
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective active site placement in Lewis acid zeolites and implications for catalysis of oxygenated compounds
Aída
Rodríguez-Fernández†
 a,
John R.
Di Iorio†
b,
Cecilia
Paris
a,
Mercedes
Boronat
a,
Avelino
Corma
a,
Yuriy
Román-Leshkov
*b and
Manuel
Moliner
*a
a,
John R.
Di Iorio†
b,
Cecilia
Paris
a,
Mercedes
Boronat
a,
Avelino
Corma
a,
Yuriy
Román-Leshkov
*b and
Manuel
Moliner
*a
aInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, 46022 València, Spain. E-mail: mmoliner@itq.upv.es
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: yroman@mit.edu
First published on 7th September 2020
Abstract
The selective incorporation of isolated framework Lewis acid sites at specific crystallographic positions in high-silica zeolites was achieved by applying a rationalized post-synthetic grafting methodology. The removal of framework Ge atoms from a Ge-BEC zeolite with low concentrations of Ge in the framework (Si/Ge ∼ 150) followed by grafting allows the synthesis of Sn-BEC zeolites with Sn atoms positionally biased into the double-4-ring (D4R) crystallographic positions of the BEC framework. Spectroscopic characterization using solid-state nuclear magnetic resonance (NMR) coupled with theoretical calculations revealed that Sn atoms preferentially form open Sn sites in the D4R of Sn-BEC. This observation was supported by IR spectra of adsorbed deuterated acetonitrile (CD3CN), a known titrant of Sn sites in zeolites. The catalytic implications of selective incorporation of open Sn sites in Sn-BEC were probed using the Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reaction. Although the MPVO turnover rates normalized by the total number of open Sn sites were comparable on Sn-BEC and a conventional Sn-Beta catalyst synthesized in fluoride media (Sn-Beta(F)), Sn-BEC demonstrated higher per gram reaction rates because of its larger fraction of open sites compared to Sn-Beta(F). These results highlight the advantage of placing active sites in targeted locations within a zeolite structure. The methodology presented here to selectively place catalytic active sites via sacrificial heteroatoms, such as Ge, can be generalized for the design of many other tetrahedrally-coordinated metal-containing zeolites.
1. Introduction
Positioning catalytically active sites in predetermined positions within the framework of a zeolite is a powerful handle to modify catalytic activity and product selectivity.1,2 The incorporation of trivalent heteroatoms, such as Al or B, into crystallographic T-site positions generates a negative charge on the zeolite framework, which must be adequately balanced in the final solid by an extra-framework inorganic or organic cation. Several direct synthetic approaches have leveraged this feature to bias the location of Al species in zeolite pores, cavities, and intersections by manipulating the size, shape and charges of the counterbalancing cations during hydrothermal synthesis.1–11 Zeolites of a given framework topology can be synthesized with varied fractions of their framework Al atoms located in pores of different size using mixtures of different structure directing agents. Accordingly, the location of Al atoms within a zeolite alters reaction rates and selectivities, as demonstrated for a variety of acid catalysed reactions using positionally biased H-MFI (e.g., olefin oligomerization,12 alkane cracking,10 and methanol conversion9,13–15) and H-FER (e.g., isomerization,4,16 alkane and alkene cracking,6 and carbonylation6) zeolites. Moreover, the synthesis of organic structure directing agents (OSDAs) that mimic known transition states has also led to the design of tailored zeolite frameworks with enhanced reactivity for toluene disproportionation, isomerization, and methanol upgrading reactions.17,18Incorporation of tetravalent heteroatoms with open coordination sites (e.g., Ti, Sn, and Zr) into crystallographic framework positions gives rise to Lewis acid sites.19–21 These catalysts are ideally suited to activate oxygenated molecules, showing exceptional selectivity and yields for the transformation of biomass-derived molecules.22–24 For example, Sn-Beta—a catalyst featuring isolated tin sites incorporated into the Beta framework—has been extensively evaluated as the paradigmatic catalyst for many reactions of bio-derived molecules, including carbohydrate isomerization and epimerization, inter and intramolecular transfer hydrogenation, and aldol condensation.25–30 However, unlike a trivalent heteroatom whose position within the zeolite framework can be directly influenced by electrostatic interactions with inorganic and organic cations, biasing the location of an isolated tetravalent Lewis acid site is difficult because charge balancing does not occur during crystallization. Consequently, directing these heteroatoms to specific crystallographic locations during zeolite synthesis has not been demonstrated using traditional synthetic approaches.
The ubiquitous use of the Beta topology to host Lewis acid sites originates from its synthetic versatility, as it is one of the few structures with large pores that can be prepared under a broad range of synthesis conditions with precise control over crystal size and heteroatom identity.31,32 The crystalline structure of Beta is formed by the intergrowth of two different polymorphs, BEA and BEB (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio), each featuring 9 distinct crystallographic tetrahedral sites (T-sites; Fig. 1a and b).33 Using extended X-ray absorption fine structure (EXAFS) to elucidate the distribution of Sn across these T-sites, Corma and co-workers proposed preferential positioning of Sn atoms at the T5 and T6 sites (Fig. 1b) in Sn-Beta synthesized with fluoride media (Sn-Beta(F)).34 Román-Leshkov et al. used solid-state magic angle spinning 31P nuclear magnetic resonance (NMR) spectroscopy to identify several well-defined signals corresponding to the adsorption of trimethylphosphine oxide (TMPO) at different Sn sites in Beta zeolites, suggesting chemical diversity of such active sites. This work established correlations between different 31P chemical shifts and the catalytic activity for the glucose isomerization and aldol condensation reactions.35 Sn atoms have also been shown to adopt different coordination within Beta zeolites, namely “open” Sn sites, which contain three bonds to framework oxygen atoms and one hydroxyl ligand (i.e., Sn(OSi)3–OH), and “closed” Sn sites, which contain four bonds to framework oxygen atoms (i.e., Sn(OSi)4).36 The number of open Sn sites has been shown to correlate with rates of Baeyer–Villiger oxidation and Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reactions.21,36 Recently, Gounder et al. have proposed that these open Sn sites are preferentially located at stacking faults present in Beta zeolites37,38 that form from the intergrowth of both polymorphs during crystallization.33,39,40 Despite these previous efforts, the precise assignment of Lewis acid sites distributed across the different T-sites in Beta zeolites remains under debate and the prospect of developing synthetic strategies to control the position of sites across 18 unique T-sites and framework defects remains obscure.
:50 ratio), each featuring 9 distinct crystallographic tetrahedral sites (T-sites; Fig. 1a and b).33 Using extended X-ray absorption fine structure (EXAFS) to elucidate the distribution of Sn across these T-sites, Corma and co-workers proposed preferential positioning of Sn atoms at the T5 and T6 sites (Fig. 1b) in Sn-Beta synthesized with fluoride media (Sn-Beta(F)).34 Román-Leshkov et al. used solid-state magic angle spinning 31P nuclear magnetic resonance (NMR) spectroscopy to identify several well-defined signals corresponding to the adsorption of trimethylphosphine oxide (TMPO) at different Sn sites in Beta zeolites, suggesting chemical diversity of such active sites. This work established correlations between different 31P chemical shifts and the catalytic activity for the glucose isomerization and aldol condensation reactions.35 Sn atoms have also been shown to adopt different coordination within Beta zeolites, namely “open” Sn sites, which contain three bonds to framework oxygen atoms and one hydroxyl ligand (i.e., Sn(OSi)3–OH), and “closed” Sn sites, which contain four bonds to framework oxygen atoms (i.e., Sn(OSi)4).36 The number of open Sn sites has been shown to correlate with rates of Baeyer–Villiger oxidation and Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reactions.21,36 Recently, Gounder et al. have proposed that these open Sn sites are preferentially located at stacking faults present in Beta zeolites37,38 that form from the intergrowth of both polymorphs during crystallization.33,39,40 Despite these previous efforts, the precise assignment of Lewis acid sites distributed across the different T-sites in Beta zeolites remains under debate and the prospect of developing synthetic strategies to control the position of sites across 18 unique T-sites and framework defects remains obscure.
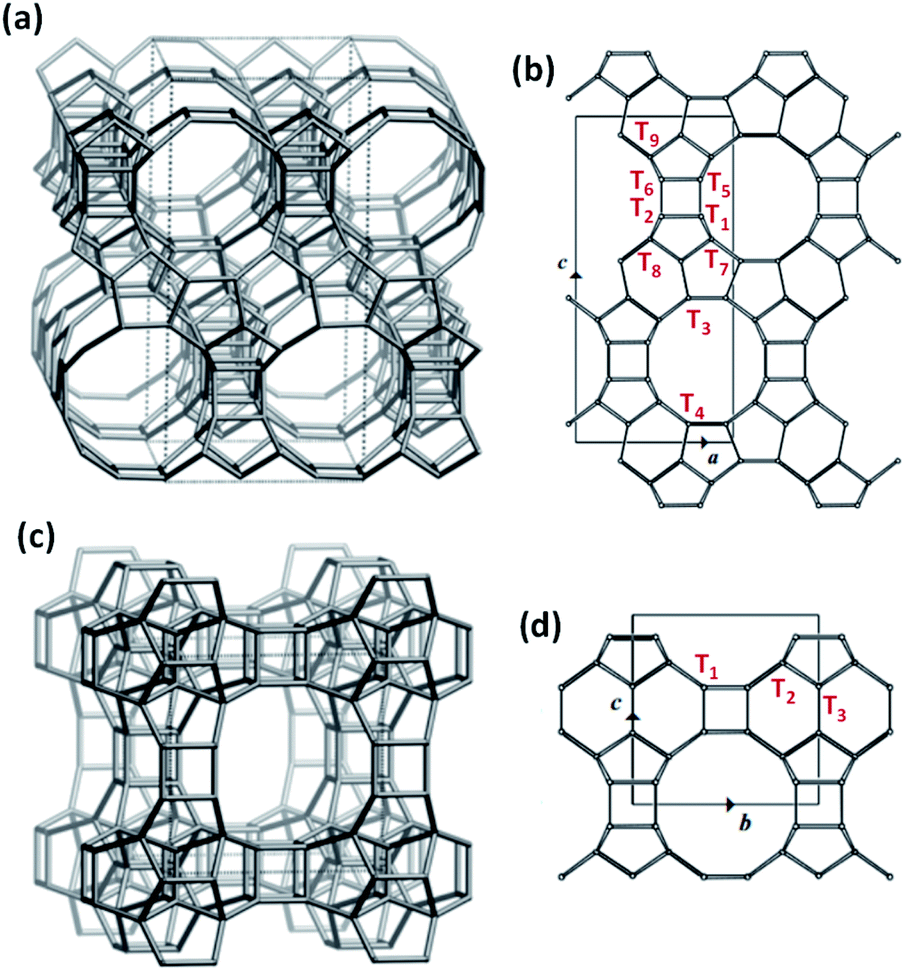 | ||
| Fig. 1 Framework structures of BEA (a) and BEC (c) and the different crystallographic T-sites in the unit cell of BEA (b) and BEC (d). | ||
A third polymorph of Beta, BEC, holds promise to overcome these limitations. The BEC framework is formed by the same zeolitic layers comprising BEA and BEB, but these layers are stacked in a different arrangement, creating a three-dimensional large pore system with straight channels (Fig. 1c). Consequently, BEC contains only three distinct crystallographic T-sites (Fig. 1d), half of which are located in the double-4-ring (D4R) secondary building units. This feature was the key to synthesizing ITQ-17, the first BEC-type zeotype obtained experimentally.41,42 Specifically, Ge heteroatoms were used in the synthesis media (Si/Ge ratio < 5) in combination with different OSDAs to crystallize BEC, surmising that Ge would be more adept than Si at accommodating the tight angles in the D4Rs. Recently, a Ge-rich Sn-BEC (Si/Ge = 5) was reported to selectively catalyze the isomerization of glucose to methyl fructoside in methanol,43 unlike Sn-Beta, which converted glucose to methyl mannoside via both isomerization and epimerization pathways under identical reaction conditions (120 °C). This difference was ascribed to the preferential occupancy of isolated Lewis acid catalytic centers in the D4Rs of the BEC structure.43 While promising, Ge-rich zeolites are hydrothermally unstable and readily undergo degermanation even in the presence of atmospheric water.44 Although the labile nature of Ge species in zeolites has been recently employed to achieve novel zeolite structures through the assembly–disassembly–organisation–reassembly (ADOR) transformation methodology45 and to create hybrid Ge-based electronic conductors,46 framework collapse via degermanation is a major impediment to the development of stable catalytic materials.
Here, we present a strategy to generate stable Sn-BEC zeolites with positionally biased Lewis acid sites and investigate the catalytic implications of this approach. Prior reports demonstrated the cooperative effect between specific OSDA molecules (Fig. 2) and F incorporation within the D4Rs to generate pure-silica BEC without requiring the presence of Ge heteroatoms.47 We build upon this strategy by generating BEC zeolites with low concentrations of framework Ge (Si/Ge > 150), hypothesizing that the limited number of Ge atoms per unit cell would be preferentially positioned in the D4Rs.47,48 These Ge atoms can then be isomorphically substituted with Sn using simple post-synthetic treatments without jeopardizing the structural integrity of the zeolite framework. Given the strain at these locations, the Sn atoms preferentially exist in an open configuration, in agreement with the observations by Gounder and co-workers where a higher proportion of open sites was detected at strained stacking fault sites between Beta polymorphs.37 NMR and FTIR spectroscopies confirmed the incorporation of Lewis acidic Sn species within the BEC framework that are located predominantly in a single crystallographic environment. Reactivity studies using MPVO as a probe reaction show that the Sn-BEC featured similar turnover rates (normalized per open Sn site) to classical Sn-Beta(F) catalysts, but showed higher reaction rates per gram of zeolite due to the larger number of open Sn sites on a mass basis. These results demonstrate that catalytic active sites can be selectively placed in targeted locations within a given zeolite structure by taking advantage of the preferential incorporation of sacrificial heteroatoms at specific crystallographic sites.
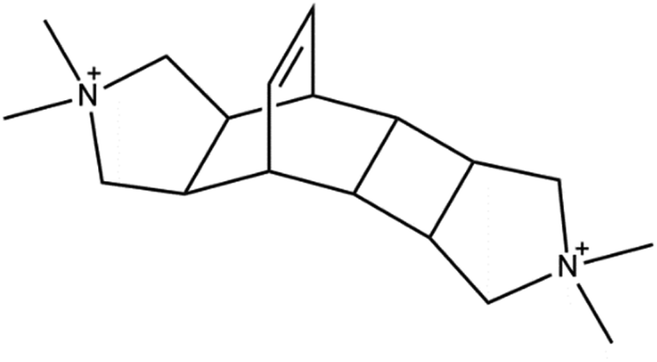 | ||
| Fig. 2 OSDA employed for the synthesis of the high-silica BEC zeolite. | ||
2. Experimental section
2.1. BEC-type zeolite syntheses
The iodide form of the organic salt was exchanged to the hydroxide form as follows: 24 mmol of the iodide salt was dissolved in water, adding 50 g of Dower-SBR Anion Exchange resin in hydroxide form (total exchange capacity 1.4 eq. l−1) and maintaining the mixture under agitation overnight. The solution was filtered to obtain the dihydroxide cation [TMTEC(OH)2] and titrated with HCl to confirm its purity.
2.2. Synthesis of Sn-Beta in fluoride media (Sn-Beta(F))
Sn-Beta(F) was synthesized following a previously reported procedure.49 Briefly, 11.65 g of an aqueous solution of tetraethylammonium hydroxide (TEAOH; Sigma-Aldrich, 40 wt%) and 8.44 g of deionized water (18.2 MΩ) were added to a perfluoroalkoxy alkane (PFA, Savillex Corp.) jar and stirred under ambient conditions for 5 minutes to homogenize the contents. Next, 12.1 g of tetraethylorthosilicate (TEOS; Sigma-Aldrich, 99 wt%) was added to the aqueous TEAOH solution, and the mixture was covered and stirred under ambient conditions for 120 minutes. Then, 0.044 g of stannous chloride (Sn(II)Cl2; Sigma-Aldrich, 98 wt%) was dissolved in ∼2 g of ethanol (Koptec, 99.5 wt%) and this mixture was added dropwise to the aqueous solution of TEAOH and TEOS. The resulting solution was left uncovered and stirred under ambient conditions to evaporate ethanol and excess water. After reaching the desired final mass, 1.30 g of hydrofluoric acid (HF; Sigma-Aldrich, 48 wt% in H2O, ≥99.99% trace metals basis) was added dropwise and the mixture was manually stirred with a PTFE spatula for 5 minutes to homogenize the contents. The resulting molar composition of the synthesis media was: 1SiO2/0.0033SnO2/0.54TEAOH/0.54HF/7.52H2O. The mixture was then seeded with 0.173 g of dealuminated Beta zeolite (5 wt% SiO2 basis), transferred to a 45 ml PTFE-lined stainless steel autoclave (4744, Parr Instruments), and heated to 160 °C under rotation (∼40 RPM) for 7 days. Solids were then recovered via centrifugation, washed repeatedly with deionized water and acetone (99.5 wt%, BDH Chemicals) until the pH of the supernatant remained constant between washes, and then dried at 100 °C overnight under ambient air. The dried solids were then heated to 580 °C under flowing air for 10 hours.2.3. Characterization
Chemical analyses were carried out in a Varian 715-ES ICP-Optical Emission spectrometer, after solid dissolution in HNO3/HCl/HF aqueous solution. Elemental analyses were performed by combustion analysis using a Eurovector EA 3000 CHNS analyzer.
The morphology of the samples was studied by field emission scanning electron microscopy (FESEM) using a ZEISS Ultra-55 microscope.
Diffuse reflectance ultraviolet-visible spectra were measured using an Agilent Cary 5000 spectrometer equipped with a UV-Vis DiffusIR environmental chamber (Pike Technologies). Zeolite powders were dehydrated by heating to 250 °C (5 °C min−1) under flowing dry N2 and held for 2 hours prior to cooling to 30 °C. Spectra were measured relative to a pure-silica Beta zeolite.
Solid-state NMR spectra were recorded at room temperature with a Bruker AV 400 MAS spectrometer. 29Si magic angle spinning (MAS) NMR spectra were recorded with a spinning rate of 5 kHz at 79.459 MHz with a 55° pulse length of 3.5 μs and repetition time of 180 s. 19F was measured at 376.28 MHz at 25 kHz with pulses of 4.5 μs corresponding to a flip angle of π/2 rad, and a recycle delay of 100 s to ensure the complete recovery of the magnetization. 29Si and 19F chemical shifts were referred to tetramethylsilane and CFCl3, respectively.
Solid-state 31P MAS NMR spectra were recorded at room temperature under magic angle spinning in a Brucker AV-400 spectrometer at 161.9 MHz with a spinning rate of 10 kHz and π/2 pulse length of 3.7 μs with spinal proton decoupling and recycle delay of 20 s. The 31P chemical shift was referred to phosphoric acid.
Prior to collection of IR spectra, each sample was heated to 400 °C (2 °C min−1) under flowing dry air (50 cm3 min−1) for 2 hours, cooled under flowing air to 30 °C, and evacuated until the pressure in the cell reached <8.0 × 10−5 torr (Edwards T-Station 75 Turbopump). Acetonitrile-d3 (CD3CN; anhydrous, ≥99.8 atom% D, Sigma-Aldrich) was purified via successive freeze–pump–thaw cycles to remove dissolved gases and volatiles, and then introduced to the cell in serial doses (1.4–7.0 × 10−7 mol per dose). An IR spectrum was measured after pressure in the cell remained constant for >30 seconds. The spectrum of the dehydrated parent zeolite was subtracted from all subsequent spectra after CD3CN dosing, which were then baseline corrected and normalized by the total area of the Si–O–Si combination and overtones modes (2100 to 1750 cm−1) of the parent zeolite sample. Assignments for CD3CN coordinated to open Sn (ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N): 2316 cm−1), closed Sn (2308 cm−1), silanol groups (2275 cm−1), and physisorbed or gas-phase CD3CN (2265 cm−1) are based on prior assignments.36,38,50 The total number of Lewis acidic Sn sites and the fraction of Sn found in and closed coordination environments were quantified using reported integrated molar extinction coefficients and deconvolution methods.38
N): 2316 cm−1), closed Sn (2308 cm−1), silanol groups (2275 cm−1), and physisorbed or gas-phase CD3CN (2265 cm−1) are based on prior assignments.36,38,50 The total number of Lewis acidic Sn sites and the fraction of Sn found in and closed coordination environments were quantified using reported integrated molar extinction coefficients and deconvolution methods.38
2.4. Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reaction between cyclohexanone and 2-butanol
Rates of the MPVO reaction between cyclohexanone and 2-butanol were measured in glass batch reactors (10 cm3, VWR) using reactant solutions containing 0.01–1 M cyclohexanone (99 wt%, Sigma-Aldrich) in 2-butanol (99.8 wt%, Sigma-Aldrich). A specific amount of catalyst (5–10 mg) and a PTFE coated magnetic stir bar were loaded into a batch reactor and sealed with a PTFE/silicone septum in an aluminum crimp top (Ace Glass). 2 cm3 of a reactant solution were added to a separate batch reactor, sealed with a PTFE/silicone septum, and both catalyst and reactant vessels were heated to 100 °C for >15 minutes. After the preheating step, the reactant solution was transferred to the catalyst vial via syringe and the reaction allowed to proceed under stirring (300 RPM) at 100 °C. Aliquots of the reactant solution were taken after ∼5 minutes, quenched in an ice bath, and filtered through a 0.2 μm PTFE syringe filter (VWR). The filtered reactant solutions were then diluted with a 2-pentanone standard solution (5 wt% in 2-butanol; 99.5 wt%, HPLC grade, Sigma-Aldrich) and injected to a gas chromatograph (7890A, Agilent Technologies) equipped with a DB-1701 column (30 m × 250 μm × 0.25 μm, Agilent Technologies) and a flame ionization detector. Initial cyclohexanol formation rates (normalized per open Sn site) were determined by extrapolating transient cyclohexanol production (moles of cyclohexanol produced gcat−1) to zero reaction time (i.e., zero conversion) using data measured at <10% conversion.2.5. Computational details
The active sites in Sn-BEC and Sn-BEA catalysts were simulated by cluster models cut out from the respective periodic crystalline structures and containing one framework Sn atom per unit cell, either bonded to four O–Si units (i.e., closed site) or with one hydrolyzed Sn–OH bond (i.e., open site). The dangling bonds that connected the cluster with the rest of the solid were saturated with H atoms, and during geometry optimization these terminal H atoms were kept fixed to maintain the zeolite structure, while all other atoms in the model were allowed to relax without restrictions. Sn was placed at T1, T2 and T3 positions in BEC, and at T4, T5 and T7 positions in BEA. The three positions in BEA were selected according to the size of the rings they belong to: 4R and 5R for T1, T2, T5 and T6; 4R, 5R and 6R for T3, T4, T8 and T9; and only 5R in the case of T7. All density functional theory (DFT) calculations were carried out using the M062X functional,51 the 6-31g(d,p) basis set for O, C, P and H atoms,52,53 and the LANL2DZ basis set and pseudopotential for Sn,54,55 as implemented in the Gaussian09 software.56 For the simulation of NMR parameters, the isotropic absolute chemical shielding constants (σ) were obtained using the gauge including atomic orbitals (GIAO) approach57,58 and the 31P chemical shifts were calculated as δ31P = σref − σ, using phosphoric acid as reference. To improve accuracy, all values were corrected with an equation obtained by fitting δ31P values at 6-31G(d,p) level against δ31P values at 6-311++G(d,p) level59 for a series of acid–base TMPO adducts.3. Results and discussion
3.1. Synthesis of Sn-containing BEC
The strategy used to generate positionally biased Sn-BEC samples using post-synthetic grafting of degermanized BEC samples is depicted in Fig. 3. Prior reports confirmed that Ge heteroatoms preferentially occupy T-sites in the D4Rs before populating other sites within the zeolite.42,44 A Si/Ge molar ratio of 150 was selected because it is similar to the Si/Sn content used in traditional hydrophobic Sn-Beta(F). PXRD patterns show that full crystallization of Ge-BEC is obtained after 4 days (Fig. 4) and FE-SEM micrographs indicate a homogeneous formation of BEC crystals with average particle sizes between 200–400 nm (see Fig. 5).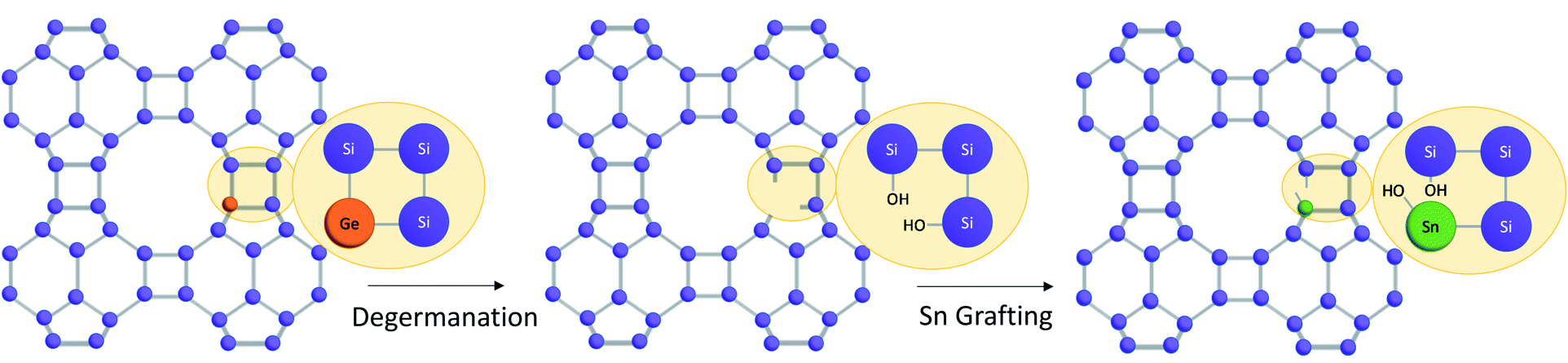 | ||
| Fig. 3 Multi-step synthesis procedure to selectively place Sn in the D4Rs of high-silica BEC zeolites. | ||
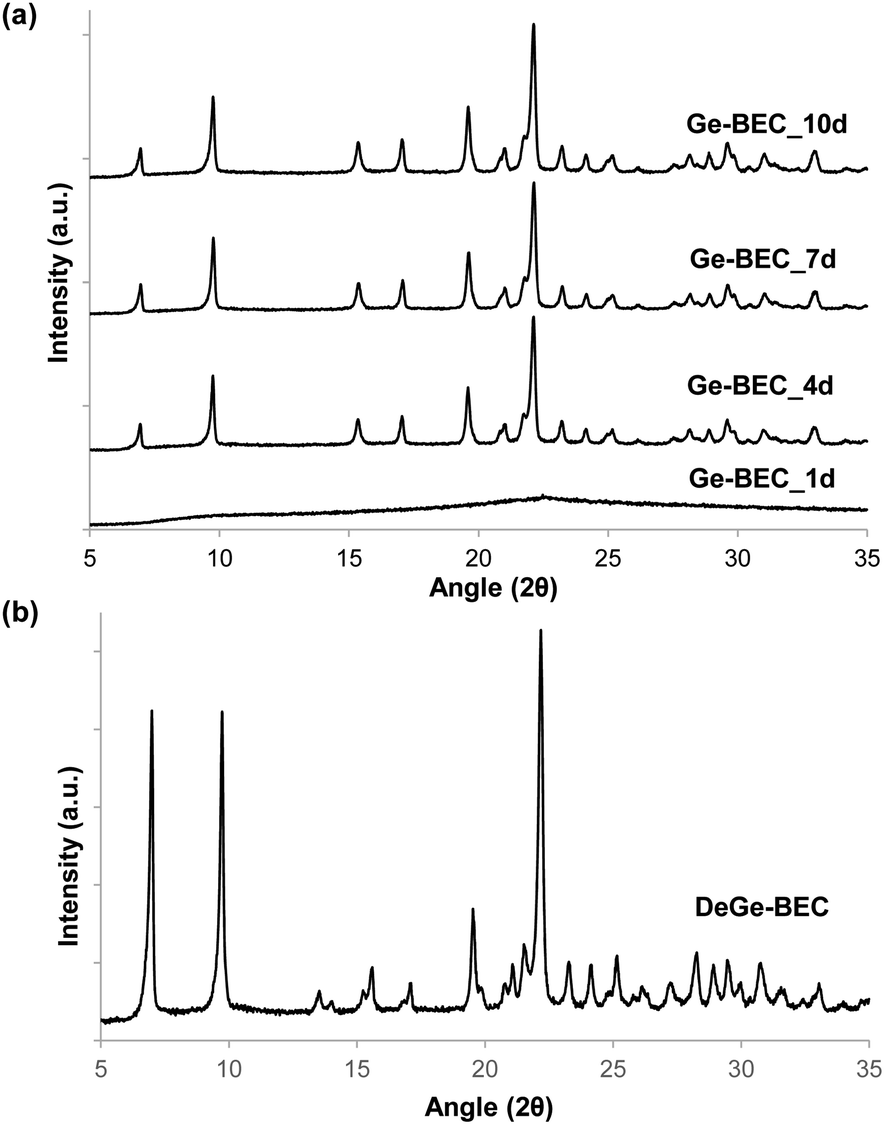 | ||
| Fig. 4 PXRD patterns of (a) Ge-BEC taken at different crystallization times (1–10 days) with a Si/Ge molar ratio of 150 and (b) degermanized BEC (DeGe-BEC). | ||
 | ||
| Fig. 5 FE-SEM images of the Ge-containing BEC materials after different crystallization times ranging from 1 (Ge-BEC_1d) to 10 (Ge-BEC_10d) days. | ||
The chemical analyses of the four samples taken at different time points during the Ge-BEC crystallization process indicate equivalent Si/Ge molar ratios ranging between 150 and 160 for all cases (Table 1). These values are analogous to those initially incorporated in the synthesis gels (Si/Ge ∼ 150). The 19F MAS NMR spectrum for the as-prepared Ge-BEC materials features two well-defined signals localized at −38 and −20 ppm (see Fig. 6). These signals have been assigned to fluoride species trapped within pure silica D4R units and D4R units containing one Ge atom, respectively.42 The ratio of the integrated areas of these two peaks remains constant at ∼10 after 4 days of synthesis time. Considering that the number of T-sites in the D4Rs is 50% of the total T-sites per unit cell (i.e., 16 of 32 T-atoms), the 19F MAS NMR data translate to ∼0.18 Ge atoms per unit cell, which corresponds to a Si/Ge molar ratio of ∼170. This value is in close agreement with the value of ∼155 measured by bulk ICP analysis (see Table 1), suggesting that Ge atoms are exclusively incorporated in the D4Rs. The 29Si MAS NMR spectrum of the as-prepared Ge-BEC_10d sample shows three signals centred at −106, −110 and −117 ppm (Fig. 7). These signals are assigned to Si atoms at the three distinct crystallographic T-sites in the BEC framework and show a T1:T2:T3 ratio of 2:1:1, in good agreement with the crystallographic information of the BEC structure.
| Sample | Si/Ge | Si/Sn | %wt Sn/TO2 |
|---|---|---|---|
| Ge-BEC_1d | 163 | — | — |
| Ge-BEC_4d | 153 | — | — |
| Ge-BEC_7d | 154 | — | — |
| Ge-BEC_10d | 155 | — | — |
| DeGe-BEC | >600 | — | — |
| Sn-BEC | >600 | 330 | 0.60 |
| Sn-Beta(F) | — | 298 | 0.73 |
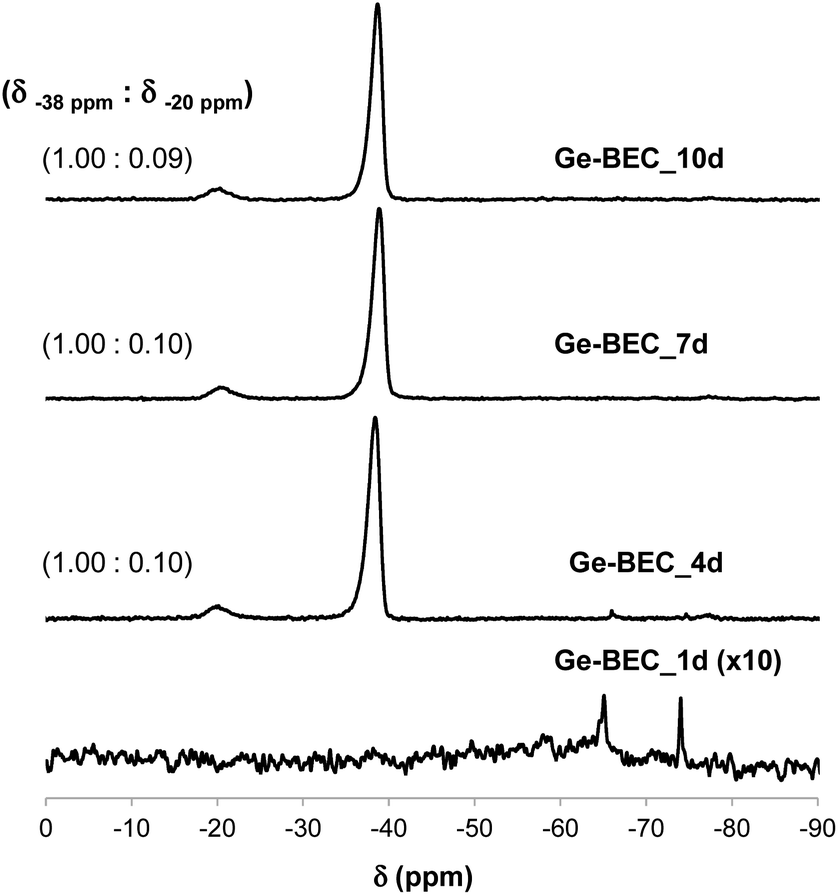 | ||
| Fig. 6 19F MAS NMR spectra of the as-prepared Ge-containing BEC materials after different crystallization times. The measured signal ratios are included for all Ge-BEC samples. | ||
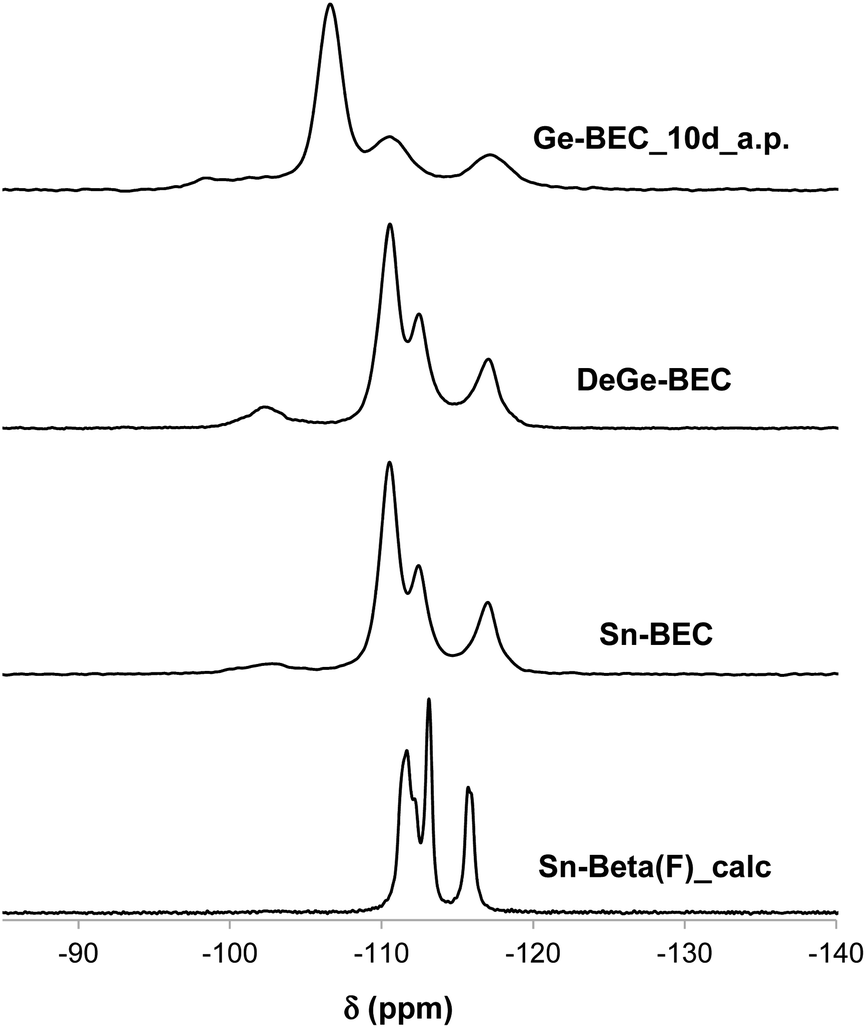 | ||
| Fig. 7 29Si MAS NMR spectra of the as-prepared Ge-BEC_10d (Ge-BEC_10d_a.p.), after the degermanization process (DeGe-BEC), and after the Sn-grafting process (Sn-BEC) and 29Si MAS NMR spectrum of the calcined Sn-Beta(F) (Sn-Beta(F)_calc). | ||
The second step in the synthesis of Sn-BEC involved the selective removal of Ge species from the BEC framework via sequential calcination and acid treatment. The resulting solid maintained its crystalline structure after degermanation, as revealed by PXRD (see DeGe-BEC in Fig. 4b). Chemical analysis of the treated solid indicated a Si/Ge molar ratio >600, confirming the removal of Ge from the original material (see DeGe-BEC in Table 1). The 29Si MAS NMR spectrum of the degermanized sample revealed a new band centred at −103 ppm (see DeGe-BEC in Fig. 7), which can be assigned to silanol defects (Si(OSi)3OH) created in the BEC structure after removal of framework Ge heteroatoms. The other 29Si chemical shifts were located at −110, −112 and −117 ppm, similar to those observed in Ge-BEC (see Fig. 7). The slight differences in peak position between both samples are attributed to framework relaxation after calcination to remove the organic SDA molecules.
Sn heteroatoms were incorporated into framework defects formed by removal of Ge via post synthetic grafting. Specifically, methyltin(IV) trichloride was grafted into the framework vacancy defects in the D4Rs of degermanized BEC using a theoretical Si/Sn molar ratio of ∼100. Chemical analysis revealed a Si/Sn molar ratio of 330, which corresponds to ∼0.6 wt% Sn/TO2 in the solid (see Table 1). Interestingly, the 29Si MAS NMR spectrum of the Sn-BEC sample showed a substantial decrease in the signal centered at −103 ppm after the Sn grafting procedure (Fig. 7), suggesting partial healing of defect sites.
Incorporation of Sn into vacancy defects of DeGe-BEC was confirmed from UV-visible spectra measured on dehydrated Sn-BEC (Fig. 8) that show an absorption feature centered near 190 nm, which is characteristic of isolated, framework Sn atoms.60 UV-Vis spectra of dehydrated Sn-BEC also show weak absorption features centered near 220 and 240 nm, which are absent in conventional Sn-Beta(F), and indicate the presence of extra-framework SnOx clusters, which may form as a result of incomplete grafting of lattice defects in DeGe-BEC by methyltin(IV) trichloride precursors. Collectively, these bulk spectroscopic data demonstrate that the synthetic methods described here give rise to framework Sn atoms that partially heal vacancy defects present in DeGe-BEC zeolites. Further optimization of the grafting procedure described here should lead to an increase in the specific reaction rate (per gram catalyst) by improving the efficiency of framework Sn incorporation.
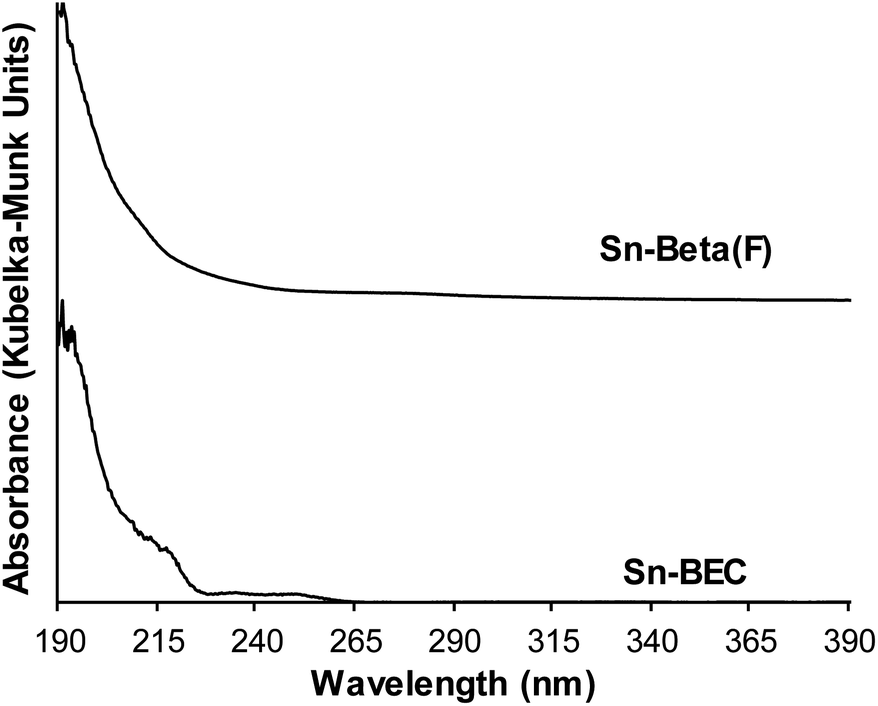 | ||
| Fig. 8 UV-visible spectra of dehydrated Sn-Beta(F) (top) and Sn-BEC (lower) zeolites. | ||
3.2. Lewis acid-site characterization
Understanding the chemical and structural environments of the grafted tin species within the BEC framework is necessary to properly assess the applicability of Sn-BEC as a heterogeneous catalyst. We used two probe molecules, TMPO and CD3CN, combined with 31P MAS NMR and FTIR spectroscopies, respectively, to shed light on the atomic environment of framework tin species. Román-Leshkov et al. recently employed solid-state 31P MAS NMR spectroscopy after adsorbing TMPO probe molecules to elucidate the identity of the Lewis acid sites in Sn-Beta (see Experimental section for details).35 Following this approach, we observed distinct 31P NMR resonance profiles for Sn-Beta(F) and Sn-BEC. At least two well-defined signals at 58.9 and 55.2 ppm appear in the 31P MAS NMR spectrum of the Sn-Beta(F) zeolite with a Si/Sn molar ratio of ∼300 when using intermediate TMPO loadings (TMPO/Sn ∼ 0.5; Fig. 9).35 In contrast, Sn-BEC presented a single 31P NMR resonance centred at 59.8 ppm at a similar TMPO loading (TMPO/Sn ∼ 0.5; Fig. 9). This result suggests the presence of Lewis acid sites with a single chemical environment within the Sn-BEC crystals. Control experiments on the degermanized BEC sample, showed only a broad signal at 47.6 ppm, which could be assigned to TMPO molecules interacting with silanol groups.61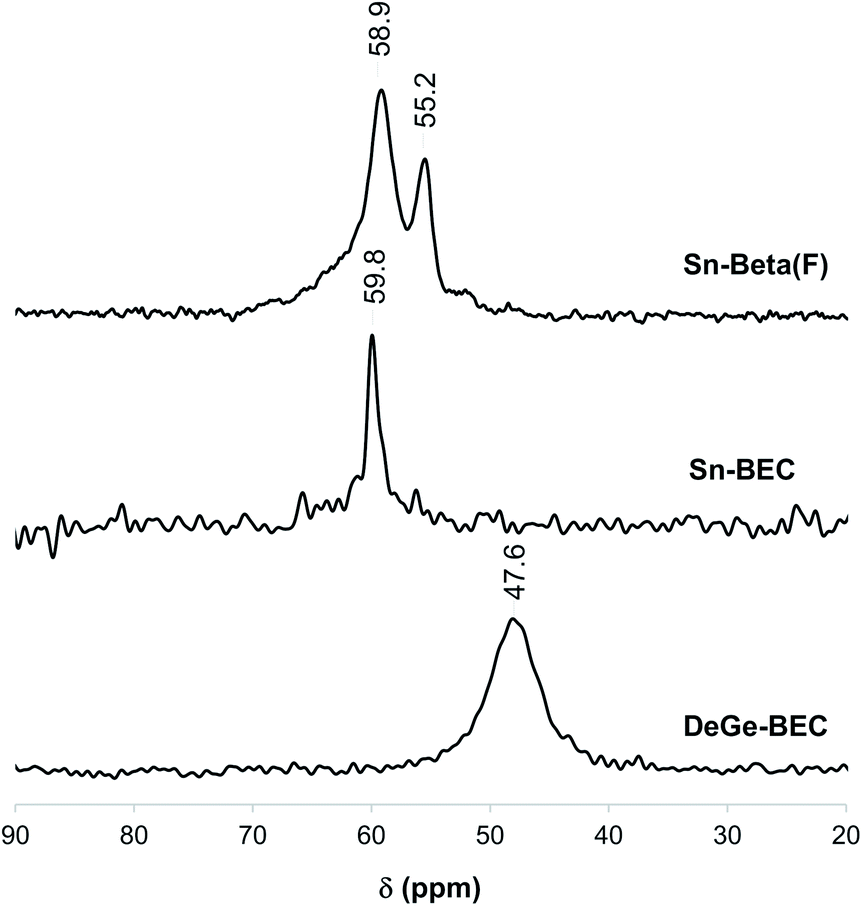 | ||
| Fig. 9 31P MAS NMR spectra of TMPO dosed on DeGe-BEC, Sn-BEC and Sn-Beta(F) at P/Sn = 0.5 loading. | ||
To correlate the experimentally observed 31P NMR resonances with specific crystallographic T-sites in BEC, we carried out a detailed DFT study aimed at evaluating the interactions of TMPO with Sn atoms placed at the three distinct T-sites in BEC. Three representative T-sites in the BEA framework (T4, T5, and T7; Fig. 1b) were also considered for comparison with BEC. Both closed and open Sn sites were considered at all T-sites, as well as silanol groups in the two zeolite structures. TMPO adsorbs on Sn sites forming stable adducts with optimized Sn–O distances of ∼2.1 Å (Fig. 10 and 11) and with the P–O bond length increasing by ∼0.04 Å with respect to the optimized value for the isolated molecule in the gas phase (1.493 Å). The interaction of TMPO with silanol groups leads to less distortion of the adsorbed TMPO, with a lengthening of the P–O bond by only 0.016 Å and calculated δ31P chemical shifts of 43 and 40 ppm in BEC and BEA, respectively (Table 2).
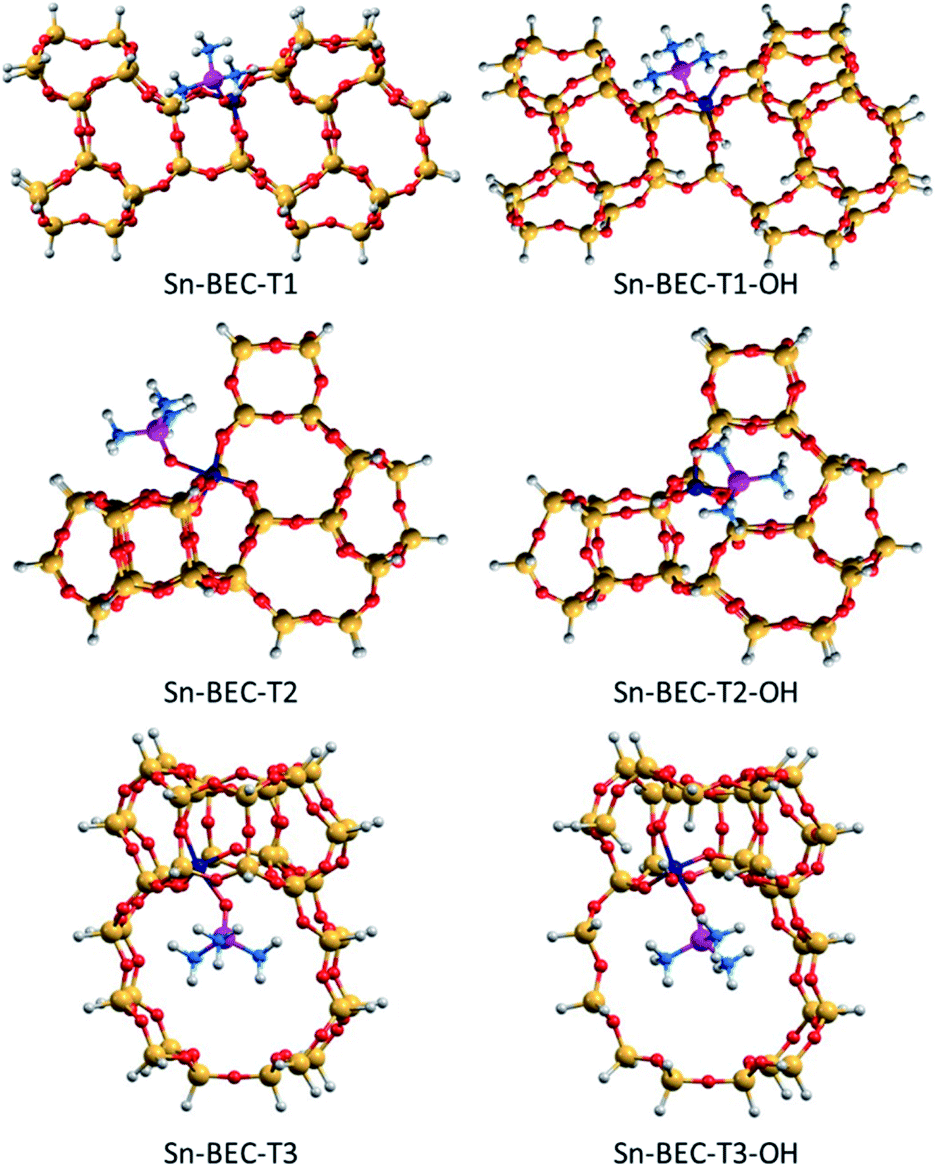 | ||
| Fig. 10 DFT optimized structures of TMPO interacting with closed and open sites of Sn-BEC. Sn, Si, O, P, C and H atoms are depicted in dark blue, yellow, red, purple, blue and white, respectively. | ||
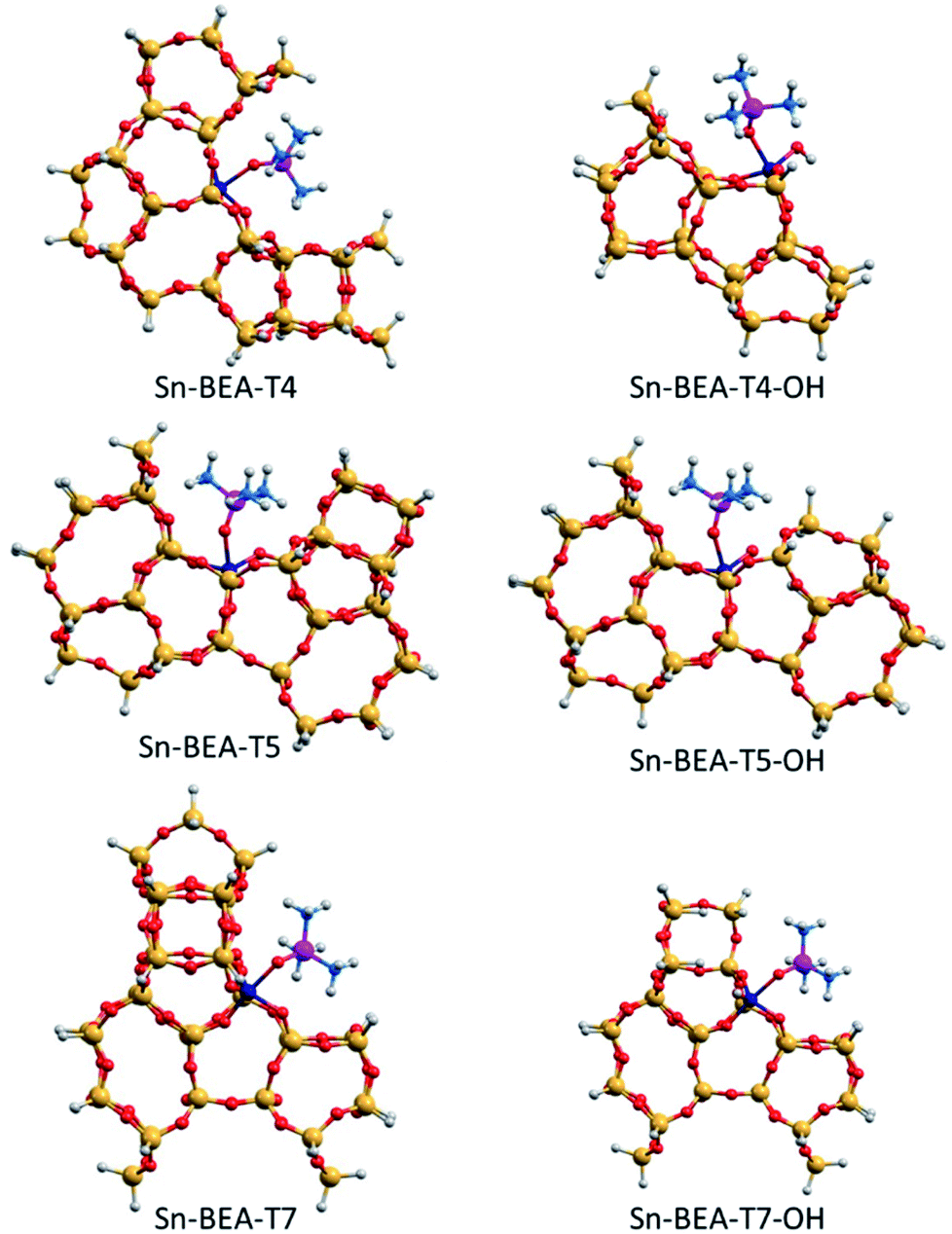 | ||
| Fig. 11 DFT optimized structures of TMPO interacting with closed and open sites of Sn-BEA. Sn, Si, O, P, C and H atoms are depicted in dark blue, yellow, red, purple, blue and white, respectively. | ||
| Catalyst | Site | δ 31P (ppm) | r(PO) (Å) | Catalyst | Site | δ 31P (ppm) | r(PO) (Å) |
|---|---|---|---|---|---|---|---|
| Sn-BEC | T1 | 61.7 | 1.539 | Sn-BEA | T4 | 54.2 | 1.530 |
| Sn-BEC | T1-OH | 60.9 | 1.537 | Sn-BEA | T4-OH | 59.0 | 1.533 |
| Sn-BEC | T2 | 65.4 | 1.542 | Sn-BEA | T5 | 57.2 | 1.533 |
| Sn-BEC | T2-OH | 67.0 | 1.542 | Sn-BEA | T5-OH | 59.5 | 1.531 |
| Sn-BEC | T3 | 62.3 | 1.536 | Sn-BEA | T7 | 60.5 | 1.532 |
| Sn-BEC | T3-OH | 60.2 | 1.532 | Sn-BEA | T7-OH | 60.9 | 1.541 |
| BEC | Si–OH | 43.1 | 1.509 | BEA | Si–OH | 40.3 | 1.509 |
As a result, the observed changes in the δ31P chemical shift allows the assignment of TMPO adsorbed to Sn at different T-sites. The calculated δ31P chemical shifts for TMPO interacting with Sn atoms at the T1 and T3 sites in Sn-BEC are similar at 61.7 and 62.3 ppm, respectively, and decrease to 60.9 and 60.2 ppm when considering open sites with a hydrolysed Sn–OH bond (see Table 2 and Fig. 10). The δ31P chemical shifts at the T2 site are noticeably larger, 65.4 and 67.0 ppm for closed and open sites, respectively, and do not match the experimental 31P MAS NMR spectrum (Fig. 9). A comparison of the calculated δ31P chemical shifts (Table 2) with the experimental 31P MAS NMR spectra (Fig. 9) suggests that Sn does not occupy the T2 position in Sn-BEC, and confirms that the experimental peak at 59.8 ppm can be mostly attributed to the placement of Sn at T1 sites. Calculated δ31P chemical shifts indicate that Sn could also occupy T3-OH open sites, but this possibility was excluded by 19F MAS NMR data that indicate the preferential occupancy of the T1 site by Ge and, thus, the placement of Sn at such sites.
Among the three T-sites considered within Sn-BEA (see Fig. 11), the closed sites at T4 and T5 lead to low δ31P chemical shifts, 54.2 and 57.2 ppm, respectively, which increase to 59.0 and 59.5 ppm, respectively, for the corresponding open sites. In contrast, the calculated δ31P chemical shifts for the open and closed Sn sites at T7 are similar and more strongly deshielded than T4 and T5 (60.9 and 60.5 ppm, respectively). The calculated δ31P chemical shifts of TMPO adsorbed at just a subset of the different T-sites present in the BEA framework, which is only one of the polymorphs that comprise conventional Sn-Beta catalysts (e.g., BEA and BEB), and the convolution of multiple chemical environments observed in experimental 31P MAS NMR spectra (Fig. 9) highlight the complexity of assigning chemical shifts in zeolites with a wide diversity of T-sites.
Lewis acidic Sn sites were also titrated by deuterated acetonitrile (CD3CN), a known probe molecule of open and closed Sn sites in zeolites.36,38 IR spectra measured after adsorption of CD3CN on Sn-containing BEC and Beta(F) zeolites at different coverages are shown in Fig. 12. At low coverages of CD3CN in both Sn-zeolites (CD3CN/Sn < 0.2) a single peak characteristic of CD3CN bound to Lewis acidic open Sn sites (ν(CN) = 2316 cm−1) is observed, which undergoes a shift in peak center with increasing coverage as CD3CN populates Lewis acidic closed Sn sites (ν(CN) = 2308 cm−1). This is consistent with prior work that has shown that CD3CN preferentially binds to open Sn sites prior to coordination at other adsorption sites in Sn-zeolites.36,38 At higher coverages (CD3CN/Sn > 0.3), peaks characteristic of CD3CN bound to SiOH groups (ν(CN) = 2275 cm−1) and physisorbed CD3CN (ν(CN) = 2265 cm−1) are observed.
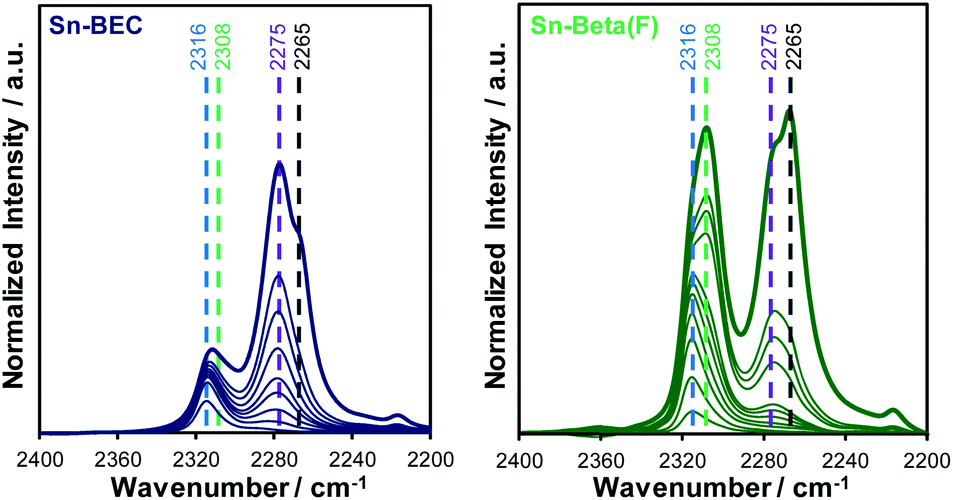 | ||
| Fig. 12 FTIR difference spectra of Sn-BEC (left) and Sn-Beta(F) (right) with increasing CD3CN coverage (0.1–0.7 μmol per dose). Dashed reference lines shown for open Sn sites (2316 cm−1), closed Sn sites (2308 cm−1), silanol groups (2275 cm−1), and gas-phase CD3CN (2265 cm−1). | ||
The total number of Lewis acid sites and the ratio of open to closed Sn sites were quantified using previously reported integrated molar extinct coefficients for CD3CN adsorbed in Sn-zeolites and these values are tabulated in Table 3.38 The fraction of Lewis acidic Sn sites (LAS/Sn = 0.81; per total Sn) and the open/closed site ratio in Sn-Beta(F) (open/closed = 0.54) are similar to the values expected for Sn-Beta zeolites synthesized in the presence of fluoride mineralizing agents.38 Sn-BEC contains a lower fraction of Lewis acidic Sn sites than Sn-Beta(F) (LAS/Sn = 0.47), likely reflecting the formation of extraframework SnOx clusters (Fig. 8), but contains a larger fraction of Sn in open configurations (open/closed = 1.40) than conventional Sn-Beta(F) (open/closed = 0.54) and Sn-Ge-BEC zeolites prepared via direct synthesis (open/closed = 0.13).37 We hypothesize that open Sn sites are preferentially formed when Sn is post-synthetically grafted into vacancy defects of degermanated BEC zeolites. This presumably reflects the inability of Sn to match the flexibility of Ge and form four bonds to framework O atoms at the T1 defect sites formed upon degermanation. Collectively, the spectroscopic and computational data of adsorbed probe molecules (i.e., TMPO and CD3CN) provide evidence that the synthetic methods described here place Sn atoms at well-defined sites in the BEC framework, a design strategy that is currently inaccessible in conventional Sn-Beta(F) zeolites.
| Species | Sn-BEC | Sn-Beta(F) |
|---|---|---|
| LAS/Sn | 0.47 | 0.81 |
| Open/closed | 1.40 | 0.54 |
3.3. Catalytic activity: Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reaction
Initial rates of the MPVO reaction, normalized per open Sn site,21 between 2-butanol and cyclohexanone to form 2-butanone and cyclohexanol were measured on both Sn-BEC and Sn-Beta(F) as a function of cyclohexanone concentration (0.01–1 M) in a 2-butanol solvent at 100 °C (Fig. 13). Cyclohexanol formation rates (per open Sn) on both samples increase linearly at low cyclohexanone concentrations (<0.1 M) before levelling off and approaching a zero-order kinetic regime at higher concentrations of cyclohexanone (>0.5 M). At low cyclohexanone concentrations, cyclohexanol formation rates are ∼3–4× larger on Sn-BEC than on Sn-Beta(F), but become similar (within 1.5×) at high cyclohexanone concentrations in the zero-order regime (>0.5 M). These data indicate that first-order free energy barriers, which reflect differences in free energy between the MPVO transition state and solution phase cyclohexanone and the 2-butanol covered site,21,62 are smaller in Sn-BEC than in Sn-Beta(F). Zero-order free energy barriers, which reflect free energy differences between the MPVO transition state and the co-adsorbed 2-butanol-cyclohexanone precursor,21 however, appear to be similar in both Sn-BEC and Sn-Beta(F) and suggest that adsorbed intermediates and the transition state they form are solvated to similar extents by the BEC and Beta frameworks. Additionally, these data indicate that cyclohexanone adsorption is likely more favourable in Sn-BEC than in Sn-Beta(F), under the conditions investigated here (2-butanol solvent, 100 °C), as differences in first-order rates reflect differences in the coupled free energy barriers of both adsorption and transition state formation. We surmise that this difference in the first-order free energy barrier reflects the preferential stabilization of the adsorbed cyclohexanone intermediate within the Sn-BEC framework due to stronger van der Waals interactions between the adsorbed species and the cylindrical BEC pore (∼0.7 nm in diameter), which is smaller in diameter than stacking faults present in Sn-Beta zeolites (∼1–1.5 nm)40 that preferentially form open Sn sites.37 This observation further highlights how turnover rates of reactions catalyzed by Lewis acidic zeolites are not dictated solely by the nature of the active site (e.g., open or closed Sn coordination), but also by the secondary solvating environment that confine such sites.63 Ultimately, these data demonstrate that open Sn sites incorporated at the T1 site in the BEC framework act as Lewis acid sites that facilitate catalytic turnover of MPVO reactions and give rise to turnover frequencies (normalized per open Sn site) comparable to conventional Sn-Beta(F) zeolites, but higher specific rates due to the higher proportion of open sites per catalyst mass.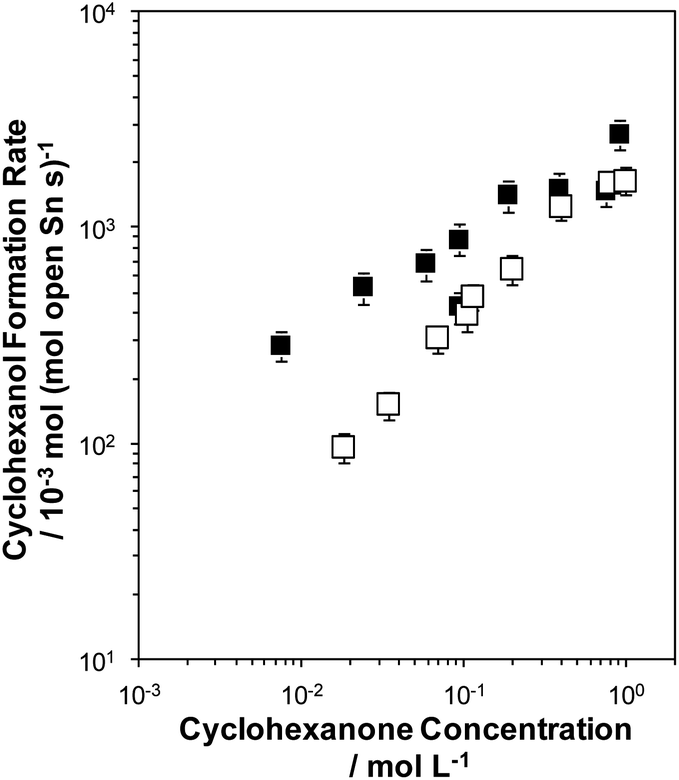 | ||
| Fig. 13 Cyclohexanol formation rates (100 °C, per open Sn site) measured on Sn-BEC (solid squares) and Sn-Beta(F) (open squares) zeolites. Error bars represent propagated experimental uncertainty. | ||
4. Conclusions
Sn-BEC zeolites were successfully synthesized with Sn atoms positionally biased into the D4R of the BEC framework via post-synthetic grafting of Sn into degermanated Ge-BEC zeolites containing low concentrations of Ge in the starting framework (Si/Ge = 155). 19F and 29Si MAS NMR spectra revealed the preferential incorporation of Ge atoms into the D4R secondary building unit of the BEC framework and showed that silanol defect sites (Si(OSi)3–OH), which serve as grafting sites for reactive Sn precursors, form upon degermanation after treatment under mild acid (0.1 M HCl). 31P MAS NMR and DFT calculations of TMPO adsorbed within Sn-BEC (TMPO/Sn = 0.5) revealed distinct chemical shifts for TMPO adsorbed at open and closed Sn sites and at the three distinct T-sites in BEC. These data indicate that Sn atoms are predominantly incorporated at the T1 site of the BEC framework and IR spectra of adsorbed CD3CN, a known titrant of open and closed Sn sites in zeolites, revealed the preferential formation of open Sn sites after post-synthetic grafting of Sn into degermanated BEC zeolites. Rates of the MPVO reaction between 2-butanol and cyclohexanone (normalized per open Sn site, 100 °C) measured on Sn-BEC are comparable to those measured on a conventional Sn-Beta(F) zeolite and show a similar kinetic dependence on cyclohexanone concentration, confirming the formation of catalytically active Sn sites in Sn-BEC. The data presented here highlight synthetic techniques that utilize sacrificial heteroatoms to selectively position catalytic active sites at specific T-sites within a given zeolite framework and demonstrate a method to control the location of adsorption and catalytic active sites in zeolites with atomic precision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Spanish Government-MINECO through ‘‘Severo Ochoa” (SEV-2016-0683), MAT2017-82288-C2-1-P (AEI/FEDER, UE) and RTI2018-101033-B-I00 (MCIU/AEI/FEDER, UE), and by Generalitat Valenciana through AICO/2019/060. JRD and YR-L thank the U.S. Department of Energy, Office of Basic Energy Sciences under Award No. DE-SC0016214 for support. ARF acknowledges the Spanish Government-MINECO for a FPU scholarship (FPU2017/01521). The Electron Microscopy Service of the UPV is acknowledged for their help in sample characterization.Notes and references
- B. C. Knott, C. T. Nimlos, D. J. Robichaud, M. R. Nimlos, S. Kim and R. Gounder, ACS Catal., 2018, 8, 770–784 CrossRef.
- J. Dědeček, E. Tabor and S. Sklenak, ChemSusChem, 2019, 12, 556–576 CrossRef.
- J. Chen, T. Liang, J. Li, S. Wang, Z. Qin, P. Wang, L. Huang, W. Fan and J. Wang, ACS Catal., 2016, 6, 2299–2313 CrossRef.
- A. B. Pinar, C. Márquez-Álvarez, M. Grande-Casas and J. Pérez-Pariente, J. Catal., 2009, 263, 258–265 CrossRef.
- A. B. Pinar, L. Gómez-Hortigüela, L. B. McCusker and J. Pérez-Pariente, Chem. Mater., 2013, 25, 3654–3661 CrossRef.
- Y. Román-Leshkov, M. Moliner and M. E. Davis, J. Phys. Chem. C, 2011, 115, 1096–1102 CrossRef.
- T. Biligetu, Y. Wang, T. Nishitoba, R. Otomo, S. Park, H. Mochizuki, J. N. Kondo, T. Tatsumi and T. Yokoi, J. Catal., 2017, 353, 1–10 CrossRef.
- E. M. Gallego, C. Li, C. Paris, N. Martín, J. Martínez-Triguero, M. Boronat, M. Moliner and A. Corma, Chem.–Eur. J., 2018, 24, 14631–14635 CrossRef.
- C. Li, A. Vidal-Moya, P. J. Miguel, J. Dedecek, M. Boronat and A. Corma, ACS Catal., 2018, 8, 7688–7697 CrossRef CAS.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303–15315 CrossRef CAS.
- J. R. Di Iorio and R. Gounder, Chem. Mater., 2016, 28, 2236–2247 CrossRef CAS.
- M. L. Sarazen, E. Doskocil and E. Iglesia, J. Catal., 2016, 344, 553–569 CrossRef CAS.
- A. J. Jones, R. T. Carr, S. I. Zones and E. Iglesia, J. Catal., 2014, 312, 58–68 CrossRef CAS.
- P. M. Kester, J. T. Miller and R. Gounder, Ind. Eng. Chem. Res., 2018, 57, 6673–6683 CrossRef CAS.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, ACS Catal., 2016, 6, 7311–7325 CrossRef CAS.
- C. Márquez-Alvarez, A. B. Pinar, R. García, M. Grande-Casas and J. Pérez-Pariente, Top. Catal., 2009, 52, 1281–1291 CrossRef.
- E. M. Gallego, M. T. Portilla, C. Paris, A. León-Escamilla, M. Boronat, M. Moliner and A. Corma, Science, 2017, 355, 1051–1054 CrossRef CAS.
- C. Li, C. Paris, J. Martínez-Triguero, M. Boronat, M. Moliner and A. Corma, Nat. Catal., 2018, 1, 547–554 CrossRef CAS.
- B. Notari, Adv. Catal., 1996, 41, 253–334 CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS.
- M. Boronat, A. Corma and M. Renz, J. Phys. Chem. B, 2006, 110, 21168–21174 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- Y. Román-Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566–1580 CrossRef.
- M. Moliner, Dalton Trans., 2014, 43, 4197–4208 RSC.
- A. Corma and M. Renz, Chem. Commun., 2004, 550–551 RSC.
- O. de la Torre, M. Renz and A. Corma, Appl. Catal., A, 2010, 380, 165–171 CrossRef CAS.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109 CrossRef.
- J. Dijkmans, W. Schutyser, M. Dusselier and B. F. Sels, Chem. Commun., 2016, 52, 6712–6715 RSC.
- R. Gounder and M. E. Davis, J. Catal., 2013, 308, 176–188 CrossRef.
- M. A. Camblor, A. Corma and S. Valencia, Chem. Commun., 1996, 2365 RSC.
- R. Martínez-Franco, C. Paris, M. E. Martínez-Armero, C. Martínez, M. Moliner and A. Corma, Chem. Sci., 2016, 7, 102–108 RSC.
- M. M. J. Treacy and J. M. Newsam, Nature, 1988, 332, 249–251 CrossRef.
- S. R. Bare, S. D. Kelly, W. Sinkler, J. J. Low, F. S. Modica, S. Valencia, A. Corma and L. T. Nemeth, J. Am. Chem. Soc., 2005, 127, 12924–12932 CrossRef.
- J. D. Lewis, M. Ha, H. Luo, A. Faucher, V. K. Michaelis and Y. Román-Leshkov, ACS Catal., 2018, 8, 3076–3086 CrossRef.
- M. Boronat, P. Concepcion, A. Corma, M. Renz and S. Valencia, J. Catal., 2005, 234, 111–118 CrossRef.
- J. S. Bates, B. C. Bukowski, J. W. Harris, J. Greeley and R. Gounder, ACS Catal., 2019, 9, 6146–6168 CrossRef.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef.
- J. M. Newsam, M. M. J. Treacy, W. T. Koetsier and C. B. De Gruyter, Proc. R. Soc. London, Ser. A, 1988, 420, 375–405 Search PubMed.
- P. A. Wright, W. Zhou, J. Pérez-Pariente and M. Arranz, J. Am. Chem. Soc., 2005, 127, 494–495 CrossRef.
- A. Corma, M. T. Navarro, F. Rey, J. Rius and S. Valencia, Angew. Chem., Int. Ed., 2001, 40, 2277–2280 CrossRef.
- G. Sastre, J. A. Vidal-Moya, T. Blasco, J. Rius, J. L. Jordá, M. T. Navarro, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2002, 41, 4722–4726 CrossRef.
- G. Zhang, P. Feng, W. Zhang, H. Liu, C. Wang, H. Ma, D. Wang and Z. Tian, Microporous Mesoporous Mater., 2017, 247, 158–165 CrossRef.
- A. Rodríguez-Fernández, F. J. Llopis, C. Martínez, M. Moliner and A. Corma, Microporous Mesoporous Mater., 2018, 267, 35–42 CrossRef.
- P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris and J. Čejka, Chem. Soc. Rev., 2015, 44, 7177–7206 RSC.
- A. Rodríguez-Fernández, P. Atienzar, C. Martínez, Y. Román-Leshkov and M. Moliner, Chem. Mater., 2019, 31, 7723–7731 CrossRef.
- M. Moliner, P. Serna, Á. Cantín, G. Sastre, M. J. Díaz-Cabanas and A. Corma, J. Phys. Chem. C, 2008, 112, 19547–19554 CrossRef.
- Á. Cantín, A. Corma, M. J. Díaz-Cabañas, J. L. Jordá, M. Moliner and F. Rey, Angew. Chem., Int. Ed., 2006, 45, 8013–8015 CrossRef.
- J. D. Lewis, S. Van de Vyver and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 9835–9838 CrossRef.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119, 525 Search PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Had and D. J. Fox, Gaussian 09., Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS.
- A. Zheng, S.-B. Liu and F. Deng, Chem. Rev., 2017, 117, 12475–12531 CrossRef CAS.
- H. Y. Luo, D. F. Consoli, W. R. Gunther and Y. Román-Leshkov, J. Catal., 2014, 320, 198–207 CrossRef CAS.
- J. W. Harris, J. S. Bates, B. C. Bukowski, J. Greeley and R. Gounder, ACS Catal., 2020, 10, 9476–9495 CrossRef CAS.
Footnote |
| † These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |