
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe noncovalent dimerization of a G-quadruplex/hemin DNAzyme improves its biocatalytic properties†
Yu
Cheng‡
ab,
Mingpan
Cheng‡
a,
Jingya
Hao
ab,
Guoqing
Jia
a,
David
Monchaud
 c and
Can
Li
*a
c and
Can
Li
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: canli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cInstitut de Chimie Moléculaire de l' Université de Bourgogne (ICMUB), CNRS UMR 6302, UBFC Dijon, 21078 Dijon, France
First published on 12th August 2020
Abstract
While many protein enzymes exert their functions through multimerization, which improves both selectivity and activity, this has not yet been demonstrated for other naturally occurring catalysts. Here, we report a multimerization effect applied to catalytic DNAs (or DNAzymes) and demonstrate that the enzymatic efficiency of G-quadruplexes (GQs) in interaction with the hemin cofactor is remarkably enhanced by homodimerization. The resulting non-covalent dimeric GQ–DNAzyme system provides hemin with a structurally defined active site in which both the cofactor (hemin) and the oxidant (H2O2) are activated. This new biocatalytic system efficiently performs peroxidase- and peroxygenase-type biotransformations of a broad range of substrates, thus providing new perspectives for biotechnological application of GQs.
Introduction
Besides its function as the repository of genetic information, DNA molecules are increasingly studied for their biocatalytic properties, often referred to as DNAzymes.1–11 Catalytic DNA usually comprises a sequence in which a tertiary structure is encoded which, upon interaction with a metal cofactor,12 accelerates chemical reactions.13–21 An interesting tertiary structure is the G-quadruplex (GQ) that folds from guanine-rich sequences through the stacking of G-quartets, i.e., arrays of four guanines associated via Hoogsteen hydrogen bonds.22–26 A GQ exhibits enticing catalytic properties notably upon interaction with hemin (Fe(III)–protoporphyrin IX), a well-known cofactor of hemoproteins.27,28 The stacking of hemin onto an accessible G-quartet of a GQ provides a catalytic GQ/hemin system that promotes peroxidase- and peroxygenase-type oxidation of different substrates in the presence of an excess of oxidants such as hydrogen peroxide (H2O2).27,29 However, despite dozens of examples and applications reported each year,12,13,30,31 the precise mechanism of these oxidations still remains unclear.Hemin can interact with both accessible G-quartets of a GQ, referred to as the 3′- and 5′-end, respectively, but is more catalytically competent at the 3′-end.29,32–35 Various catalytic intermediates have been postulated to explain this observation, but a firm demonstration is still expected.36,37 Efforts have been invested to optimize the hemin binding site via the modification of the adjacent nucleobases or the surrounding loops of the 3′-end.32,38–44 However, most if not all optimizations have been attempted with monomeric GQs.45 We previously demonstrated that multimeric GQs (comprising several covalently linked GQ units) display better catalytic performance than the corresponding monomeric GQs.46–48 This improvement originates from the formation of a high-activity hemin binding site located at the interface between two consecutive GQ units. In these assemblies, hemin is sandwiched in between the 3′-end of a GQ unit and the 5′-end of the other GQ unit (Fig. 1A).46–48
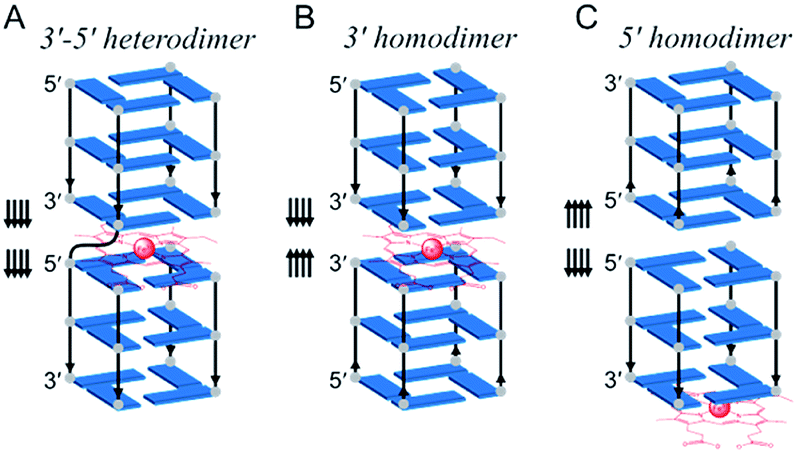 | ||
| Fig. 1 Schematic representation of dimeric GQ interaction with hemin: (A) a covalent multimeric GQ considered as a 3′–5′ heterodimeric GQ;46 two non-covalent GQ assemblies that interact via their 3′-ends (3′–3′ homodimeric GQ, B) and their 5′-ends (5′–5′ homodimeric GQ, C). The arrows indicate the strand orientation from the 5′- to 3′-direction; only the structural core of GQs is represented for clarity. | ||
Here, we hypothesize that providing hemin with a binding pocket formed by two 3′-ends can enhance its catalytic proficiency. To do so, we decided not to use covalent multimeric GQs (that require long sequences, i.e., being poorly efficient and having expensive syntheses, and for which only 5′–3′ interfaces have been reported) but we designed 3′–3′ stacked non-covalent dimeric GQs (along with 5′–5′ dimers as controls), resulting from the self-association of two blunt-ended GQs with naked 3′-ends (and 5′-ends, respectively, Fig. 1B and C). In this original biocatalytic system, hemin nestles in between two 3′-ends (Fig. 1B) where it acquires a high catalytic activity that allows the resulting system to perform a broad variety of biotransformations in a highly efficient manner.
