BBIm2+·2[PF6−] | 40 |
| 7 | Acr-Mes+·[ClO4−] instead of BBIm2+·2[PF6−] | 46 |
| 8 | 365 nm LED lamp (60 W) instead of Xe lamp | 67 |
| 9 | 405 nm LED lamp (60 W) instead of Xe lamp | 35 |
|
||
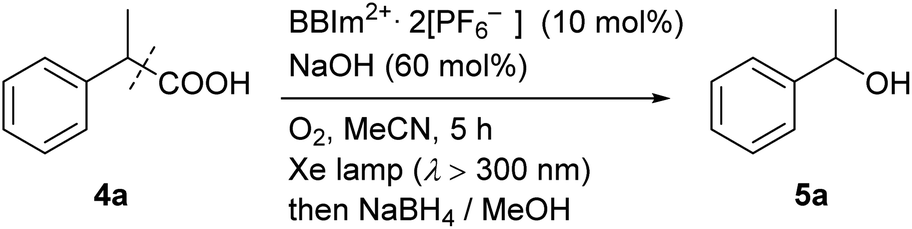
During the course of our investigation of photocatalytic decarboxylative hydroxylation reactions using BBIm2+·2[PF6−], we found that the tertiary benzylic carboxylic acid 6a underwent decarboxylative dimerisation to give 7a under identical conditions. Although this type of decarboxylative dimerisation reaction has already been reported, including Kolbe electrolysis34a and reactions using a stoichiometric amount of an oxidant, such as S2O8−,34b and Hg(II),34c and a heterogeneous a Pt–TiO2 photocatalyst,14b,c this is the first example of the use of an organic photoredox catalyst, to the best of our knowledge. Further optimization for this dimerisation was performed using 6a as a substrate (Table 3). The yield of 7a was increased when the reaction was carried out under an atmosphere of air rather than oxygen (entries 1 and 2). Finally, shortening the reaction time to 3 h resulted in a better yield, because the undesired decomposition of 7a could be suppressed (57%, entry 3).35 The use of the monocation 1+·[Br−] or Acr-Mes+·[ClO4−]16a,32 as a catalyst resulted in lower yields of 4% and 33%, respectively (entries 4 and 5). These results again underscore the prominent activity of this dicationic catalyst for the oxidation of unactivated organic acids.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Atmosphere | Time (h) | NMR yields (%) | |
| 7a | 6a | ||||
a Reaction conditions: 6a (0.15 mmol), cat. (0.015 mmol), NaOH (0.090 mmol), in acetonitrile/H2O (6.0 mL), xenon lamp (500 W) irradiation, RT.
b The reaction was conducted in CH3CN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1).
|
|||||
| 1b | BBIm2+·2[PF6−] | O2 | 18 | 23 | 0 |
| 2b | BBIm2+·2[PF6−] | Air | 18 | 47 | 1 |
| 3 | BBIm2+·2[PF6−] | Air | 3 | 57 | 9 |
| 4 | 1 +·[Br−] | Air | 3 | 4 | 0 |
| 5 | Acr-Mes+·[ClO4−] | Air | 3 | 33 | 0 |
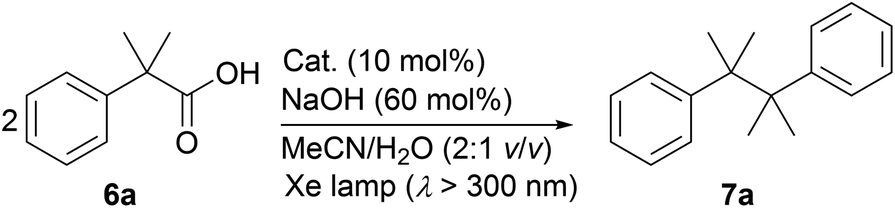
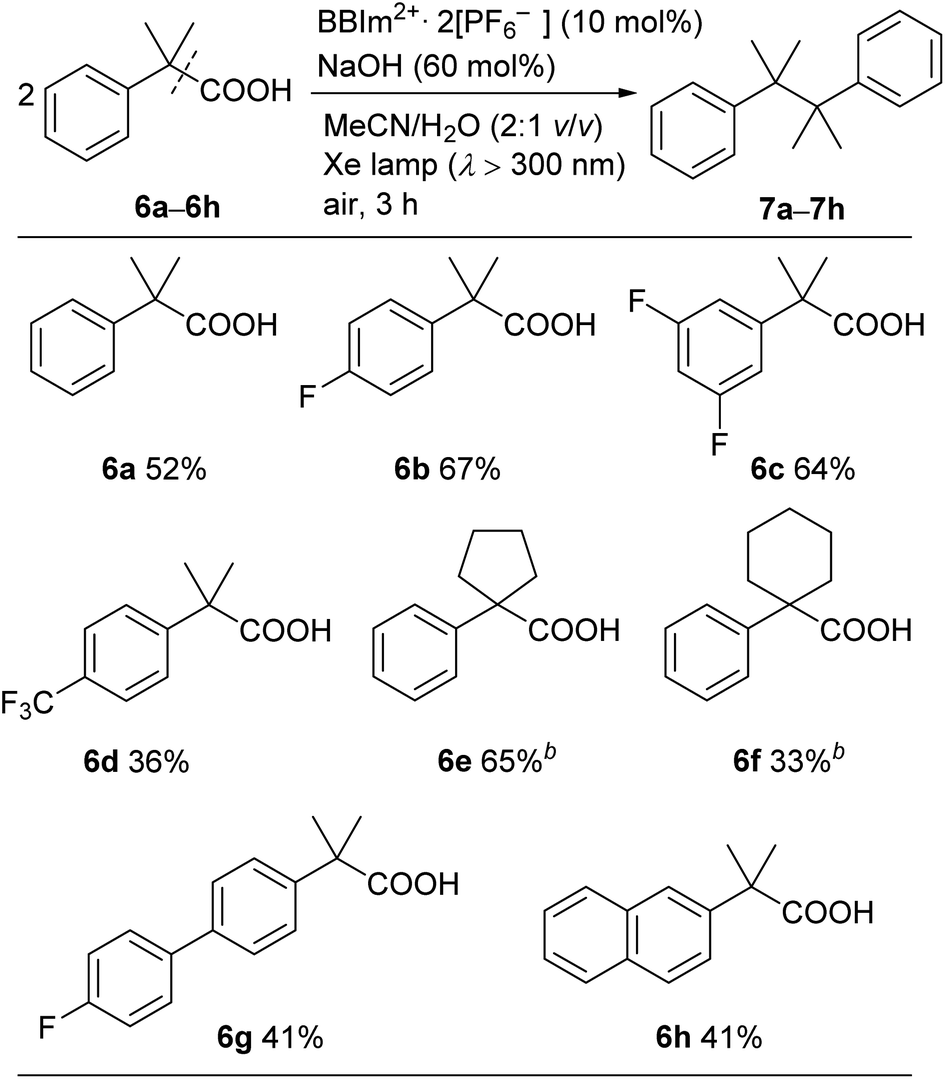
A range of tertiary benzylic carboxylic acids (6a–6h) could be oxidatively dimerised by BBIm2+·2[PF6−] under photoirradiation to be converted to the corresponding dimers (Table 4). Benzylic carboxylic acids bearing a fluoro group on the phenyl ring, such as 6b and 6c, effectively underwent dimerisation. The introduction of a trifluoromethyl group, as in 6d, decreased the yield of the dimerised product. Phenylacetic acids bearing a spiro ring system at the benzylic position, such as 6e and 6f, gave the corresponding dimers 7e and 7f. π-Extended derivatives, as represented by 6g and 6h, also served as good substrates for decarboxylative dimerisation. Aliphatic and secondary benzylic carboxylic acids failed to give any decarboxylative dimerisation products under these conditions, but alcohols, as in Table 2, were formed as products instead.
To elucidate the mechanism for this photoinduced decarboxylation reaction, quenching measurements36 were performed by fluorescence and triplet–triplet (T–T) absorption spectroscopies. Electron transfer from tBuCOO− to the singlet excited state of BBIm2+ (1BBIm2+*) is energetically favorable because the free energy change is significantly negative: ΔGet = Eox − Ered − 1E* = 1.02 − (−0.50) − 3.58 = −2.06 eV. However, no fluorescence quenching was observed when tBuCOO− was added to an acetonitrile solution containing BBIm2+. The fluorescence lifetime of BBIm2+ was determined to be 0.4 ns by time-resolved fluorescence measurements, which is too short to quench the low concentration of tBuCOO− (0–0.2 mM) under the present experimental conditions.37 It therefore appears that the reactive species may be the triplet excited state of BBIm2+ (3BBIm2+*) rather than the short-lived singlet excited state. The free energy change for the electron transfer from tBuCOO− to 3BBIm2+* was estimated to be ΔGet = Eox − Ered − 3E* = 1.02 − (−0.50) − 1.91 = −0.39 eV, where 3E* was determined from the phosphorescence spectrum (λmax = 641 nm) observed in an ethanol glass at 77 K. The negative ΔGet value indicates that the electron transfer from tBuCOO− to 3BBIm2+* is energetically feasible. To observe the electron transfer process between tBuCOO− and 3BBIm2+*, the T–T absorption measurements were carried out by nanosecond laser flash spectroscopy (Fig. 3a). A typical T–T absorption at λmax = 580 nm was detected taken at 0.8 ms after laser excitation at 355 nm due to the absorption of BBIm2+. The T–T absorption decays obeying first-order kinetics with the rate constant (kT) of 1.9 × 102 s−1. tBuCOO− was added to an acetonitrile solution of BBIm2+·2[PF6−] and the decay time profile was monitored at 580 nm. The observed decay rate constant (kobs) increased with increasing concentration of tBuCOO−, as shown in Fig. 3b. Interestingly, a saturated dependence was observed, rather than the typical linear relationship observed for a simple quenching mechanism. The saturated behavior revealed that photoinduced electron transfer occurred with the formation of an ion pair complex composed of the 3BBIm2+* and tBuCOO−, in competition with bimolecular electron transfer from tBuCOO− to 3BBIm2+* and triplet decay of 3BBIm2+* to the ground state without electron transfer (Scheme 4).38 The association constant (Kassoc) for the formation of an encounter complex 3BBIm2+*·[tBuCOO−][PF6−] and the rate constant (kET) for intra-complex photoinduced electron transfer from tBuCOO− to 3BBIm2+* were estimated to be 5.8 × 105 M−1 and 3.4 × 102 s−1, respectively, by curve fitting using eqn (1).
| kobs − kT = kETKassoc[tBuCOO−]/[1 + Kassoc[tBuCOO−]] | (1) |
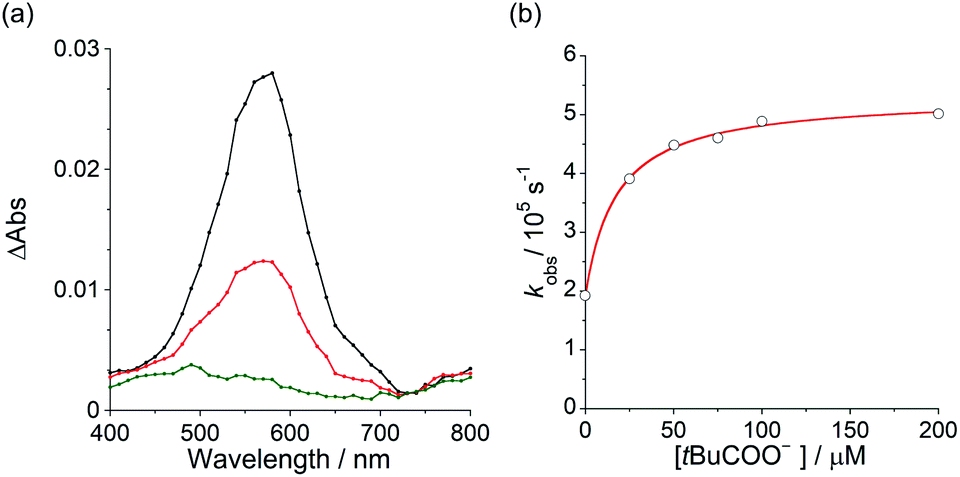 | ||
| Fig. 3 (a) Transient absorption spectra of BBIm2+ taken at 0.8 ms (black), 5 ms (red), 20 ms (green) after laser excitation at 355 nm in deaerated MeCN. (b) Dependence of decay rate constant of triplet–triplet absorption of 3BBIm2+*on the concentration of pivalate. | ||
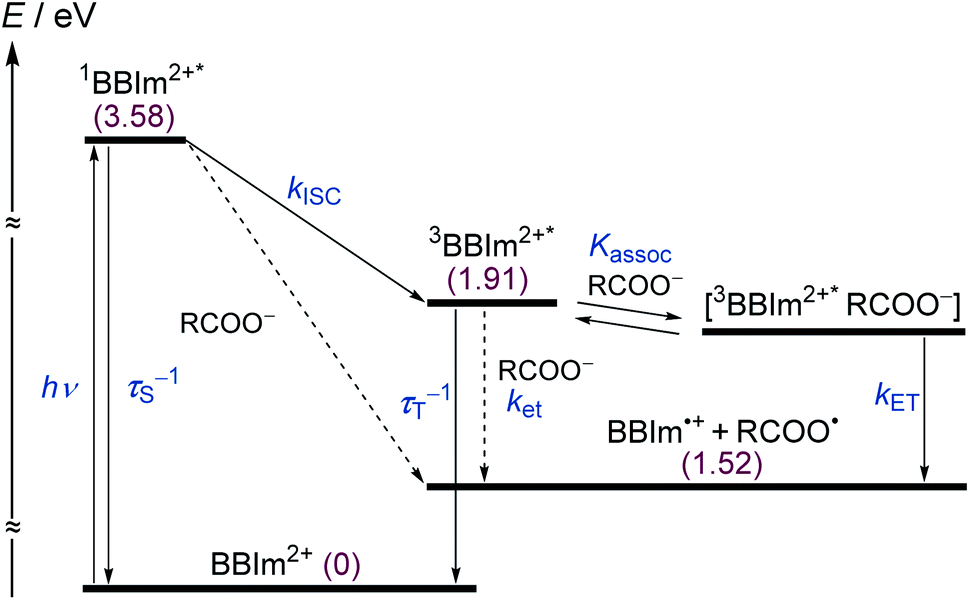 | ||
| Scheme 4 Energy diagram of BBIm2+. R = tBu, τS = fluorescence lifetime, τT = phosphorescence lifetime, kISC = rate constant for intersystem crossing, ket = rate constant for electron transfer from tBuCOO− to 3BBIm2+*, Kassoc = association constant for the formation of an encounter complex 3BBIm2+*·[tBuCOO−][PF6−], kET = rate constant for intra-complex photoinduced electron transfer from tBuCOO− to 3BBIm2+*. Numbers in parentheses refer to energy levels of the species relative to BBIm2+ in the ground state. | ||
The electrochemical and optical properties of BBIm2+·2[PF6−] are summarised in Table 5 and an energy diagram of BBIm2+ is shown in Scheme 4. The photoirradiation initially generates 1BBIm2+*, which is not quenched with tBuCOO− by electron transfer under the experimental conditions. Intersystem crossing, which is in competition with the fluorescence decay bound for the ground state, results in the formation of 3BBIm2+*. An intimate encounter complex between 3BBIm2+* and the anionic tBuCOO− substrate is formed, and electron transfer in the complex then occurs to produce a radical pair (BBIm˙+/tBuCOO˙). Based on the saturated behavior shown in Scheme 3b, an outer sphere electron transfer process from tBuCOO− to 3BBIm2+* is presumably significantly slower than the intra-complex electron transfer. No back electron transfer from BBIm˙+ to tBuCOO˙ occurs due to the rapid spontaneous decarboxylation of tBuCOO˙ leading to the products.
| E red/V | 1 E*/eV |
|
3 E*/eV |
|
λ max/nm | λ em/nm | τ S/ns |
|---|---|---|---|---|---|---|---|
a All potentials in V vs. SCE. Ered = ground state reduction potential, 1E* = singlet excitation energy, 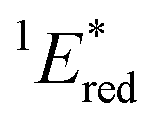 = excited singlet state reduction potential, 3E* = triplet excitation energy, = excited singlet state reduction potential, 3E* = triplet excitation energy, 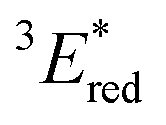 = excited triplet state reduction potential. λmax = absorption peak, λem = emission peak excited at 290 nm. τS = fluorescence lifetime. = excited triplet state reduction potential. λmax = absorption peak, λem = emission peak excited at 290 nm. τS = fluorescence lifetime.
|
|||||||
| −1.02 | +3.58 | +2.56 | +1.91 | +0.86 | 290 | 428 | 0.4 |
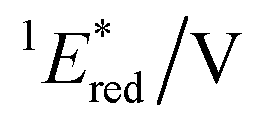
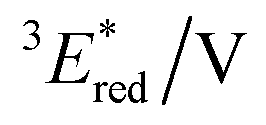
A plausible reaction mechanism for decarboxylative hydroxylation and dimerisation is shown in Scheme 5. Photoredox catalyst BBIm2+ is initially excited by photoirradiation and subsequent intersystem crossing generates 3BBIm2+*, which oxidises the carboxylate Avia single electron transfer (SET) to give the carboxyl radical B and the radical cation BBIm˙+. The carboxyl radical B rapidly releases CO2 to produce the alkyl radical C. Under an O2 atmosphere, the generated radical C is captured by oxygen39 to generate peroxyl radical D [Ered (tBuO2˙/tBuO2−) = +0.50 V vs. SCE],16a,38 which is subsequently reduced by the radical cation BBIm˙+ (Ered = −1.02 V vs. SCE) to form the peroxyl anion E with the regeneration of BBIm2+. Anion E is prone to undergo disproportionation to generate a mixture of an alcohol and a ketone, which was isolated as a single alcohol product by treatment with NaBH4 in this study. When the reaction is performed under an atmosphere of air, in which the oxygen concentration is relatively low, two alkyl radicals C undergo radical–radical coupling to form dimer G before it is captured by oxygen.40 In this case, SET from the radical cation BBIm˙+ to oxygen [Ered(O2/O2˙−) = −0.86 V vs. SCE]41 regenerates the catalyst BBIm2+ with the concomitant formation of superoxide (O2˙−), which is eventually reduced to hydrogen peroxide.33
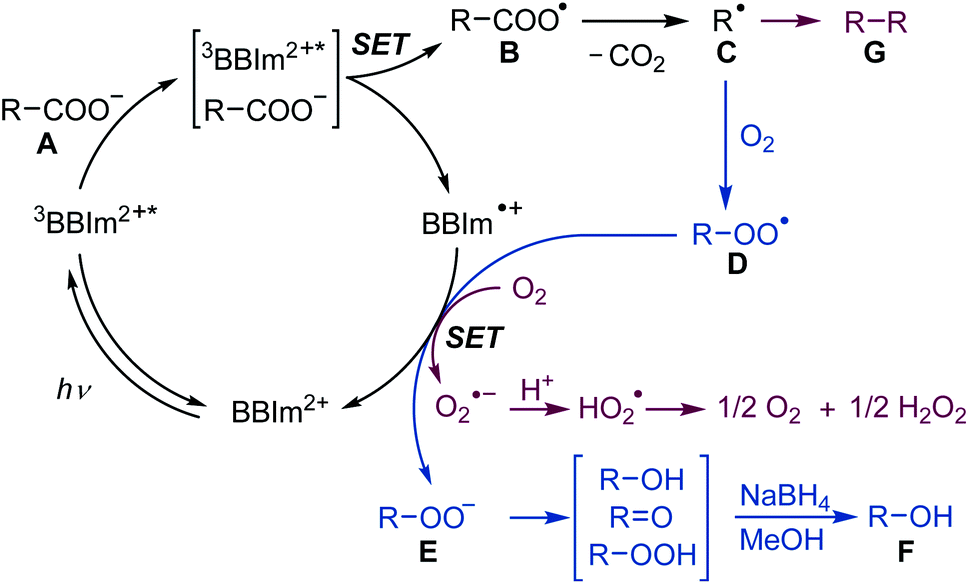 | ||
| Scheme 5 Proposed mechanism for the photocatalytic hydroxylation under O2 (blue) and dimerisation of carboxylic acids under an atmosphere of air (red). SET: single electron transfer. | ||
The key to the successful application of BBIm2+ in photocatalytic decarboxylative transformation of benzylic carboxylic acids is its dicationic nature, which serves to reduce the activation barrier of the SET from carboxylate by lowering the reorganization energy of the polar solvents.31b,c When an electronically neutral photocatalyst (PC) oxidises a monoanionic substrate (S−), a radical anion of PC (PC˙−) and a neutral radical (S˙) are generated (Scheme 6a). In this process, the charges of the both components changes (PC: neutral → negative; S: negative → neutral), which requires the solvent molecules around PC and S to be reorganised to form polarity-matched clusters. Similarly, a large solvent reorganization energy would be expected to be required when a monocationic PC is used (Scheme 6b, PC: positive → neutral; S: negative → neutral). In contrast, the reorganization energy becomes lower when a dicationic PC is used, since the sign of the charge of PC remains unchanged (Scheme 6c).31c
 | ||
| Scheme 6 Comparison of the reorganization of polar solvents during single electron transfer processes from monoanionic substrate (S−) to (a) neutral, (b) monocationic, and (c) dicationic photocatalysts (PCs). Arrows represent the dipole moments of the solvents. | ||
Conclusions
In conclusion, we report on the development of a novel dicationic photoredox catalyst BBIm2+, the photophysical and electrochemical properties of which indicate that it has a strong oxidation ability in the excited state. In fact, BBIm2+ was demonstrated to be a viable organic photocatalyst for promoting the decarboxylative hydroxylation and dimerisation of unactivated carboxylic acids. The striking feature of BBIm2+ is its dicationic nature, which allows it to form an intimate ion pair with an anionic substrate, resulting in an energetically favorable SET process. Further development of new cationic organic photocatalysts and their application to otherwise difficult oxidative transformations is now ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Scientific Research on Innovative Area “Hybrid Catalysis” (20H04818, 20H04819) and Grant-in-Aid for Early-Career Scientists (19K15564) from MEXT, Japan. MT wishes to thank the Hoansha Foundation for their financial support. We also thank Prof. Ikeda (Osaka Prefecture University) and Prof. Asahara (Osaka University) for helpful discussion and Prof. Minakata, Prof. Takeda, Prof. Kida, and Prof. Mori (Osaka University) for their assistance with the fluorescence lifetime measurements.Notes and references
