
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMononuclear ruthenium(II) theranostic complexes that function as broad-spectrum antimicrobials in therapeutically resistant pathogens through interaction with DNA†
Kirsty L.
Smitten
ab,
Eleanor J.
Thick
a,
Hannah M.
Southam
b,
Jorge
Bernardino de la Serna
cd,
Simon J.
Foster
b and
Jim A.
Thomas
*a
aDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: james.thomas@sheffield.ac.uk
bDepartment of Molecular Biology and Biotechnology, The University of Sheffield, Western Bank, Sheffield, UK
cNational Heart and Lung Institute, Faculty of Medicine, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
dResearch Complex at Harwell, Rutherford Appleton Laboratory, Central Laser Facility, United Kingdom Research and Innovation, OX11 0FA, UK
First published on 30th July 2020
Abstract
Six luminescent, mononuclear ruthenium(II) complexes based on the tetrapyridophenazine (tpphz) and dipyridophenazine (dppz) ligands are reported. The therapeutic activities of the complexes against Gram-negative bacteria (E. coli, A. baumannii, P. aeruginosa) and Gram-positive bacteria (E. faecalis and S. aureus) including pathogenic multi- and pan-drug resistant strains were assessed. Estimated minimum inhibitory and bactericidal concentrations show the activity of the lead compound is comparable to ampicillin and oxacillin in therapeutically sensitive strains and this activity was retained in resistant strains. Unlike related dinuclear analogues the lead compound does not damage bacterial membranes but is still rapidly taken up by both Gram-positive and Gram-negative bacteria in a glucose independent manner. Direct imaging of the complexes through super-resolution nanoscopy and transmission electron microscopy reveals that once internalized the complexes' intracellular target for both Gram-negative and Gram-positive strains is bacterial DNA. Model toxicity screens showed the compound is non-toxic to Galleria mellonella even at exposure concentrations that are orders of magnitude higher than the bacterial MIC.
Introduction
Most medicinal research involving RuII polypyridyl complexes is focused on their potential as anti-cancer therapeutics or – due to their luminescent properties – imaging probes.1–7 Yet the first studies to investigate the medical potential of such species, conducted in the 1950s by the Dwyer group, focused on their antimicrobial effects.8,9 This work centered around mononuclear, tris(bidentate), inert derivatives of the [Ru(phen)3]2+cation (phen = 1,10-phenanthroline) in which the effect of lipophilicity on activity against bacteria was explored. The parent compound investigated was shown to be inactive against all bacteria strains investigated; however, the addition of methyl groups improved this activity. This led to the identification of a derivative that displayed promising activity against Gram-positive bacteria strains. Yet, as this lead showed lower activities than commercial antibiotics of the time, no further development in this area occurred for decades.In the ensuing years, antimicrobial resistant (AMR) has emerged as a growing global threat to public health.10–12 Infections with pathogens that display extensive and even pan-drug resistance can lead to high mortality rates.13–15 Although certain Gram-positive pathogens such as methicillin resistant Staphylococcus aureus (MRSA),10,11 are much publicized as AMR pathogens, in general, Gram-negative bacteria are a higher priority problem.12,13 For example, other members of the ESKAPE group of serious hospital acquired infections, such as Pseudomonas Aeruginosa, Acinetobacter baumannii and members of the Enterobacteriaceae, are Gram-negative and these latter three bacteria have been identified as Priority 1 pathogens in the recent WHO list for research and development.14
As treatment with last-line drugs, such as carbapenems, increasingly fail15–18 the search for new antimicrobials has been reinvigorated. In the context of this developing health crisis, the potential of metal complexes,19–21 and in particular RuII systems, as antimicrobials has recently been revisited.22 A common strategy to enhance the activity of such entities is the construction of oligonuclear structures related to the original lead identified by Dwyer. For example, Collins and Keene, have developed a series of oligonuclear analogues, in which the original [Ru(phen)3]2+d6-metal centers are tethered together by flexible methylene-based linkers of varying lengths. This approach produced a set of complexes with considerably lowered MICs compared to mononuclear analogues.23 Although the activity and imaging properties of these compounds have been explored, their mechanism of action is still being investigated. A recent report indicated that the ability of these compounds to span the membrane may account for their antimicrobial activity;24 however, another mode of action has also been hypothesized. Intracellular polar accumulation of the complexes has been attributed to binding to ribosomes causing subsequent condensation of polysomes.25 Apart from two mononuclear derivatives that are more active against Gram-negative species, but are also cytotoxic toward eukaryotes,26,27 all these systems show their highest activity against Gram-positive strains.
In ongoing studies, we have developed oligonuclear RuII polypyridyl complexes as eukaryotic cell probes, as well as therapeutics and theranostics for cancer cell lines.3,7,28,29 This work led to the identification of a dinuclear system [{Ru(phen)2}2(tpphz)]4+, (phen = 1,10-phenanthroline, tpphz = tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine) that can be used as a DNA imaging probe for super-resolution STED nanoscopy.30 More recently we have extended these studies to produce more lipophilic derivatives of the STED probe that are therapeutically active against bacterial pathogens. By incorporating ancillary ligands such as 3,4,7,8-tetramethyl-1,10-phenanthroline, TMP, we synthesized complexes such as [{Ru(TMP)2}2(tpphz)]4+ that retained their low toxicity to eukaryotic cells. Yet, in contrast to the complexes reported by Collins and Keene and others these complexes display high active against Gram-negative bacteria but are less active against Gram-positive S. aureus. Studies revealed that these complexes bind to and consequently damage Escherichia coli bacterial membranes,31 although the therapeutic effect is less pronounced in S. aureus due to binding to specific cationic components of the Gram-positive cell wall.32
Herein, we report on the activity of mononuclear derivatives of the dinuclear antimicrobial – Fig. 1. Surprisingly, we find these systems are highly active in both Gram-positive and Gram-negative bacteria, we think this is due to the lower cationic charge in comparison to the dinuclear derivative resulting in increased compound uptake in Gram-positive strains. The most promising lead is a broad band antimicrobial that, displays high-activity even against multi-drug resistant pathogens but is nontoxic toward the commonly employed insect model, Galleria mellonella. By exploiting its intrinsic multimodal imaging capabilities, we have confirmed that once internalized the compound is active through binding to cellular DNA.
 | ||
| Fig. 1 Structures of the mononuclear RuII complexes relevant to this report. | ||
Results and discussion
Cell free studies
In previous studies, involving related complexes and eukaryotic cells aimed at creating anticancer leads, it was found the RuII(tpphz) fragment was vital for bioactivity, as changing the ligand to dppz resulted in complete loss of activity toward cancer cells.33,34 To investigate the importance of the tpphz ligand on the potential antimicrobial activity of complexes 12+–42+, dppz-based analogues, 52+ and 62+ were also synthesized.Previously reported complexes 12+ and 52+ were synthesized through an established procedure,35 complexes 22+, 32+, 42+ and 62+ were synthesized and characterized through related methods36 – see ESI† for details. All these complexes were synthesized as hexafluorophosphate salts and converted to chloride salts via anion metathesis for cell-free and in-cell studies. In MeCN, the hexafluorophosphate salts all display the expected intense RuII → tpphz 3MLCT emission centered at ≈ 670 nm and, as expected, the corresponding emission from the chloride salts is greatly diminished in water.
Many previous studies have demonstrated the relationship between lipophilicity and hydrophobicity as a factor in live-cell uptake of bioactive substrates.12–15 Log P for all four tpphz complexes – determined from octanol–water partition using the shake flask procedure (Table 1) – reveals that none of the compounds are truly lipophilic. However, a comparison of Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P for 12+–62+ indicates that relative lipophilicity increases with the number of methyl groups attached to the ancillary ligand. In addition, the tpphz ligand increases lipophilicity of the complexes relative to dppz.
P for 12+–62+ indicates that relative lipophilicity increases with the number of methyl groups attached to the ancillary ligand. In addition, the tpphz ligand increases lipophilicity of the complexes relative to dppz.
P data 12+–42+
As previous studies have reported 12+ binds to duplex DNA with a high affinity33 – we investigated the interaction of 42+ and DNA in cell-free conditions. On addition of CT-DNA, buffered aqueous solutions of 42+ showed a distinctive increase in emission that is typical of a DNA light-switch effect which was used to construct saturation binding curves – see ESI.† Complexes that exhibit DNA light-switch properties have an increase in emission intensity in solvent inaccessible environments due to vibrational/isomerization pathways being blocked when the dyes bind to DNA. Fits of the data to the McGhee–von-Hippel model for non-cooperative binding37 produced a binding constant Kb: 2.2 × 107 M−1 and site size of 2.5 base pairs. Although this affinity is higher than the parent complex33 – probably due to the enhanced hydrophobicity and extended aromatic surfaces of 42+ – it is entirely consistent with values obtained for other related metallo-intercalators such as [{Ru(bpy)2}2(tpphz)]4+ (Kb: 5 × 107 M−1).34,38 Perhaps more significantly, its DNA binding affinity is an order of magnitude higher than that of groove-binding [{Ru(TMP)2}2(tpphz)]4+.31
Antimicrobial activity
In previous studies by the Thomas group, the cell uptake properties of the dinuclear derivative [{Ru(TMP)2}2(tpphz)]4+ were studied.31 It was found that it was readily internalized by both Gram-positive (Staphylococcus aureus) and Gram-negative (Escherichia coli) bacteria. In the present studies, all the complexes were screened against a panel of Gram-positive and Gram-negative bacteria, this specific panel was chosen as the strains are all listed in The WHO Priority Pathogen list.Gram-positive members of the initial panel comprised of a gastrointestinal, vancomycin-resistant, strain of Enterococcus faecalis (V583) and a non-pathogenic wild-type strain of S. aureus, (SH1000). The Gram-negative species included a wildtype E. coli (MG1655), and pathogenic ST131 E coli strain (EC958), a multi-drug resistant strain of A. baumannii (AB184) and a pan-drug resistant, clinical isolate strain of P. aeruginosa, (PA2017) taken from a patient at the University of Surrey. EC958 was chosen as it is an emerging pathogen of concern which causes urinary tract infections and exhibits high levels of multi-drug resistance through production of CTX-M extended spectrum beta lactamase. V583 was selected for comparison as it also causes urinary tract infections and exhibits resistance to the antibiotic vancomycin.39–41
Both, A. baumannii and P. aeruginosa were selected as they belong to WHO's Priority 1: critical class for developing new antibiotics,14 and the PA2017 clinical isolate was found to be pan-drug resistant. In addition, AB184 is the most prevalent clonal group of A. baumannii in the United Kingdom. The Minimum Inhibitory Concentration, MIC), of the six complexes were obtained in both nutrient-limited glucose defined or chemically defined minimal media (MM) and nutrient rich Mueller-Hinton II (MH-II) – Table 2. This is the lowest compound concentration required to inhibit bacterial cell proliferation. Although nutrient-rich growth media, like MH-II, are used as a standard in clinical assessments of antimicrobials, nutrient limited environments represent relevant biological conditions during different stages of infection in vivo. Apart from V583 and SH1000, glucose defined MM was used but, as Gram-positive bacteria cannot grow in these conditions, chemically defined MM was used for these bacteria. As observed in previous studies, all the complexes show increased activity in MM. This observation can be attributed to the complexes interacting with components within the more chemically complex MH-II media. Consistent with this hypothesis, at highest concentrations (256–512 μg mL−1) 22+ was seen to precipitate out of MH-II.
| MG1655 | EC958 | V583 | SH1000 | PA2017 | AB184 | |
|---|---|---|---|---|---|---|
| a Control = ampicillin except SH1000, where oxacillin was used. | ||||||
| Defined medium values | ||||||
| 12+ | 17.5 | 34.9 | 69.8 | 69.8 | 34.9 | 17.5 |
| 22+ | 33.9 | 33.9 | 135.6 | 33.9 | 16.9 | 16.4 |
| 32+ | 8.2 | 16.5 | 65.8 | 32.9 | 16.4 | 8.5 |
| 42+ | 3.9 | 3.9 | 31.1 | 7.8 | 7.8 | 7.8 |
| 52+ | 344.2 | 344.2 | 688.4 | 688.4 | 688.4 | 344.2 |
| 62+ | 303.4 | 303.4 | 606.8 | 303.4 | 606.8 | 606.8 |
| Controla | 5.0 | >512 | 7.5 | 5.0 | >512 | >512 |
| MH-II values | ||||||
| 12+ | 139.7 | 279.5 | 279.5 | 139.7 | 279.5 | 69.9 |
| 22+ | 271.1 | 271.1 | 542.3 | 271.1 | 271.1 | 135.6 |
| 32+ | 65.8 | 263.3 | 263.3 | 97.5 | 195.0 | 65.8 |
| 42+ | 31.1 | 62.2 | 62.2 | 38.9 | 62.2 | 15.6 |
| 52+ | 344.2 | 344.2 | 688.4 | 688.4 | 688.4 | 688.4 |
| 62+ | 303.4 | 303.4 | 606.8 | 606.8 | 606.8 | 606.8 |
| Controla | 10.0 | >512 | 5.0 | 5.0 | >512 | >512 |
Through the series of RuII(tpphz)-based complexes, an increase in lipophilicity correlates with increased activity. This trend – which is seen across all strains and is consistent with studies on dinuclear analogues31 – results in the most lipophilic compound, 42+, showing the highest activity. Interestingly, the two dppz-based complexes, which exhibit a lower lipophilicity than their tpphz analogues, display much higher MICs, even when more lipophilic ancillary ligands are employed, illustrating that the RuII(tpphz) moiety is required for high activity.
Estimates of minimum bactericidal concentrations, MBC, for 12+–62+ were also obtained, and this data is likewise summarized in Table 3. This is lowest compound concentration required to cause bacterial cell death. Trends are similar to the MICs, in that the most lipophilic compounds have the lowest MBC, resulting in 42+ showing a lower MBC against MG1655 than ampicillin in MM. An increase in MBC between MM and MH-II is again observed. In defined media the MBC of all six compounds lies within a 4-fold magnitude of the MIC and therefore they can be classified as bactericidal. Again, as expected from the MIC data, 52+ and 62+ are not active. One notable observation that emerged from these data is that, apart from the vancomycin resistant E. faecalis strain, in which MIC/MBC concentration are around ×4 higher, complex 42+ shows similar activity across the panel of bacteria, whether they are Gram-positive or Gram-negative, therapeutically sensitive or resistant. Indeed, the complex retains its high active against the pan-drug resistant PA2017-strain of P. aeruginosa, which – even in its wild-type form – is notoriously difficult to treat due to its multiple resistance mechanisms.42 The activity against S. aureus was particularly striking.