Results and discussion
Formation of a non-covalent dimeric GQ–DNAzyme system
An approach to control the dimerization of GQs is to use DNA sequences that fold into tetramolecular GQs (that is, with a single stretch of guanines comprising 3 to 5 guanines) with various 3′- and 5′-ends.49–51 The 3′-homodimeric GQ system was built with 5′-TTAGn-3′ (n = 3–5), displaying a blunt 3′-G-quartet, along with 5′-TTAGnT-3′, 5′-TTAGnTT-3′ and 5′-TTAG3p-3′ (corresponding to the 5′-TTAGn-3′ sequence with a 3′ phosphorylated end) as controls since the presence of dTs (or phosphate groups) on the 3′-end is likely to preclude GQ dimerization. We experimentally established this by non-denaturing polyacrylamide gel electrophoresis (PAGE) (Fig. 2A and B): in a GQ-promoting K+-rich buffer (100 mM KCl), 5′-TTAG3-3′ was predominantly found as a dimer (64% versus 36% as a monomer), its 3′-homodimeric nature having been previously established by nuclear magnetic resonance (NMR) spectroscopy.52,53 Conversely, 5′-TTAG3TT-3′ was found as a monomer only,52 and both 5′-TTAG3T-3′ and 5′TTAG3p-3′ were found as mixtures of monomeric and dimeric forms. We confirmed this by NMR for 5′-TTAG3-3′, 5′-TTAG3T-3′ and 5′-TTAG3TT-3′ (Fig. S1†). The 5′-homodimeric GQ system was built using 5′-(G3T)4-3′, 5′-T(G3T)4-3′ and 5′-TT(G3T)4-3′. These sequences were selected since it was previously demonstrated that 5′-(G3T)4-3′ self-assembles into a 5′-homodimer (PDB 2LE6), 5′-TT(G3T)4-3′ exists as a monomer only (PDB 2LK7) and 5′-T(G3T)4-3′ as a mixture of monomeric and dimeric forms.54,55 To further demonstrate the generality of this approach, other sequences were studied (a total of 23 sequences, Table S1†), whose GQ folding ability was assessed by circular dichroism (CD, Fig. S2†)56 and dimerization was assessed by PAGE (Fig. S3†).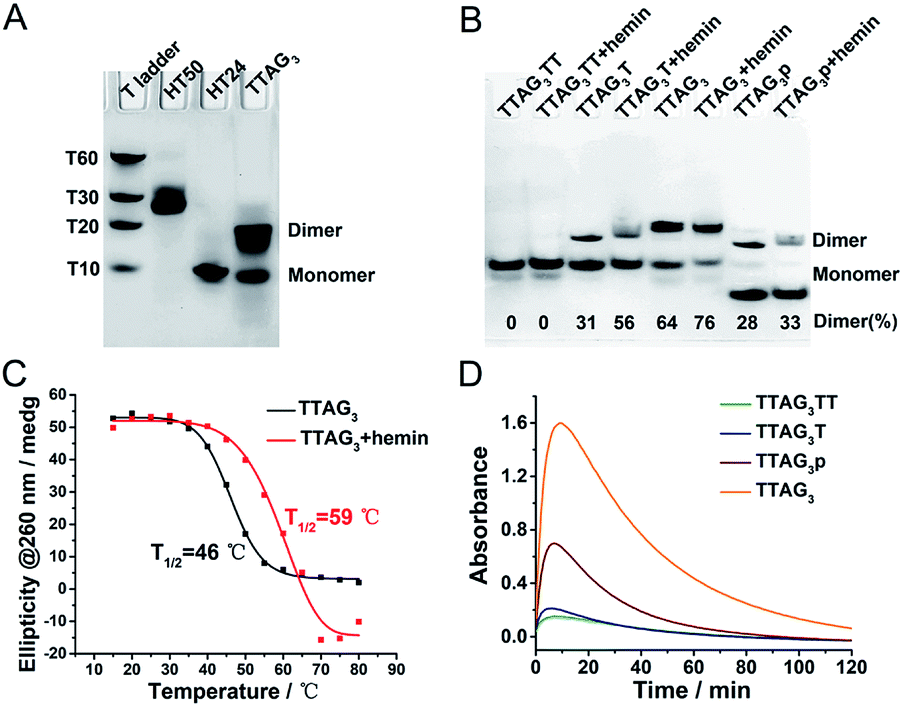 | ||
Fig. 2 (A) PAGE analysis of the dimerization of 5′-TTAG3-3′ in 100 mM KCl. Oligonucleotides HT50 (5′-(TTAG3)8TT-3′)57 and HT24 (5′-T2G3(TTAG3)3A-3′, PDB# 2GKU)58 and poly(dT)s were used as migration markers for dimeric and monomeric GQs, respectively. (B) PAGE analysis of the dimerization of 5′-TTAG3TT-3′, 5′-TTAG3T-3′, 5′-TTAG3-3′, and 5′-TTAG3p-3′ (50 μM), with and without hemin (100 μM), in 40 mM HEPES buffer (pH 7.5) with 100 mM KCl. The percentage of the dimer is given at the bottom of the gel. (C) CD-melting profiles at 260 nm of 5′-TTAG3-3′ (10 μM) in the absence (black line) and presence of hemin (5 μM, red line) in 40 mM HEPES buffer (pH 7.5) with 100 mM KCl and 0.05% (v/v) Triton X-100 (Fig. S4†). (D) Catalytic oxidation of ABTS (2 mM, absorbance at λ = 414 nm as a function of the time) promoted by H2O2 (0.6 mM) in the presence of GQs (0.25 μM) and hemin (0.5 μM) in 40 mM HEPES buffer (pH 7.5, optimized by pH-dependent catalytic reactions, Fig. S6†) with 100 mM KCl and 0.05% (v/v) Triton X-100, at 25 °C. The initial reaction velocities (V0) of oxidations catalyzed by 5′-TTAG3TT-3′, 5′-TTAG3T-3′, 5′-TTAG3p-3′ and 5′-TTAG3-3′ are V0 = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 054, 5333, 1624 and 1054 nM s−1, respectively. 054, 5333, 1624 and 1054 nM s−1, respectively. | ||
Next, the influence of the presence of hemin was studied by both PAGE and CD (Fig. 2B and S2†): hemin was found not to disturb GQ dimerization and was even able to promote and stabilize the GQ dimer. As an example, the proportion of the dimeric form of 5′-TTAG3-3′ increases from 64 to 76% (Fig. 2B) and the melting temperature (assessed by CD-melting experiment) increases by 13 °C (Fig. 2C) after the addition of hemin. This strong GQ dimer/hemin interaction thus opened the way towards the use of these new catalytic systems for performing peroxidation-like reactions. The oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) by H2O2 was used as a model reaction, given that it is a firmly established model reaction and it provides a readily monitorable output (appearance of a characteristic absorbance at λ = 414 nm corresponding to the product ABTS˙+). To assess the performance of these new catalytic systems over a relatively long period of time (2 h), we first used a high concentration of the substrate (2 mM, Fig. 2D). We found that the relationship between homodimerization and catalytic activity was straightforward, with initial velocities (V0) of 11054, 5333, 1624 and 1054 nM s−1 for 5′-TTAG3-3′, 5′-TTAG3p-3′, 5′-TTAG3T-3′ and 5′-TTAG3TT-3′, respectively (of note is that after 10 min of reaction, the absorbance started to decrease due to the degradation of ABTS˙+ under the conditions used). 5′-TTAG3-3′, which is found mostly as a 3′-homodimer, is thus 11-fold more catalytically competent than 5′-TTAG3TT-3′, which exists as a monomer only.
We then compared the performances of the 3′- and 5′-homodimers by decreasing the ABTS concentration (0.5 mM, Fig. 3). Only small variations in catalytic performances were monitored with the 5′-homodimers (Fig. 3A), with V0 = 140, 172 and 142 nM s−1 for 5′-(G3T)4-3′, 5′-T(G3T)4-3′ and 5′-TT(G3T)4-3′, respectively, which are roughly 2-fold lower than that of 5′-TTAG3-3′ (V0 = 288 nM s−1, Fig. 3B). The generality of this approach was demonstrated through using dAs instead of dTs as nucleobases that disrupt GQ dimerization: again the presence of dAs had drastic consequences on the catalytic activity of the 3′-homodimers,32,40,42 with 5′-TTAG3-3′ being far more efficient than 5′-TTAG3A-3′ and 5′-TTAGnAA-3′ (V0 = 288, 47 and 18 nM s−1, respectively, Fig. 3B), while it barely affected those of the 5′-homodimers (V0 = 140, 185 and 187 nM s−1 for 5′-(G3T)4-3′, 5′-A(G3T)4-3′ and 5′-AA(G3T)4-3′, respectively, Fig. 3C).
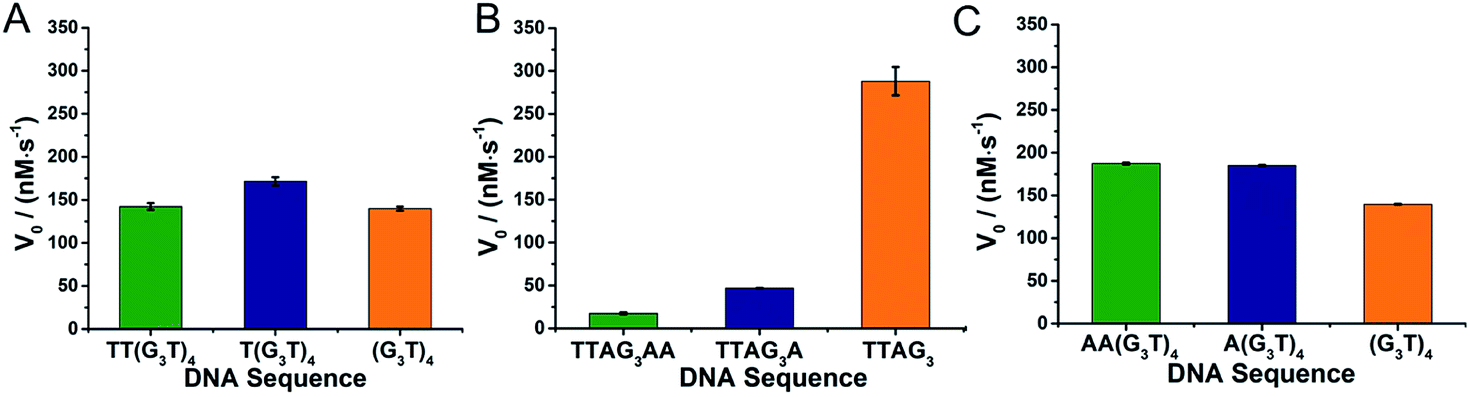 | ||
| Fig. 3 Catalytic activities of non-covalent dimeric GQ–DNAzymes formed from 5′-homodimers with dTs on their 5′-ends (A, 5′-TT(G3T)4-3′, 5′-T(G3T)4-3′ and 5′-(G3T)4-3′), 3′-homodimers with dAs on their 3′-ends (B, 5′-TTAG3AA-3′, 5′-TTAG3A-3′ and 5′-TTAG3-3′) and 5′-homodimers with dAs on their 5′-ends (C, 5′-AA(G3T)4-3′, 5′-A(G3T)4-3′ and 5′-(G3T)4-3′). The reactions were performed with GQs (0.25 μM) and hemin (0.5 μM) in the presence of ABTS (0.5 mM) and H2O2 (0.5 mM) in 40 mM HEPES (pH 7.5) with 100 mM KCl and 0.05% (v/v) Triton X-100, at 25 °C. | ||
Potassium promotes GQ dimerization and DNAzyme activity
We then decided to further investigate GQ dimerization via1H-NMR. The analysis of 5′-TTAG3-3′ was performed in the presence of increasing amounts of KCl (from 0 to 300 mM, Fig. 4A, B and S1A†). The 1H-NMR signals between 10 and 12 (the so called imino protons) are characteristic of the guanines embedded in G-quartets. Our results confirmed first that the GQ was not folded without K+; they also showed that monomeric GQs are favoured at low-concentration of K+ and that an increase in K+ concentration favoured dimeric GQs. This was further demonstrated by size exclusion chromatography (SEC)-HPLC (Fig. 4C)59 and PAGE experiments (Fig. 4D). A comparative quantitative analysis of these two techniques showed remarkable agreement in the percentage of dimeric GQs promoted by K+ (Fig. 4E), with >75% of the dimeric form in the presence of 300 mM KCl. We also verified the behaviour of both 5′-TTAG3T-3′ and 5′-TTAG3TT-3′ under similar conditions: as expected, the monomeric form was found to be predominant for both sequences even at 300 mM KCl concentration (Fig. S1B and C†). This K+-promoted dimerization had also strong consequences in terms of biocatalysis. Indeed, varying the KCl concentration from 0 to 500 mM strongly impacted the catalytic performance of 5′-TTAG3-3′ but not that of 5′-TTAG3T-3′ and 5′-TTAG3TT-3′: in the presence of 500 mM KCl, 5′-TTAG3-3′ was found to be 41- and 10-fold more catalytically active than 5′-TTAG3TT-3′ and 5′-TTAG3T-3′, respectively (Fig. 4F). The straightforward relationship between the V0 values and the percentage of dimeric GQs is unambiguously demonstrated in Fig. 4G, without being a simple linear correlation (Fig. 4H). Of note is that we demonstrated the specificity of the K+ cations by replacing them with Li+, Na+, NH4+ or Mg2+, which resulted in a decrease of both GQ dimerization and GQ–DNAzyme activities (Fig. S5†). We also performed these experiments at pH 7.5 and 25 °C since these conditions were found to be optimal for GQ–DNAzyme catalysis (Fig. S6†).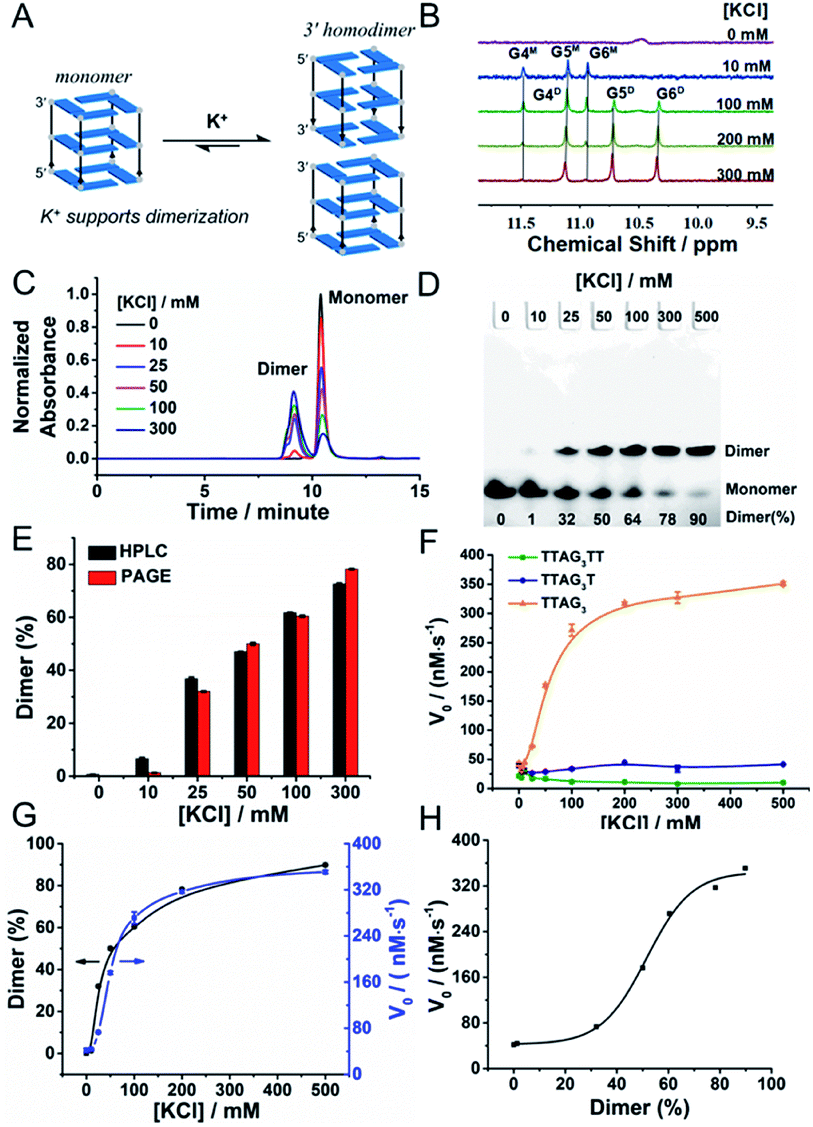 | ||
| Fig. 4 Schematic representation (A) of the K+-promoted GQ dimerization of 5′-TTAG3-3′, 1H-NMR spectra (B) of 5′-TTAG3-3′ (GQ, 100 μM) at different K+ concentrations (0, 10, 100, 200 and 300 mM), in 10% (v/v) D2O/H2O, pH/pD 7.5. The imino protons were assigned according to previously published articles;27,42 ‘M’ and ‘D’ stand for the monomeric and dimeric GQs, respectively. Guanines are numbered according to their position in the d(T1T2A3G4G5G6) sequence. SEC-HPLC (C) and PAGE analysis (D) of 5′-TTAG3-3′ at different K+ concentrations (0 to 500 mM). (E) Comparison of the percentage of the dimeric form of 5′-TTAG3-3′ obtained by both SEC-HPLC and PAGE at different K+ concentrations. Catalytic activities (V0, F) of non-covalent dimeric GQ–DNAzymes formed from 5′-TTAG3-3′, 5′-TTAG3T-3′ and 5′-TTAG3TT-3′ as a function of the K+ concentration. Comparison (G) and relationship (H) of the percentage of the dimeric GQ formed from 5′-TTAG3-3′ and the catalytic activity of the 5′-TTAG3-3′/hemin DNAzyme (V0) at different K+ concentrations. | ||
We finally assessed whether the 5′-TTAG3-3′ dimer withstood dilution, in order to operate in the low micromolar concentration range routinely used for DNAzyme-type experiments. SEC-HPLC and PAGE analyses showed that the percentage of the dimeric GQ did not change with dilution (Fig. S7A–C and Table S2†). The optimal concentration for GQ–DNAzyme experiments was thus determined (Fig. S7D†): the catalytic activity was found to increase rapidly with the GQ concentration in the 0–2.5 μM range and to reach a plateau after 5.0 μM. This concentration was thus chosen for subsequent studies.
Kinetics and scope of non-covalent dimeric GQ–DNAzymes
The kinetics of GQ–DNAzymes with 3′-homodimers were studied via the measurement of V0 values for reactions performed with increasing H2O2 concentration (0 to 6 mM). The kinetic curves of 5′-TTAG3-3′, 5′-TTAG3T-3′, 5′-TTAG3TT-3′ and 5′-TTAG3p-3′ are seen in Fig. 5A and B (those of 5′-TTAG4-3′, 5′-TTAG4T-3′ and 5′-TTAG4TT-3′ as well as 5′-TTAG5-3′, 5′-TTAG5T-3′ and 5′-TTAG5TT-3′ in Fig. S8A†). Michaelis–Menten fitting gave the kinetic parameters kcat, Km and kcat/Km (summarized in Fig. S8B and Table S3†): the blunt-end dimeric GQs 5′-TTAGn-3′ (n = 3, 4 or 5) were found to be far more efficient (kcat/Km between 1.2 and 1.6 s−1 mM−1, Fig. 5C) than any other sequences, with kcat/Km between 4.3- and 5.4-fold higher than those of the monomeric GQs 5′-TTAGnTT-3′ (n = 3, 4 or 5). The best results were obtained with 5 μM 5′-TTAG3-3′ (denoted by * in Fig. 5C), with kcat = 8.2 s−1, Km = 3.8 mM and kcat/Km = 2.2 s−1 mM−1, which represent a 82-fold (kcat) and 7.4-fold (kcat/Km) enhancement as compared to those of monomeric 5′-TTAG3TT-3′. These results positioned the 3′-homodimeric system in the upper range of the reported kcat examples (1.1, 4.2, 4.3 and 2.3 s−1 for PS2.M,60 c-Myc,60 EAD2 (ref. 60) and c-Myc3C-2A,38 respectively), but still lower than that of horseradish peroxidase (HRP, 330 s−1),28 and in the high range of kcat/Km values (e.g., 2.1 s−1 mM−1 for c-Myc3C-2A),38 but still far lower than that of HRP (104 s−1 mM−1).28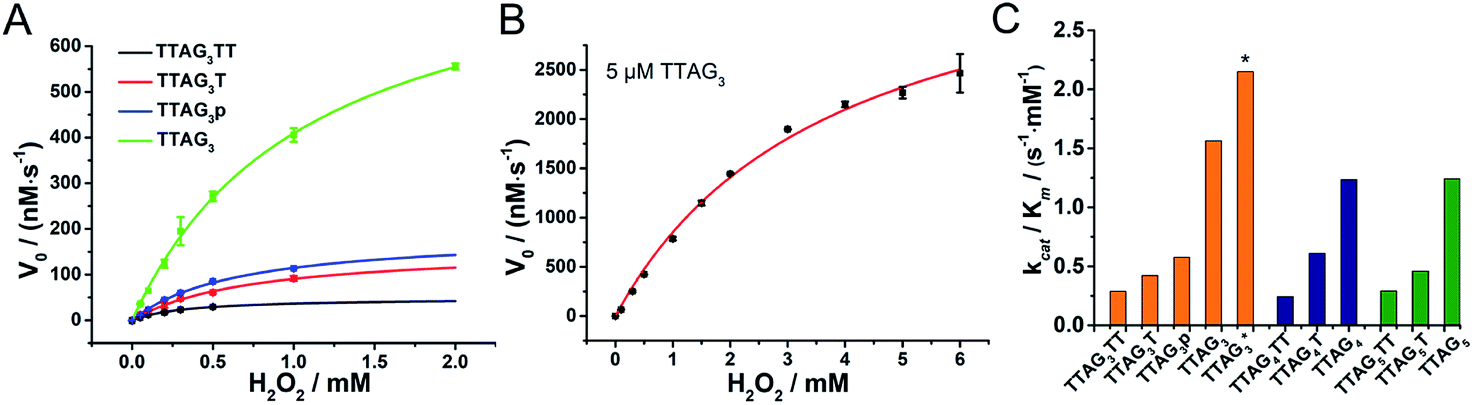 | ||
| Fig. 5 Michaelis–Menten analysis of non-covalent dimeric GQ–DNAzymes from 5′-TTAG3TT-3′, 5′-TTAG3T-3′, 5′-TTAG3p-3′ and 5′-TTAG3-3′ at 0.25 μM concentration (A) and from 5′-TTAG3-3′ at 5 μM concentration (B). Catalytic efficiency (kcat/Km values, C) of 3′-homodimeric DNAzymes formed from GQs (0.25 μM; * data obtained with 5 μM GQ) and hemin (0.5 μM) in the presence of ABTS (0.5 mM) at increasing H2O2 concentrations (0 to 6.0 mM) in 40 mM HEPES (pH 7.5) with 100 mM KCl and 0.05% (v/v), at 25 °C. | ||
To further investigate the scope of applications of this new system, we studied the catalytic oxidations of eight different substrates (Fig. 6), i.e., luminol,61 TMB,62L-tyrosine,63D-tyrosine,63L-dopa,63 dopamine,63 NADH64 and thioanisole.29 The logic behind this selection is three-fold: first, luminol and TMB are two classical substrates for DNAzyme-based biosensors;13 second, L/D-tyrosine, L-dopa, dopamine and NADH are naturally occurring substrates whose oxidation products are implicated in oxidative stress;36,65,66 third, most of these substrates are an illustrative panel of organic contaminants from industrial activities. Oxidative enzymes such as HRP are indeed used for remediation purposes,67 allowing for the treatment of wastewaters and contaminated soils, with this enzyme being effective with a wide range of substrates,68 from organic dyes69 to phenolic contaminants.70 This series of dyes (luminol, ABTS and TMB), phenolic (tyrosine) and biphenolic compounds (dopamine) and a thiol derivative (thioanisole)71 was used to assess the efficiency of both 3′-homodimers (5′-TTAG3-3′, 5′-TTAG3T-3′ and 5′-TTAG3TT-3′) and 5′-homodimers (5′-(G3T)4-3′, 5′-T(G3T)4-3′ and 5′-TT(G3T)4-3′) as tools for degrading environmental pollutants. We found that the 5′-TTAG3-3′ dimer strongly accelerated all these oxidations, while the dT-containing GQ and the monomeric GQs only moderately (Fig. S9†). We also checked whether some of these catalysts were asymmetric but no inductions of chirality were observed for L- and D-tyrosine (Fig. S9.3 and S9.4†), and thioanisole (see chiral HPLC traces in the ESI†), which is not unexpected given the lack of a chiral environment provided by the homodimers.
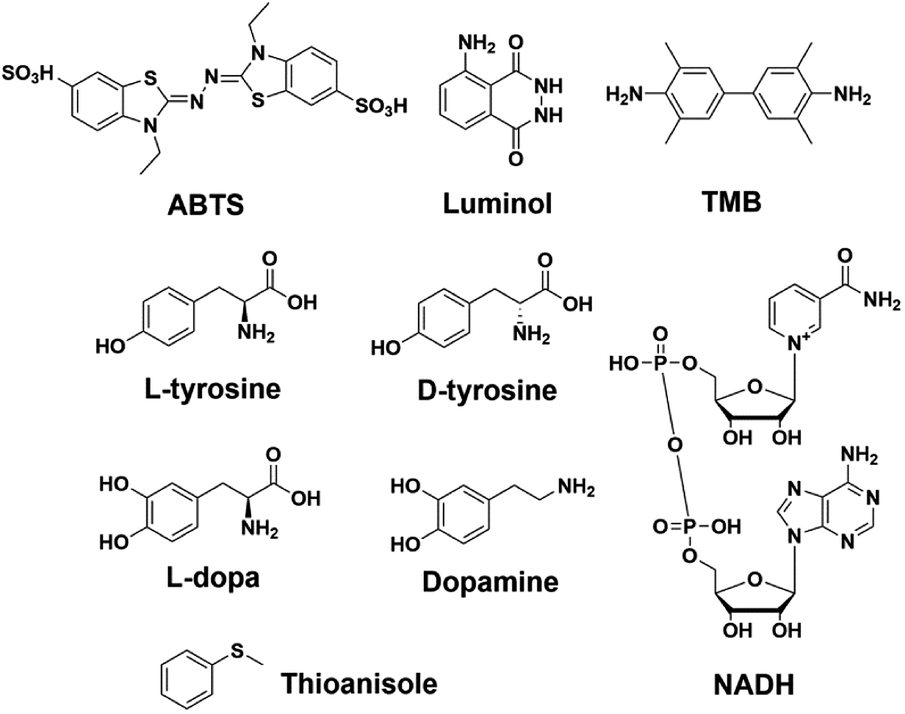 | ||
| Fig. 6 Different substrates used for assessing the scope of non-covalent dimeric GQ–DNAzymes. | ||
Model of the non-covalent dimeric GQ–DNAzyme
The actual structure of the DNAzyme system formed from the dimerization of the GQ formed using 5′-TTAG3-3′, and sandwiching hemin, was further investigated by SEC-HPLC. This analysis allows for both discrimination of the monomeric and dimeric GQs (absorbance of the DNA at 260 nm) and identification of the GQ/hemin complex (absorbance of hemin at 404 nm). In the absence of hemin, the results seen in Fig. 7A show that the monomeric and dimeric GQs can be easily identified; after addition of hemin, the signatures seen in Fig. 7B demonstrate both the coexistence of the monomeric and dimeric GQs (absorbance at 260 nm) in the presence of hemin (meaning that it does not affect the equilibrium, in line with the PAGE results seen in Fig. 2B) and the exclusive formation of the dimeric GQ/hemin complex (absorbance at 404 nm). These results indicated that hemin binds to the dimeric GQs only; we can thus postulate that the preferred hemin binding site is the cleft formed at the 3′–3′ interface (Fig. 1B).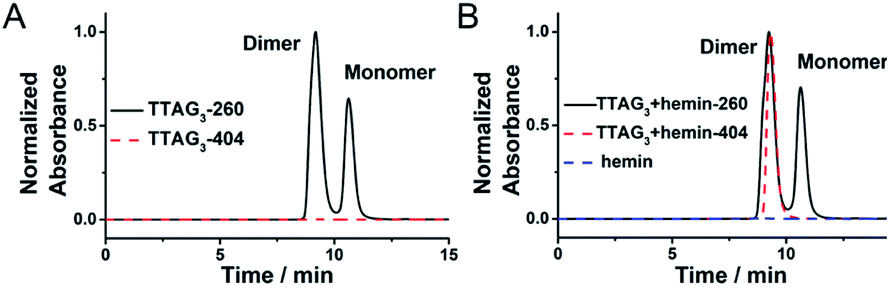 | ||
| Fig. 7 SEC-HPLC profiles of 5′-TTAG3-3′ (10 μM) in the absence (A) and presence (B) of hemin (2.0 μM), monitored at 260 nm (DNA absorbance, solid line) and 404 nm (hemin absorbance, dashed line) in 40 mM HEPES (pH 7.5) with 100 mM KCl. | ||
To further demonstrate that the 5′-end is not involved in the catalytic properties of the resulting GQ/hemin complexes, a series of 3′-homodimers was designed with various 5′-ends (5′-TAG3-3′, 5′-AAG3-3′, 5′-TTG3-3′ and 5′-ATG3-3′). The dimerization of these sequences was checked by PAGE (Fig. S3C†). We also found that these sequences displayed similar catalytic activities (Fig. S10A†), in line with the primary role of the nature of their 3′-ends only. As controls, we disrupted the G-tract of 5′-TTAG3-3′ (both 5′-TTAGCG-3′ and 5′-TTACCG-3′, Fig. S3D†); the resulting complexes did not exhibit catalytic activity (Fig. S10B†). Finally, we compared the catalytic proficiencies of the non-covalent dimer formed using 5′-TTAG3-3′ and those of the covalent multimers 46AG,46 70AG46 and HT50:57 the results seen in Fig. S10C and Table S4† confirmed the far better properties, at least 22-fold higher catalytic efficiency of the non-covalent dimeric GQ precatalysts as compared to their covalently linked counterparts.
Altogether, this wealth of data lends credence to a model in which hemin becomes catalytically active once sandwiched in between the two 3′-ends of a homodimeric GQ (Fig. 1B). This hypothesis is further substantiated by recent molecular-level simulations that scrutinized GQ dimerization and highlighted the positive roles played by small-molecule ligands in the stability of the GQ/ligand/GQ assemblies at their 3′ interfaces.51 We decided to keep on investigating the hemin/GQ interactions via UV-Vis titrations, and compare our results with those of known GQ–DNAzymes and enzymes (Table S5†): the evolution of the characteristic UV-Vis bands such as Soret, E and D bands (Fig. S11†) indicates that the coordination of the iron atom with the dimeric GQ is of high spin and the six-coordination (HS/6C)-type, similar to what was described with PS2.M and MetMb.28,60 The negligible changes of these UV-Vis bands after addition of the monomeric control 5′-TAG3TT-3′ and 5′-TAG3-3′ at low potassium concentration demonstrated again the privileged binding of hemin to dimeric GQs (Fig. S11†). The apparent dissociation constant Kd decreased with the steric hindrance on the 3′-end (Kd = 5.96, 3.91 and 2.84 μM for 5′-TTAG3T-3′, 5′-TTAG3p-3′ and 5′-TTAG3-3′, respectively, Table S4 and Fig. S12†), and with increasing K+ concentrations (Kd = 5.68, 4.16, 2.84 and 0.91 μM for 5′-TTAG3-3′ at 25, 50, 100 and 300 mM K+, respectively, Table S4 and Fig. S12†), thus further accrediting the hypothesis whereby dimeric GQs provide a better binding pocket to hemin than monomeric GQs.
We next measured the formation rate of reactive intermediates (high-valent iron species) via the H2O2-mediated oxidative degradation of GQ/hemin complexes monitored via the time-dependent evolution of the Soret band in the absence of a reducing substrate (Fig. S14†).66 GQ/hemin complexes formed from 10 selected sequences (Fig. 8A) were tested: the initial degradation velocities (Vd) determined indicated that complexes from the 5′-TTAG3-3′ and 5′-TTAG4-3′ series were degraded more quickly than the corresponding monomers, with the notable exception of the 5′-TTAG5-3′ series (vide infra). To further illustrate this, we plotted the Vd values versus the kcat/Km determined above: the representation seen in Fig. 8B shows that the most catalytically competent sequences experience faster degradation. The case of 5′-TTAG5-3′ is interesting since it exhibits a high catalytic activity and a low degradation rate, thereby opening new avenues towards the design of ever more competent and robust GQ–DNAzyme systems.
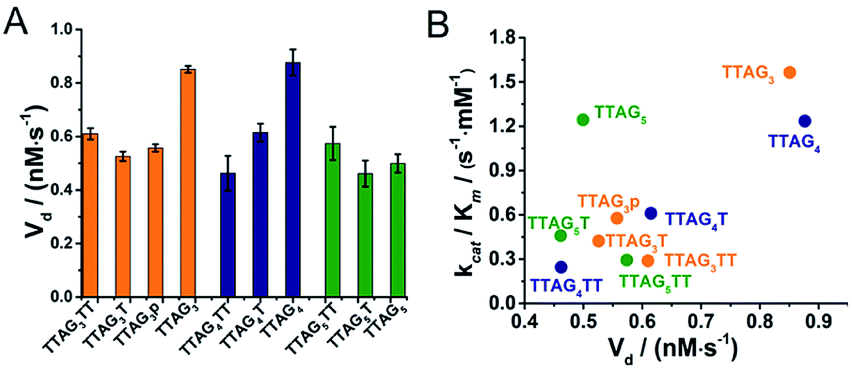 | ||
| Fig. 8 Oxidative degradation of 3′-homodimeric GQ–DNAzymes monitored through the initial apparent degradation velocities (Vd, A) and the correlation between Vd and kcat/Km values (B). Reactions were performed with GQs (0.25 μM) and hemin (0.5 μM) in the presence of H2O2 (0.5 mM) in 40 mM HEPES (pH 7.5) with 100 mM KCl and 0.05% (v/v) Triton X-100. | ||
A mechanism of why and how GQ dimerization enhances the DNAzyme activity of the resulting complexes can be built in agreement and on the basis of previously published models28,39,62,65,66,72,73 (Fig. 9): at the 3′–3′ interface, one of the guanines can flip out from the proximal G-quartet (a ‘wobbling’ guanine)36,39,66,74 to coordinate axially to the iron atom and activate hemin while the distal G-quartet interacts with iron-bound H2O and H2O2. This H-bonding contributes to the heterolysis of O–H via an electron-withdrawing activation. The orchestrated action of both proximal and distal G-quartets uniquely provides a finely tuned hemin binding site, close to what is found in hemoproteins, which is more defined than in monomeric GQs in which one activator is the G-quartet and the other one a structurally labile adjacent nucleobase32,38,39,42 or externally added nucleotides.62,75,76 The presence of two G-quartets uniquely allows for a concerted mechanism (Fig. 9, right red box) that enhances the catalytic efficiency of the resulting complex via a concomitant activation of the cofactor and the oxidant.
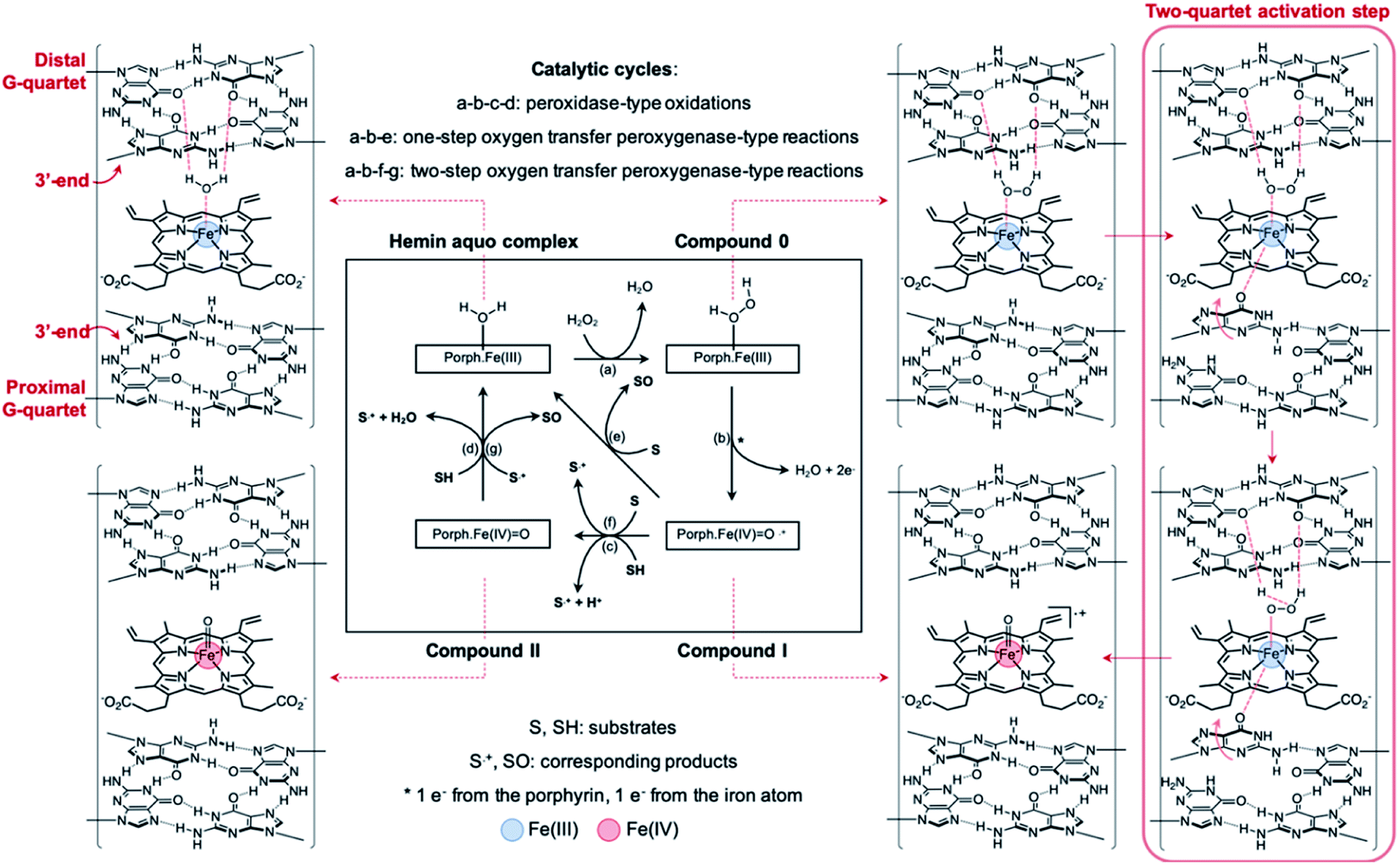 | ||
| Fig. 9 Postulated mechanism in which distal and proximal G-quartets concomitantly activate both hemin and H2O2 to enhance the catalytic proficiency of non-covalent dimeric GQ–DNAzyme degradation systems. | ||
Conclusion
We report here a new prototypic GQ–DNAzyme system comprising two GQ units that self-assemble in a non-covalent manner. We demonstrate that dimeric GQs provide a more defined binding pocket to hemin than monomeric GQs, in which all reaction partners (cofactor and oxidant) are concomitantly activated. The sequence used to validate this proof-of-concept derives from the human telomeric sequence 5′-TTAGGG-3′, in which the 3′-end was left naked on purpose, to make the resulting tetramolecular GQ self-assemble to form a 3′-homodimer, in light of the capacity of blunt-ended GQs to interact with each other, drawn in like magnets through hydrophobic, electrostatic and π-stacking interactions. This new system referred to as the non-covalent dimeric GQ–DNAzyme exhibits high catalytic efficiency, with kcat values up to 8 s−1 for the ABTS oxidation. This system contributes to a new strategy to design efficient GQ–DNAzymes, which can be used to catalyse various types of biotransformations: the scope and versatility of this system were demonstrated via a selection of substrates that are representative of organic pollutants of anthropogenic origin. This provides interesting perspectives for the use of DNA catalysts as biosourced tools for green chemistry purposes and potential sustainable solutions to critical environmental concerns.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Jean-Louis Mergny (Paris, France), Dr Jun Zhou (Nanjing University, China) and Dr Sheng-mei Lu (Dalian Institute of Chemical Physics, China) for their helpful comments on our manuscript. This work was supported by the National Major Scientific Instruments and Equipment Development Project of the National Natural Science Foundation of China, NSFC Grant (No. 2152780065), the National Key R&D Program of China (2017YFB0702800), the CNRS, the Agence Nationale de la Recherche (No. ANR-18-CE07-0017-03), the Université de Bourgogne and Conseil Régional de Bourgogne (PARI) and the European Union (PO FEDER-FSE Bourgogne 2014/2020 programs).Notes and references
- K. Kruger, P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling and T. R. Cech, Cell, 1982, 31, 147 CrossRef CAS.
- C. Guerrier-Takada, K. Gardiner, T. Marsh, N. Pace and S. Altman, Cell, 1983, 35, 849 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223 CrossRef CAS.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153 RSC.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948 CrossRef CAS.
- S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595 CrossRef CAS PubMed.
- M. Liu, D. Chang and Y. Li, Acc. Chem. Res., 2017, 50, 2273 CrossRef CAS.
- D. Morrison, M. Rotherbroker and Y. Li, Small Methods, 2018, 2, 1700319 CrossRef.
- M. Hollenstein, Curr. Opin. Chem. Biol., 2019, 52, 93 CrossRef CAS PubMed.
- M. Cepeda-Plaza and A. Peracchi, Org. Biomol. Chem., 2010, 18, 1697 RSC.
- L. Ma and J. Liu, iScience, 2020, 23, 100815 CrossRef CAS PubMed.
- J. L. Mergny and D. Sen, Chem. Rev., 2019, 119, 6290 CrossRef CAS PubMed.
- Y. Xing, X. Liu, Q. Pu, M. Wu and J. X. Zhao, ACS Appl. Bio Mater., 2018, 1, 1019 CrossRef CAS.
- C. E. McGhee, K. Y. Loh and Y. Lu, Curr. Opin. Biotechnol., 2017, 45, 191 CrossRef CAS PubMed.
- A. Rioz-Martinez and G. Roelfes, Chem. Biol., 2015, 25, 80 CAS.
- D. J.-F. Chinnapen and D. Sen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 65 CrossRef CAS PubMed.
- C. Wang, G. Jia, J. Zhou, Y. Li, Y. Liu, S. Lu and C. Li, Angew. Chem., Int. Ed., 2012, 51, 9352 CrossRef CAS PubMed.
- M. Cheng, Y. Li, J. Zhou, G. Jia, S. M. Lu, Y. Yang and C. Li, Chem. Commun., 2016, 52, 9644 RSC.
- M. Cheng, J. Hao, Y. Li, Y. Cheng, G. Jia, J. Zhou and C. Li, Biochimie, 2018, 146, 20 CrossRef CAS.
- Y. Cheng, M. Cheng, J. Hao, G. Jia and C. Li, ChemBioChem, 2018, 19, 2233 CrossRef CAS.
- H. Ibrahim, P. Mulyk and D. Sen, ACS Omega, 2019, 4, 15280 CrossRef CAS.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627 CrossRef CAS.
- R. Hansel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279 CrossRef CAS PubMed.
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650 CrossRef.
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123 CrossRef.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459 CrossRef CAS PubMed.
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505 CrossRef CAS PubMed.
- P. Travascio, A. J. Bennet, D. Y. Wang and D. Sen, Chem. Biol., 1999, 6, 779 CrossRef CAS.
- L. C. Poon, S. P. Methot, W. Morabi-Pazooki, F. Pio, A. J. Bennet and D. Sen, J. Am. Chem. Soc., 2011, 133, 1877 CrossRef CAS PubMed.
- E. Golub, R. Freeman and I. Willner, Anal. Chem., 2013, 85, 12126 CrossRef CAS PubMed.
- J. Chen, Y. Zhang, M. Cheng, J. L. Mergny, Q. Lin, J. Zhou and H. Ju, Mikrochim. Acta, 2019, 186, 786 CrossRef CAS PubMed.
- W. Li, Y. Li, Z. Liu, B. Lin, H. Yi, F. Xu, Z. Nie and S. Yao, Nucleic Acids Res., 2016, 44, 7373 CrossRef CAS PubMed.
- K. Saito, H. Tai, H. Hemmi, N. Kobayashi and Y. Yamamoto, Inorg. Chem., 2012, 51, 8168 CrossRef CAS PubMed.
- T. Ai, Q. Yang, Y. Lv, Y. Huang, Y. Li, J. Geng, D. Xiao and C. Zhou, Chem.–Asian J., 2018, 13, 1805 CrossRef CAS PubMed.
- Y. Yamamoto, H. Araki, R. Shinomiya, K. Hayasaka, Y. Nakayama, K. Ochi and T. Shibata, Biochemistry, 2018, 57, 5938 CrossRef CAS PubMed.
- P. Travascio, P. K. Witting, A. G. Mauk and D. Sen, J. Am. Chem. Soc., 2001, 123, 1337 CrossRef CAS PubMed.
- H. Shimizu, H. Tai, K. Saito, T. Shibata, M. Kinoshita and Y. Yamamoto, Bull. Chem. Soc. Jpn., 2015, 88, 644 CrossRef CAS.
- T. Chang, H. Gong, P. Ding, X. Liu, W. Li, T. Bing, Z. Cao and D. Shangguan, Chem.–Eur. J., 2016, 22, 4015 CrossRef CAS PubMed.
- J. Chen, Y. Zhang, M. Cheng, Y. Guo, J. Šponer, D. Monchaud, J. L. Mergny, H. Ju and J. Zhou, ACS Catal., 2018, 8, 11352 CrossRef CAS.
- Y. Guo, J. Chen, M. Cheng, D. Monchaud, J. Zhou and H. Ju, Angew. Chem., Int. Ed., 2017, 56, 16636 CrossRef CAS PubMed.
- W. Li, S. Chen, D. Xu, Q. Wen, T. Yang and J. Liu, Chem.–Eur. J., 2018, 24, 14500 CrossRef CAS PubMed.
- J. Chen, Y. Guo, J. Zhou and H. Ju, Chem.–Eur. J., 2017, 23, 4210 CrossRef CAS PubMed.
- E. Golub, H. B. Albada, W. C. Liao, Y. Biniuri and I. Willner, J. Am. Chem. Soc., 2016, 138, 164 CrossRef CAS PubMed.
- M. Cheng, J. Zhou, G. Jia, X. Ai, J. L. Mergny and C. Li, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1913 CrossRef CAS PubMed.
- A. Ponce-Salvatierra, K. Wawrzyniak-Turek, U. Steuerwald, C. Höbartner and V. Pena, Nature, 2016, 529, 231 CrossRef CAS PubMed.
- L. Stefan, F. Denat and D. Monchaud, J. Am. Chem. Soc., 2011, 133, 20405 CrossRef CAS PubMed.
- Y. Li, G. Jia, C. Wang, M. Cheng and C. Li, ChemBioChem, 2015, 16, 618 CrossRef CAS PubMed.
- Y. Li, C. Wang, J. Hao, M. Cheng, G. Jia and C. Li, Chem. Commun., 2015, 51, 13174 RSC.
- S. Kolesnikova, M. Hubalek, L. Bednarova, J. Cvacka and E. A. Curtis, Nucleic Acids Res., 2017, 45, 8684 CrossRef CAS PubMed.
- C. J. Lech, B. Heddi and A. T. Phan, Nucleic Acids Res., 2013, 41, 2034 CrossRef CAS PubMed.
- M. Kogut, C. Kleist and J. Czub, PLoS Comput. Biol., 2019, 15, e1007383 CrossRef CAS PubMed.
- Y. Kato, T. Ohyama, H. Mita and Y. Yamamoto, J. Am. Chem. Soc., 2005, 127, 9980 CrossRef CAS PubMed.
- Y. Wang and D. J. Patel, Biochemistry, 1992, 31, 8112 CrossRef CAS PubMed.
- N. Q. Do and A. T. Phan, Chem.–Eur. J., 2012, 18, 14752 CrossRef CAS PubMed.
- N. Q. Do, K. W. Lim, M. H. Teo, B. Heddi and A. T. Phan, Nucleic Acids Res., 2011, 39, 9448 CrossRef CAS PubMed.
- R. Del Villar-Guerra, J. O. Trent and J. B. Chaires, Angew. Chem., Int. Ed., 2018, 57, 7171 CrossRef CAS.
- L. Petraccone, C. Spink, J. O. Trent, N. C. Garbett, C. S. Mekmaysy, C. Giancola and J. B. Chaires, J. Am. Chem. Soc., 2011, 133, 20951 CrossRef CAS PubMed.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963 CrossRef CAS PubMed.
- E. Largy and J. L. Mergny, Nucleic Acids Res., 2014, 42, 149 CrossRef PubMed.
- X. Cheng, X. Liu, T. Bing, Z. Cao and D. Shangguan, Biochemistry, 2009, 48, 7817 CrossRef CAS PubMed.
- Y. Xiao, V. Pavlov, R. Gill, T. Bourenko and I. Willner, ChemBioChem, 2004, 5, 374 CrossRef CAS PubMed.
- L. Stefan, F. Denat and D. Monchaud, Nucleic Acids Res., 2012, 40, 8759–8772 CrossRef CAS PubMed.
- A. M. Rojas, P. A. Gonzalez, E. Antipov and A. M. Klibanov, Biotechnol. Lett., 2007, 29, 227 CrossRef CAS PubMed.
- E. Golub, R. Freeman and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 11710 CrossRef CAS.
- D. Sen and L. C. Poon, Crit. Rev. Biochem. Mol. Biol., 2011, 46, 478 CrossRef CAS PubMed.
- J. Wang, M. Cheng, J. Chen, H. Ju, D. Monchaud, J.-L. Mergny and J. Zhou, Chem. Commun., 2020, 56, 1839 RSC.
- N. Durán and E. Esposito, Appl. Catal., B, 2000, 28, 83 CrossRef.
- T. Rasheed, M. Bilal, F. Nabeel, M. Adeel and H. M. N. Iqbal, Environ. Int., 2019, 122, 52 CrossRef CAS PubMed.
- Q. Husain, Rev. Environ. Sci. Biotechnol., 2009, 9, 117 CrossRef.
- G. R. Lopes, D. C. G. A. Pinto and A. M. S. Silva, RSC Adv., 2014, 4, 37244 RSC.
- S. Zhao, H. Xi, Y. Zuo, S. Han, Y. Zhu, Z. Li, L. Yuan, Z. Wang and C. Liu, J. Hazard. Mater., 2019, 367, 91 CrossRef CAS PubMed.
- Y. Li and D. Sen, Chem. Biol., 1998, 5, 1 CrossRef CAS.
- X. Yang, C. Fang, H. Mei, T. Chang, Z. Cao and D. Shangguan, Chem.–Eur. J., 2011, 17, 14475 CrossRef CAS PubMed.
- A. F. Voter, Y. Qiu, R. Tippana, S. Myong and J. L. Keck, Nat. Commun., 2018, 9, 4201 CrossRef.
- D.-M. Kong, J. Xu and H.-X. Shen, Anal. Chem., 2010, 82, 6148 CrossRef CAS PubMed.
- L. Stefan, D. Duret, N. Spinelli, E. Defrancq and D. Monchaud, Chem. Commun., 2013, 49, 1500 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02907f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |