
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of the fungal glyceraldehyde-3-phosphate dehydrogenase inhibitor heptelidic acid and mechanism of self-resistance†
Yan
Yan‡§
 a,
Xin
Zang‡
b,
Cooper S.
Jamieson
c,
Hsiao-Ching
Lin¶
a,
K. N.
Houk
c,
Jiahai
Zhou
*b and
Yi
Tang
*ac
a,
Xin
Zang‡
b,
Cooper S.
Jamieson
c,
Hsiao-Ching
Lin¶
a,
K. N.
Houk
c,
Jiahai
Zhou
*b and
Yi
Tang
*ac
aDepartment of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095, USA. E-mail: yitang@ucla.edu
bState Key Laboratory of Bio-organic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai 200032, China. E-mail: jiahai@mail.sioc.ac.cn
cDepartment of Chemistry and Biochemistry, University of California Los Angeles, CA 90095, USA
First published on 19th August 2020
Abstract
Overcoming resistance to bioactive small molecules is a significant challenge for health care and agriculture. As a result, efforts to uncover the mechanisms of resistance are essential to the development of new antibiotics, anticancer drugs and pesticides. To study how nature evolves resistance to highly potent natural products, we examined the biosynthesis and mechanism of self-resistance of the fungal glyceraldehyde-3-phosphate dehydrogenase (GAPDH) inhibitor heptelidic acid (HA). HA is a nanomolar inhibitor of GADPH through the covalent modification of the active site cysteine thiol. The biosynthetic pathway of HA was elucidated, which uncovered the enzymatic basis of formation of the epoxide warhead. Structure–activity relationship study using biosynthetic intermediates established the importance of the fused lactone ring system in HA. The molecular basis of HA inhibiting human GAPDH was illustrated through the crystal structure of Hs-GAPDH covalently bound with HA. A GAPDH isozyme HepG encoded in the HA cluster was characterized to be less sensitive to HA, and therefore contribute to self-resistance for the producing host. Comparison of the crystal structures of human GAPDH and HepG showed mutations both within and remote to the active site can contribute to resistance of inactivation, which was confirmed through mutagenesis. Due to the critical role GAPDH plays in aerobic glycolysis and other cellular functions, knowledge of HA mode of action and self-resistance mechanism could accelerate the development of improved inhibitors.
Introduction
The rapid emergence of resistance to bioactive small molecules has become a major challenge for health care and agriculture.1 Once effective antibiotics, antifungal, anticancer drugs and pesticides are gradually becoming obsolete due to the rise of resistant strains that have acquired mutations in the cellular targets.2,3 Therefore, uncovering and overcoming the resistance mechanisms are of paramount importance for civilization. One approach that can provide insights into how nature evolves resistance in essential housekeeping enzymes is by examining how microbial hosts self-protect against biosynthesized, potent natural products (NPs). Often referred to as the chemical agent in microbial warfare, NPs are synthesized by microbes including both bacteria and fungi, to target essential housekeeping enzymes in antagonistic organisms. To protect against the NPs, the producing hosts have developed different strategies, including the ingenious use of a mutated and functional version of the housekeeping enzyme that is insensitive to the NP, and hence self-resistant.4,5 Gene encoding the self-resistance enzyme is frequently colocalized with the biosynthetic gene cluster (BGC) of a NP, and has been used as a predictive tool for the bioactivity of the NP synthesized by the BGC.6,7 Clearly, nature has evolved self-resistance enzymes from housekeeping enzymes to become resistant to potent NPs. Understanding the molecular differences between sensitive and resistant enzymes of the same catalytic function can therefore provide immense information on the origins and mechanisms of resistance.The central metabolism of microorganisms is a frequent target of NPs. Notably, both bacteria and fungi have evolved terpenoids to inhibit the highly conserved glyceraldehyde-3-phosphate dehydrogenase (GAPDH),8,9 which catalyses the sixth step of glycolysis to convert D-glyceraldehyde 3-phosphate (GAP) to D-glycerate 1,3-bisphosphate (1,3-DPG) (Fig. 1A).10 The mechanism of GAPDH catalysis has been intensively studied and involves a catalytic cysteine in the active site that participates covalently in the oxidative phosphorylation of GAP with the concomitant reduction of NAD+ to NADH.10–12 As an example of convergent evolution, the fungal-derived heptelidic acid (HA, 1, also known as koningic acid and avocettin)8,13 and the bacterial-derived pentalenolactone (2)14 are both potent GAPDH inhibitors that use epoxide warheads to covalently target the thiolate of the cysteine (Fig. 1B and C).15,16 The potent inhibition of human GAPDH (Hs-GAPDH)17 by HA has been used to limit glycolytic flux associated with the Warburg physiology in human cancer cell lines18 and in activated immune cells.19 While the structures of GADPH from different organisms,20–23 including E. coli, yeast, rat and human, have been solved to reveal the overall tetrameric fold and the catalytic environment surrounding the cysteine, there is no structure of GAPDH bound with a specific covalent inhibitor such as HA to date.
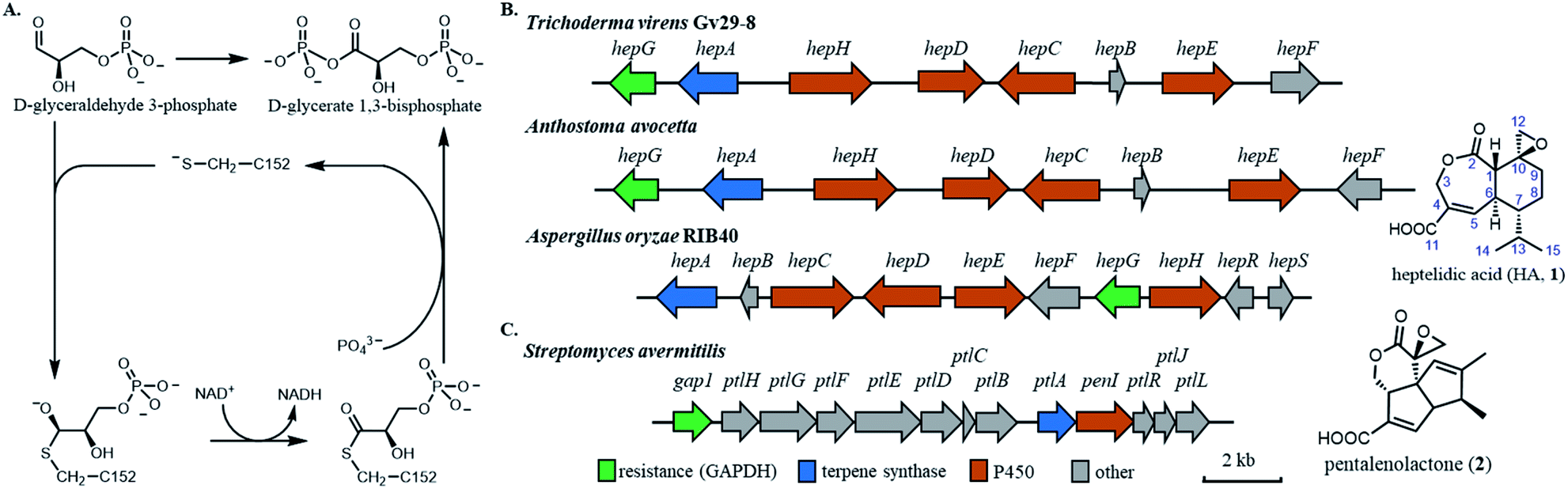 | ||
| Fig. 1 Biosynthetic gene clusters (BGCs) of fungal and bacterial derived GAPDH inhibitors. (A) The reaction mechanism of human GAPDH. (B) BGCs of heptelidic acid from three producing strains (left), and chemical structure of heptelidic acid (1). (C) BGC of pentalenolactone (left) and chemical structure of pentalenolactone (2). | ||
The chemical structures of HA and pentalenolactone contain similar features, such as the exo-epoxide warhead, fused lactone and α,β-unsaturated carboxylic acid. However, the differences in the ring systems clearly indicate divergent biosynthetic pathways that start with the formation of different terpene scaffolds. The biosynthetic pathway of 2 is well-studied by Cane and co-workers.24–29 During the course of these work, a GAPDH homolog Gap1, that is 64% identical to housekeeping enzyme in S. avermitilis, was found in the BGC (Fig. 1C). This enzyme was shown to be insensitive to 2, and was proposed to be the self-resistance enzyme.24 The BGC of HA was recently reported from the fungal producer Aspergillus oryzae (Fig. 1B), which contains a sesquiterpene cyclase and set of cytochrome P450 enzymes.29 The fungal BGC also contains a GAPDH homolog HepG that is 72% identical to the housekeeping GAPDH in A. oryzae. The IC50 of HA towards A. oryzae HepG was ∼60 times higher compared to that of the housekeeping GAPDH,29 implying HepG may also play a self-resistance role during the biosynthesis of HA. Similarly, two GAPDH isozymes from Trichoderma koningii, a different producer of HA, were cloned and shown to have different sensitivity to HA.30
To understand how HA inhibits Hs-GAPDH, and identify mutations that may increase resistance to HA, we obtained the crystal structures of Hs-GAPDH covalently modified with HA, as well as that of HepG. We also delineated the biosynthetic pathway of HA and used the biosynthetic intermediates to probe the structural–activity relationship in Hs-GAPDH inhibition. These studies enabled us to identify key mutations in Hs-GAPDH and HepG that play critical roles in conferring resistance.
Results
Characterization of heptelidic acid biosynthetic pathway
The HA BGC encodes a sesquiterpene synthase (hepA), four P450s (hepC, hepD, hepE and hepH) and a putative antibiotic biosynthesis monooxygenase (hepB) (Fig. 1B and Table S1†). The functions of two transcription factors (hepR and hepS) in regulating HA biosynthesis have been verified.29 Bioinformatics analysis showed HA BGC is conserved in other producing strains, including Trichoderma virens Gv29-8 and Anthostoma avocetta (Fig. 1B).13,31 To elucidate the functions of the biosynthetic enzymes, and isolate potential biosynthetic intermediates, we chose to work with the T. virens hep BGC. This is due to our ability to observe production of HA when the strain was grown in potato dextrose broth (PDB) at 28 °C while shaking at 250 rpm (Fig. 2A). When the culturing condition was switched to stationary incubation, we observed the appearance of three new metabolites 3–5 with molecular weights that indicate these may be biosynthetic intermediates of HA (Fig. 2A). These new compounds were isolated, and their structure were determined by NMR (Fig. 2D, S3 and Table S5†). All three compounds are decalin terpenoids that are less oxidized compared to HA. Based on the levels of oxidative modifications, we reason the exo-olefin containing 3 is the earliest intermediate of the three, and can be epoxidized to 4. Oxidation of the C2 hydroxyl group in 4 gives the ketone 5, which can be ring-expanded to 1 by Baeyer–Villiger oxidation. These modifications are proposed to be catalysed by cytochrome P450s enzymes encoded in the hep BGC, requiring molecular oxygen as the oxidant.32,33 The emergence of 3–5 under stationary culture may hence be a result of lower aeration of the stationary culture compared to under shaking conditions.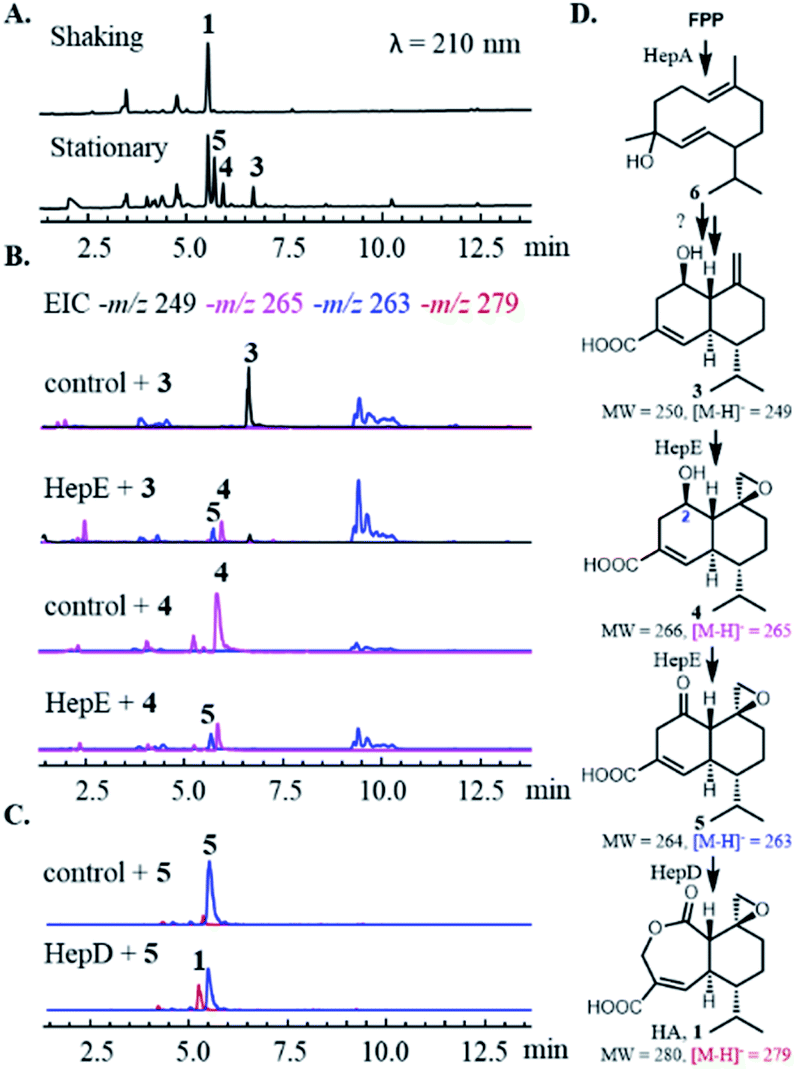 | ||
| Fig. 2 Characterization of heptelidic acid biosynthesis. (A) Fermentation of T. virens (top) in shaking and stationary (bottom) culturing conditions. (B) In vivo bioconversion of 3 and 4 using S. cerevisiae expressing HepE and cytochrome P450 reductase. The control strain is S. cerevisiae only expressing cytochrome P450 reductase. (C) In vitro biochemical assay of 5 using microsomal fractions of S. cerevisiae expressing HepD and cytochrome P450 reductase. The control assay is performed using microsome of S. cerevisiae only expressing cytochrome P450 reductase. (D) The proposed biosynthetic pathway of heptelidic acid. | ||
We then characterized the biosynthetic enzymes in the BGC, starting with the terpene cyclase HepA. The FLAG-tagged HepA was obtained upon heterologous expression in Saccharomyces cerevisiae RC01. In the presence of 1 mM farnesyl pyrophosphate (FPP) and 5 mM MgCl2, HepA was able to convert FPP to 6 as the only product, which has the identical GC-MS spectrum to germacrene D-4-ol (Fig. S2†).34 The result is consistent with previous isotopically labelled acetate incorporation experiments, which indicated HA is derived from a germacrene-type sesquiterpene precursor.35 The same compound 6 was observed in the culturing media of Saccharomyces cerevisiae expressing HepA (Fig. S2B†).