- Selected reviews: (a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (b) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS; (c) H. Huang, K. Jia and Y. Chen, ACS Catal., 2016, 6, 4983–4988 CrossRef CAS; (d) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS; (e) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- (a) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS; (b) Y. N. Ogibin, L. K. Rakhmatullina and G. I. Nikishin, Russ. Chem. Bull., 1975, 24, 2608–2613 CrossRef; (c) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489–519 CrossRef CAS; (d) M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC; (e) J. Kan, S. Huang, J. Lin, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 2199–2203 CrossRef CAS; (f) R. Xia, M.-S. Xie, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2014, 16, 444–447 CrossRef CAS.
- (a) D. H. R. Barton, B. Lacher and S. Z. Zard, Tetrahedron, 1987, 43, 4321–4328 CrossRef CAS; (b) D. Crich and L. Quintero, Chem. Rev., 1989, 89, 1413–1432 CrossRef CAS; (c) D. H. R. Barton and M. Ramesh, Tetrahedron Lett., 1990, 31, 949–952 CrossRef CAS; (d) M. F. Saraiva, M. R. C. Couri, M. L. Hyaric and M. V. Almeida, Tetrahedron, 2009, 65, 3563–3572 CrossRef CAS; (e) X. Xu, P. Li, Y. Huang, C. Tong, Y. Yan and Y. Xie, Tetrahedron Lett., 2017, 58, 1742–1746 CrossRef CAS.
- (a) M. Dinda, C. Bose, T. Ghosh and S. Maity, RSC Adv., 2015, 5, 44928–44932 RSC; (b) I. B. Krylov, V. A. Vil' and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92–146 CrossRef; (c) Z. Guo, X. Jiang, C. Jin, J. Zhou, B. Sun and W. Su, Synlett, 2017, 28, 1321–1326 CrossRef CAS; (d) Y. Lv, K. Sun, W. Pu, S. Mao, G. Li, J. Niu, Q. Chen and T. Wang, RSC Adv., 2016, 6, 93486–93490 RSC; (e) X. Xu, P. Li, Y. Huang, C. Tong, Y. Yan and Y. Xie, Tetrahedron Lett., 2017, 58, 1742–1746 CrossRef CAS.
- (a) D. R. G. Brimage, R. S. Davidson and P. R. Steiner, J. Chem. Soc., Perkin Trans. 1, 1973, 3563–3572 Search PubMed; (b) S. Fukuzumi, T. Kitano and T. Tanaka, Chem. Lett., 1989, 18, 1231–1234 CrossRef.
- Y. Kurauchi, N. Nobuhara and K. Ohga, Bull. Chem. Soc. Jpn., 1986, 59, 897–905 CrossRef CAS.
- A. G. Griesbeck, N. Hoffmann and K.-D. Warzecha, Acc. Chem. Res., 2007, 40, 128–140 CrossRef CAS.
- (a) J. Libman, J. Am. Chem. Soc., 1975, 97, 4139–4141 CrossRef CAS; (b) K. Tsujimoto, N. Nakao and M. Ohashi, J. Chem. Soc., Chem. Commun., 1992, 366–367 RSC; (c) H. Yokoi, T. Nakano, W. Fujita, K. Ishiguro and Y. Sawai, J. Am. Chem. Soc., 1998, 120, 12453–12458 CrossRef CAS.
- A. John, M. A. Hillmyer and W. B. Tolman, Organometallics, 2017, 36, 506–509 CrossRef CAS.
- Selected reviews: (a) J. M. R. Narayanama and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (d) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (e) G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- (a) A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS; (b) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- Selected examples: (a) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS; (b) A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS; (c) S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 CrossRef CAS; (d) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 CrossRef CAS; (e) L. Candish, E. A. Standley, A. G.-S. S. Mukherjee and F. Glorius, Chem.–Eur. J., 2016, 22, 9971–9974 CrossRef CAS.
- Decarboxylation reactions of alkyl carboxylic acids using heterogeneous TiO2-based photocatalysts have been reported: (a) D. K. Ellison, P. C. Trulove and R. T. Iwamoto, Tetrahedron, 1986, 42, 6405–6410 CrossRef CAS; (b) D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580–13583 CrossRef CAS; (c) D. W. Manley and J. C. Walton, Org. Lett., 2014, 16, 5394–5397 CrossRef CAS.
- H. Mokbel, D. Anderson, R. Plenderleith, C. Dietlin, F. Morlet-Savary, F. Dumur, D. Gigmes, J.-P. Fouassier and J. Lalevée, Polym. Chem., 2017, 8, 5580–5592 RSC.
- (a) K. Suga, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2006, 110, 3860–3867 CrossRef CAS; (b) H.-T. Song, W. Ding, Q.-Q. Zhou, J. Liu, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2016, 81, 7250–7255 CrossRef CAS; related reactions using cerium salts (c) R. A. Sheldon and J. K. Kochi, J. Am. Chem. Soc., 1968, 90, 6688–6698 CrossRef CAS; (d) V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 RSC; (e) S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, J. Am. Chem. Soc., 2020, 142, 5668–5675 CrossRef CAS.
- (a) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS; (b) L. Candish, L. Pitzer, A. Gómez-Suarez and F. Glorius, Chem.–Eur. J., 2016, 22, 4753–4756 CrossRef CAS.
- Selected examples: (a) Y. Yoshimi, J. Photochem. Photobiol., A, 2017, 342, 116–130 CrossRef CAS; (b) Y. Yoshimi, T. Itou and M. Hatanaka, Chem. Commun., 2007, 5244–5246 RSC; (c) K. Nishikawa, Y. Yoshimi, K. Maeda, T. Morita, I. Takahashi, T. Itou, S. Inagaki and M. Hatanaka, J. Org. Chem., 2013, 78, 582–589 CrossRef CAS.
- The photocatalytic decarboxylative transformation of aromatic carboxylic acids was recently achieved: S. Kubosaki, H. Takeuchi, Y. Iwata, Y. Tanaka, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2020, 85, 5362–5369 CrossRef CAS.
- For recent reviews of the molecular design of organic photocatalyst, see (a) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS; (b) J. Mateos, F. Rigodanza, A. Vega-Peñaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyó, M. Bonchio and L. Dell'Amico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS; (c) A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006416.
- (a) J. Santamaria, R. Ouchabane and J. Rigaudy, Tetrahedron Lett., 1989, 30, 2927–2928 CrossRef CAS; (b) J. Santamaria, M. T. Kaddachi and J. Rigaudy, Tetrahedron Lett., 1990, 31, 4735–4738 CrossRef CAS.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS.
- H. Cheng, X. Wangb, L. Chang, Y. Chen, L. Chu and Z. Zuo, Sci. Bull., 2019, 64, 1896–1901 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS.
- S. Zhang, J. Zhang, Y. Zhang and Y. Deng, Chem. Rev., 2017, 117, 6755–6833 CrossRef CAS.
- M. Villanueva, A. Coronas, J. García and J. Salgado, Ind. Eng. Chem. Res., 2013, 52, 15718–15727 CrossRef CAS.
- D. Rottschäfer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef.
- H. Baier, A. Kelling, R. Jackstell and H.-J. Holdt, Z. Anorg. Allg. Chem., 2013, 639, 1731–1739 CrossRef CAS.
- Electric potential values were determined vs. Fc/Fc+ as a reference and converted to vs. SCE using Fc/Fc+ = +0.38 vs. SCE: N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS . As for the positive potential side of cyclic voltammogram for BBIm2+·2[PF6−], no significant peak was observed. See Fig. S6b in ESI.†.
-
1
E* = [1/(λabs[nm] × 8067 × 10−7) + 1/(λem[nm] × 8067 × 10−7)] × 1/2 = 3.58 eV. λabs = 290 nm, λem = 428 nm for BBIm2+.
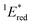 = 1E* + Ered = +3.58 + (−1.02) = +2.56 V vs. SCE.
= 1E* + Ered = +3.58 + (−1.02) = +2.56 V vs. SCE. 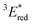 = 3E* + Ered = +1.91 + (−1.02) = +0.89 V vs. SCE, where 3E* was determined from the phosphorescence spectrum (λmax = 641 nm) observed in an ethanol glass at 77 K.
= 3E* + Ered = +1.91 + (−1.02) = +0.89 V vs. SCE, where 3E* was determined from the phosphorescence spectrum (λmax = 641 nm) observed in an ethanol glass at 77 K. - (a) P. B. Merkel, P. Luo, J. P. Dinnocenzo and S. Farid, J. Org. Chem., 2009, 74, 5163–5173 CrossRef CAS; (b) K. Ohkubo, T. Kobayashi and S. Fukuzumi, Opt. Express, 2012, 20, A360–A365 CrossRef CAS; (c) S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459–8467 CrossRef CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS.
- The quantum yields for the decarboxylative hydroxylation reaction of 4a with photocatalysts BBIm2+·2[PF6−], 1+·[Br−] and Acr-Mes+·[ClO4−] were determined using a 365 LED lamp (60 W) as the light source under the same absorbance condition [abs. at 365 nm = 1.0]. The quantum yield for the photocatalytic oxygenation of p-xylene with Acr-Mes+·[ClO4−] was used as a reference value: K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC . Yield/irradiation time profiles are shown in Fig. S4 and the quantum yields were summarized in Table S2 in ESI.†.
- (a) H. Kolbe, Ann. Chem. Pharm., 1848, 64, 339–341 CrossRef; (b) F. Minisci, E. Vismara, G. Morini, F. Fontana, S. Levi and M. Serravalle, J. Org. Chem., 1986, 51, 476–479 CrossRef CAS; (c) M. H. Habibi and S. Farhadi, Tetrahedron Lett., 1999, 40, 2821–2824 CrossRef CAS.
- A. Albini and S. Spreti, J. Chem. Soc., Perkin Trans. 2, 1987, 1175–1179 RSC.
- H. Boaz and G. K. Rollefson, J. Am. Chem. Soc., 1950, 72, 3435–3443 CrossRef CAS.
- Notes: (a) The intensity of the fluorescence of BBIm2+ apparently increased with increasing amount of added sodium pivalate, which is likely due to a significant structural change by complexation between BBIm2+ and pivalate. We were able to isolate BBIm2+·[tBuCOO−][PF6−], which was immediately formed by mixing BBIm2+·2[PF6−] and sodium pivalate in CH3CN; (b) The yield of photocatalytic decarboxylative dimerisation was not significantly affected by the concentration of BBIm2+, suggesting that the short-lived 1BBIm2+* is not an active species for the reaction; (c) In addition to the short life time, another possible reason for the absence of fluorescence quenching by tBuCOO− in spite of the highly negative driving force is that the process is located in the Marcus inverted region: R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- T. N. Das, T. Dhanasekaran, Z. B. Alfassi and P. Neta, J. Phys. Chem. A, 1998, 102, 280–284 CrossRef CAS.
- 18O labeling experiments were performed to confirm the origin of oxygen atom in 5a. When the photocatalytic hydroxylation reaction was conducted in 18O2-saturated acetonitrile using 4a as a substrate, [18O]5a was detected by HRMS (Fig. S3a in ESI†). Similarly, when the reaction was performed in non-labeled O2-saturated acetonitrile, only [16O]5a was detected and no [18O]5a was observed (Fig. S3b in ESI†). This result indicates that the oxygen in the 5a product is derived from O2.
- The use of water decreased the oxygen concentration in the system, which increased the yield of the dimerisation product. Indeed, when the reaction was conducted in acetonitrile using 6a as a substrate, the yield of the dimer decreased to 24% and hydroxylation proceeded in 41% yield. There is a difference in the solubility of oxygen between water (1.3 mM) and acetonitrile (13 mM), see (a) I. Nakanishi, S. Fukuzumi, T. Konishi, K. Ohkubo, M. Fujitsuka, O. Ito and N. Miyata, J. Phys. Chem. B, 2002, 106, 2372–2380 CrossRef CAS; (b) S. Fukuzumi, S. Fujita, T. Suenobu, H. Yamada, H. Imahori, Y. Araki and O. Ito, J. Phys. Chem. A, 2002, 106, 1241–1247 CrossRef CAS; (c) S. Fukuzumi, M. Ishikawa and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1989, 1037–1045 RSC.
- D. T. Sawyer, T. S. Calderwood, K. Yamaguchi and C. T. Angelis, Inorg. Chem., 1982, 22, 2577–2583 CrossRef.
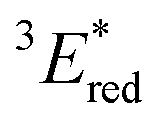
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1992124. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03958f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |