
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of pentasubstituted 2-aryl pyrroles from boryl and stannyl alkynes via one-pot sequential Ti-catalyzed [2 + 2 + 1] pyrrole synthesis/cross coupling reactions†
Yukun
Cheng
 ,
Channing K.
Klein
and
Ian A.
Tonks
*
,
Channing K.
Klein
and
Ian A.
Tonks
*
Department of Chemistry, University of Minnesota – Twin Cities, 207 Pleasant St SE, Minneapolis, Minnesota 55455, USA. E-mail: itonks@umn.edu
First published on 8th September 2020
Abstract
Multisubstituted pyrroles are commonly found in many bioactive small molecule scaffolds, yet the synthesis of highly-substituted pyrrole cores remains challenging. Herein, we report an efficient catalytic synthesis of 2-heteroatom-substituted (9-BBN or SnR3) pyrroles via Ti-catalyzed [2 + 2 + 1] heterocoupling of heteroatom-substituted alkynes. In particular, the 9-BBN-alkyne coupling reactions were found to be very sensitive to Lewis basic ligands in the reaction: exchange of pyridine ligands from Ti to B inhibited catalysis, as evidenced by in situ11B NMR studies. The resulting 2-boryl substituted pyrroles can then be used in Suzuki reactions in a one-pot sequential fashion, resulting in pentasubstituted 2-aryl pyrroles that are inaccessible via previous [2 + 2 + 1] heterocoupling strategies. This reaction provides a complementary approach to previous [2 + 2 + 1] heterocouplings of TMS-substituted alkynes, which could be further functionalized via electrophilic aromatic substitution.
Introduction
Pyrroles are important heterocycles found in diverse applications from pharmaceuticals to conducting materials.1–6 However, their ubiquity belies significant challenges in the facile synthesis of highly substituted pyrrole cores. Many of the synthetic routes such as the Paal–Knorr condensation require multi-steps backbone synthesis, which add difficulties to late-stage substituent diversification.7,8 Multicomponent reactions provide a shortcut to the construction of structures with high complexity. A series of pioneering studies has been reported by Odom on Ti-catalyzed multicomponent pyrrole synthesis based on hydroamination and iminoamination.9–11 Following a slightly different strategy, we recently developed a multicomponent [2 + 2 + 1] Ti-catalyzed pyrrole forming reaction that yields the heterocycle in a single step.12 Chemo- and regioselective intermolecular reactions can be achieved via the heterocoupling of trialkylsilyl-protected alkynes, which selectively engage in migratory insertion into a key azatitanacyclobutene [2 + 2] cycloadduct intermediate.13 (Fig. 1, top).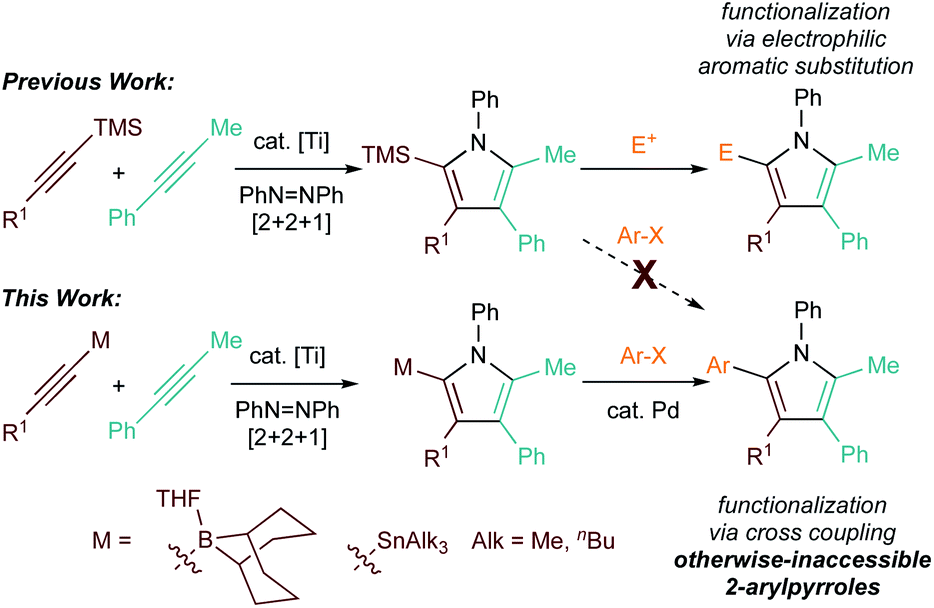 | ||
| Fig. 1 Heterocoupling strategies for selective [2 + 2 + 1] pyrrole synthesis. | ||
Although the TMS-substituted pyrrole heterocoupling products were good candidates for further diversification through electrophilic aromatic substitution of the electron-rich silylpyrrole, we were not able to directly install aryl groups into either the 2- or 5-position around the pyrrole. This limitation arises from the polarization of the Ti-imido bond in [2 + 2] cycloaddition, as well as the limited utility of TMS-substituted arenes in Csp2–Csp2 bond forming reactions. Thus, we envisioned that the development of other heteroatom-substituted alkyne heterocoupling reactions would lead to alternative strategies for pyrrole diversification.
Given the enormous library of well-established group to metal-catalyzed cross coupling reactions, we were interested in the direct synthesis of pyrroles with heteroatoms that could potentially serve as good transmetallation partners.14–21 Herein, we report the application of B-alkynyl 9-borabicyclo[3,3,1]nonanes and alkynyl stannanes in selective Ti-catalyzed [2 + 2 + 1] pyrrole synthesis (Fig. 1, bottom). These reactions provide efficient methods for the construction of versatile polysubstituted pyrrole building blocks, and also provide the opportunity for further diversification into otherwise-inaccessible 2-arylpyrroles through one-pot alkyne cyclization/Suzuki coupling reactions. Preliminary results with other heteroatom-substituted alkynes such as B-alkynyl pinacolborane and copper acetylides are also presented.
Results and discussion
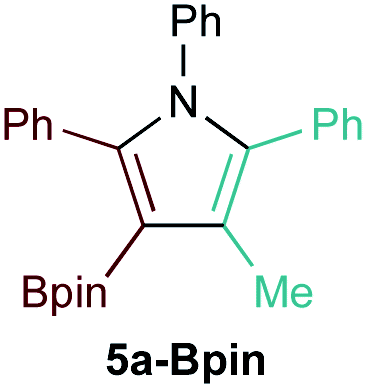
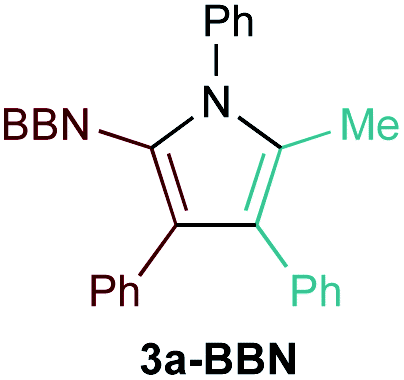
First, several potential heteroatom-substituted alkynes were examined as candidates for the [2 + 2 + 1] reaction, focusing on heteroatomic groups that could later be good transmetallation partners in cross-coupling catalysis (Table 1). The functional groups involved in the initial screen included boronic acid pinacol ester (Table 1, entry 1, 1a-Bpin) and the THF adduct of 9-borabicyclo[3,3,1]nonane (Table 1, entry 2, 1a-BBN), SnMe3 (Table 1, entry 3, 1a-SnMe3), and Cu (Table 1, entry 4, 1a-Cu). Initial reaction conditions were based off from previously successful conditions for TMS-substituted alkyne substrates,13 using chloride-based Ti catalysts that are typically most robust for [2 + 2 + 1] reactions.22,23 All new heteroatom-substituted reactions resulted in significantly lower yields than the corresponding TMS-substituted alkyne reactions, highlighting the challenges of conserving a reactive transmetallating agent through another organometallic transformation.|
|
||||
|---|---|---|---|---|
| Entry | X | Product | Yield | Selectivityb |
| a Conc. = 0.2 M. b Selectivity with respect to all heterocoupling pyrrole regioisomer products. Selectivity = 3a-M/(4a-M + 5a-M). In parenthesis: selectivity with respect to all possible pyrrole products. Selectivity in parenthesis = 3a-M/(4a-M + 5a-M + homocoupled products of 2). c Selectivities calculated for major heterocoupling product 5a-M instead of 3a-M. d t = 16 h. e t = 20 h. f Other pyrrole products cannot be quantified due to their low yield and peak overlapping. | ||||
| 1d |

|
|
19% | 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (1.1:1)c :1 (1.1:1)c |
| 2e |

|
|
7% | 22.3:1 (12.5:1) |
| 3e |
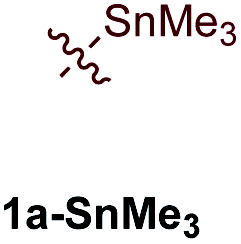
|
|
51% | 6.4:1 (4.5:1) |
| 4d |

|
|
7% | n.d.f |

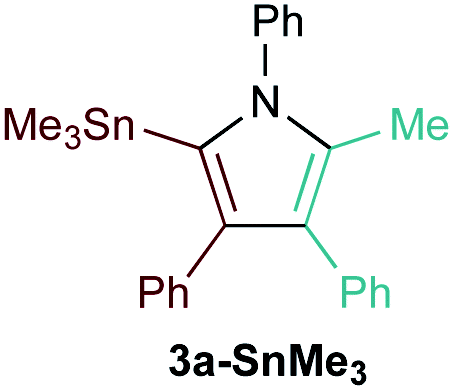
The reaction of PhCCBpin (1a-Bpin) with PhCCMe yielded 3-Bpin-substituted pyrrole 5a-Bpin as the major product of the reaction (Table 1, entry 1); however, the heterocoupling selectivity with respect to 3a-Bpin and 4a-Bpin (2.5:1) and overall selectivity toward 5a-Bpin (1.1:1) was poor. Additionally, there was obvious decomposition of the Bpin moiety, indicated by the observation of white Ti-oxo precipitates. This leads us to speculate that oxophilic Ti may be transmetallating or otherwise reacting with the Bpin B–O bonds.24 Further optimization attempts with Bpin-substituted alkynes were unsuccessful (Fig. S11†). Although alkynyl borates exhibited compatibility issues with our catalytic system, a recent report from Schafer has demonstrated a borane-functionalized alkyne as a hydroamination substrate in Ti hydroamination catalysis.25 Thus, we next examined PhCC-BBN·THF (1a-BBN) (Table 1, entry 2). Although 1a-BBN gave a low yield of 3a-BBN as the major product, both the heterocoupling selectivity and overall selectivity of the reaction were very high. Retention of the 9-BBN moiety in this reaction was encouraging, given the diverse modes of reactivity and transmetallation of the boryl unit with transition metals and unsaturated species.26–31 In fact, there are no reports of organometallic reactions of 9-BBN-substituted alkynes that retain the 9-BBN group. Similar to 1a-BBN, reaction of PhCCSnMe3 (1a-SnMe3) resulted in the chemo- and regioselective formation of 3a-SnMe3 (Table 1, entry 3). 3a-SnMe3 is also stable to aqueous workup, making stannyl alkynes another attractive candidate class for optimization and method development. Regioselectivity in these reactions results from the polarized C–C triple bond (Fig. 2).32–34 In the case of 9-BBN, polarization is a result of the B mesomeric effect,26 while for SnMe3 polarization results from hyperconjugation between σSn-R and 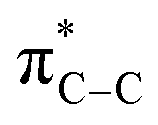 in a manner similar to TMS-protected alkynes.13
in a manner similar to TMS-protected alkynes.13
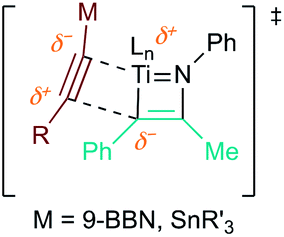 | ||
| Fig. 2 Alkyne polarization results in high regioselectivity for 2nd insertion into the putative azatitanacyclobutadiene intermediate. | ||
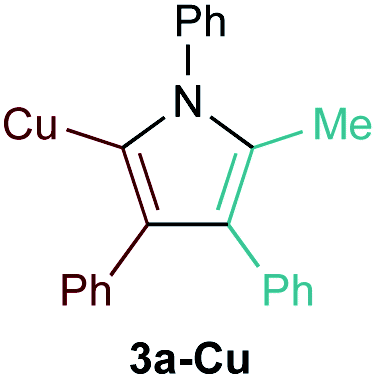
[PhCCCu]n (1a-Cu) exhibited excellent regioselectivity for the formation of 3a-Cu (Table 1, entry 4; Fig. S10†); however, the yield and overall chemoselectivity for the heterocoupled product was very low owing to the insolubility of polymeric 1a-Cu. Further, significant protodecupration occurred in all attempts with 1a-Cu, hampering potential utility. Despite these initial challenges with Cu, a recent report from Schafer on Ti-catalyzed hydroamination of NHC-Cu alkynes indicates that alkynylcuprates could yet be good candidates for [2 + 2 + 1] pyrrole synthesis.35
Having identified 9-BBN- and Sn-substituted alkynes as potential heterocoupling candidates, we next optimized these reactions and explored their substrate scope. Optimization experiments for PhCC-BBN·THF (1a-BBN) are presented in Table 2, while optimization of PhCC-SnMe3 (1a-SnMe3) are presented in Table S2.† Increasing Ti catalyst loading to 10% and changing the solvent from PhCF3 to C6D5Br resulted in significant increases to the yield of 3a without erosion of the overall selectivity. Under these optimized conditions, the reactions were completed within 0.5 h (Table 2, entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | % [Ti]b | Solvent | [py] equiv.c | Yieldd (selectivitye) |
| a Conc. = 0.2 M. b [PhNNPh] was adjusted coordinatingly to the change in [Ti] to keep the nitrene equivalent as 1, on basis of the relationship [nitrene] = [Ti] + 2[PhNNPh]. c Total equivalent of pyridine in the reaction. d Yield determined by GC-FID. e Selectivity with respective to all possible pyrrole products. f t = 0.5 h. | ||||
| 1 | 5 | PhCF3 | 0.2 | 7% (12.5:1) |
| 2 | 5 | C6D5Br | 0.2 | 22% (6.2:1) |
| 3 | 10 | C6D5Br | 0.4 | 74% (17.1:1) |
| 4 | 15 | C6D5Br | 0.6 | 65% (13.2:1) |
| 5 | 10 | PhCH3 | 0.4 | 67% (19.6:1) |
| 6 | 10 | PhCF3 | 0.4 | 55% (15.7:1) |
| 7 | 10 | PhOCH3 | 0.4 | 20% (9.6:1) |
| 8f | 10 | C6D5Br | 0.4 | 66% (22.7:1) |

Surprisingly, the yield of 3a dropped from 74% to 65% upon increasing the catalyst loading from 10 mol% to 15 mol% (Table 2, entries 3 and 4). We hypothesized that B and Ti may be undergoing dative ligand (THF or py) exchange and that the resulting B-L/Ti-L speciation may be impacting catalysis. Thus, several experiments were conducted where the L donor identities and molar ratios were changed (Fig. 3). First, reaction of 1a-BBN with pyridine-free catalyst [TiCl2(NPh)]n (Fig. 3A) resulted in dramatically lower yields, indicating that pyridine is needed for productive catalysis (in part, at least, due to catalyst solubility). Excess 1a-BBN (Fig. 3B) resulted in a lower yield of 3a, and monitoring by 11B NMR (Fig. S73†) indicated that remaining 1b had abstracted pyridine from the catalyst forming PhCC-BBN·py (1a-BBN-py). Reaction of preformed 1a-BBN-py resulted in very slow conversion to 3a (Fig. 3C). In situ11B NMR analysis of the optimized reaction of 1a-BBN (Table 2, entry 8) and the reaction of 1a-BBN-py are shown in Fig. 4. In both cases, 1a-BBN-py is evident at t = 0 and is not fully consumed at the end of the reaction at t = 0.5 h. These results indicate that a careful stoichiometric balance of pyridine must be struck with these Lewis acidic substrates: the Ti catalyst needs py bound for productive catalysis, but py-bound 1a-BBN-py undergoes significantly slower reaction than THF-bound 1a-BBN or free PhCC-BBN (Fig. 3D).
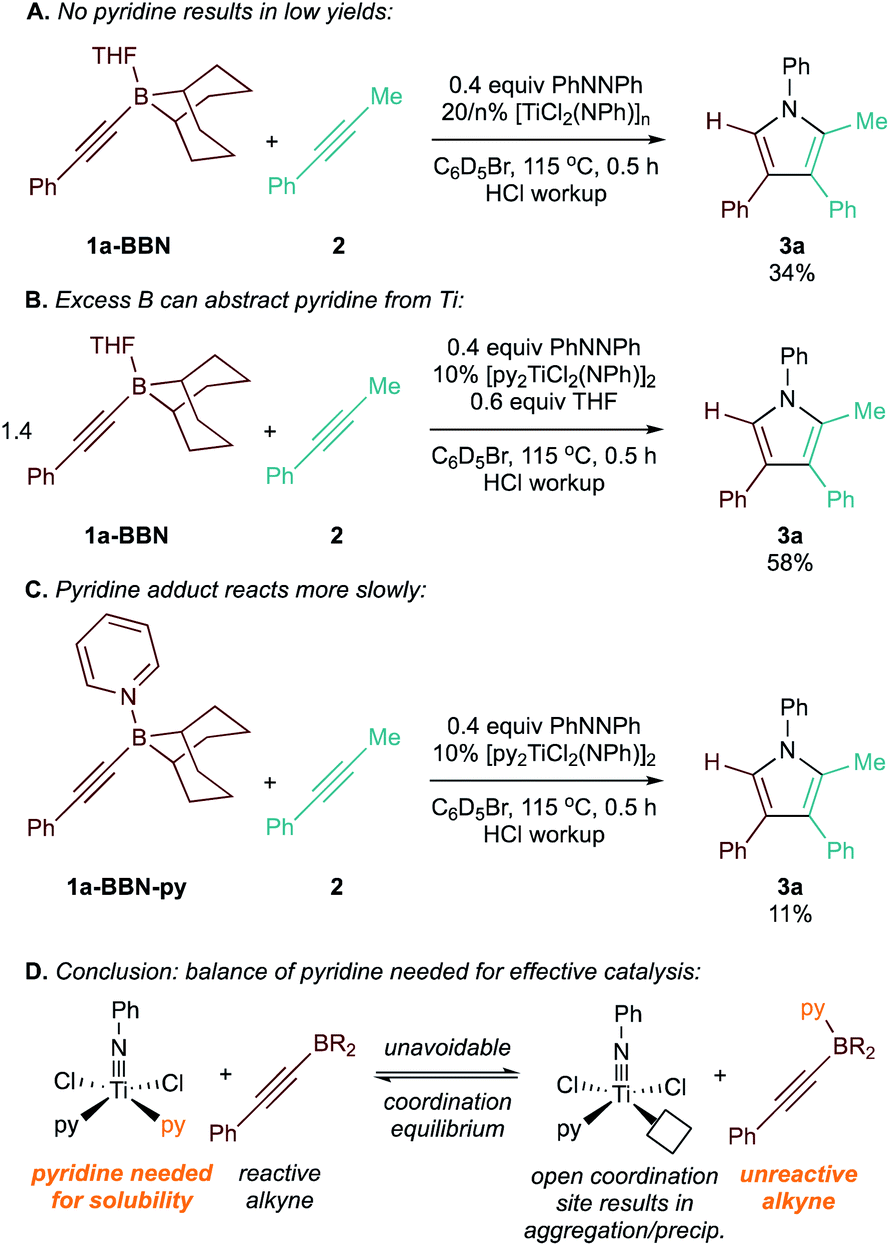 | ||
| Fig. 3 Control reactions studying the effect of L donor on the Ti-catalyzed [2 + 2 + 1] heterocoupling of 1a-BBN with 2. (A) Reaction with 0 equiv. pyridine. (B) Excess 1a-BBN acts as a pyridine scavenger. (C) Pyridine-bound 1a-BBN-py reacts significantly slower than 1a-BBN. (D) Schematic demonstrating pyridine coordination equilibrium effects. | ||
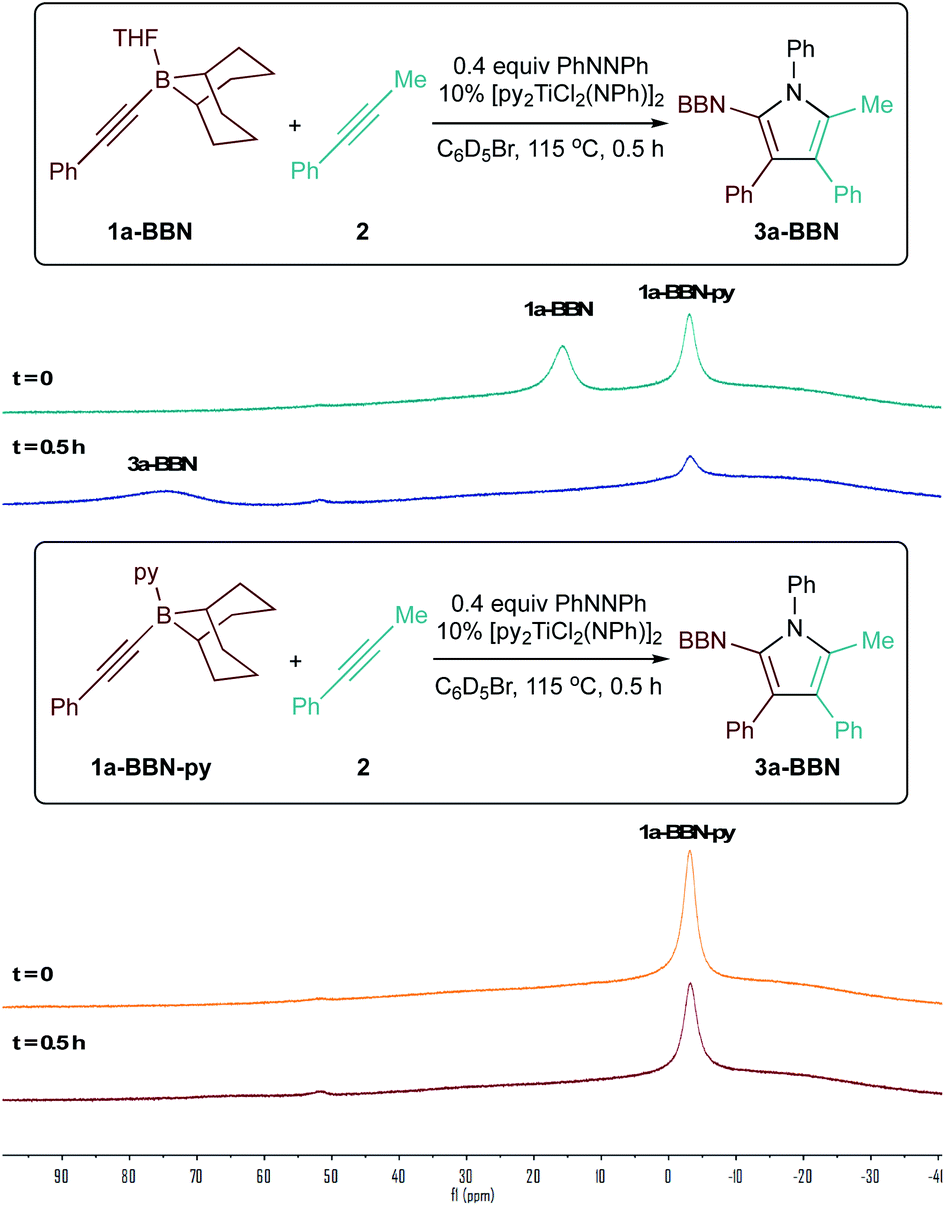 | ||
| Fig. 4 11B NMR study demonstrating pyridine-bound 1a-BBN-py (bottom) reacts more slowly than 1a-BBN (top). | ||
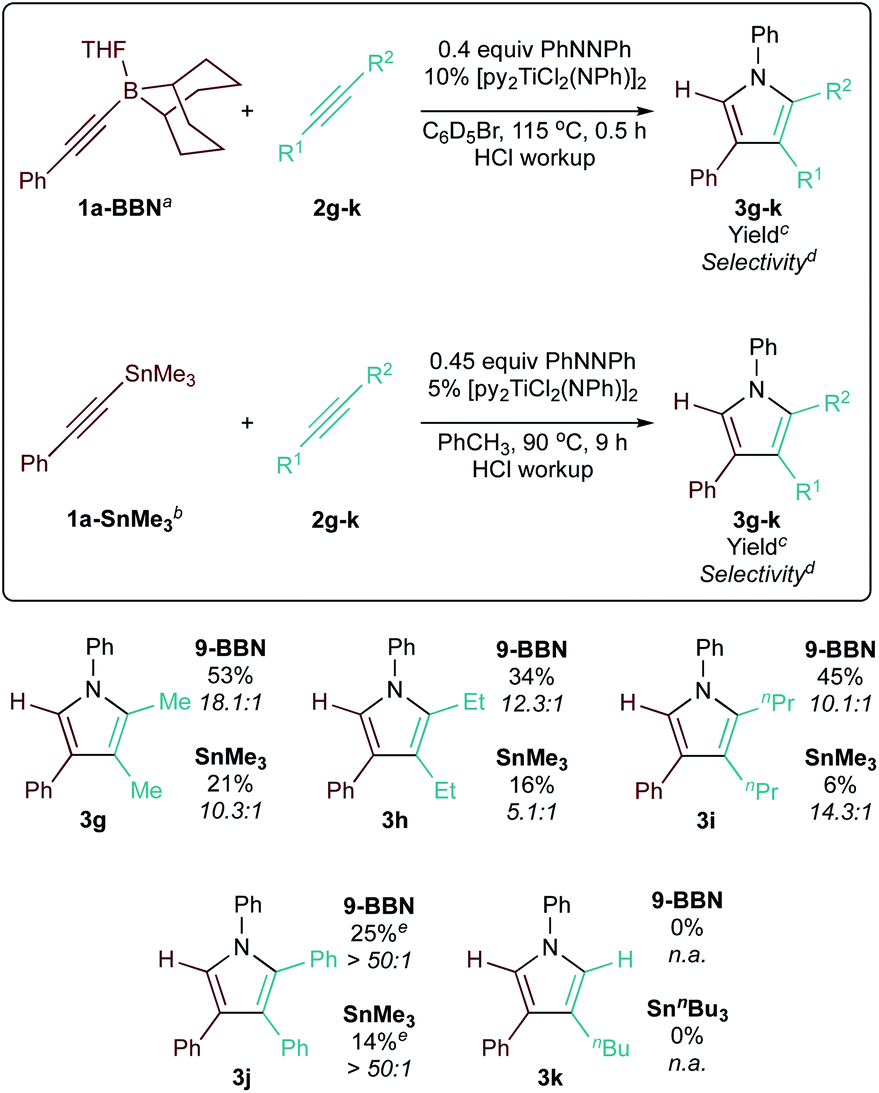
Next, a small scope of 9-BBN-substituted and SnR3-substituted alkynes was examined in heterocoupling with 2 (Table 3). Reactions of the alkynes examined resulted in good selectivity and yield of the corresponding 2-borylpyrroles and 2-stannylpyrroles, which were hydrolyzed with HCl in methanol to simplify analysis. Neither electronics or sterics on the arylalkyne significantly impacted yield and selectivity: electron-rich (1b-BBN, 1b-SnMe3) and electron-deficient (1c-BBN, 1c-SnMe3) arylalkynylboranes reacted equally well, as did the more sterically encumbered o-tolyl-alkynylborane (1d-BBN, 1d-SnMe3). Lastly, the reaction of alkyl-substituted alkynes nBuCC-BBN·THF (1e-BBN) and MeCC-SnnBu3 (1f-SnnBu3) were also highly selective.

Further, we investigated the [2 + 2 + 1] heterocoupling reactions with different hydrocarbon alkynes (Table 4). Various symmetric internal alkynes (2g–j) demonstrated productive heterocoupling reactivity. The non-polarized nature of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of these symmetric alkynes led to lower reactivity in [2 + 2] cycloaddition, resulting in lower yields. However, these less reactive alkynes were also less prone to competitive insertion chemistry, contributing to the higher overall selectivity (>50:1 in the case of 3j) of these reactions. Lastly, a terminal alkyne (2k) was also tested, which resulted in mostly alkyne trimerization instead of productive reactivity (see ESI pg S126†).
C bond of these symmetric alkynes led to lower reactivity in [2 + 2] cycloaddition, resulting in lower yields. However, these less reactive alkynes were also less prone to competitive insertion chemistry, contributing to the higher overall selectivity (>50:1 in the case of 3j) of these reactions. Lastly, a terminal alkyne (2k) was also tested, which resulted in mostly alkyne trimerization instead of productive reactivity (see ESI pg S126†).
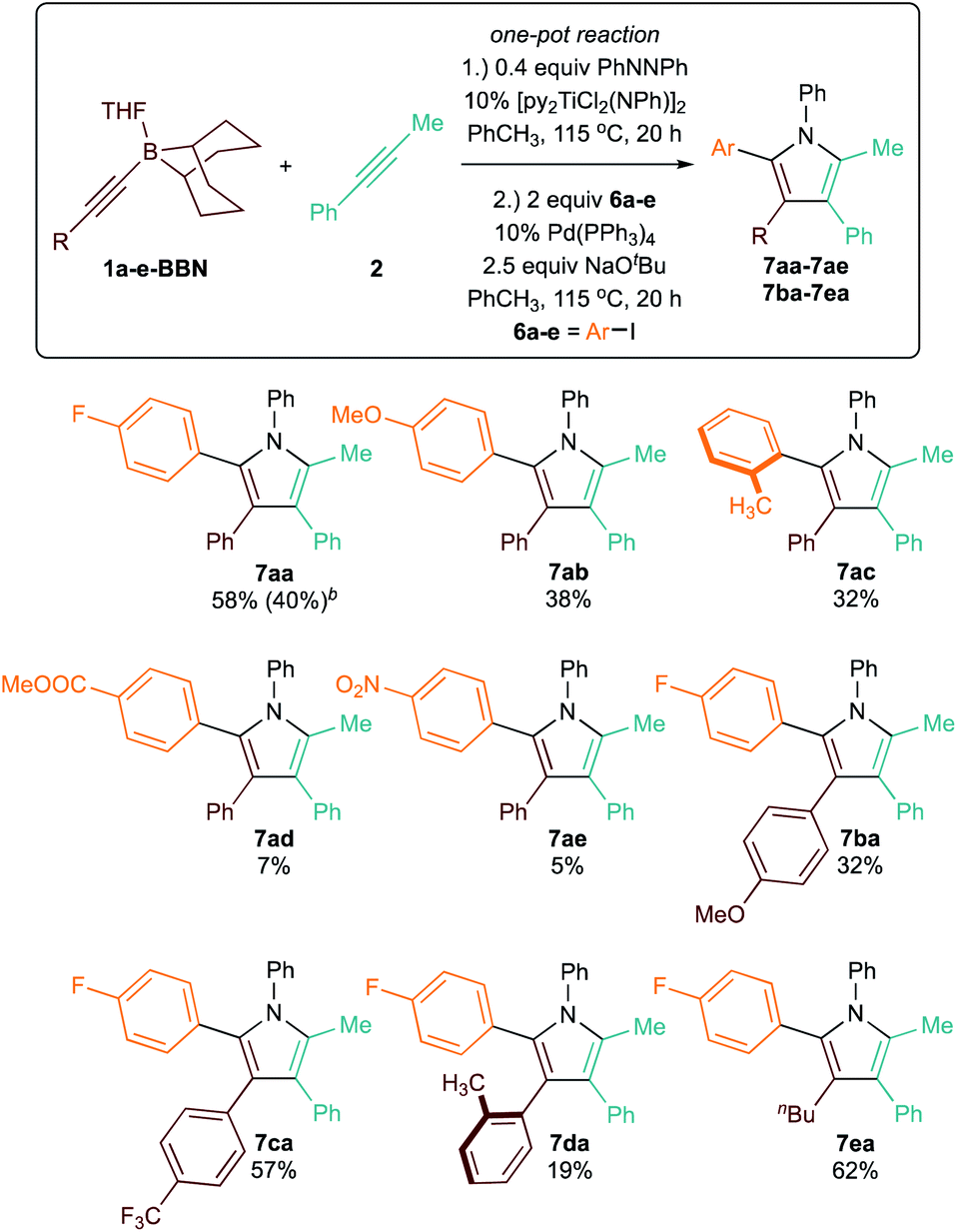
Though 9-BBN is frequently used in Suzuki cross coupling reactions between Csp3-9-BBN and various C–X electrophiles,36–40 the sp2–sp2 Suzuki cross coupling of aryl-9-BBN nucleophiles is rare.41 Nonetheless, we sought to develop a one-pot sequential [2 + 2 + 1] pyrrole synthesis and arylation procedure (Table 5). Reaction of 1a-BBN with 2 in PhCH3in situ produces 3a-BBN; after formation of the pyrrole, addition of p-iodofluorobenzene (6a), 10% Pd(PPh3)4, and 2.5 equiv. NaOtBu generates the pentasubstituted pyrrole product 7aa in good (58%) overall yield. Since these Ti redox catalytic reactions are tolerant of aryl halide functional groups, the reaction can also be carried in the desired aryl halide solvent in similar overall yield (40% for 7aa) and shorter [2 + 2 + 1] reaction time. This one-pot procedure provides convenient access to unsymmetrical pentasubstituted 2-aryl pyrroles that cannot be accessed via previous [2 + 2 + 1] heterocoupling protocols, which could only install aryl groups at the N-, 3-, and 4-positions. Further exploration on the scope of the one-pot pyrrole synthesis/arylation revealed that productive chemistry can be performed on a broad scope of alkynylborane substrates and aryl halides, giving moderate to good yields over the two-step sequence. In general, the substrate scope revealed limited effect on the yield of [2 + 2 + 1] step (as seen in Table 3), but large effects on the arylation step. For example, the arylation step is very sensitive to steric hindrance: formation of 7da, which requires transmetallation42 of a sterically encumbered 3-tolyl-2-(9-BBN)pyrrole, resulted in large amount of protodeborylated 3d (Fig. S123†) and only 19% 7da. In contrast, in the formation of 7ac (where the aryl and tolyl groups are transposed, resulting in a less bulky 3-tolyl-2-(9-BBN)pyrrole), there was a smaller amount of protodeborylated 3a observed (Fig. S141†). Similarly, the arylation to form 7ea is much higher yielding, with only trace amount of 3e formed (Fig. S130†). The aryl ether substrate 6b underwent coupling to form 7ab with moderate yield, although some demethoxylation was evident. Other oxygenated substrates such as 6d and 6e were poor cross coupling partners. Although nitro groups and esters are commonly tolerated in Suzuki reactions, 11B NMR spectroscopic evidence indicates that deleterious chemistry with the 9-BBN group may be taking place (Fig. S150†). In addition to sp2–sp2 Suzuki cross coupling, we also attempted Csp3–C–X cross couplings with aryl-9-BBN. These reactions are also rarely studied, although Fu has several demonstrations with aryl and vinyl 9-BBN substrates.18,43 Unfortunately, rapid protodemetallation of the 9-BBN pyrrole was observed in all attempts.
Finally, given that 9-BBN and Sn alkynes undergo coupling with similar chemo- and regioselectivity to TMS-protected alkynes,13 intramolecular competition experiments were conducted to determine the relative directing ability of the two functional groups compared to TMS (Fig. 5). There are few points of comparison for the regioselectivity of insertion into these types of doubly-functionalized alkynes. Studies of protodemetallation of TMS–CC–M (M = Si, Ge, Sn) indicate that β-hyperconjugative stabilization of putative vinyl carbocation intermediates increases Si < Ge ≪ Sn,44 which could potentially also stabilize the building δ+ on the β-C during 1,2 insertion of the alkyne into the Ti–C bond of the azatitanacyclobutadiene intermediate. If this were the dominant mechanism of regiocontrol, Sn would be a stronger director than Si. Reaction of TMSCC-BBN·THF (1g-BBN) with 2 resulted in formation of 10% 3g-BBN and 25% 4g-BBN (Fig. 5, top), while reaction of TMSCCSnnBu3 (1g-SnnBu3) with 2 resulted in the formation of 6% 3g-SnnBu3 and 12% 4g-SnnBu3. Thus, TMS is a stronger directing group for insertion than both 9-BBN and SnnBu3 (Fig. 5, bottom). Thus, the β-stabilization from the alkyne substituent does not play a dominant role in determining relative selectivity, and other factors such as the relative strength of the forming Ti–CSi bond vs. Ti–CM bond may also be involved: for example, Micalizio has demonstrated that insertion into 2-silyl-3-stannyltitanacyclopropenes will occur on the Ti–CSn bond compared to the Ti–CSi bond.45,46
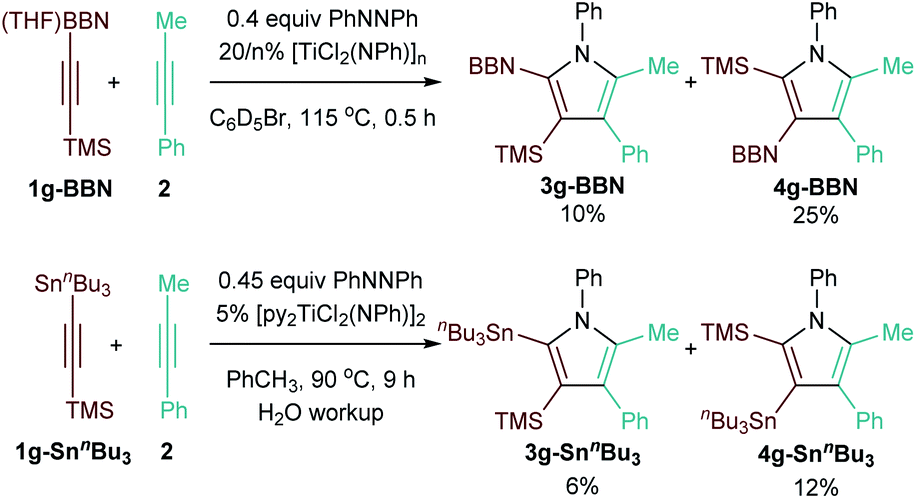 | ||
| Fig. 5 Directing group strength comparisons. | ||
Conclusions
In summary, both alkynyl boranes and stannanes are efficient alkyne heterocoupling partners in titanium-catalyzed [2 + 2 + 1] pyrrole synthesis, generating the corresponding heteroatom-substituted pyrroles with high chemo- and regioselectivity. The resulting products are candidates for further functionalization through cross coupling, as demonstrated by a one-pot sequential [2 + 2 + 1] boryl pyrrole synthesis/Suzuki coupling reaction. These one-pot sequential reactions provide access to unique, highly decorated pentasubstituted pyrroles that are otherwise inaccessible via [2 + 2 + 1] heterocoupling protocols or classical pyrrole synthetic strategies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Institutes of Health (R35GM119457), and the Alfred P. Sloan Foundation (I. A. T. is a 2017 Sloan Fellow). Instrumentation for the University of Minnesota Chemistry NMR facility was supported from a grant through the National Institutes of Health (S10OD011952). We thank B. J. Foley and Prof. O. V. Ozerov (Texas A&M University) for providing an iridium catalyst used for the synthesis of Bpin-substituted alkynes.Notes and references
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS
.
- M. K. Kathiravan, A. B. Salake, A. S. Chothe, P. B. Dudhe, R. P. Watode, M. S. Mukta and S. Gadhwe, The biology and chemistry of antifungal agents: a review, Bioorg. Med. Chem., 2012, 20, 5678–5698 CrossRef CAS
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Biological formation of pyrroles: nature's logic and enzymatic machinery, Nat. Prod. Rep., 2006, 23, 517 RSC
- P. S. Sharma, A. Pietrzyk-Le, F. D'Souza and W. Kutner, Electrochemically synthesized polymers in molecular imprinting for chemical sensing, Anal. Bioanal. Chem., 2012, 402, 3177–3204 CrossRef CAS
- A. Loudet and K. Burgess, BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS
-
G. W. Gribble, in Comprehensive Heterocyclic Chemistry II, Elsevier, 1996, pp. 207–257 Search PubMed
- V. Estévez, M. Villacampa and J. C. Menéndez, Recent advances in the synthesis of pyrroles by multicomponent reactions, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Transition Metal-Mediated Synthesis of Monocyclic Aromatic Heterocycles, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS
- B. Ramanathan, A. J. Keith, D. Armstrong and A. L. Odom, Pyrrole Syntheses Based on Titanium-Catalyzed Hydroamination of Diynes, Org. Lett., 2004, 6, 2957–2960 CrossRef CAS
- E. Barnea, S. Majumder, R. J. Staples and A. L. Odom, One-Step Route to 2,3-Diaminopyrroles Using a Titanium-Catalyzed Four-Component Coupling, Organometallics, 2009, 28, 3876–3881 CrossRef CAS
- C. M. Pasko, A. A. Dissanayake, B. S. Billow and A. L. Odom, One-pot synthesis of pyrroles using a titanium-catalyzed multicomponent coupling procedure, Tetrahedron, 2016, 72, 1168–1176 CrossRef CAS
- Z. W. Gilbert, R. J. Hue and I. A. Tonks, Catalytic formal [2+2+1] synthesis of pyrroles from alkynes and diazenes via TiII/TiIV redox catalysis, Nat. Chem., 2016, 8, 63–68 CrossRef CAS
- H. C. Chiu and I. A. Tonks, Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2+2+1] Pyrrole Synthesis, Angew. Chem., Int. Ed., 2018, 57, 6090–6094 CrossRef CAS
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS
- S. Kotha, K. Lahiri and D. Kashinath, Recent applications of the Suzuki–Miyaura cross-coupling reaction in organic synthesis, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS
- F. Liron, C. Fosse, A. Pernolet and E. Roulland, Suzuki–Miyaura Cross-Coupling of 1,1-Dichloro-1-alkenes with 9-Alkyl-9-BBN, J. Org. Chem., 2007, 72, 2220–2223 CrossRef CAS
- Z. Lu and G. C. Fu, Alkyl-Alkyl Suzuki Cross-Coupling of Unactivated Secondary Alkyl Chlorides, Angew. Chem., Int. Ed., 2010, 49, 6676–6678 CrossRef CAS
- S. L. Zultanski and G. C. Fu, Nickel-Catalyzed Carbon–Carbon Bond-Forming Reactions of Unactivated Tertiary Alkyl Halides: Suzuki Arylations, J. Am. Chem. Soc., 2013, 135, 624–627 CrossRef CAS
- I. Nakamura and Y. Yamamoto, Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS
-
V. Farina, V. Krishnamurthy and W. J. Scott, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1997, pp. 1–652 Search PubMed
- G. Evano, N. Blanchard and M. Toumi, Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS
- Z. W. Davis-Gilbert, X. Wen, J. D. Goodpaster and I. A. Tonks, Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2 + 2 + 1] Pyrrole Synthesis from Alkynes and Azobenzene, J. Am. Chem. Soc., 2018, 140, 7267–7281 CrossRef CAS
- X. Y. See, B. Reiner, X. Wen, T. A. Wheeler, C. Klein, J. Goodpaster and I. Tonks, Iterative Supervised Principal Component Analysis-Driven Ligand Design for Regioselective Ti-Catalyzed Pyrrole Synthesis, 2020, DOI:10.26434/chemrxiv.12284378.v1.
- H.-C. Chiu, X. Y. See and I. A. Tonks, Dative Directing Group Effects in Ti-Catalyzed [2+2+1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling, ACS Catal., 2019, 9, 216–223 CrossRef CAS
- H. Hao, K. A. Thompson, Z. M. Hudson and L. L. Schafer, Ti-Catalyzed Hydroamination for the Synthesis of Amine- Containing p-Conjugated Materials, Chem.–Eur. J., 2018, 24, 5562–5568 CrossRef CAS
- S.-W. Leung and D. A. Singleton, Reactions of Alkynyldihaloboranes with 1,3-Dienes. 1,4-Alkynylborations and Stepwise Diels–Alder Reactions, J. Org. Chem., 1997, 62, 1955–1960 CrossRef CAS
- M. Yamaguchi and I. Hirao, A synthesis of alkynyl azacycloalkanes by the coupling reaction of alkynyl boranes and lactams, Tetrahedron Lett., 1983, 24, 1719–1722 CrossRef CAS
- F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, Borole Formation by 1,1-Carboboration, J. Am. Chem. Soc., 2014, 136, 68–71 CrossRef CAS
- G. Kehr and G. Erker, 1,1-Carboboration, Chem. Commun., 2012, 48, 1839–1850 RSC
- O. Ekkert, O. Tuschewitzki, C. G. Daniliuc, G. Kehr and G. Erker, Reaction of strongly electrophilic alkenylboranes with phosphanylalkynes: rare examples of intermolecular 1,1-alkenylboration reactions, Chem. Commun., 2013, 49, 6992 RSC
- G. Kehr and G. Erker, Advanced 1,1-carboboration reactions with pentafluorophenylboranes, Chem. Sci., 2016, 7, 56–65 RSC
- D. A. Singleton and S. W. Leung, An unprecedented electronic preference for the ‘meta’ product in Diels-Alder reactions of ethynyldialkylboranes. [(Trimethylsilyl)ethynyl]-9-BBN as a reactive and versatile dienophile, J. Org. Chem., 1992, 57, 4796–4797 CrossRef CAS
- M. A. Silva, S. C. Pellegrinet and J. M. Goodman, A DFT Study on the Regioselectivity of the Reaction of Dichloropropynylborane with Isoprene, J. Org. Chem., 2003, 68, 4059–4066 CrossRef CAS
- C. Cauletti, C. Furlani, G. Granozzi, A. Sebald and B. Wrackmeyer, The .sigma.-.pi. interactions in alkynyltin(IV) compounds studies by UV photoelectron spectroscopy and pseudopotential ab initio calculations, Organometallics, 1985, 4, 290–295 CrossRef CAS
- H. Han and L. L. Schafer, Ti Catalyzed Hydroamination: A Direct Functionalization of Cu Acetylide, 2019, DOI:10.26434/chemrxiv.11455269.v1.
- N. Miyaura, T. Ishiyama, H. Sasaki, M. Ishikawa, M. Sato and A. Suzuki, Palladium-catalyzed inter- and intramolecular cross-coupling reactions of B-alkyl-9-borabicyclo[3.3.1]nonane derivatives with 1-halo-1-alkenes or haloarenes. Syntheses of functionalized alkenes, arenes, and cycloalkenes via a hydroboration-coupling sequence, J. Am. Chem. Soc., 1989, 111, 314–321 CrossRef CAS
- T. Ishiyama, S. Abe, N. Miyaura and A. Suzuki, Palladium-Catalyzed Alkyl-Alkyl Cross-Coupling Reaction of 9-Alkyl-9-BBN Derivatives with Iodoalkanes Possessing β-Hydrogens, Chem. Lett., 1992, 21, 691–694 CrossRef
- A. Fürstner and A. Leitner, General and User-friendly Method for Suzuki Reactions with Aryl Chlorides, Synlett, 2001, 2001, 0290–0292 CrossRef
- S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, A Rationally Designed Universal Catalyst for Suzuki–Miyaura Coupling Processes, Angew. Chem., Int. Ed., 2004, 43, 1871–1876 CrossRef CAS
- S. L. Zultanski and G. C. Fu, Catalytic Asymmetric γ-Alkylation of Carbonyl Compounds via Stereoconvergent Suzuki Cross-Couplings, J. Am. Chem. Soc., 2011, 133, 15362–15364 CrossRef CAS
- To the best of our knowledge, there are only three reports on sp2–sp2 Suzuki cross coupling of aryl-9-BBN nucleophiles:
(a) M. Ishikura, I. Oda and M. Terashima, A Simple and Regioselective Preparation of 2- or 3-Substituted Quinoline Derivatives via Dialkylquinolylboranes, Heterocycles, 1985, 23, 2375 CrossRef CAS
- A. J. J. Lennox and G. C. Lloyd-Jones, Selection of boron reagents for Suzuki–Miyaura coupling, Chem. Soc. Rev., 2014, 43, 412–443 RSC
- M. R. Netherton, C. Dai, K. Neuschütz and G. C. Fu, Room-Temperature Alkyl–Alkyl Suzuki Cross-Coupling of Alkyl Bromides that Possess β Hydrogens, J. Am. Chem. Soc., 2001, 123, 10099–10100 CrossRef CAS
- C. Dallaire and M. A. Brook, The β-Effect with Vinyl Cations: Kinetic Study of the Protiodemetalation of Silyl-, Germyl-, and Stannylalkynes, Organometallics, 1993, 12, 2332–2338 CrossRef CAS
- H. Mizoguchi and G. C. Micalizio, Synthesis of Highly Functionalized Decalins via Metallacycle-Mediated Cross-Coupling, J. Am. Chem. Soc., 2015, 137, 6624–6628 CrossRef CAS
- G. C. Micalizio and H. Mizoguchi, The Development of Alkoxide-Directed Metallacycle-Mediated Annulative Cross-Coupling Chemistry, Isr. J. Chem., 2017, 57, 228–238 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. See DOI: 10.1039/d0sc01576h |
| This journal is © The Royal Society of Chemistry 2020 |