
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-catalyzed stereoselective tandem ring-opening amination/cyclization of vinyl γ-lactones: access to caprolactam diversity†
Jianing
Xie
 a,
Xuetong
Li
a and
Arjan W.
Kleij
*ab
a,
Xuetong
Li
a and
Arjan W.
Kleij
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007–Tarragona, Spain. E-mail: akleij@iciq.es
bCatalan Institute of Research and Advanced Studies (ICREA), Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 6th August 2020
Abstract
A stereoselective amination/cyclization cascade process has been developed that allows for the preparation of a series of unsaturated and substituted caprolactam derivatives in good yields. This conceptually novel protocol takes advantage of the easy access and modular character of vinyl γ-lactones that can be prepared from simple precursors. Activation of the lactone substrate in the presence of a suitable Pd precursor and newly developed phosphoramidite ligand offers a stereocontrolled ring-opening/allylic amination manifold under ambient conditions. The intermediate (E)-configured ε-amino acid can be cyclized using a suitable dehydrating agent in an efficient one-pot, two-step sequence. This overall highly chemo-, stereo- and regio-selective transformation streamlines the production of a wide variety of modifiable and valuable caprolactam building blocks in an operationally attractive way.
Introduction
Lactam derivatives play an important role in the development of new pharmaceuticals1 and are useful as a starting point for synthetic polyamides,2 with caprolactam being the most prominent monomer known to date.3 Classic synthesis of lactams is achieved using methods such as the Beckmann rearrangement of oximes,4 the Schmidt reaction between an azide and a carbonyl compound,5 and through cyclization of amino acids or similar precursors.6 Apart from the easy availability of stoichiometric methodologies, catalytic formation of lactams has materialized as a more sustainable alternative within the synthetic community fueled by the need for a higher atom and resource economy.7 Efficient methodologies have been developed by Fujita, Yamaguchi et al. who reported on a Rh-catalyzed oxidative N-heterocyclization of amino alcohols giving primarily access to five- and six-membered lactams,8 whereas Hong and coworkers showed that direct lactam synthesis can be achieved using lactones and amines as reagents under Ir-catalysis.9 Other approaches that rely on the use of prefunctionalized substrates have also been recently disclosed.10Caprolactams represent key motifs of many natural products and other biologically active compounds (Scheme 1, top),11 and often serve as versatile synthetic intermediates for bicyclic amino compounds and azepane ring systems.12 Despite the recent progress noted for the catalytic synthesis of the more common five- and six-membered lactams,4b,8–10,13 the scope of functional and modular seven-membered analogues remains surprisingly limited. Therefore, designing new synthetic procedures that can streamline the preparation of such caprolactam building blocks can create new incentives for their use as synthons in drug-development programs and the creation of new polyamide monomers. With this challenge in mind, we decided to contemplate on combining our expertise in the area of allylic substitution chemistry14 and merging this with vinyl γ-lactones as modular substrates as outlined in Scheme 1.
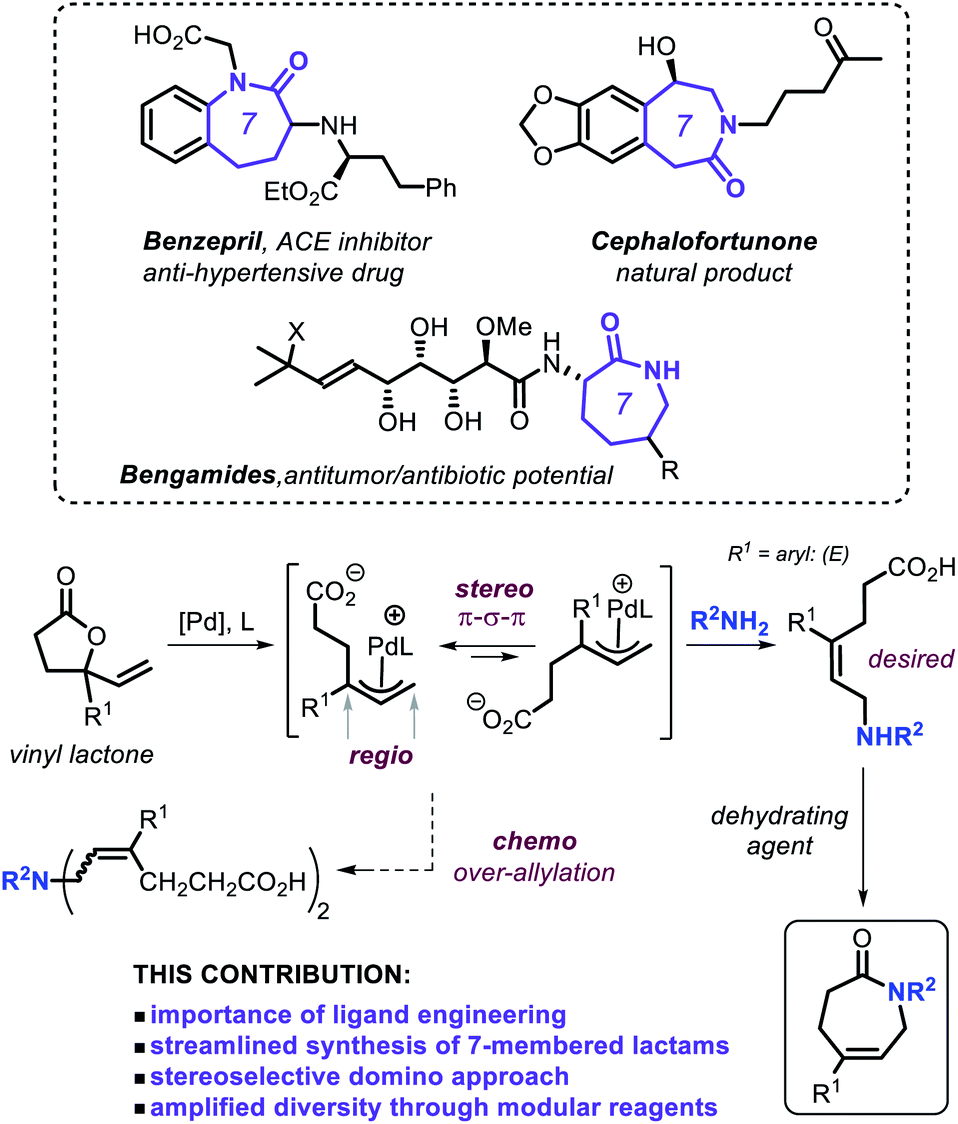 | ||
| Scheme 1 Approach towards the stereoselective domino synthesis of caprolactams. | ||
Non-strained, stable γ-lactones have so far only sporadically emerged as versatile and readily accessible substrates under suitable catalytic conditions.15 More specifically, the use of vinyl γ-lactones (Scheme 1)16 may offer a tangible way to prepare ε-amino acid intermediates through allylic amination under Pd catalysis. In the overall manifold, proper ligand engineering17 is crucial to control simultaneously the chemo-, regio- and stereo-selectivity under ambient conditions in order to generate the (E)-configured ε-amino acid intermediate (Scheme 1) en route to the caprolactam target.18 Here we present a general and practically attractive approach for the preparation of a library of functional caprolactams through a novel and formal tandem ring-opening allylic amination/cyclization process, which is controlled by a newly developed, readily accessible phosphoramidite-based Pd catalyst.
Results and discussion
At the onset of the screening studies, we considered to use previously reported successful phosphorus ligands14,17 to examine their efficacy to generate Pd-catalysts derived from Pd2(dba)3·CHCl3 (see the ESI†). We were pleased to find that the presence of phosphoramidite ligand L1 proved to be highly beneficial for the ring-opening allylic amination step under ambient conditions. Furthermore, the in situ rapid cyclization of the intermediate (E)-ε-amino acid towards the lactam target using the well-known reagent EDC19 was combined in a one-pot two step sequence. Our screening then focused on producing the intermediate ε-amino acids 1 in the most efficient way from lactone A using phosphoramidite L1 (Table 1, entries 1–6) as to favor the formation of the desired lactam 3.20 The type of solvent has a significant effect on the outcome of the tandem process. The use of both DCM and MeOH gave access to moderate yields of 3 while minimizing the formation of the double allylated byproduct 2. The presence of highly polar solvents such as DMSO and DMF or THF, however, resulted in unproductive processes, whereas the utilization of ACN lowered the chemo-selectivity of the process. It is important to note that in the absence of L1 (entry 7), there was no formation of any product pointing at the crucial role of the ligand.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | Solvent | Yieldb of 1 (%) |
E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z-1c :Z-1c |
1/2c | Yieldb of 3 (%) |
| a Lactone A (0.20 mmol), PhNH2 (0.30 mmol, 1.5 equiv.), solvent (0.20 mL), Pd2(dba)3·CHCl3 (2.0 mol%), L (8.0 mol%), rt, 12 h; then EDC (0.30 mmol), 1 h. b Determined by 1H NMR analysis in CDCl3 using CH2Br2 as an internal standard. c Determined by 1H NMR analysis. d PhNH2 (2.0 equiv.). e Pd2(dba)3·CHCl3 (3.0 mol%), L1 (12.0 mol%). f Solvent (0.30 mL). | ||||||
| 1 | L1 | DCM | 83 | 82:18 |
16:1 |
66 |
| 2 | L1 | THF | 0 | — | — | — |
| 3 | L1 | ACN | 67 | 89:11 |
5:1 |
59 |
| 4 | L1 | MeOH | 78 | 82:18 |
17:1 |
64 |
| 5 | L1 | DMF | 0 | — | — | — |
| 6 | L1 | DMSO | 0 | — | — | — |
| 7 | — | DCM | 0 | — | — | — |
| 8 | L2 | DCM | 85 | 77:23 |
16:1 |
65 |
| 9 | L3 | DCM | 82 | 61:39 |
>20:1 |
53 |
| 10 | L4 | DCM | <1 | — | — | — |
| 11 | L5 | DCM | 0 | — | — | — |
| 12 | L6 | DCM | 0 | — | — | — |
| 13 | L7 | DCM | 79 | 96:4 |
10:1 |
76 |
| 14 | L8 | DCM | 80 | 97:3 |
10:1 |
77 |
| 15 | L9 | DCM | 79 | 94:6 |
13:1 |
74 |
| 16 | L10 | DCM | <1 | — | — | — |
| 17 | L11 | DCM | 0 | — | — | — |
| 18 | L12 | DCM | 82 | 91:9 |
15:1 |
75 |
| 19d,e | L8 | DCM | 88 | 93:7 |
>20:1 |
82 |
| 20d,e,f | L8 | DCM | 86 | 98:2 |
>20:1 |
84 |
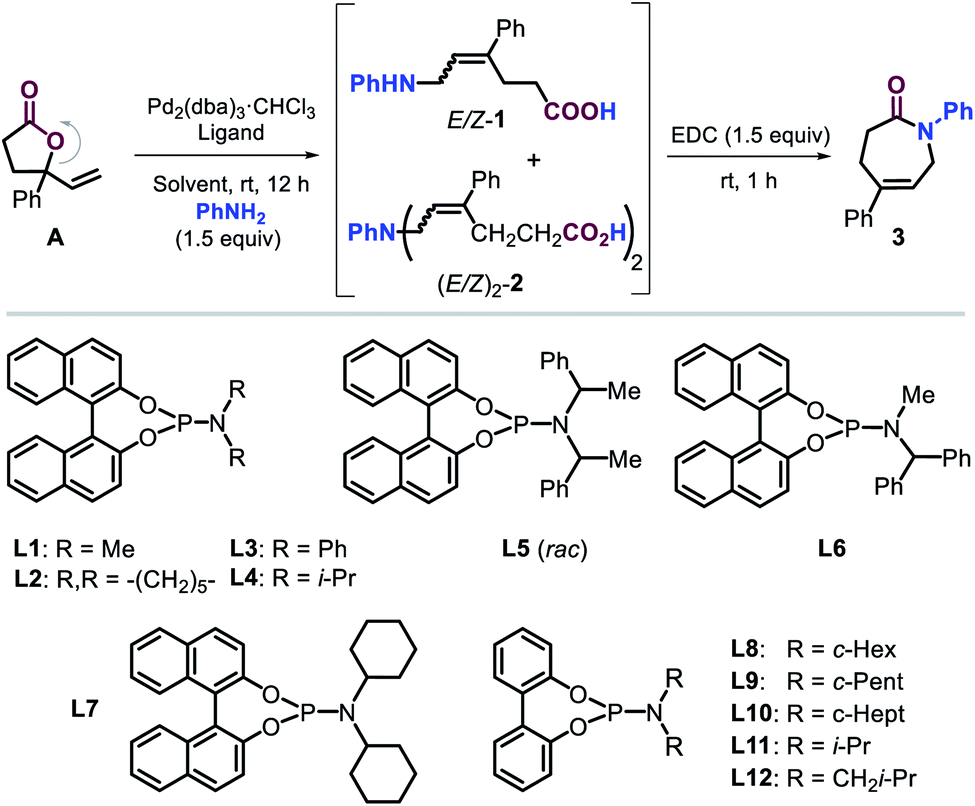
A variety of phosphoramidite ligands L2–L12 (entries 8–18) were then scrutinized under the conditions of entry 1, illustrating the critical influence of the N-substitution and biaryl backbone on the chemo-, regio- and stereoselectivity of this catalytic process. For instance, too bulky N-substituents (L4–L6, entries 10–12) do not permit the formation of 3, whereas ligands equipped with piperidine and phenyl (L2 and L3) provided good yields of intermediate 1 with high chemoselectivity and moderate E/Z ratios (entries 8 and 9). To our delight, the introduction of cyclohexyl groups (L7, entry 13) further increased the E-stereoselective formation of 1 and thus the yield of 3 while retaining good chemo-selectivity. Interestingly, a binaphthyl ligand backbone was not essential to achieve high selectivity, which became clear from studying the biphenyl based ligands L8–L12 and the steric effect of their N-substituents (entries 15–18). Cyclopentyl and i-Bu substituted ligands L9 and L12 provided productive and rather selective catalysts for the formation of 3 though with slightly lower yields and stereoselectivity as compared to L8. The reaction did not proceed with ligands comprising of cycloheptyl or i-Pr groups (L10 and L11). These results indicate that steric modulation of the N-substituent of the ligand is key to the success of the formation of caprolactam 3. From all ligands studied, the novel ligand L8 (entry 14) provided the best combination of yield (for 1 and 3) and selectivity features. A further increase in the yield of 3 in the presence of L8 was finally obtained by increasing the amount of catalyst, aniline and solvent (entries 19 and 20) providing the caprolactam product in 84% yield and with high stereo- (E/Z = 98:2) and chemo-selectivity (1:2 > 20:1).
With the optimized conditions of entry 20 in Table 1, we then set out to examine the scope of this new transformation (Scheme 2, compounds 3–28) focusing first on variation of the vinyl lactone reagent. Lactones with para-substituted aryl substituents were productive substrates giving the caprolactam derivatives 3–10 in high isolated yields (>80% in most cases) and with excellent stereo- (typically with E/Z ratios >95:5), regio- and chemo-selectivity control. The presence of meta-, ortho, para-di- or 3,5-di-substituted aryl groups (11–15; 71–90%, E/Z ≥ 96:4) did not have any significant effect on the outcome of the tandem process, and the desired products could be easily formed and isolated. Larger aromatic surfaces such as those present in lactam products 16 and 17 are also tolerated, while this new protocol furthermore allows the introduction of heteroaromatic fragments (18–22) in the final products.
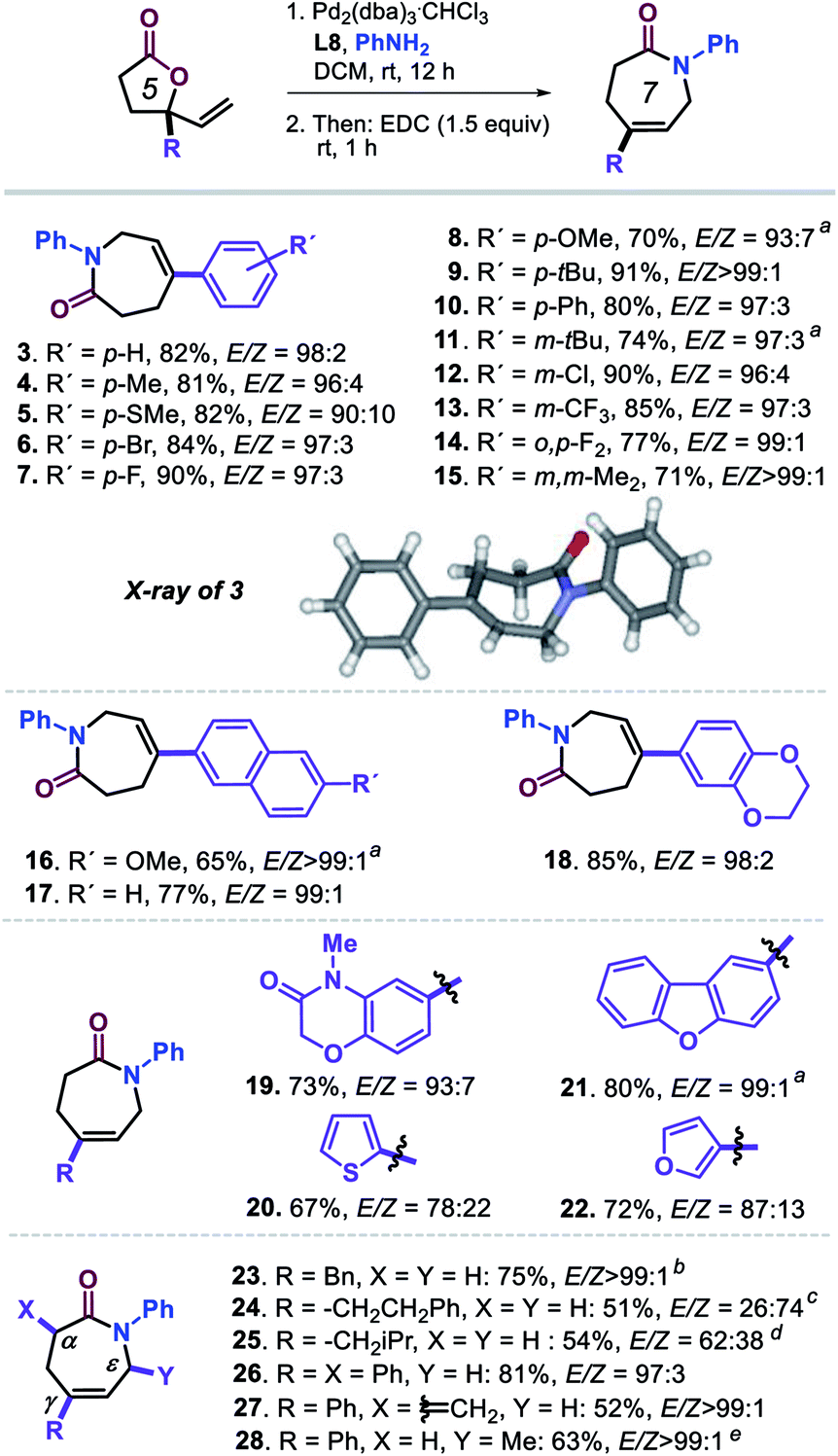 | ||
| Scheme 2 Scope in vinyl-substituted γ-lactone substrates. All reactions were performed under the optimized conditions (Table 1, entry 20). Isolated yields are reported, and the reported E/Z ratios for the intermediate ε-amino acids were determined by 1H NMR; the inset shows the molecular structure determined for 3 by X-ray analysis, see footnote 21. aUsing DCM/EtOH (1:1 v/v) as solvent. bL1 (12.0 mol%), MeOH (0.30 mL), 24 h. cUsing L1 (12.0 mol%), MeOH, 24 h, 30 °C; note that the same stereoisomer is formed though with a different E/Z priority assignment, and thus in this case the Z isomer is required towards lactam formation. dUsing P(OPh)3 (12.0 mol%), MeOH, 24 h. eL1 (12.0 mol%), MeOH, 60 °C. | ||
In the case of 20 and 22 bearing a 2-thienyl and 3-furyl group, respectively, the observed stereoselectivity was significantly lower which may be ascribed to the potential of these groups to interact with the Pd-catalyst while affecting the π–σ–π equilibrium as depicted in Scheme 1. Finally, introducing other γ-substituents in the lactam product such as different alkyl substituents (23–25; 54–75%),22 and the introduction of additional α-positioned groups (26, 81%; 27, 52%) were also feasible preserving excellent stereoselectivity. The methylidene substituted product 27 could act as potential Michael acceptor for late-stage modification towards the formation of biologically active compounds. The preparation of 28 (63%) additionally demonstrates that also lactones with substituted vinyl groups are productive substrates.23
Our attention was then on further widening the scope of caprolactam products by combining vinyl lactone A and various amine reaction partners (Scheme 3, compounds 29–49). A range of substituted anilines could be coupled to ring-opened lactone A under the optimized conditions providing overall good to excellent yields of caprolactams 29–40 under high stereocontrol. The use of larger (hetero)aromatic amines (41–44) gave access to lactam products with N-indole, N-1,3-benzodioxole, N-benzofuran and N-naphthyl groups. The di-lactam derivative 45 could be prepared in appreciable yield (69%) despite the more challenging nature of this formal tandem bis allylic amination/cyclization process.
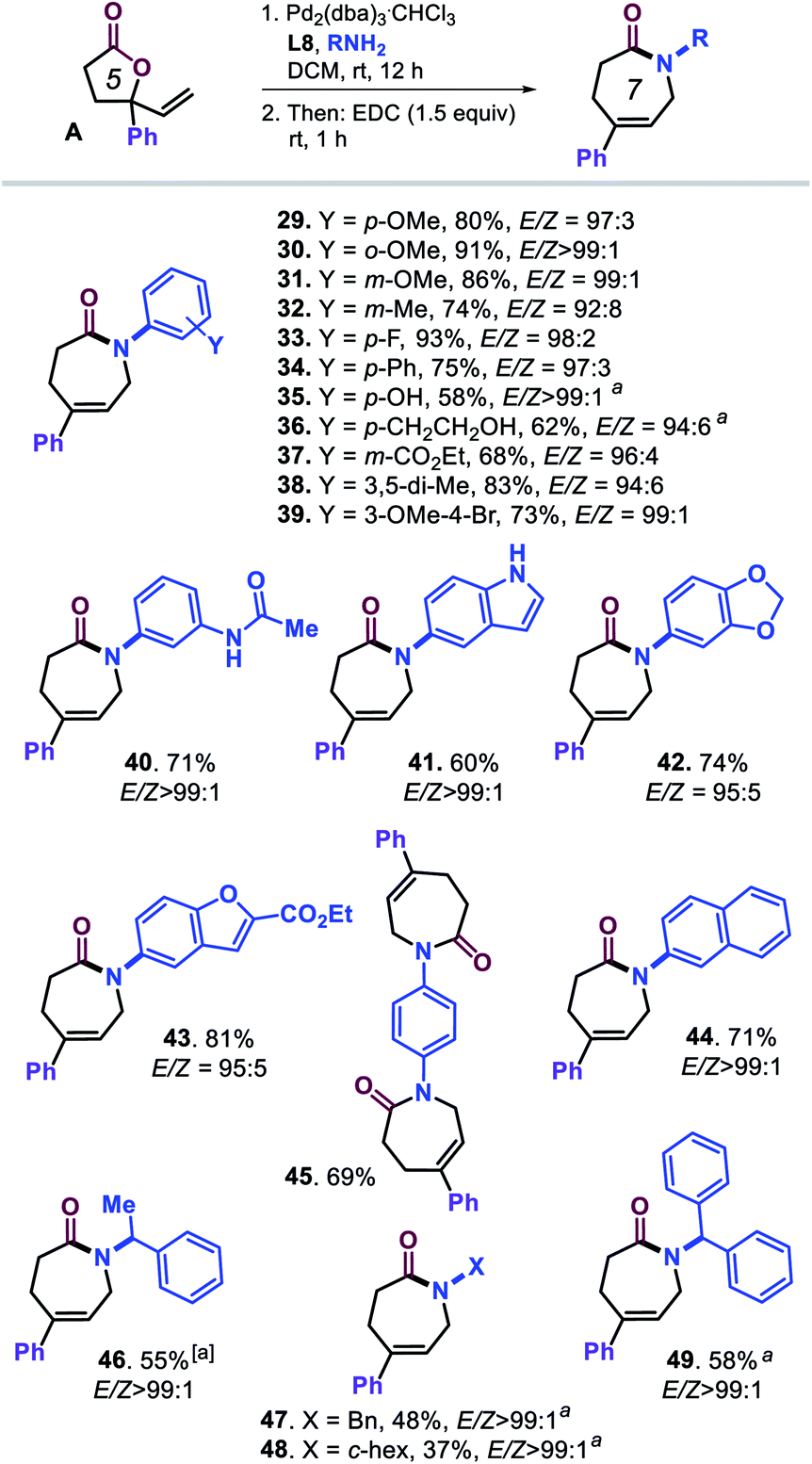 | ||
| Scheme 3 Scope in amine reagents. All reactions were performed under the optimized conditions (Table 1, entry 20). Isolated yields are reported, and the E/Z ratios for the intermediate ε-amino acids were determined by 1H NMR, though in the preparation 45, the E/Z selectivity could not be determined accurately. aUsing DCM/EtOH (1:1 v/v) as solvent. | ||
Lastly, we tested several, more nucleophilic aliphatic amines to challenge the chemo-selectivity of our developed protocol. As may be expected, substantially lower yields for 46–49 were obtained (37–58%) though under excellent stereocontrol (E/Z > 99:1) in a mixed solvent system consisting of DCM/EtOH. The 1H NMR analysis of the crude products indeed indicated that larger amounts of bis-allylated amines of type 2 had been formed.24
In order to examine whether the stereoselectivity indeed plays an important role prior to the cyclization step (Scheme 1), we compared the yield of lactam 3 attained from a configurationally pure sample of 1 (E/Z > 99:1) and one being a 69:31 E/Z mixture (Scheme 4a). From the results, it may be concluded that the maximum yield unquestionably depends on the amount of E-1 that is initially formed, and also molecular models strongly suggest that the (Z) isomer of 1 is unable to cyclize towards a lactam product. These results reinforce the assumption that stereocontrol in the first step is vital to the success of the overall manifold. The use of a simple ligand structure (cf., L8) shows that the presence of a chiral ligand is not a requisite. This is demonstrated by the fact that both (rac)- and enantiopure (S)-L1 give virtually the same stereocontrol (for 1) and lead to a similar yield of 3 (Scheme 4b), thus being consistent with the results described in Table 1.
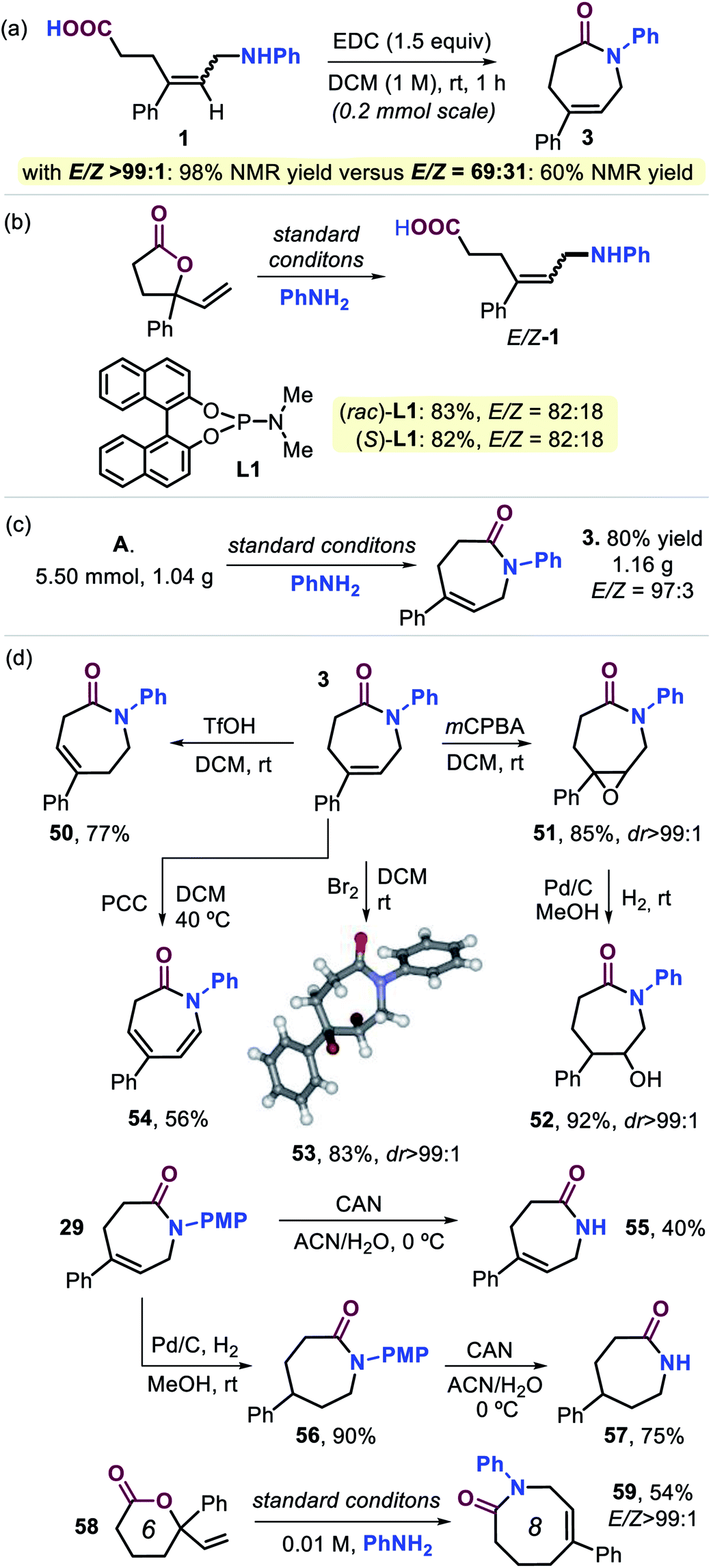 | ||
| Scheme 4 (a and b) Control experiments. (c) Gram-scale preparation of 3. (d) Product diversification studies based on 3 and 29. For details, see the ESI.† | ||
The practicality of this new procedure for the preparation of functional seven-membered lactams is illustrated by the scale-up of compound 3 (Scheme 4c), and product diversification studies using 3 and 29 were also initiated (Scheme 4d). After establishing that scaling up does not influence the stereo-outcome or yield of the product (cf., Scheme 2 and 3), several post-modifications were carried out. Triflic acid mediated double bond isomerization in 3 was feasible to produce 50 in good yield, whereas epoxidation afforded 51 with excellent diastereoselectivity. The latter compound could be reduced by using Pd-catalyzed hydrogenation leading to 52 in high yield. Dibromination of 3 gives 53 (83%, with its molecular structure confirmed by X-ray analysis, see Scheme 4),21 whereas pyridinium chlorochromate (PCC) assisted oxidation of 3 furnished the conjugated, bis-unsaturated caprolactam derivative 54. In order to provide free lactams potentially useful as a new type of monomer for polyamide synthesis or pharmaceutical precursors, compound 29 was treated with cerium ammonium nitrate (CAN) to afford 55 in 40% yield. Alternatively, standard hydrogenation of 29 (56, 90%) followed by deprotection gave easy access to free lactam 57 in 75% yield. Finally, six-membered vinyl lactone 58 could be transformed into eight-membered lactam 59 (54%) with high stereocontrol, and this implies that the developed cascade process may be suitable for the preparation of an even wider range of larger ring lactams.25
Conclusions
In summary, this work provides a new, efficient and practical approach for valuable caprolactam scaffolds that involves (after ring-opening of vinyl γ-lactones) a Pd-mediated stereo- and regio-selective allylic amination followed by a cyclization step affording the lactam target. This formal tandem process relies on the use of a new, easily prepared phosphoramidite ligand (L8) that allows to control the various process selectivity features and, as such, the yield of the final product under ambient conditions. The scale-up and diversification studies show that more functional scaffolds may be created that can be of great value to build up molecular complexity from this new library of readily accessible caprolactam building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the CERCA Program/Generalitat de Catalunya, ICREA, and the Spanish MINECO (CTQ-2014–60419-R) and AGAUR (2017-SGR-232) for financial support. Dr Eduardo C. Escudero-Adán is thanked for measuring the X-ray molecular structures of 3 and 53. J. X. and X. L. thank the Chinese Research Council (CSC) for funding their predoctoral fellowships (2016-06200061 and 2019-06870036).Notes and references
- For reviews of different ring-size, pharma-relevant lactams:
(a) R. Fernandes, P. Amador and C. Prudêncio, Rev. Med. Microbiol., 2013, 24, 7 CrossRef
; (b) U. Nubbemeyer, Top. Curr. Chem., 2001, 216, 125 CrossRef CAS
-
(a) M. Winnacker, Biomater. Sci., 2017, 5, 1230 RSC
-
(a) R. Kumar, S. Shah, P. P. Das, G. G. Bhabavanbhai, A. Al Fatesh and B. Chowdhury, Catal. Rev.: Sci. Eng., 2019, 61, 516 CrossRef CAS
-
(a) A. H. Blatt, Chem. Rev., 1933, 12, 215 CrossRef CAS
-
(a) S. Lang and J. A. Murphy, Chem. Soc. Rev., 2006, 35, 146 RSC
- For some examples:
(a) C. Xie, J. Song, H. Wu, Y. Hu, H. Liu, Z. Zhang, P. Zhang, B. Chen and B. Han, J. Am. Chem. Soc., 2019, 141, 4002 CrossRef CAS PubMed
-
(a) M. Szostak and J. Aubé, Chem. Rev., 2013, 113, 5701 CrossRef CAS PubMed
- K.-I. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785 CrossRef CAS PubMed
- K. Kim and S. H. Hong, J. Org. Chem., 2015, 80, 4152 CrossRef CAS PubMed
-
(a) H. F. Motiwala, C. Fehl, S.-W. Li, E. Hirt, P. Porubsky and J. Aubé, J. Am. Chem. Soc., 2013, 135, 9000 CrossRef CAS PubMed
- For examples:
(a) H. Hotoda, M. Furukawa, M. Daigo, K. Murayama, M. Kaneko, Y. Muramatsu, M. M. Ishii, S.-I. Miyakoshi, T. Takatsu, M. Inukai, M. Kakuta, T. Abe, T. Harasaki, T. Fukuoka, Y. Utsui and S. Ohya, Bioorg. Med. Chem. Lett., 2003, 13, 2829 CrossRef CAS PubMed
-
(a)
Amide Linkage: Selected Structural Aspects in “Chemistry, Biochemistry, and Materials Science”, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley, New York 2000 Search PubMed
-
(a) G. A. Molander, Acc. Chem. Res., 1998, 31, 603 CrossRef CAS
- For a selection of stereo- and enantio-selective allylic aminations:
(a) A. Cai, W. Guo, L. Martínez-Rodríguez and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 14194 CrossRef CAS PubMed
-
(a) W. Guo, J. E. Gómez, L. Martínez-Rodríguez, N. A. G. Bandeira, C. Bo and A. W. Kleij, ChemSusChem, 2017, 10, 1969 CrossRef CAS PubMed
-
(a) B. M. Trost and T. P. Klun, J. Am. Chem. Soc., 1979, 101, 6756 CrossRef CAS
- For the crucial role of the ancillary ligand in Pd-catalyzed allylic substitution:
(a) L. Hu, A. Cai, Z. Wu, A. W. Kleij and G. Huang, Angew. Chem., Int. Ed., 2019, 58, 14694 CrossRef CAS PubMed
- The chemo-selectivity issue here is related to the occurrence of over-allylation of the primary amine reagent as indicated in Scheme 1. As for the stereoselectivity, the (E) configuration of the intermediate ε-amino acid is deemed favorable for intramolecular dehydration affording the caprolactam product whereas the (Z) intermediate will likely follow an intermolecular dehydrative pathway.
- EDC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, was selected as it represents a practical and standard dehydrative reagent/carboxylic acid activator. See for instance: J. Sheehan, P. Cruickshank and G. Boshart, J. Org. Chem., 1961, 26, 2525 CrossRef CAS
- For more extensive data from the screening phase, please refer to the ESI.† In these reactions the virtual complete linear regio-selectivity in the allylic amination step was unexpected as phosphoramidite ligands have recently been shown to direct allylic amination and sulfonation towards the branched products, see ref. 14b, c and 17b. A further control experiment using L8 in a previously reported allylic amination reaction (ref. 14a) indeed shows that a 15:85 regioisomeric mixture of branched and linear allylic amine is obtained, see the ESI† for details.
- For more details, see CCDC 1998235 (3) and CCDC 2001102 (53).
- Martin et al. reported previously a Pd-catalyzed allylic alkylation process involving an γ-alkyl-γ-vinyl substituted lactone and a carbon based nucleophile
affording the allylic alkylation product with a 1:1 E/Z ratio, see: B. L. Ashfeld and S. F. Martin, Org. Lett., 2005, 7, 4535 CrossRef CAS PubMed
- The use of a lactone substrate devoid of additional substitution (R1, Scheme 1) was also tried but failed to produce the caprolactam product under the optimized conditions. See details and comments in the ESI.†.
- Bis-allylation of primary aliphatic amines has been observed before, see:
(a) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613 CrossRef CAS PubMed
- Attempts to prepare a nine-membered lactam derivative gave only access to low yield (<10%) of product, see the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1998235 (3) and 2001102 (53). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03647a |
| This journal is © The Royal Society of Chemistry 2020 |