| MG1655 | EC958 | V583 | SH1000 | PA2017 | AB184 | |
|---|---|---|---|---|---|---|
| a Control = ampicillin except SH1000 where oxacillin was used. | ||||||
| Defined medium values | ||||||
| 12+ | 34.9 | 17.5 | 279.5 | 69.8 | 34.9 | 34.9 |
| 22+ | 33.9 | 17.0 | 271.2 | 33.9 | 16.9 | 16.9 |
| 32+ | 8.2 | 16.5 | 131.7 | 32.9 | 32.9 | 8.2 |
| 42+ | 3.9 | 3.9 | 31.1 | 7.8 | 7.8 | 7.8 |
| 52+ | 344.2 | 688.4 | 688.4 | 688.4 | 688.4 | 344.2 |
| 62+ | 303.4 | 606.8 | 606.8 | 606.8 | 606.8 | 303.4 |
| Controla | 10.0 | >512 | 7.5 | 10.0 | >512 | >512 |
| MH-II values | ||||||
| 12+ | 139.7 | 297.5 | 558.9 | 297.5 | 297.5 | 69.9 |
| 22+ | 542.3 | 271.2 | 542.3 | 542.3 | 271.1 | 135.6 |
| 32+ | 131.7 | 131.7 | 263.4 | 131.7 | 195.0 | 65.8 |
| 42+ | 62.2 | 124.5 | 124.5 | 62.2 | 62.2 | 38.9 |
| 52+ | 344.2 | 688.4 | 688.4 | 688.4 | 688.4 | 344.2 |
| 62+ | 303.4 | 606.8 | 606.8 | 606.8 | 606.8 | 303.4 |
| Controla | 10.0 | >512 | 7.5 | 10.0 | >512 | >512 |
Our previous studies on the dinuclear analogue of 42+ showed that while it is very active against all Gram-negative bacteria it has been tested against, including resistant pathogens, its activity against even the wild-type S1000 strain of S. aureus is comparatively low. Furthermore, subsequent screening against BH1CC, a mecA positive methicillin resistant strain of S. aureus, revealed a further large decrease in bactericidal activity, indicating that S. aureus possesses innate and/or acquired resistance to the complex.32
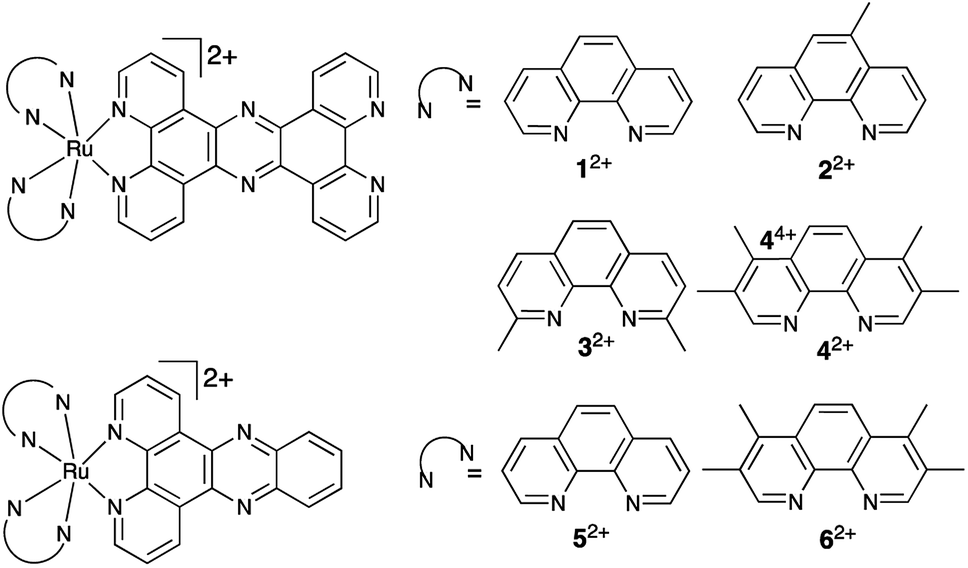
Time-kill assays were conducted to determine the rate of cell death from CFU mL−1 – Fig. 2 – OD600 time-kill assays given in the ESI.†42+ caused a significant cell death in EC958, MG1655 and SH1000 within 30 minutes of incubation. At concentrations above the MIC, cell death is observed in all strains, with exponential cell death occurring at the highest concentrations. Concentration discrepancies between the MBC and the time-kill assays can be accounted for by the different conditions of the experiments, the MBC is conducted stationary in a 96-well plate over 24 hours, whereas the time-kill is conducted with 90% aeration and rotation over six hours.
 | ||
| Fig. 2 Time-kill kinetic assays. Complex 42+ induces dose-dependent killing of E. coli EC958 (I), MG1655 (II) and SH1000 (III) planktonic cultures in vitro. The complex was added at various concentrations below and exceeding the MIC of 42+ in defined media (GDMM and CDM). Killing was determined by monitoring the number of colony forming units (CFU) per mL at time intervals up to 6 h post treatment. Error bars represent three independent biological repeats ± standard deviation (SD). | ||
Given that the MBC concentration of 42+ in SH1000 is entirely comparable to those obtained with the tested Gram-negative bacteria, we went on to investigate its activity in a clinical isolate displaying AMR as well as the BH1CC MRSA strain. These experiments led to MBC values of 7.8 μM and 11.7 μM respectively, indicating that, in stark contrast to its dinuclear analogue, even resistant strains of S. aureus display little or no increased tolerance toward 42+. These observations further indicate that the complex has a different action mechanism to the dinuclear complexes. To further investigate this issue, we used ICP-AES to explore internalization of 42+ by Gram-positive and Gram-negative bacteria.
Cell uptake studies
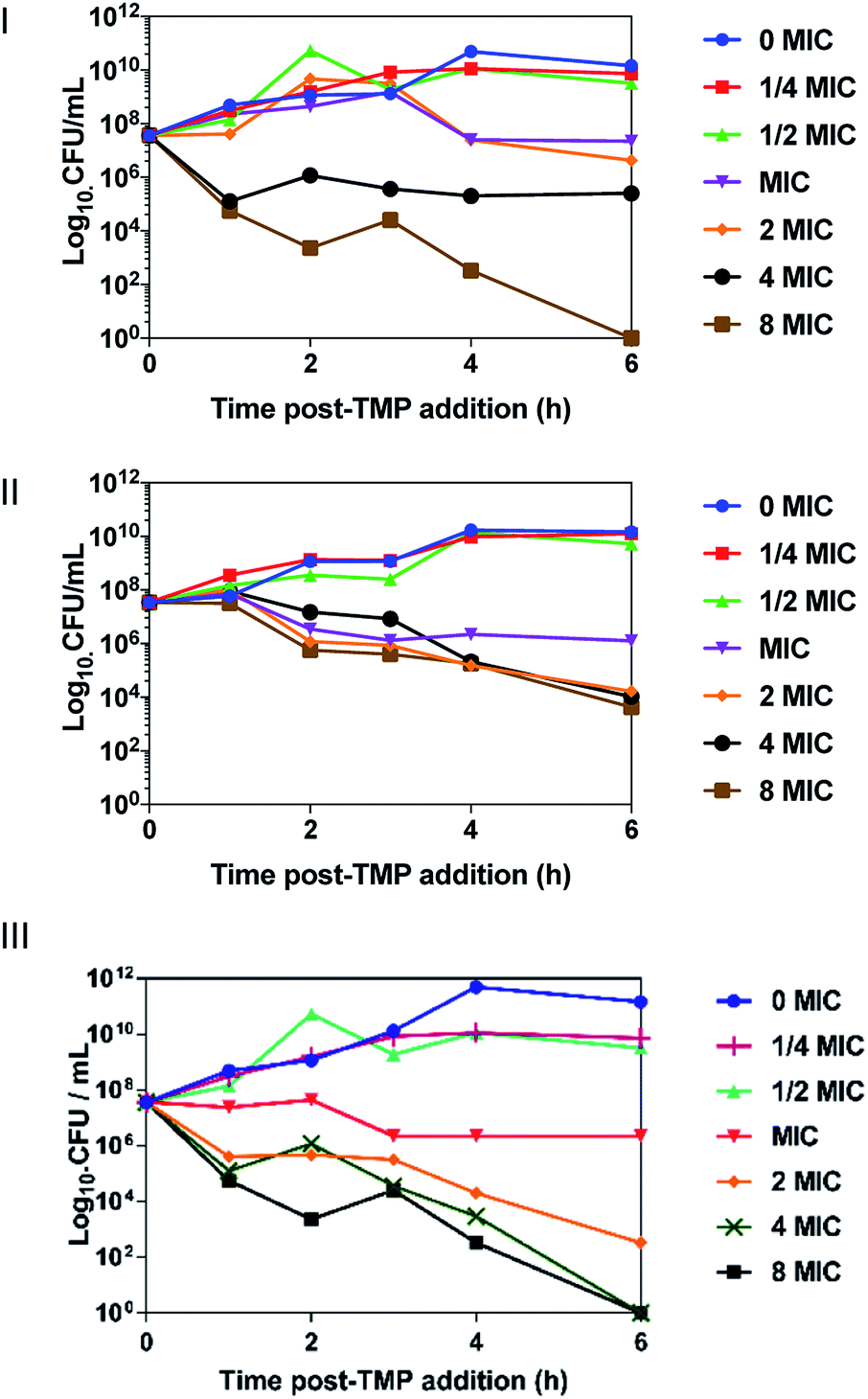
First, experiments on the uptake of 42+ by pathogenic EC958 cells were conducted over an hour, as time-kill assay showed bactericidal effects within this Gram-negative species over this exposure time. Accumulation of ruthenium was measured by ICP-AES in the absence and presence of glucose, Fig. 3, and iron content of the cells (a trace element in all cells) was quantified as a control. ICP-AES detects both intracellular accumulation and strongly membrane–bound complexes.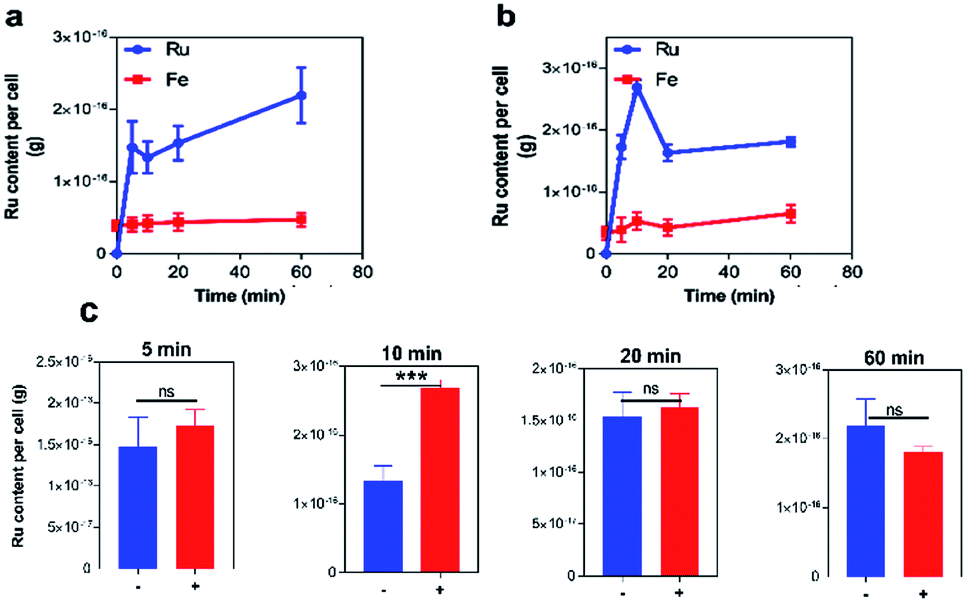 | ||
| Fig. 3 Accumulation experiments. ICP-AES data for the uptake of ruthenium by E. coli EC958 in the absence (a) and presence (b) of glucose after exposure to 42+. Comparison between uptake in the absence and presence of glucose at each time-point (c). Ru (blue) and Fe (red) levels per cell are expressed as metal (g) per cell. Fe levels were calculated as a control. Condition: concentration of 42+ = 3.9 μM. Cells were washed with 0.5%(v/v) nitric acid to remove unbound complex. After washing the whole cell is dissolved for metal content measurement. Error bars represent three independent biological repeats ± SD. | ||
Although negligible changes in CFU mL−1 indicated that the compound did not cause the cells to lyse during the experiment, in the presence of glucose, an initial large accumulation of the complex was observed at both the 5- and 10 minute time-points, followed by a drop in Ru content at 20 minutes. Interestingly, in the absence of glucose, this efflux does not occur, suggesting an energy-dependent transport mechanism is active in transporting 42+ out of cells. Although significant differences in Ru content is initially observed in the presence or absence of glucose, after 60 minutes intracellular Ru content is virtually undifferentiated. Similar to the time-kill assay, these observations suggest that cell function is affected by 42+ within an hour of exposure.
Conversely, similar experiments with Gram-positive S. aureus cells shows no evidence of efflux with rapid initial uptake being observed in all conditions (see ESI†), so that peak intracellular concentration of the complex is reached within 5 minutes.
More interestingly, a comparison on the uptake of 42+ with its dinuclear complex, [{Ru(TMP)2}2(tpphz)]4+, revealed that – in the absence or presence of glucose – complex 42+ is taken up at an approximately three times higher concentrations than its dinuclear analogue (see ESI†). Again, this observation suggests that the two complexes are internalized by bacteria through different mechanisms. To investigate this issue further, tests on membrane function in treated cells were carried out.
As an ATP release assay using the dinuclear compound had previously revealed that it causes membrane damage within both Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria,31,32 similar experiments were carried out with 42+ and the EC958 strain of E coli – Fig. 4. Significantly, when compared with a compound-free control (0 MIC) and a positive control using polymyxin (4 μg mL−1), this membrane damage assay revealed no significant ATP release indicating that, unlike the dinuclear parent compound, the mechanism of action of 42+ does not involve membrane disruption. Interestingly, the amount of extracellular ATP in the 42+ treated system is lower than the control at all time points, vide infra. Retention of intracellular ATP has been linked to DNA damage as cells require more ATP when they are repairing their DNA. Further support for this hypothesis came from studies on membrane polarization.
 | ||
| Fig. 4 ATP release membrane damage assay. Determination of 42+-induced ATP released from E. coli EC958 cells. Extracellular [ATP] (nM) quantified with recombinant luciferase and D-luciferin, with ATP released measured on a luminometer for samples exposed to 0 (control), 1.95, and 3.9 μM (MIC) of 42+ over a period of two hours. A 3 star significant difference is observed between 0 and 1 MIC. Error bars represent three biological repeats ± SD. | ||
The probe DiOC2(3) was used to determine whether 42+ has an effect on membrane potential as its emission shifts from green to red on any increase in potential and the extent of the shift is proportional to the membrane potential change – see ESI.† Although bacteria treated with 42+ do produce some detectable color change in the emission of DiOC2(3) it is far less pronounced than observed for [{Ru(TMP)2}2(tpphz)]4+, providing evidence that the mononuclear complex has a considerably less disruptive effect on bacterial membranes. To probe the interaction of 42+ with bacteria in more detail a range of imaging techniques were then employed.
Imaging studies
In a previous report, we demonstrated that this class of compounds display excellent compatibility with super resolution microscopy techniques.5–7,22 Luminescence at specific intracellular targets indicates localization of the complex as, thanks to the DNA light-switch effect, emission from 42+ only occurs when the compound is bound to solvent inaccessible environments. Hence, structured illumination microscopy (SIM) – which provides sub-diffraction limited resolutions of ∼100 nm (ref. 43) – was first used to identify intracellular targets of 42+ within EC958 cells.As observed in Fig. 5, these images confirm that 42+ is readily taken up by the EC958 strain of E. coli, with the compound producing a distinctive localization pattern which is quite different to that observed by [{Ru(TMP)2}2(tpphz)]4+. Whereas the dinuclear complex eventually localizes at the poles of E. coli cells after 20–30 minutes exposure, within 10 minutes of incubation with 42+ a pattern of emission that is highly characteristic of bacterial DNA staining is observed.
 | ||
| Fig. 5 Super resolution microscopy – SIM – imaging, Z-stack 3D projections. Localization of 42+ in E. coli EC958 cells visualized through SIM at 10 min (I), 20 min (II), and 60 min (III). 3D luminescent intensity surface plots are given for selected cells (a–c) showing peak emission within the centre of the cell where DNA is localized. (IV) Cell filamentation studies using the Image J measurement tool with cell measurements given in μm. Cells imaged using the emission of 42+ on excitation at 450 nm. After treatment with 3.6 μM 42+, cells were washed with PBS before fixing with paraformaldehyde (4%). | ||
This hypothesis was confirmed by co-staining with the well-established DNA-targeting, fluorescent probe DAPI, which resulted in a Pearson's coefficient of 0.69 for co-localization with 42+ – see ESI.† Pearson's correlation coefficient is measured from −1 to +1, values of 0.7 and above are considered a strong correlation, indicating an appreciable correlation between DAPI and 42+. Using Alexa Fluor NHS-ester 405 – a probe that is impermeable to non-compromised bacterial membranes – the possibility that 42+ displays membrane localization was also explored. These experiments showed no correlation between the staining pattern of 42+ and NHS–ester (see ESI†). This observation is consistent with the membrane studies described above, and confirms that 42+ does not bind to, or strongly interact with, the cell wall and membrane of bacteria. The cellular localization properties of 42+ are strikingly different from [{Ru(TMP)2}2(tpphz)]4+ which is externally bound in both Gram-negative and Gram-positive bacteria and only internalizes after inducing membrane damage.31
Taken together, the imaging studies and membrane assays imply that complex 42+ is intrinsically cell permeant in both Gram-positive and Gram-negative bacteria and, once internalized, it preferentially binds to DNA.
The SIM images also provide further evidence that 42+ binds to DNA within bacterial cells, as treated EC958 cells display characteristic filamentous growth – Fig. 5. Whilst healthy E. coli cells are typically 2.0 μm in length, after an hour of exposure treated cells display a ×3 increase in cell length. Moreover, cells exhibit multi-nucleation, these morphologies are commonly observed on exposure to antibiotics, like ampicillin, that disrupt DNA replication.44
As with E coli, treated Gram-positive bacteria reveal that DNA is targeted by 42+ – Fig. 6. For example, 3D-surface plots of treated S. aureus (SH1000) reveal maximum luminescent intensity at the centre of the bacterial cells where DNA is localized. Colocalization with DAPI is again observed – Fig. 6 C–E. In fact, a Pearson's coefficient of 0.895 provides even stronger evidence of colocalization between 42+ and DAPI in S. aureus. Although cell division and DNA replication are evident in these images, they were taken at sub-MIC concentrations.
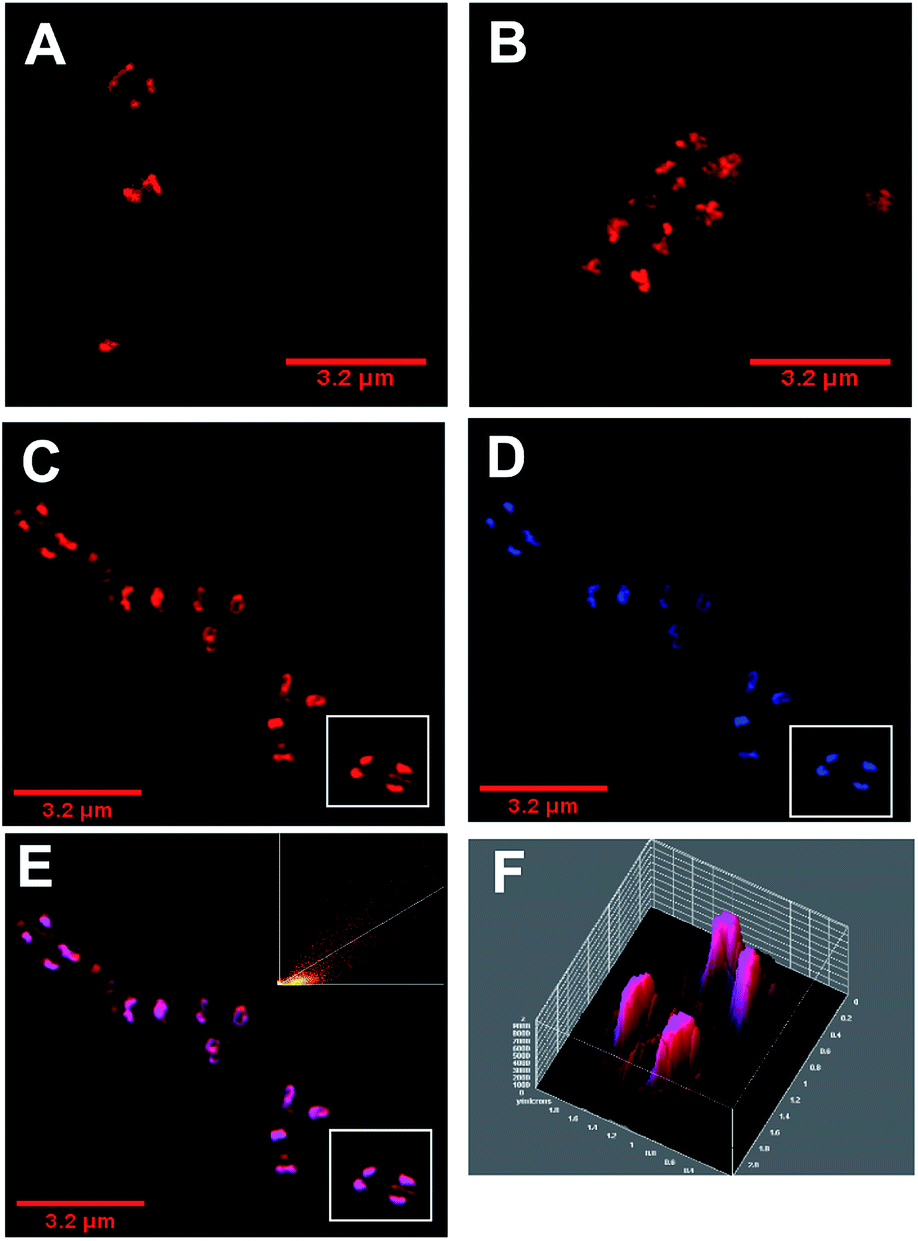 | ||
| Fig. 6 Super resolution microscopy – SIM – imaging. Localization of 42+ in S. aureus SH1000 cells visualized through SIM at (A) 20 min and (B) 60 min. (C–E) Co-staining localization of 42+ after 60 minutes incubation. Following fixation with PFA (4%) cells were stained for 5 minutes with; (C) 42+; (D) DAPI (300 nm), (E), overlay of 42+ and DAPI with colocalization scatter plot (F) 3D surface plot intensity for the co-stain overlay for selected cells in white box shown in C–E. Cells imaged using the emission of 42+ on excitation at 488 nm using A568 filter and the emission of DAPI on excitation at 405 nm using the DAPI filter. After treatment with 3.6 μM 42+, cells were washed with PBS before fixing with paraformaldehyde (4%). | ||
To probe DNA binding at higher resolutions 3D-STED nanoscopy was employed – Fig. 7. Higher resolution techniques were employed to see the DNA structure more clearly. From the resultant images, intracellular DNA content at each stage of cellular mitosis could be clearly visualized in dividing cells, again confirming 42+ targets bacterial DNA. The difference in localization between internalized 42+ and [{Ru(TMP)2}2(tpphz)]4+ can be attributed to the considerably higher DNA binding affinity of the mononuclear complex compared to the dinuclear complex.
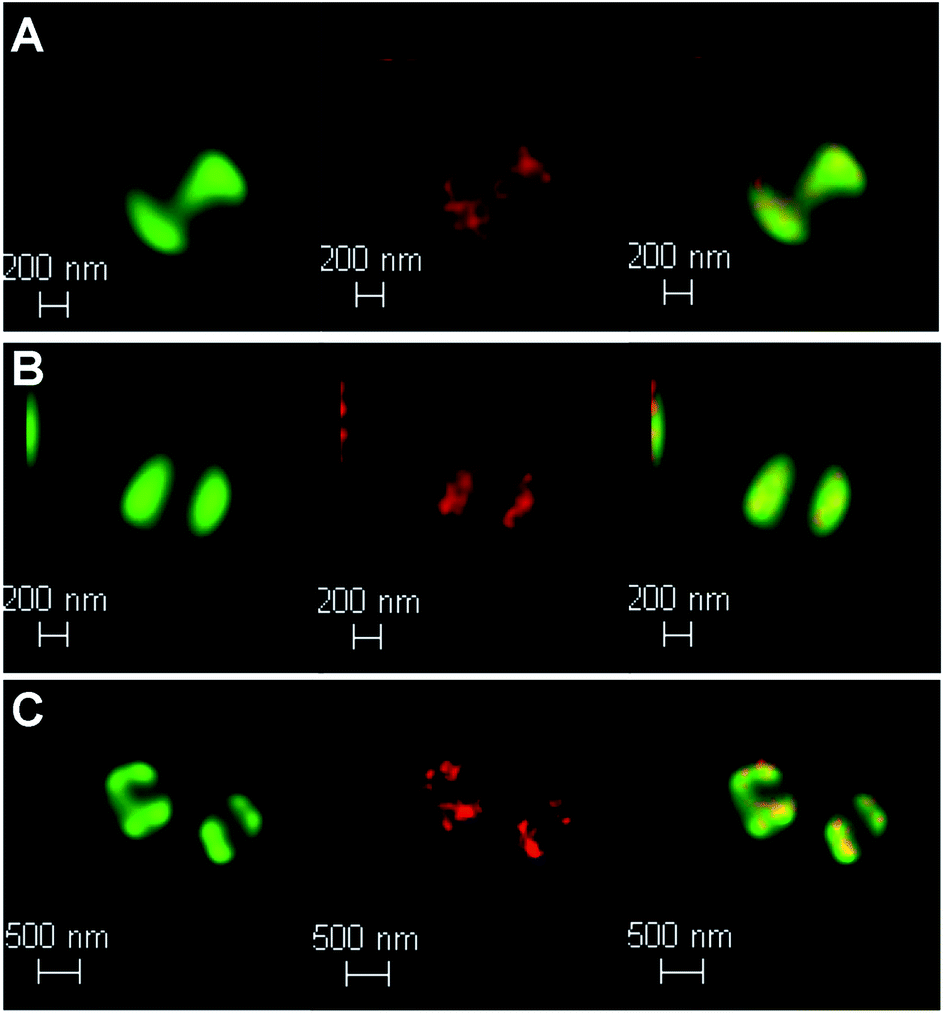 | ||
| Fig. 7 Localization of 42+ in S. aureus SH1000 cells visualized through laser scanning confocal microscopy, d-LCSM, (green, left) and stimulated emission depletion nanoscopy, STED, (red, middle) and overlay image (right) at; (A) 20 min, (B) 60 min and (C)120 min. Cells imaged using the emission of 42+ on excitation at 470 nm with a white light laser and a 470 nm notch filter. STED effect was obtained by employing a 770 nm depletion laser, and a 780 nm vortex phase plate. Both d-LCSM and d-STED images were processed using Hyugens software (SVI). Cells were treated with the same conditions as Fig. 6. | ||
Given that the complex binds to bacterial DNA, it was postulated that this interaction is responsible for its therapeutic action. Indeed, as noted in the previously described ATP release assay (Fig. 4), bacteria treated with 42+ retained ATP even more than the control. This is a known indicator of bacterial DNA damage, as cells have a high demand for ATP as they repair such damage.45
To assess whether exposure of EC958 cells to 42+ causes DNA damage and mutagenesis, an Ames fluctuation test was employed – Fig. 8. The results were compared to natural mutagenic rates as a negative control and exposure to UV light for 15 minutes as a positive control. Although treatment with 42+ does not produce effects as pronounced as those of UV-light, the complex clearly induces increased DNA mutagenesis at its MIC concentration and above, denoting that DNA damage plays a role in its mechanism of action.
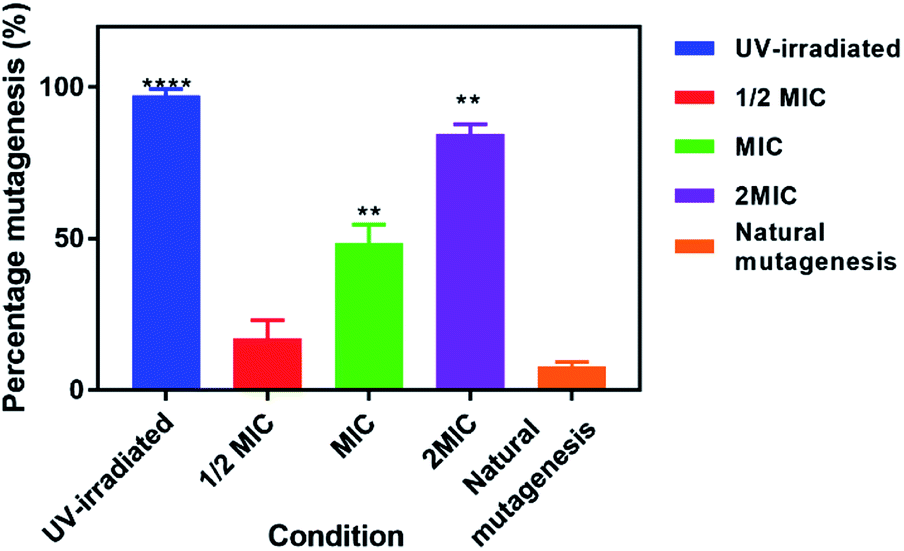 | ||
| Fig. 8 Ames test the fluctuation method. Percentage mutagenesis was measured by treated cells with 42+ (0-2MIC) and incubating with histidine. Bromocresol purple indicator was added, and the percentage color change from purple to yellow measured after incubation for 48 hours. A positive control with cells irradiated with UV-light is added for comparison. | ||
As transmission electron microscopy (TEM) provides yet higher resolutions, this technique was also used to probe the interaction of 42+ and bacterial DNA in both E. coli – Fig. 9 – and S. aureus (ESI†). As 42+ incorporates an electron-dense RuII center it functions as an EM contrast stain in itself; therefore, conventional stains such as osmium tetroxide, or uranyl acetate were not required in these studies. Hence, any intracellular contrast staining in the figure is solely due to 42+, allowing its localization to be directly visualized.
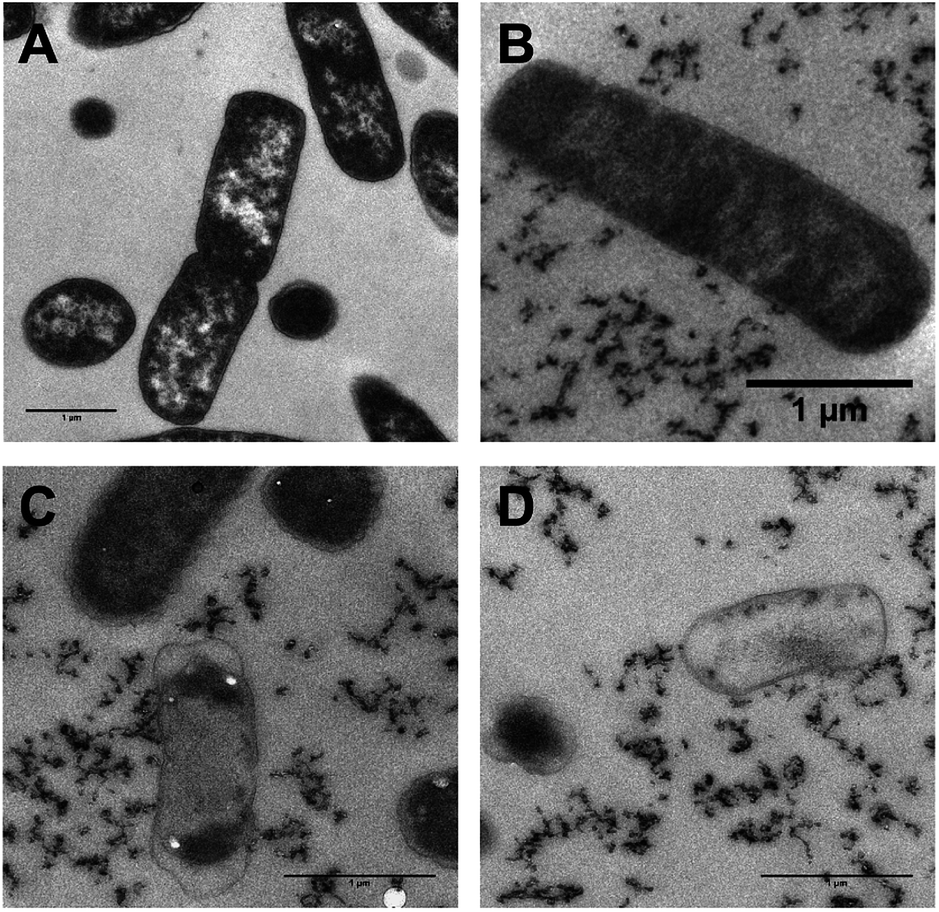 | ||
| Fig. 9 Transmission electron microscopy –TEM – imaging. Localization of 42+ in E. coli EC958 cells following treatment with MIC concentrations of 42+ (A) 0 min control (B) 10 min after exposure (C), 60 min (D) 120 min. Cell leakage/debris is clearly revealed in B and C, although the images in C and D reveal membranes that are still intact. Cells were fixed with glutaraldehyde and the 0 min control was stained with osmium tetroxide, uranyl and led acetate. Bar = 1 μm in all images. | ||
Whilst, these TEM images confirm the compound binds cellular DNA, staining is also evident in the cytoplasm and at the polar regions of the cell – Fig. 9. This full pattern of localization is not observed in optically-based images as – thanks to the DNA light-switch effect – emission from 42+ only occurs when it is bound in the solvent inaccessible environment provided by DNA.
Interestingly after 10 minutes of exposure, cell debris begins to be observed – Fig. 9B–D. As the studies discussed above indicate that 42+ is not active through membrane disruption, this observation suggests that treated cells begin to die very soon after exposure. After 60 minutes many cells display shrinkage. However, in contrast to its dinuclear equivalent exposure to 42+ does not appear to cause cell lysis; although contents have leaked from the dead cell – see Fig. 9C and D – they still possess intact membranes, suggesting osmotic effects cause the cytoplasmic shrinkage. Treated S. aureus cells also exhibit similar morphology with cytoplasmic shrinkage and loss of shape being observed – see ESI.†
Given these mononuclear complexes show high activity against bacteria cell lines, their toxicity toward eukaryotes was then explored. In this study the compounds were tested against Galleria mellonella larvae, Fig. 10. As its physiology and immune system is surprisingly similar to mammals this insect model is increasingly employed as an in vivo model, especially in toxicity screening where it produces results that are comparable to more commonly used mammalian models.46,47 A toxicity screen conducted with 42+ used concentrations above the MIC in complex media and at all concentrations the larvae survived for the entire study (120 hours). Given an average larvae weight of 300 mg a concentration of 80 mg kg−1 is 120 μg mL−1.
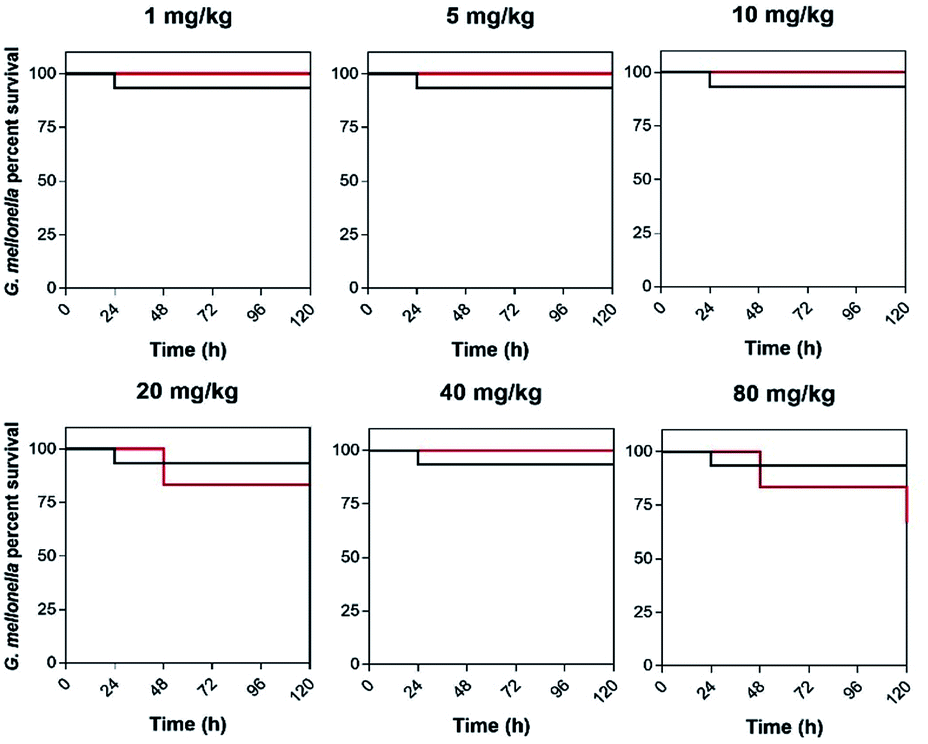 | ||
| Fig. 10 Kaplan–Meier survival curves. Determination of 42+toxicity in the insect model Galleria mellonella. Larvae were injected with 10 μL of water (control), or 42+ (0–80 mg kg−1, 0–120 μg mL−1) The larvae were incubated at 37.5 °C and live/dead scores were conducted at 120 hours. | ||
In addition to live/dead scoring, activity tests and melanisation scores were determined – see ESI.† From these graphs it is clear there is no difference in melanisation and activity in the treated conditions relative to the controls. Melanisation only began to occur at 72 hours, in a small percentage of larvae, and they still remained active for the entire experiment. Like its dinuclear parent complex, 42+ accumulates in the larval hemolymph and this could even be confirmed by eye as the hemolymph became distinctly dyed red at higher concentrations. These experiments confirm that 42+ can be administered to Galleria larvae at concentrations well above the required dose to treat bacterial infections without any detectable deleterious effects in this model organism.
Conclusions
In a medical context, the emergence of therapeutically resistant pathogens is developing into a perfect storm.48–50 Aided by overuse of antibiotics and more frequent international travel, AMR has evolved and become globally disseminated, so that multidrug and even pan-drug resistant pathogens are becoming increasingly common. And yet, at the same time, the antibiotic development pipeline has failed to identify new lead molecular architectures. This situation is particularly acute for Gram-negative infections as no new class of antibiotic has been approved for these pathogens in over fifty years.51It has been noted that while natural product-based treatments for Gram-positive bacteria are relatively common, three out of the four classes of broad-spectrum antibacterials effective against Gram negative pathogens are derived from synthetic structures. It has been suggested that this is because the structural requirements for broad spectrum activity are relatively incompatible with the biosynthesis of natural products and, due to this, unnatural synthetic systems should be investigated more closely.13,52 Metal complexes like 42+ containing ligands with extended aromatic systems meet many of the criteria determined to enhance uptake into Gram-negative bacteria, in that they are polar, amphiphilic, rigid, possess low globularity, and incorporate amine sites.33,34 In fact, most of these criteria are also met by the previously reported dinuclear analogues of 42+; yet, the activity of these complexes are greatly lowered against Gram-positive pathogens such as therapeutically resistant S. aureus, whilst complex 42+ maintains its activity even in an MRSA strain. This divergence is down to the different uptake mechanisms of [{Ru(TMP)2}2(tpphz)]4+ and 42+ by Gram-positive bacteria. In wild type S. aureus the dinuclear complex initially binds to the bacterial cell surface and only internalizes 20 minutes after exposure as a consequence of cell membrane damage, but its uptake and therapeutic activity in resistant MRSA strains is hampered by irreversible binding to anionic teichoic and lipoteichoic acid residues which make up a higher content of the cell wall in these strains.32 In contrast, it seems the lower charged, more lipophilic, complex 42+ is membrane permeant because it does not strongly interact with such residues, but once internalized binds with high affinity to bacterial DNA. As 42+ binds DNA and its closely related analogues has shown phototoxic effects in eukaryotic cells it is expected the compound may exhibit phototoxic antimicrobial activity. Future work will look at studying the phototoxic antimicrobial effects across a library of bacteria strains. In addition, we will use the Galleria model to develop an in vivo antimicrobial efficacy profile.
It has been suggested that DNA is a promising but underexploited target for antimicrobials.53,54 Although a number of organic and inorganic leads have been reported, only two DNA binding antimicrobials, metronidazole55 and nitrofurantoin,56 are in current clinical use. In this context, the lead compound described herein is particularly notable as it displays potent broad-spectrum activity yet exhibits low toxicity to a eukaryotic model.
An advantage of this relatively simple structure is its synthetic accessibility. Recently reported derivatives of natural products showing activities that are entirely comparable to 42+ are only available through intensive multi-step syntheses12,51,57 requiring specialized reagents that may be challenging to scale up. Conversely, the modular synthesis of complex 42+ can be accomplished from readily available starting materials in very few steps, allowing facile scale-up. Using this approach, a variety of derivatives of this structure, aimed at optimizing activity, can easily be envisioned. Cell-based studies to investigate the therapeutic action mechanism of 42+ and its derivatives in more detail are underway and will form the basis of future reports.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KS is grateful to the BBSRC for a PhD studentship through the White Rose Structural Biology DTP. HMS and RKP also acknowledge support from a BBSRC grant (BB/M022579/1). Imaging work was performed at the Wolfson Light Microscopy Facility, using a DeltaVision/GE OMX optical microscope, funded by the MRC grant MK/K0157531/1. ICP-AES was performed by Neil Bramall at the Faculty of Science Mass Spectrometry Centre, University of Sheffield. TEM was performed with Christopher Hill at the Electron Microscope Facility, University of Sheffield.Notes and references
- E. Meggers, Curr. Op. Chem. Biol., 2007, 11, 287–292 CrossRef CAS PubMed.
- F. R. Keene, J. A. Smith and J. G. Collins, Coord. Chem. Rev., 2009, 253, 2021–2035 CrossRef CAS.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- K. K.-W. Lo, W.-K. Hui, C.-K. Chung, K. H.-K. Tsang, D. C.-M. Ng, N. Zhu and K.-K. Cheung, Coord. Chem. Rev., 2005, 249, 1434–1450 CrossRef CAS.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- H. K. Saeed, S. Sreedharan and J. A. Thomas, Chem. Commun., 2020, 56, 1464–1480 RSC.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190–191 CrossRef CAS PubMed.
- F. P. Dwyer, A. Shulman, G. M. Laycock and S. Dixson, Aust J Exp Biol Med Sci, 1969, 47, 203–218 CrossRef CAS PubMed.
- F. D. Lowy, J. Clin. Invest., 2003, 111, 1265–1273 CrossRef CAS PubMed.
- S. Deresinski, Clin. Infect. Dis., 2005, 40, 562–573 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- M. F. Richter and P. J. Hergenrother, Ann. N. Y. Acad. Sci., 2018, 24, 1–21 Search PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, F. R. Burkert, O. Cars, G. Cornaglia, S. Gandra, C. G. Giske, D. A. Goff, M. Guzman Blanco, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, K. Leder, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, A. Pan, D. L. Paterson, M. Paul, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, G. E. Thwaites, J. W. van der Meer, S. Vega, A. Wechsler-Fördös, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect Dis, 2018, 18, 318–327 CrossRef PubMed.
- P. Nordmann, L. Dortet and L. Poirel, Trends Mol. Med., 2012, 18, 263–272 CrossRef CAS PubMed.
- D. L. Paterson and P. N. A. Harris, Lancet Infect Dis, 2015, 16, 1–2 Search PubMed.
- L. Poirel, A. Jayol and P. Nordmann, Clin. Microbiol. Rev., 2017, 30, 557–596 CrossRef CAS PubMed.
- X. Yao, Y. Doi, L. Zeng, L. Lv and J.-H. Liu, Lancet Infect Dis, 2016, 16, 288–289 CrossRef CAS PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- A. Regiel-Futyra, J. M. Dąbrowski, O. Mazuryk, K. Śpiewak, A. Kyzioł, B. Pucelik, M. Brindell and G. Stochel, Coord. Chem. Rev., 2017, 351, 76–117 CrossRef CAS.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- A. I. Ramos, T. M. Braga and S. S. Braga, Mini Rev. Med. Chem., 2012, 12, 227–235 CrossRef CAS PubMed.
- F. Li, J. G. Collins and F. R. Keene, Chem. Soc. Rev., 2015, 44, 2529–2542 RSC.
- D. K. Weber, M.-A. Sani, M. T. Downton, F. Separovic, F. R. Keene and J. G. Collins, J. Am. Chem. Soc., 2016, 138, 15267–15277 CrossRef CAS PubMed.
- F. Li, E. J. Harry, A. L. Bottomley, M. D. Edstein, G. W. Birrell, C. E. Woodward, F. R. Keene and J. G. Collins, Chem. Sci., 2014, 5, 685–693 RSC.
- A. K. Gorle, M. Feterl, J. M. Warner, S. Primrose, C. C. Constantinoiu, F. R. Keene and J. G. Collins, Chem.–Eur. J., 2015, 21, 10472–10481 CrossRef CAS PubMed.
- X. Liu, B. Sun, R. E. M. Kell, H. M. Southam, J. A. Butler, X. Li, R. K. Poole, F. R. Keene and J. G. Collins, ChemPlusChem, 2018, 83, 643–650 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock, J. A. Weinstein and J. A. Thomas, Angew. Chem., Int. Ed., 2014, 53, 3367–3371 CrossRef CAS PubMed.
- S. Sreedharan, M. R. Gill, E. Garcia, H. K. Saeed, D. Robinson, A. Byrne, A. Cadby, T. E. Keyes, C. Smythe, P. Pellett, J. Bernardino de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2017, 139, 15907–15913 CrossRef CAS PubMed.
- K. L. Smitten, H. M. Southam, J. Bernardino de la Serna, M. R. Gill, P. J. Jarman, C. G. W. Smythe, R. K. Poole and J. A. Thomas, ACS Nano, 2019, 13, 5133–5146 CrossRef CAS PubMed.
- K. L. Smitten, S. D. Fairbanks, C. C. Robertson, J. Bernardino de la Serna, S. J. Foster and J. A. Thomas, Chem. Sci., 2020, 11, 70–79 RSC.
- M. R. Gill, H. Derrat, C. G. W. Smythe, G. Battaglia and J. A. Thomas, Chembiochem, 2011, 12, 877–880 CrossRef CAS PubMed.
- P. J. Jarman, F. Noakes, S. Fairbanks, K. Smitten, I. K. Griffiths, H. K. Saeed, J. A. Thomas and C. Smythe, J. Am. Chem. Soc., 2018, 141, 2925–2937 CrossRef PubMed.
- M. R. Gill, P. J. Jarman, S. Halder, M. G. Walker, H. K. Saeed, J. A. Thomas, C. Smythe, K. Ramadan and K. A. Vallis, Chem. Sci., 2018, 9, 841–849 RSC.
- J. Bolger, A. Gourdon, E. Ishow and J.-P. Launay, Inorg. Chem., 1996, 35, 2937–2944 CrossRef CAS.
- J. D. J. McGhee and P. H. P. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS PubMed.
- D. A. Lutterman, A. Chouai, Y. Liu, Y. Sun, C. D. Stewart, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2008, 130, 1163–1170 CrossRef CAS PubMed.
- I. T. Paulsen, L. Banerjei, G. Myers, K. E. Nelson, R. Seshadri, T. D. Read, D. E. Fouts, J. A. Eisen, S. R. Gill, J. F. Heidelberg, H. Tettelin, R. J. Dodson, L. Umayam, L. Brinkac, M. Beanan, S. Daugherty, R. T. DeBoy, S. Durkin, J. Kolonay, R. Madupu, W. Nelson, J. Vamathevan, B. Tran, J. Upton, T. Hansen, J. Shetty, H. Khouri, T. Utterback, D. Radune, K. A. Ketchum, B. A. Dougherty and C. M. Fraser, Science, 2003, 299, 2071–2074 CrossRef CAS PubMed.
- M.-D. Phan, K. M. Peters, S. Sarkar, S. W. Lukowski, L. P. Allsopp, D. G. Moriel, M. E. S. Achard, M. Totsika, V. M. Marshall, M. Upton, S. A. Beatson and M. A. Schembri, PLoS Genet., 2013, 9, e1003834 CrossRef PubMed.
- N. K. Petty, N. L. Ben Zakour, M. Stanton-Cook, E. Skippington, M. Totsika, B. M. Forde, M. D. Phan, D. Gomes Moriel, K. M. Peters, M. Davies, B. A. Rogers, G. Dougan, J. Rodriguez-Bano, A. Pascual, J. D. D. Pitout, M. Upton, D. L. Paterson, T. R. Walsh, M. A. Schembri and S. A. Beatson, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 5694–5699 CrossRef CAS PubMed.
- P. D. Lister, D. J. Wolter and N. D. Hanson, Clin. Microbiol. Rev., 2009, 22, 582–610 CrossRef CAS PubMed.
- L. Shao, P. Kner, E. H. Rego and M. G. L. Gustafsson, Nat. Methods, 2011, 8, 1044–1046 CrossRef CAS PubMed.
- D. J. Dwyer, D. M. Camacho, M. A. Kohanski, J. M. Callura and J. J. Collins, Mol. Cell, 2012, 46, 561–572 CrossRef CAS PubMed.
- M. A. Osley, T. Tsukuda and J. A. Nickoloff, Mutat. Res., 2007, 618, 65–80 CrossRef CAS PubMed.
- N. Ramarao, C. Nielsen-Leroux and D. Lereclus, J. Visualized Exp., 2012, e4392 Search PubMed.
- A. R. Brochado, A. Telzerow, J. Bobonis, M. Banzhaf, A. Mateus, J. Selkrig, E. Huth, S. Bassler, J. Zamarreño Beas, M. Zietek, N. Ng, S. Foerster, B. Ezraty, B. Py, F. Barras, M. M. Savitski, P. Bork, S. Göttig and A. Typas, Nature, 2018, 559, 259–263 CrossRef CAS PubMed.
- C. Walsh, Nature, 2000, 406, 775–781 CrossRef CAS PubMed.
- K. H. Luepke, K. J. Suda, H. Boucher, R. L. Russo, M. W. Bonney, T. D. Hunt and J. F. Mohr III, Pharmacotherapy, 2016, 37, 71–84 CrossRef PubMed.
- D. M. Shlaes and P. A. Bradford, Pathog. Immun., 2018, 3, 19–43 CrossRef PubMed.
- P. A. Smith, M. F. T. Koehler, H. S. Girgis, D. Yan, Y. Chen, Y. Chen, J. J. Crawford, M. R. Durk, R. I. Higuchi, J. Kang, J. Murray, P. Paraselli, S. Park, W. Phung, J. G. Quinn, T. C. Roberts, L. Rougé, J. B. Schwarz, E. Skippington, J. Wai, M. Xu, Z. Yu, H. Zhang, M.-W. Tan and C. E. Heise, Nature, 2018, 561, 189–194 CrossRef CAS PubMed.
- M. F. Richter and P. J. Hergenrother, Chem, 2017, 3, 10–13 CAS.
- A. Bolhuis, L. Hand, J. E. Marshall, A. D. Richards, A. Rodger and J. Aldrich-Wright, Eur. J. Pharm. Sci., 2011, 42, 313–317 CrossRef CAS PubMed.
- A. Bolhuis and J. R. Aldrich-Wright, Bioorg. Chem., 2014, 55, 51–59 CrossRef CAS PubMed.
- D. I. Edwards, J. Antimicrob. Chemother., 1977, 3, 43–48 CrossRef CAS PubMed.
- L. Sandegren, A. Lindqvist, G. Kahlmeter and D. I. Andersson, J. Antimicrob. Chemother., 2008, 62, 495–503 CrossRef CAS PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 1–19 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03410j |
| This journal is © The Royal Society of Chemistry 2020 |