To assign catalytic functions to the P450 enzymes in the hep BGC, cDNAs of hepC, hepD, hepE, hepH, as well as the T. virens electron transfer partner cytochrome P450 reductase (CPR) were obtained. The cytochrome P450s were individually expressed in S. cerevisiae RC01 together with the CPR, all under the adh2 promoter.36 Compounds 3–6 were then supplied to the yeast strains as substrates for biotransformation. After 12 h, the cultures were extracted with ethyl acetate and subjected to HPLC-MS analysis. Unexpectedly, none of the strains were able to modify 6 into a more oxidized substrate, such as 3. Repeated attempts with coexpression of multiple P450s, addition of other hep genes, and the use of P450-containing microsomal fractions were also unsuccessful, thereby precluding the determination of the initial oxidative steps. We proposed that unidentified or unclustered enzymes may be involved in modification of 6, or 6 may be an off-pathway shunt product formed under in vitro and yeast assay conditions.
With 3 as the substrate, only the yeast strain expressing HepE was able to transform the compound into the epoxide 4, and further oxidize into the ketone 5 (Fig. 2B). Directly supplying 4 led to transformation by HepE into 5, thereby establishing HepE is a bifunctional P450 in oxidizing the upper periphery of the terpene (Fig. 2D). When 5 was supplied to the yeast strains expressing one of the other P450s, however, the proposed final oxidation of 5 to HA was not detected. We suspected that formation of HA under in vivo conditions would inhibit yeast GADPH and arrest metabolism. In addition, any HA produced would be covalently bound to GADPH and preclude detection from the organic extract.15 Therefore, we performed the assays using the corresponding yeast microsomal fractions containing the individual P450s and CPR. We observed HepD-containing microsomes were able to convert 5 into HA (Fig. 2C). This result is therefore consistent with previous isotope labelling experiment, which suggested a Baeyer–Villiger oxidation reaction is required to generated the lactone ring of HA.35 Although P450-mediated Baeyer–Villiger oxidation has precedence in catalysing castasterone to brassinolide in biosynthesis of the plant NP brassinosteroid, HepD represents the first microbial P450 discovered to catalyze this reaction.37 In comparison, formation of the lactone in 2 is catalysed by a FAD-dependent monooxygenase PntE that is more commonly associated with Baeyer–Villiger reactions.28 It's worth noting that as in PntE, HepD catalyses migration of the less substituted methylene from the Criegee intermediate associated with Baeyer–Villiger oxidation, indicating enzymatic control of regioselectivity.38
Inhibition kinetics of HA and biosynthetic intermediates
In the first step of GAPDH catalysis, the deprotonated thiolate of C152 attacks the carbonyl of GAP to form a hemithioacetal intermediate, which is then oxidized to the thioester via hydride transfer to the cofactor NAD+. Following cofactor exchange in which NADH is replaced with a new molecule of NAD+, the high-energy thioester is then attacked by inorganic phosphate to release 1,3-DPG (Fig. 1A). Upon binding of HA, a proposed attack of the thiolate on the exo-epoxide leads to covalent and irreversible inactivation of GADPH.15,16 The structures of both HA and 2 contain a lactone ring formed by the Baeyer–Villiger reaction, hinting that the ester oxygen atom plays an important role in binding to GADPH. Furthermore, it is intriguing that the overall size of HA, with its 6,7-bicyclic ring system (15 carbons), is considerably bulkier than the linear substrate GAP (3 carbons).To assess the structural–activity-relationship of HA binding to GAPDH, we assayed the inhibition parameters of HA, 4 and 5 on Hs-GAPDH (accession number: NP_001276674.1) using the Kitz–Wilson method.39 Purified Hs-GAPDH was first incubated with the compound individually at various concentrations (Fig. S4A†). This was followed by a 20-fold dilution of the GAPDH–inhibitor mixture and initiating the GAPDH reaction via addition of GAP. Reaction progress was measured by the appearance of NADH with UV at λ = 340 nm. All three epoxide-bearing compounds showed time-dependent, irreversible inhibition of Hs-GAPDH. Semilog plots of the fraction of residual GAPDH activity against the concentration of the inhibitors showed pseudo-first-order inactivation (Fig. S5†). The binding constant KI, the maximum potential rate of covalent bond formation kinact, and the overall inactivation rate constant kinact/KI that indicates the rate to generate covalent GAPDH–inhibitor complex, of each inhibitor were determined and are shown in Table 1.
| Compound | GAPDH | K I (mM) | k inact (min−1) | k inact/KI (min−1 mM−1) | K M, GAP (mM) | k cat, GAP (min−1) | k cat/KM (min−1 mM−1) |
|---|---|---|---|---|---|---|---|
| 1 | Hs-GAPDH | 0.039 ± 0.005 | 0.81 ± 0.04 | 21 | 0.24 ± 0.02 | 1300 ± 100 | 5400 |
| 5 | Hs-GAPDH | 0.15 ± 0.03 | 0.19 ± 0.01 | 1.3 | |||
| 4 | Hs-GAPDH | 4.7 ± 0.5 | 0.057 ± 0.002 | 0.012 | |||
| 1 | Tv-GAPDH | 1.5 ± 0.1 | 0.31 ± 0.01 | 0.21 | 1.1 ± 0.2 | 6900 ± 1300 | 6300 |
| 1 | HepG | 1.2 ± 0.4 | 0.11 ± 0.01 | 0.09 | 0.28 ± 0.05 | 2500 ± 400 | 8900 |
| 1 | Hs-GAPDH-T177A | 0.032 ± 0.006 | 0.19 ± 0.01 | 5.8 | 0.039 ± 0.08 | 770 ± 40 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 1 | Hs-GAPDH-A232S-F233V | 0.060 ± 0.012 | 0.42 ± 0.04 | 7.0 | 0.49 ± 0.21 | 460 ± 120 | 940 |
| 1 | Hs-GAPDH-L203A | 0.16 ± 0.06 | 0.23 ± 0.05 | 1.4 | 0.19 ± 0.02 | 1000 ± 100 | 5300 |
| 1 | HepG-A201L | 0.047 ± 0.006 | 0.20 ± 0.01 | 4.3 | 0.26 ± 0.05 | 1300 ± 200 | 5000 |
Hs-GAPDH is rapidly inhibited by HA with a KI of 40 μM compared to the KM of 240 μM towards GAP. The kinact/KI of inhibition of Hs-GAPDH by 5 is 16-fold lower than HA, with nearly 4-fold increase in KI and 4-fold decrease in kinact. Therefore, the inserted lactone oxygen atom in HA has a significant effect on inhibitor binding and subsequent inactivation. A more significant, 1750-fold decrease in kinact/KI of 4 compared to HA, which corresponds to over 100-fold decrease compare to that of 5, was observed. This indicates the importance of the C2 ketone group in binding to GAPDH, in addition to serving as a prerequisite functional group for the Baeyer–Villiger oxidation.
We next assayed the properties of the proposed self-resistance enzyme HepG29 in the hep BGC. We cloned and expressed both HepG and the putative housekeeping copy of GAPDH in T. virens (Tv-GAPDH, accession number: XP_013958680.1), which share 73% sequence identity (Fig. S12†). HA showed time-dependent and irreversible inhibition towards both T. virens enzymes (Fig. S6 and S7†), indicating HA can bind and form covalent adduct with the cysteine residues. However, the overall inhibition rate constants kinact/KI of HepG and Tv-GAPDH are 233 and 100-fold lower than that of Hs-GAPDH, respectively (Table 1). In the absence of HA, both enzymes exhibited similar kcat/KM (with respect to GAP) to that of Hs-GAPDH (Table 1). This indicates the amino acid residue(s) responsible for decreased sensitivity of HA in T. virens enzymes may be orthogonal to those responsible for the catalytic efficiency of GAPDH (Table 1).
Crystal structure of Hs-GAPDH and heptelidic acid complex
To understand the mechanism of action of HA, we solved the crystal structure of HA-bound Hs-GAPDH complex at 1.82 Å resolution through molecular replacement method (Fig. 3A and Table S6†). Similar to the previously reported human liver GAPDH crystal structure (PDB: 1ZNQ, RMSD = 0.304 Å for 332 Cα atoms in subunit O), the complex contains four identical subunits (chain O, P, Q and R) arranged as homotetramer in an asymmetric unit, in which chain O and chain Q are in holo form and each has a NAD+ cofactor in the active site (Fig. 3A).40 Each subunit consists of an N-terminal NAD+-binding domain (residues 1–150 and 314–335) and a C-terminal catalytic domain (residues 149–313). The N-terminal NAD+-binding domain has an α/β dinucleotide-binding fold, while the C-terminal catalytic domain consists of eight-stranded mixed parallel β-sheets connected by either short α-helices or turns. A molecule of HA with occupancy of 0.8 and 0.7 in chain O and chain Q, respectively, is observed together with a NAD+ cofactor in the active sites (Fig. 3A). The exo-epoxide ring of HA is opened by the C152 thiolate to form a thioether linkage, which verifies the mode of action of the inhibitor (Fig. 3B and C). The conformation of NAD+ cofactor in Hs-GAPDH bound with HA is identical to the reported holo human liver GAPDH, suggesting that inhibitor binding does not alter the conformation of NAD+ (Fig. S13A†).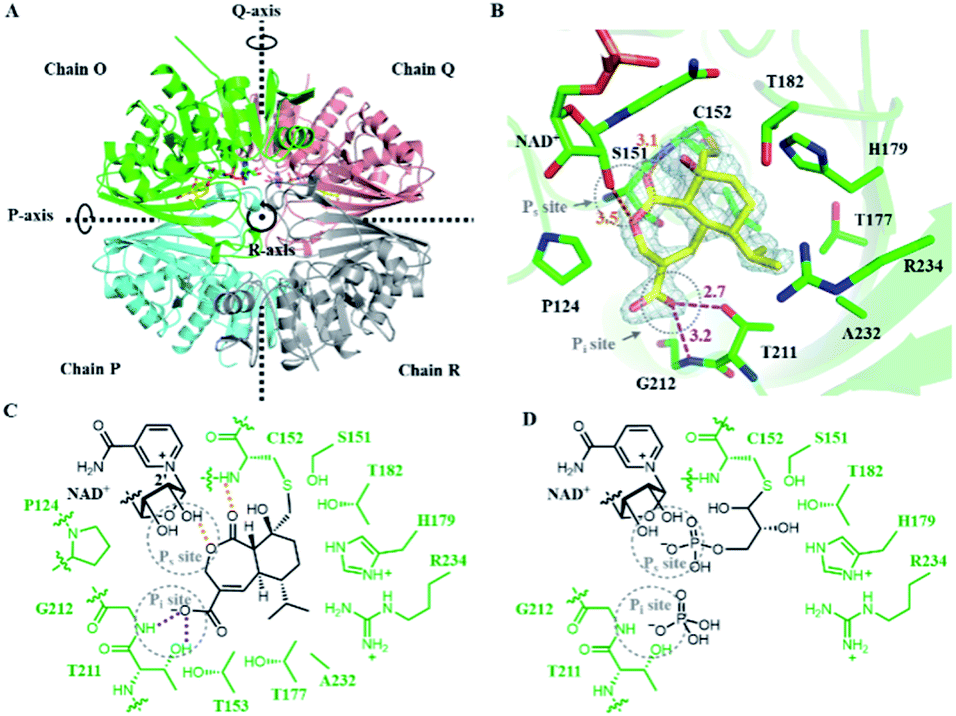 | ||
| Fig. 3 Crystal structure of Hs-GAPDH with HA bound in the active sites. (A) Overview of the Hs-GAPDH homotetramer bound with HA (PDB: 6M61, 1.82 Å). Chain O is in green, chain P is in cyan, chain Q is in salmon, chain R is in grey, and HA is shown in yellow. (B) View of the active site in Hs-GAPDH–HA complex. 2Fo–Fc (light gray mesh, contoured at 1.0σ) electron density map of HA and C152 are shown in grey mesh. The distances (Å) between the atoms involved in HA binding is labelled in orange (hydrogen bond) and purple (ion–dipole interaction). (C) Detailed interactions between the active site of Hs-GAPDH and inhibitor HA. Hydrogen bond and charge–dipole interactions are shown with orange and purple dashed lines, respectively. (D) The proposed model of Hs-GAPDH bound with native substrate GAP. | ||
In addition to the covalent thioether adduct, the oxidized portion of HA interacts extensively with the two anion recognition sites in Hs-GAPDH that bind to inorganic phosphate and the phosphate group of GAP (Fig. 3C and D).11 The Pi site is formed by the 209–215 loop (Fig. 3B and D), and in previous GAPDH structures, binds inorganic phosphate when adopting conformation A, while switching to conformation B that can bind the C3 phosphate group of the thioester after hydride transfer to NAD+.21,41 In the structure of Hs-GAPDH–HA complex, the 209–215 loop has 0.175 Å RMSD for 7 Cα atoms to conformation A (PDB: 1NQA) and 1.189 Å RMSD for 7 Cα atoms to conformation B (PDB: 1DC4), which indicates this loop adopts conformation A in binding to HA (Fig. S13B and C†). The carboxylate of HA forms an anion–dipole interaction with hydroxyl of T211 and backbone amide of G212 (Fig. 3B and C), both of which are involved in binding to inorganic phosphate.11 The α,β-unsaturated double bond conjugated with the carboxylate further lowers the pKa of HA and facilitates the formation of carboxylate anion. The Ps site binds the phosphate group in GAP prior to oxidative thioester formation, and also after cofactor exchange, to allow inorganic phosphate binding in the Pi site (Fig. 3D). In the HA-bound structure, a key interaction in the Ps site is the hydrogen bond between the ester oxygen atom in the lactone ring in HA and 2′-hydroxyl group of ribose adjacent to the nicotinamide in the NAD+ cofactor (Fig. 3B and C).11 In addition, the backbone amide of C152 also participates in hydrogen-bonding to the lactone C2 carbonyl of HA (Fig. 3B and C). These observed interactions are consistent with the results of inhibition assays performed with 4 and 5: the seven-membered lactone ring and the C2 carbonyl both provide binding energy to the HA–GAPDH interactions. Collectively, the carboxylate group mimics the inorganic phosphate in the Pi site and the lactone replaces the phosphate group in GAP in interaction with the Ps site, and synergistically position the inhibitor in the proper orientation in the active site for the thiolate attack on the epoxide (Fig. 3C and D).
Structure of HepG implies the resistance mechanism at molecular level
While catalytically comparable to Hs-GAPDH, the more resistant HepG has 31-fold increase in KI, and 7-fold decrease in kinact towards HA. The crystal structure of HepG was determined at 2.06 Å resolution (Fig. 4A and Table S6†). Each subunit of the tetrameric HepG contains a NAD+ cofactor, and the overall structure of HepG is highly similar to Hs-GAPDH, with identity of 66.4% and RMSD of 0.449 Å for 305 core Cα atoms of chain O. Compared to Hs-GAPDH, the conformation of NAD+ cofactor, catalytic residues (C150 and H177) and anion recognition site residues (T209 and G210) in the active site of HepG are nearly all the same (Fig. 4B).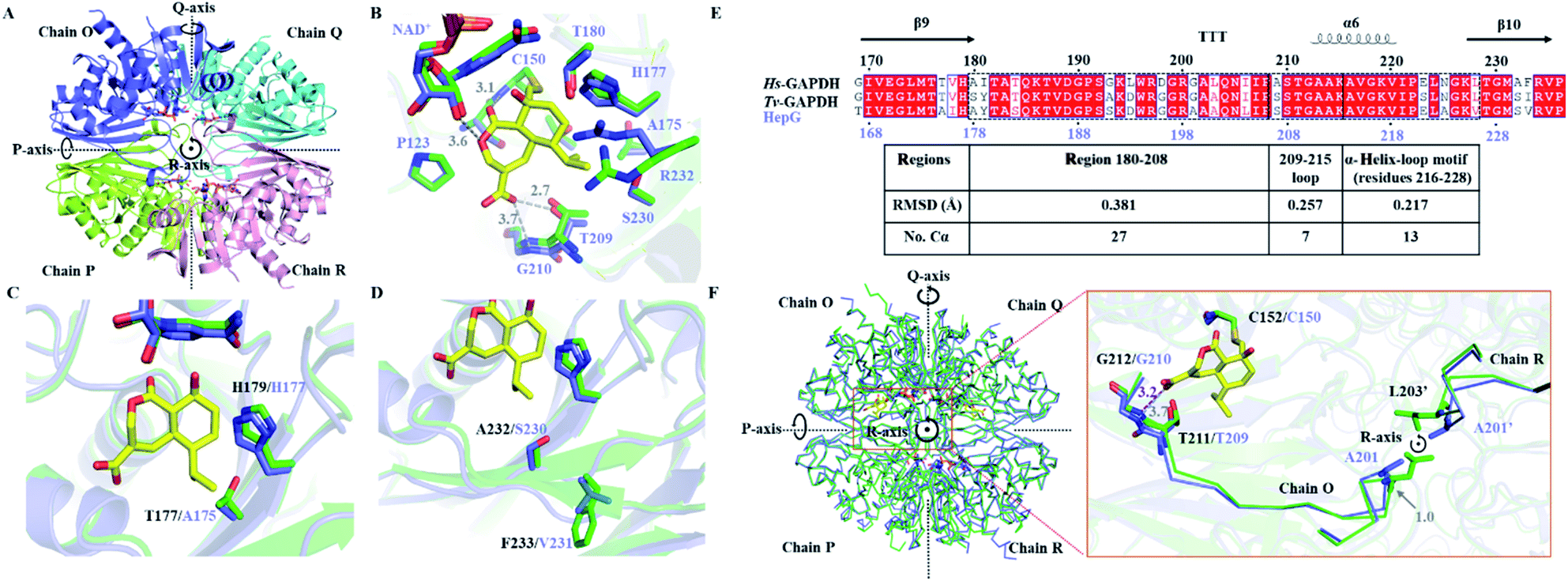 | ||
| Fig. 4 Comparing the crystal structure of Hs-GAPDH and HepG. (A) Overview of the HepG homotetramer (PDB: 6M5X, 2.10 Å). Chain O is in purple, chain P is in light green, chain Q is in cyan, and chain R is in pink. (B) View of the superimposed active site of Hs-GAPDH (green) and HepG (purple) in chain O. The carbon backbone of HA is shown in yellow. The distances (Å) between HA and HepG are shown in grey dashed lines. The residue numbers of HepG are labelled in purple. (C) Superimposed HA binding site of Hs-GAPDH and HepG with T177 of Hs-GAPDH and A175 of HepG shown. (D) Superimposed HA binding site of Hs-GAPDH and HepG with S232-F233 in Hs-GAPDH and A230-V231 in HepG shown. (E) Sequence alignment and secondary structures near loop 209–215 (α-helix is labelled with helix, β-sheet is labelled with arrow, and β-turn is labelled with “T”). The RMSD of corresponding motifs in the black dashed boxes are shown in the table below. (F) Superimposed structures of homotetrameric Hs-GAPDH (green) and HepG (purple). Zoomed in view near loop 209–215 is shown on the right. The main chain of residues 203–212 in Hs-GAPDH and 201–210 in HepG are shown as ribbons. Residues A201 in HepG and L203 in Hs-GAPDH from chain R are labelled as A201′ and L203′ respectively. | ||
Next, we sought to identify unique amino acid residues in HepG, when introduced into the Hs-GAPDH, could lead to increased resistance towards inactivation by HA. Guided by sequence alignment and structural comparisons, potential residues in Hs-GAPDH were mutated. The first candidate residue is T177 in Hs-GAPDH, which is conserved in nearly all GAPDH sequences except the substitution of alanine in HepG and the more resistant copy from T. koningii.30 T177 and the general base H179, which is proposed to deprotonate C152 to generate the reactive thiolate,42 are located on the same side of the β-sheet although no directly hydrogen bonding between the two side chains is observed (Fig. 4C). In HepG, the T177A substitution results in slight change in orientation of the imidazole side chain of H179, which could affect the kcat and kinact of the enzyme. The Hs-GAPDH-T177A mutant was constructed, and the inhibitory effect of HA against the mutant was measured (Fig. S4D and S8†). As predicted, kinact was decreased by ∼4 fold, while the kcat is decreased by 1.7 fold (Table 1). While the KI remained unchanged for the mutant, the KM of the mutant towards GAP unexpectedly decreased by 6.2-fold, resulting in an overall 4-fold increase in kcat/KM (Table 1). Hence, the T177A mutation in Hs-GAPDH increases resistance to HA through enhanced catalytic efficiency towards GAP. The imidazole side chain of the H179 is also proposed to interact with the phosphate group in GAP, which may be enhanced by the nearby T177A mutation.21 However, compared to HepG, the Hs-GAPDH-T177A mutant remains ∼64-fold more sensitive to HA, which indicates the change from Thr to Ala is not the primary determinant of resistance in HepG.
We also identified A232 that is conserved in GAPDH active site is substituted with serine in HepG. A232 interacts with the hydrocarbon portion of HA, and replacement with serine could lower the binding affinity of HA. The next residue F233 of which the side chain points away from the active site is substituted by valine in HepG, which may also affect the active site geometry and interaction with HA (Fig. 4D). Both residues were mutated to give a double mutant Hs-GAPDH-A232S-F233V (Fig. S4E and S9†). Although the KI towards HA increased by 1.5-fold after mutation, the KM towards GAP is also increased by 2-fold, and the overall inactivation rate constant kinact/KI remained similar. Therefore these two residues are not determinants of resistance in HepG (Table 1).
We next examined if resistance to HA may be increased from subtle changes in the active site conformation caused by more remote mutations. The Hs-GAPDH–HA complex structure was superimposed to HepG and distance between HA and key residues in Pi and Ps sites of HepG were measured (Fig. 4B). In the Ps site in HepG, the distance between lactone oxygen of HA and 2′-OH of ribose moiety of the NAD+ cofactor is slightly increased from 3.4 Å in Hs-GAPDH to 3.6 Å (Fig. 3B and 4B). In the Pi site, the distance between carboxylate of HA and side chain hydroxyl of T209 is essentially unchanged (2.7 Å in Hs-GAPDH vs. 2.8 Å in HepG), and the ion–dipole interaction is maintained. The most significant difference is the distance between carboxylate of HA and backbone amide of G210, which is increased from 3.2 Å in Hs-GAPDH to 3.7 Å in HepG. This corresponds to a significant disruption to this critical ion–dipole interaction between HA and Pi site (Fig. 3B and 4B). Therefore, we hypothesized that weakening the interaction with HA in the Pi site may account for increased resistance towards HA.
The mutation L203A in Hs-GAPDH confers resistance against HA
Given T209 and G210 in Pi site are conserved among all GAPDH sequences, we examined if changes in surrounding secondary structures near the 209–215 loop may lead to the observed differences between Hs-GAPDH and HepG. The N-terminus of this loop is directly connected to a region defined by residues 180–208, while the C-terminus is connected to an α-helix (α6)-loop motif (residues 216–228) (Fig. 4E). Superimposing crystal structures of both Hs-GAPDH–HA complex and HepG shows the 209–215 loop of the Pi site has RMSD of 0.257 Å for 7 Cα atoms, the C-terminal α-helix-loop motif exhibits RMSD of 0.217 Å for 13 Cα atoms, and the N-terminal region 180–208 shows RMSD of 0.381 Å for 27 Cα atoms (Fig. 4E). This suggests the subtle structural differences of Pi sites between Hs-GAPDH and HepG may result from the altered conformation of the N-terminal region. In particular, the Cα atoms of two residues, L203 and I181 in Hs-GAPDH showed RMSD deviation of 1.0 Å each to the corresponding residues A201 and Y179 in HepG. These distances are significantly greater than the RMSD of the entire region 180–208 (0.381 Å). Interestingly, L203 is well-conserved among primate GAPDH, while in other eukaryotic and microbial GAPDHs, the position is occupied primarily by alanine (Fig. S12†). Therefore, we propose these two residues, in particular L203, may contribute to the high sensitivity of Hs-GAPDH to HA.The single-point mutants I181Y and L203A of Hs-GAPDH were constructed and purified (Fig. S4F†). Whereas the I181Y mutation made the enzyme completely inactive, the L203A mutant has the same kcat/KM as the wild type (Fig. S4G and S10†). However, the L203A mutant showed a 16-fold decrease of the overall inhibition rate constant kinact/KI, contributed from 4-fold increase in KI and ∼4-fold decrease in kinact (Table 1). To test if the opposite mutation, A201L, in HepG can make the enzyme more sensitive to inhibition by HA, the mutant was constructed and assayed (Fig. S4H and S11†). While the catalytic efficiency of HepG A201L mutant is comparable to the wild type HepG, the mutant is significantly more sensitive towards HA inhibition with an overall 48-fold increase in inhibition rate constant kinact/KI (Table 1). In particular, the binding affinity of HA towards the mutant is drastically enhanced, as seen in the ∼25-fold decrease in KI which approaches that of the Hs-GAPDH. Overall, interchanging the identity of this residue led to a complete switch of relative sensitivity/resistance between the two enzymes, with the Hs-GAPDH-L203A mutant now more resistant towards HA than the HepG-A201L mutant.
At the secondary and tertiary structure levels, residue L203 in Hs-GAPDH is part of a β-turn region (residues 202–204), which is located on the peripheral of the globular Hs-GAPDH monomer (Fig. 4F). At the quaternary structure level, motif 180–208 is located in the centre of the homotetramer (Fig. 4F) and is in close proximity to the same region from other monomers. The L203 residues from Hs-GAPDH monomers O and R are located close to each other with a Cα atoms distance of 6.9 Å, and can have steric clashes from certain rotamers of the leucine side chains. On the other hand, residues A201 from HepG monomers O and R are separated by Cα distance of 6.5 Å (Fig. 4F), and the smaller side chains of alanine do not result in steric clashes. In Hs-GAPDH, mutation of L203 to alanine relieves the potential steric interactions, and results in conformation change of the motif 180–208. As T209 and G210 are directly connected to this N-terminal region, conformation change to the 180–208 region can be relayed to the Pi site, and lead to disruption of the anion–dipole binding interactions with HA as proposed in HepG. As a result, L203A mutation in Hs-GAPDH caused significant increases in KI of HA and the opposite is observed in the HepG A201L mutant. The interchanging mutations in both enzymes do not affect the catalytic properties, which indicates the binding affinities of inorganic phosphate in both Pi sites are not compromised.
Discussion
In this study, we investigated biosynthesis of the fungal GAPDH inhibitor HA and uncovered the self-resistance mechanism in its fungal producer at molecular level. Given the central role of GAPDH in metabolism,43 both bacterial and fungi have evolved terpene NPs as inhibitors. The potential use of GAPDH inhibitor in cancer and inflammatory disease treatment has received attention in recent years.19,44 Locasale and coworkers demonstrated that targeting the Warburg effect by inhibition of GAPDH using HA was effective in suppressing the growth of highly glycolytic cancer cell lines in vitro and in orthotopic tumor models.18 By targeting GAPDH in activated immune cells, Snyder et al. recently showed HA can suppress glycolysis and immune responses.19 Besides its role in glycolysis, GAPDH has been shown to be involved in other cellular processes such as regulation of apoptosis, transcription activation and vesicle transportation.45–47 For example, Ryu and coworkers showed that GAPDH can bind Rheb and inhibits mTORC1 signaling under low glucose concentrations, thereby correlating glycolytic flux and mTORC1 control of cell growth.48 The dimer and tetramer interfaces of GAPDH have also been shown to bind the adenine–uridine rich elements involved in posttranscriptional regulation of mRNA with submicromolar affinity.49 Therefore, understanding the mode of inhibition of GAPDH by HA and molecular basis of resistance can be useful for improving inhibitor design.The multistep mechanism of GAPDH involves the binding of one cofactor (NAD+) and two substrates (GAP and Pi). The order and the locations of substrate binding in the active site have been postulated based on numerous structures and biochemical analysis.11,21,40,41 NAD+ is bound first, followed by entry of GAP, formation of the hemithioacetal and oxidation to the thioester. In these steps, the phosphate of GAP binds to the Ps site and interact with the 2′-OH of NAD+. After thioester is formed, the GAP phosphate flips into the Pi site, which allows the NADH to be exchanged with NAD+. The GAP phosphate then flips back to the Ps site to enable inorganic phosphate to bind to the Pi site and attack the thioester to form 1,3-DPG. The Pi site can adopt two conformations to bind either the phosphate from GAP or inorganic phosphate. The overall active site is therefore spacious and dynamic during catalysis. From the Hs-GAPDH structure complexed with HA, it is evident that HA binds to both Pi and Ps site through the furnished functional groups. The interactions of the carboxylate with the Pi site and the lactone with the Ps site anchor the inhibitor in this large active site and properly position the epoxide within attack range from the thiolate. This rationalizes the relatively larger size of HA compared to GAP, as well the common structural features shared between HA and pentalenolactone.
The observed binding mode of HA showcases the ingenuity of nature in evolving potent natural product inhibitors for GAPDH. The biosynthesis of HA and pentalenolactone both start with the synthesis of multicyclic hydrocarbon scaffolds derived from isoprene building blocks. A collection of oxidative enzymes then precisely modify the hydrocarbon at selective positions to install polar (lactone), charged (carboxylate) and reactive (epoxide) functionalities to target the various sites in the active site of GAPDH. Our biotransformation and biochemical assays suggest a possible oxidative sequence as shown in Fig. 2. Starting with the carboxylate intermediate 3, each subsequent biosynthetic transformation increases the inhibition potency. Modification of 3 to 4 enables covalent modification by the thiolate, while modification of 4 to 5, then to 1, allows binding of HA to the Ps site. One may therefore consider these enzymatic transformations to reflect the evolutionary steps that nature took to arrive at the end-product HA. Because we are not able to reconstitute the initial oxidation of 6, alternative oxidative sequence may possibly be employed in the biosynthesis of 2, given some fungal P450s have substrate promiscuity. Although the oxidation steps in biosynthesis of HA differ from those of pentalenolactone, in which the epoxide formation is the last step, the closely mirrored biosynthetic pathways between the two molecules is a stunning example of convergent evolution.
While the kinetic parameters (kcat, KM) of HepG and Hs-GAPDH are comparable, the KI of HepG towards HA is ∼30-fold higher and the kinact is ∼8 fold slower. Comparison of crystal structure of HepG and Hs-GAPDH revealed both active site and remote residues contribute to the difference in sensitivity towards HA between Hs-GAPDH and HepG. The T177A mutation in the active site was shown to selectively decrease the Hs-GAPDH KM towards GAP. Both kcat and kinact are decreased for the mutant, indicating the threonine residue which is close to the general base H179, influences catalytic turnover. Endo et al. previously isolated two GAPDH variants from T. koningii and noted the more resistant copy has an alanine at this position as in HepG, while the more sensitive copy has a threonine as in Hs-GAPDH.50 However, two factors point to this mutation not being the primary determinant of sensitivity displayed by Hs-GAPDH: (1) the KI of the Hs-GAPDH-T177A mutant towards HA remained unchanged; and (2) the Tv-GAPDH housekeeping enzyme which is 100-fold more resistant than Hs-GAPDH, also contains a threonine at the corresponding position.
This led us to examine differences in the Pi and Ps binding sites between the enzymes to identify the large difference in KI. The shift of G210 in the Pi pocket of HepG disrupts the critical anion–dipole interaction with the carboxylate of HA, and weakens the overall binding of HA in the active site by “loosening” one end of the interactions. We were able to trace the cause of this structural difference to a flexible loop at the N-terminal of the Pi loop, and pinpoint L203 as a key determinant of resistance. The importance of this residue was validated by mutagenesis in both Hs-GAPDH and HepG, which led to a dramatic reversal in the KI values toward HA. We speculate that such subtle differences in active sites may be exploited in the design of selective inhibitors of microbial GAPDH over Hs-GAPDH in the search for new antibacterial or antifungal candidates.
Lastly, our data show the housekeeping GAPDH, Tv-GAPDH, is also significantly less sensitive to HA compared to Hs-GAPDH, and the kinact/KI is only 2.3-fold higher than that of HepG. Because HA is a covalent and irreversible inhibitor, the host may had evolutionary pressure to evolve its housekeeping copy to also be more resistant, while still requiring the resistant copy in HepG. The increased resistance displayed by HepG provides further protection to the host, before HA is transported to the outside of the cell, possibly by the MFS efflux pump HepF. The less sensitivity measured for the housekeeping GAPDH here is in contrast to previous reports in which housekeeping GAPDH from A. oryzae and T. koningii are much more sensitive towards HA. For example, Endo and coworkers showed the kinact/KI of housekeeping GAPDH in T. koningii was ∼160-fold greater than the proposed resistant isozymes.30 While Koyama and coworkers showed the IC50 of HA towards the housekeeping GAPDH is 60-fold lower than the HepG homolog in A. oryzae. These differences in the level of sensitivity displayed by the housekeeping GAPDH may therefore reflect the different extent of evolution in conferring self-resistances towards HA. While Tv-GAPDH may indeed have evolved to be more resistant to HA, the different methodology used for the assays in the prior reports can also contribute to the differences. As HA is a covalent inhibitor that irreversibly inactivates the enzyme, the values of potency of reversible binding (KI) and maximum potential rate of inactivation (kinact) measured by the Kitz–Wilson method are more representative quantitative measurements of inhibition. On the other hand, IC50 values are strongly affected by both preincubation time between enzyme and inhibitor, and the overall time of the assay.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr John M. Billingsley, Dr Yang Hai and Dr Mengbin Chen for helpful discussions. This work was supported by NIH (1R35GM118056) to Y. T. Work in the J. Z. lab is supported by MOST (2018YFA0901900), CAS (121731KYSB201800) and SMSTC (19XD1404800). We thank the staff of beamline BL18U1 of Shanghai Synchrotron Radiation Facility for access and help with the X-ray data collection. We thank Professor Jianhua Gan of Fudan University for suggestions about refinement of protein structure.Notes and references
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- A. S. Lee, H. de Lencastre, J. Garau, J. Kluytmans, S. Malhotra-Kumar, A. Peschel and S. Harbarth, Nat. Rev. Dis. Primers, 2018, 4, 18033 CrossRef PubMed.
- N. Vasan, J. Baselga and D. M. Hyman, Nature, 2019, 575, 299–309 CrossRef CAS PubMed.
- Y. Yan, N. Liu and Y. Tang, Nat. Prod. Rep., 2020, 37, 879–892 RSC.
- K. H. Almabruk, L. K. Dinh and B. Philmus, ACS Chem. Biol., 2018, 13, 1426–1437 CrossRef CAS PubMed.
- Y. Yan, Q. Liu, X. Zang, S. Yuan, U. Bat-Erdene, C. Nguyen, J. Gan, J. Zhou, S. E. Jacobsen and Y. Tang, Nature, 2018, 559, 415–418 CrossRef CAS PubMed.
- X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS PubMed.
- A. Endo, K. Hasumi, K. Sakai and T. Kanbe, J. Antibiot., 1985, 38, 920–925 CrossRef CAS PubMed.
- S. Hartmann, J. Neeff, U. Heer and D. Mecke, FEBS Lett., 1978, 93, 339–342 CrossRef CAS.
- E. Racker and I. Krimsky, Nature, 1952, 169, 1043–1044 CrossRef CAS PubMed.
- S. Moniot, S. Bruno, C. Vonrhein, C. Didierjean, S. Boschi-Muller, M. Vas, G. Bricogne, G. Branlant, A. Mozzarelli and C. Corbier, J. Biol. Chem., 2008, 283, 21693–21702 CrossRef CAS PubMed.
- B. A. Orsi and W. W. Cleland, Biochemistry, 1972, 11, 102–109 CrossRef CAS PubMed.
- Y. Itoh, K. Kodama, K. Furuya, S. Takahashi, T. Haneishi, Y. Takiguchi and M. Arai, J. Antibiot., 1980, 33, 468–473 CrossRef CAS PubMed.
- Y. Ogawa and H. Yonehara, Tetrahedron Lett., 1969, 10, 2737–2740 CrossRef.
- M. Kato, K. Sakai and A. Endo, Biochim. Biophys. Acta, 1992, 1120, 113–116 CrossRef CAS.
- D. E. Cane and J.-K. Sohng, Biochemistry, 1994, 33, 6524–6530 CrossRef CAS PubMed.
- L. Ercolani, B. Florence, M. Denaro and M. Alexander, J. Biol. Chem., 1988, 263, 15335–15341 CAS.
- M. V. Liberti, Z. Dai, S. E. Wardell, J. A. Baccile, X. Liu, X. Gao, R. Baldi, M. Mehrmohamadi, M. O. Johnson, N. S. Madhukar, A. A. Shestov, I. I. C. Chio, O. Elemento, J. C. Rathmell, F. C. Schroeder, D. P. McDonnell and J. W. Locasale, Cell Metab., 2017, 26, 648–659 CrossRef CAS PubMed.
- M. D. Kornberg, P. Bhargava, P. M. Kim, V. Putluri, A. M. Snowman, N. Putluri, P. A. Calabresi and S. H. Snyder, Science, 2018, 360, 449–453 CrossRef CAS PubMed.
- W. D. Mercer, S. I. Winn and H. C. Watson, J. Mol. Biol., 1976, 104, 277–283 CrossRef CAS.
- M. Yun, C.-G. Park, J.-Y. Kim and H.-W. Park, Biochemistry, 2000, 39, 10702–10710 CrossRef CAS PubMed.
- F. Ferreira-da-Silva, P. J. B. Pereira, L. Gales, M. Roessle, D. I. Svergun, P. Moradas-Ferreira and A. M. Damas, J. Biol. Chem., 2006, 281, 33433–33440 CrossRef CAS PubMed.
- J. Frayne, A. Taylor, G. Cameron and A. T. Hadfield, J. Biol. Chem., 2009, 284, 22703–22712 CrossRef CAS PubMed.
- C. N. Tetzlaff, Z. You, D. E. Cane, S. Takamatsu, S. Omura and H. Ikeda, Biochemistry, 2006, 45, 6179–6186 CrossRef CAS PubMed.
- Z. You, S. Omura, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 6566–6567 CrossRef CAS PubMed.
- R. Quaderer, S. Omura, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 13036–13037 CrossRef CAS PubMed.
- Z. You, S. Omura, H. Ikeda and D. E. Cane, Arch. Biochem. Biophys., 2007, 459, 233–240 CrossRef CAS PubMed.
- M.-J. Seo, D. Zhu, S. Endo, H. Ikeda and D. E. Cane, Biochemistry, 2011, 50, 1739–1754 CrossRef CAS PubMed.
- Y. Shinohara, I. Nishimura and Y. Koyama, Biosci., Biotechnol., Biochem., 2019, 83, 1506–1513 CrossRef CAS PubMed.
- K. Sakai, K. Hasumi and A. Endo, Eur. J. Biochem., 1990, 193, 195–202 CrossRef CAS PubMed.
- G. A. Cordell, Chem. Rev., 1976, 76, 425–460 CrossRef CAS.
- T. Noguchi, S. Fujioka, S. Choe, S. Takatsuto, F. E. Tax, S. Yoshida and K. A. Feldmann, Plant Physiol., 2000, 124, 201–210 CrossRef CAS PubMed.
- H. Zeng, G. Yin, Q. Wei, D. Li, Y. Wang, Y. Hu, C. Hu and Y. Zou, Angew. Chem., Int. Ed., 2019, 58, 6569–6573 CrossRef CAS PubMed.
- Y. Yoshikuni, V. J. J. Martin, T. E. Ferrin and J. D. Keasling, Chem. Biol., 2006, 13, 91–98 CrossRef CAS PubMed.
- R. D. Stipanovic and C. R. Howell, Tetrahedron, 1983, 39, 1103–1107 CrossRef CAS.
- M.-C. Tang, H.-C. Lin, D. Li, Y. Zou, J. Li, W. Xu, R. A. Cacho, M. E. Hillenmeyer, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2015, 137, 13724–13727 CrossRef CAS PubMed.
- T.-W. Kim, J.-Y. Hwang, Y.-S. Kim, S.-H. Joo, S. C. Chang, J. S. Lee, S. Takatsuto and S.-K. Kim, Plant Cell, 2005, 17, 2397–2412 CrossRef CAS PubMed.
- K. Chen, S. Wu, L. Zhu, C. Zhang, W. Xiang, Z. Deng, H. Ikeda, D. E. Cane and D. Zhu, Biochemistry, 2016, 55, 6696–6704 CrossRef CAS PubMed.
- Z. D. Parsons and K. S. Gates, Methods Enzymol., 2013, 528, 129–154 CAS.
- S. A. Ismail and H. W. Park, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 1508–1513 CrossRef CAS PubMed.
- C. Didierjean, C. Corbier, M. Fatih, F. Favier, S. Boschi-Muller, G. Branlant and A. Aubry, J. Biol. Chem., 2003, 278, 12968–12976 CrossRef CAS PubMed.
- M. Reis, C. N. Alves, J. Lameira, I. Tuñón, S. Martí and V. Moliner, Phys. Chem. Chem. Phys., 2013, 15, 3772–3785 RSC.
- C. Nicholls, H. Li and J.-P. Liu, Clin. Exp. Pharmacol. Physiol., 2012, 39, 674–679 CrossRef CAS PubMed.
- S. Ganapathy-Kanniappan, R. Kunjithapatham and J.-F. Geschwind, Oncotarget, 2012, 3, 940–953 CrossRef PubMed.
- A. Tarze, A. Deniaud, M. Le Bras, E. Maillier, D. Molle, N. Larochette, N. Zamzami, G. Jan, G. Kroemer and C. Brenner, Oncogene, 2007, 26, 2606–2620 CrossRef CAS PubMed.
- D. Zala, M.-V. Hinckelmann, H. Yu, M. M. Lyra da Cunha, G. Liot, F. P. Cordelières, S. Marco and F. Saudou, Cell, 2013, 152, 479–491 CrossRef CAS PubMed.
- D.-M. Chuang, C. Hough and V. V. Senatorov, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 269–290 CrossRef CAS PubMed.
- M. N. Lee, S. H. Ha, J. Kim, A. Koh, C. S. Lee, J. H. Kim, H. Jeon, D.-H. Kim, P.-G. Suh and S. H. Ryu, Mol. Cell. Biol., 2009, 29, 3991–4001 CrossRef CAS PubMed.
- M. R. White, M. M. Khan, D. Deredge, C. R. Ross, R. Quintyn, B. E. Zucconi, V. H. Wysocki, P. L. Wintrode, G. M. Wilson and E. D. Garcin, J. Biol. Chem., 2015, 290, 1770–1785 CrossRef PubMed.
- H. Watanabe, K. Hasumi, Y. Fukushima, K. Sakai and A. Endo, Biochim. Biophys. Acta, 1993, 1172, 43–48 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03805a |
| ‡ These authors contribute equally. |
| § Current address: Key Laboratory of Tropical Marine Bio-resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology, Chinese Academy of Sciences, 164 West Xingang Road, Guangzhou 510301, China. Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), 1119 Haibin Rd. Nansha District, Guangzhou, 511458, China. |
| ¶ Current address: Institute of Biological Chemistry, Academia Sinica, Taipei 115, Taiwan. |
| This journal is © The Royal Society of Chemistry 2020 |