
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation triggers guest dissociation during reorganization of an FeII4L6 twisted parallelogram†
Alex J.
Plajer‡
,
Felix J.
Rizzuto‡
 ,
Larissa K. S.
von Krbek§
,
Yohan
Gisbert
,
Víctor
Martínez-Agramunt
and
Jonathan R.
Nitschke
*
,
Larissa K. S.
von Krbek§
,
Yohan
Gisbert
,
Víctor
Martínez-Agramunt
and
Jonathan R.
Nitschke
*
Department of Chemistry, University of Cambridge, Lensfield Rd, Cambridge, CB2 1EW, UK. E-mail: jrn34@cam.ac.uk
First published on 9th September 2020
Abstract
A three-dimensional FeII4L6 parallelogram was prepared from ferrocene-containing ditopic ligands. The steric preference of the bulky ferrocene cores towards meridional vertex coordination brought about this new structure type, in which the ferrocene units adopt three distinct conformations. The structure possesses two distinct, bowl-like cavities that host anionic guests. Oxidation of the ferrocene FeII to ferrocenium FeIII causes rotation of the ferrocene hinges, converting the structure to an FeII1L1+ species with release of anionic guests, even though the average charge per iron increases in a way that would ordinarily increase guest binding strength. The degrees of freedom exhibited by these new structures – derived from the different configurations of the three ligands surrounding a meridional FeII center and the rotation of ferrocene cores – thus underpin their ability to reconfigure and eject guests upon oxidation.
Introduction
Metal–organic self-assembly has emerged as a powerful tool to generate complex three-dimensional structures.1–3 The construction principles of such architectures take into account the symmetries of their subunits, which must connect in such a way to generate the overall superstructure. Coordinative and ligand stereochemistries also impact upon structure,4–15 although controlling these design elements can be challenging.16,17 For example, the factors that affect the stereochemical configuration of ligands around octahedral metal ions in the assembly of discrete three-dimensional architectures are complex. In the case of mononuclear compounds with sterically undemanding ligands, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of fac:mer isomers is statistically favored; however, multimetallic polyhedra often favor the formation of fac corners exclusively due to the high-symmetry structures that result. Such high symmetry structures often incorporate a smaller number of components and thus incur a lower entropic penalty to assembly. In contrast, asymmetrical mer vertices lead to assemblies that are larger and more complex than their threefold-symmetric fac analogues.8,17–21 Such assemblies may be better able to mimic the recognition processes of biological receptors, which often bind complex, asymmetrical guests.22
:3 ratio of fac:mer isomers is statistically favored; however, multimetallic polyhedra often favor the formation of fac corners exclusively due to the high-symmetry structures that result. Such high symmetry structures often incorporate a smaller number of components and thus incur a lower entropic penalty to assembly. In contrast, asymmetrical mer vertices lead to assemblies that are larger and more complex than their threefold-symmetric fac analogues.8,17–21 Such assemblies may be better able to mimic the recognition processes of biological receptors, which often bind complex, asymmetrical guests.22
The phenomenon of mer or fac coordination is exclusive to octahedral coordination geometries around metal vertices. Other geometries, such as square planar motifs, lead to the formation of entirely different polyhedra,23–27 while tetrahedral metal-binding pockets can lead to helicates or interlocked structures.28–34 Other metal coordination motifs that are tied to their own rules of isomerism could lead to new families of metal–organic polyhedra.
Among such motifs, metallocenes35–44 can display conformational isomerism by rotation of the cyclopentadiene ligands with respect to one another. The different eclipsed and staggered conformations of metallocenes offer a large range of possible dihedral angles between covalently-connected substituents, rendering such building blocks attractive in the synthesis of low-symmetry metal–organic assemblies.
We thus envisioned that the incorporation of metallocene ligands into supramolecular capsules with octahedral metal ion vertices could enable the formation of low-symmetry products. Metallocenes are furthermore redox-active, enabling potential stimuli-responsive behavior42,45–53 that may be used for different host–guest interactions,45,47,54 such as redox-triggered guest ejection or structural reconfiguration.55,56
Results and discussion
Here we show the use of a ferrocene-containing ditopic ligand to generate a low-symmetry M4L6 assembly, with electrochemical, magnetic, and optical functionality. Ferrocene is sterically demanding and can rotate, adopting conformers along the Cp–FeII–Cp axis. This flexibility enables a single ligand to adopt multiple rotational conformations, creating a new structure type: twisted parallelogram 1 (Fig. 1). | ||
| Fig. 1 (a) Subcomponent self-assembly of 1 from A, 2-formylpyridine, and FeII. Geometrical and stereochemical elements leading to the formation of 1 are highlighted. (b and c) Frameworks defined by ligand scaffold (grey), mer-FeII vertices (purple), and ferrocene FeII centers (light pink). Views down and perpendicular to the C2 axis (left to right). | ||
The reaction of A,57 2-formylpyridine, and FeII(PF6)2 in CD3CN (Fig. 1a) gave rise to a 1H NMR spectrum with six sets of ligand environments per diimine ligand LA (Fig. S1–S4†). All signals gave the same rate of diffusion by 1H DOSY NMR spectroscopy, consistent with the formation of a single species (Fig. S12†). Two-dimensional NMR spectra suggested that the structure contained two distinct mer coordination environments (Fig. S6–S11†). Low and high resolution ESI mass spectrometry corresponded to an FeII4L6 assembly (Fig. S15 and S16†).
Employing preformed ligand Lazo (a congener of LA with diazo, instead of diimine, coordinating units) with FeII(NTf2)2 during assembly resulted in the formation of a product 1azo that appeared isostructural to 1 by NMR and ESI-MS (Fig. 2a, ESI Section 2.2†). Single crystals of 1azo suitable for X-ray diffraction studies were grown. The crystal structure confirms the formation of an FeII4L6 assembly with meridional coordination vertices (Fig. 2). As observed in solution, there are two distinct mer coordination environments in the solid state, imparting approximate C2 symmetry. The two enantiomers co-crystallized, and are related by inversion symmetry in the crystal. They are composed of alternating Δ and Λ pyridyl-imine FeII centers that frame a parallelogram. The overall scaffold can be visualized as two connected, twisted diamonds (Fig. 1c).
 | ||
| Fig. 2 Crystal structure of 1azo. (a) Lazo assembled with FeII ions to form 1azovia guest templation. Crystal structure (right) is viewed down the C2 axis with each magnetically-unique ligand displayed in a different color. (b) With two Tf2N− anions (depicted in beige) binding in distinct pockets (c) View perpendicular to the C2 axis, with two Tf2N− anions. The remaining anions, disorder and solvent are not shown for clarity. | ||
Of the many possible rotational conformers of LA, only three were observed in 1: four of the six ferrocene units accommodate a slightly staggered conformation, with the phenylene substituents spread by 59° and 126°, respectively, and two are in an eclipsed conformation, with phenylene units 72° apart (Fig. 1a, bottom). This new structure type is enabled by the bulky but flexible ferrocene linkers; mer-coordinated rigid ditopic linkers have been observed to form barrel-like or cubic assemblies instead.19,21,58–60 The formation of 1 thus illustrates how the conformational flexibility associated with bis-coordinated metallocenes may lead to the formation of otherwise inaccessible supramolecular structure types.
Assembly 1 shows two distinct binding pockets for guests in the solid state, within which two anions are found. These pockets are defined by ferrocene subunits, which extend above and below the plane of the four mer-coordinated FeII vertices (Fig. 2b and c).
The 19F NMR spectrum of 1 showed two sets of PF6− resonances, suggesting binding of PF6− within 1 in slow exchange on the NMR timescale (Fig. S13†). We infer the presence of suitably-shaped anions to lock the rotation of the ferrocene units in 1 into a single configuration. When FeII(OTf)2 was used instead of the hexafluorophosphate salt, a broader 1H NMR spectrum was observed, with ESI mass spectra confirming the presence of assembly 1 (Fig. S14 and S17†). PF6− was thus necessary to generate a sharp NMR spectrum, which corresponds to the solid-state configuration in Fig. 2. Due to the broad 1H NMR spectra of the assemblies formed with anions other than PF6−, we were unable to perform titrations that would provide accurate binding affinities for guests within 1.
Having confirmed the unique geometry of 1, we investigated its electrochemical properties. Ferrocene is a well-studied one electron reductant (E = 0.4 V vs. SCE; 0 V vs. Fc/Fc+).61 Cyclic voltammetry experiments conducted in 0.1 M nBu4PF6/CH3CN electrolyte revealed one redox process centered at E1 = 0.3 V, and one irreversible process at E2 = 1.1 V vs. Fc/Fc+ (Fig. S18 and S19†). We assigned these processes to the oxidation of the ferrocene units, and to the oxidation of the FeII vertices, respectively. The observation that all ferrocene cores undergo oxidation simultaneously suggests there is no electronic communication between metallocene cores within 1.
To assess the structural effects of oxidizing the ferrocene cores within 1, we added 6 equivalents of [NO]PF6 (Fig. 3a) (a 1e− oxidant, E = 0.87 V vs. Fc/Fc+), which can only oxidize the ferrocene units, and not the FeII vertices,61 to a solution of 1 in CH3CN (Fig. 3). In contrast to the case for 1, where six ligand environments were observed in the diamagnetic region (ca. 0 to 10 ppm, Fig. 3b and S2†) of the NMR spectrum, the wide sweep 1H NMR spectrum (ca. −60 to 300 ppm) of the oxidized product showed only one set of ligand environments (Fig. 3c and S23†). This spectrum was consistent with the formation of a highly-symmetric, paramagnetic species. High-resolution ESI-MS confirmed the formation of [FeL]3+ complex 2 (Fig. S27†). The spectra of complex 2 suggest the adoption of a conformation of the ferrocene moieties in which the phenylene linkers stack on top of each other (Fig. 3a). High resolution MS-MS studies on 2 furthermore revealed that the z = +1 peak of 2 fragmented only into the oxidised ferrocene ligand LA; no protonated neutral ligand was observed (Fig. S28†). These observations indicated that the FeII(pyridylimine) vertices were not oxidised (i.e., they remained as FeII)62 following the oxidation of ferrocene to ferrocenium.
 | ||
| Fig. 3 (a) Oxidation-triggered rearrangement of 1 into mononuclear complex 2 with ejection of the PF6− guests. (b) Aromatic region of the 1H NMR (700 MHz, CD3CN, 298 K) spectrum of 1. Six distinct environments were identified; imine signals are marked with purple parallelograms. (c) Partial wide-sweep 1H NMR (400 MHz, CD3CN, 298 K) spectrum of 2. Only one ligand environment remained after oxidation of 1; imine signal marked with a blue dot. (d) UV-vis spectral changes of 1 during chemical oxidation with [NO]PF6 to generate 2. Arrows indicate the direction of spectral progression. | ||
Attempted chemical reduction via treatment of 2 with 6 equivalents of tetrakis(dimethylamine)ethylene reductant did not result in the clean regeneration of 1. All paramagnetic NMR signals disappeared, but no clean NMR spectrum corresponding to 1 was observed, although some degree of reversibility was indicated by UV-vis titration (Fig. S33†). We infer that small amounts of free, insoluble FeIII products were generated during oxidation (and potential reaction with NO radicals), hampering reassembly.
To assess the electronic and optical effects of ferrocene oxidation, we titrated [NO]PF6 into a CH3CN solution of 1 and followed the UV-vis spectral progressions upon oxidation (Fig. 3d). The most pronounced change in the UV-vis spectra was the loss of the FeII(pyridylimine)3 MLCT bands at 500–600 nm, which brings about a color change from the purple of 1 to the orange of 2. A shifting crossover point was observed during the titration, as opposed to a regular isosbestic point. This observation indicated that the conversion of 1 into 2 was not a simple process, suggesting the presence of intermediates.
Similar spectral progressions were observed by UV-vis spectroelectrochemical measurements. An applied potential of +0.7 V vs. Ag+/Ag resulted in a decrease of the MLCT band, which regenerated upon relieving the applied potential (Fig. S30 and S31†). Unlike experiments involving chemical oxidation, which were allowed to equilibrate before collection of spectra, spectroscopy during electrochemical oxidation was done continuously, giving rise to differences in spectral progression between these two experiments. Both optical spectroscopy experiments suggest that the mer-FeII metal centers transition from low to high spin63 upon oxidation of the ferrocene centres of 1.
Taken together, our NMR, MS-MS, optical and electrochemical data suggest that the FeII center in 2 is high spin. Mononuclear 2 thus contains high spin pyridylimine-ligated FeII (S = 2) and a low spin ferrocenium FeIII (S = 1/2). Loss and regeneration of the FeII MLCT band upon oxidation and reduction, respectively, along with MS charge and stoichiometry evidence, suggest that the pyridylimine FeII center switches from low to high spin upon oxidation of the ferrocene units. The overall S = 5/2 spin of the system was confirmed using the Evans method (ESI Section 4.3†).
Upon oxidation, we propose that 1 is destabilized by Coulombic repulsion between the FeIII ferrocenium centres, and between the ferrocenium centres and FeII vertices, causing the disassembly of 1 to mononuclear complexes of 2. This disassembly process occurs in multiple steps, leading to the observed lack of an isosbestic point in the UV-vis titration – we hypothesize that a fast oxidation process causes a rapid change in the UV-vis spectra, followed by rearrangement and guest release, which is slow and manifests as a gradual transition in the UV-vis spectra (Fig. 3d). We previously observed that Coulombic repulsion between cationic ligands can prevent self-assembly.64,65
The disassembly process also destroys the pockets that bind PF6− anions, leading to their ejection. 19F NMR spectroscopy after oxidation shows only one PF6− signal, at the chemical shift of the free anion, indicating that 2 does not interact strongly with PF6− (Fig. S24†). This observation appears counterintuitive: oxidation of 1 should render the assembly more positively charged and make anion binding more favorable. Here, however, destruction of the anion-binding cavities in 1 led to guest ejection in spite of the increased Coulombic attraction anticipated between the more highly-charged iron centers of oxidized 1 and PF6−. This guest ejection via oxidative structural rearrangement thus provides a new mode of control over host–guest chemistry in metal–organic assemblies.
Intrigued by the ability of our ferrocene cores to dictate the mer stereochemistry of tris-pyridylimine metal vertices, we investigated the self-assembly of mono-functional ferrocene-aniline subcomponent B into mononuclear complexes (Fig. 4). The assembly of FeII ions, 2-formylpyridine and B produced mer-3 as the exclusive product (Fig. 4). ESI-MS reflected the formation of a discrete FeIILB3 complex, while 1H and 13C NMR showed three distinct ligand environments for mer-3 (Fig. S36 and S37†), as well as confirming the low-spin character of the complex. No equilibration to the statistical mixture (1:3 fac:mer) was observed over the range 233–300 K.
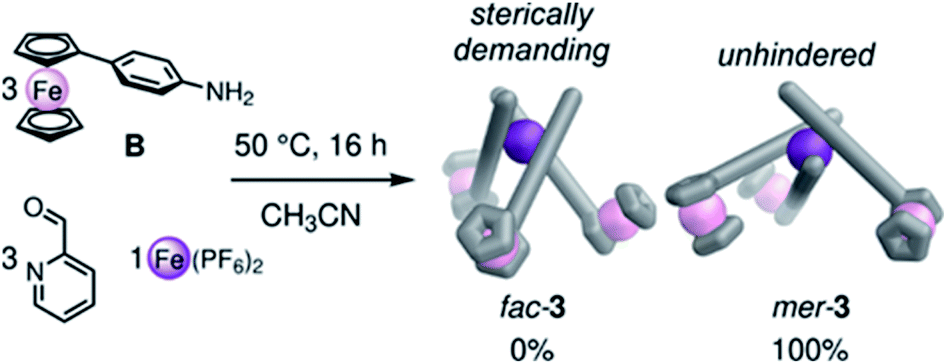 | ||
| Fig. 4 The assembly of B, 2-formylpyridine and FeII subcomponents led to the exclusive formation of mer-3. | ||
Building on observations from other systems,19 we infer that this preference for mer stereochemistry results from steric clash between the ferrocene units in the fac complex in some of their rotational configurations, disfavoring its formation. In the mer structure, rotation of the ferrocene units is unhindered and steric repulsion is minimized, as seen in the MM3 molecular models presented in Fig. 4.
Conclusions
A bulky, axially-flexible ferrocene substituent can thus be used to design low-symmetry assemblies with meridional coordination stereochemistry. The new structure type of assembly 1 can be formed because of the ease of rotation of ferrocenes. Our strategy of using metallocenes of different symmetries may grant access to even larger and more complex assemblies in the future. The oxidation-driven reassembly and guest release demonstrated herein may also be put to use in larger systems, where the anions may be more complex and biologically relevant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (695009) and the UK Engineering and Physical Sciences Research Council (EPSRC, EP/P027067/1). The authors thank the Cambridge University Chemistry NMR facility for performing some NMR experiments. AJP and FJR thank the Cambridge Trust (Vice Chancellor Scholarship for AJP and Cambridge Australia Scholarship for FJR). L. K. S. v. K. acknowledges the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship. VMA thanks UJI and GVA (ACIF/2017/189) for financial support. We thank Diamond Light Source for time on beamline I-19 (MT15768-3).Notes and references
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- E. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120(16), 8987–9014 CrossRef CAS.
- Q. Shi, X. Zhou, W. Yuan, X. Su, A. Neniškis, X. Wei, L. Taujenis, G. Snarskis, J. S. Ward, K. Rissanen, J. de Mendoza and E. Orentas, J. Am. Chem. Soc., 2020, 142, 3658–3670 CrossRef CAS.
- C. Tan, J. Jiao, Z. Li, Y. Liu, X. Han and Y. Cui, Angew. Chem., Int. Ed., 2018, 57, 2085–2090 CrossRef CAS.
- C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. Dean Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS.
- F. J. Rizzuto, P. Pröhm, A. J. Plajer, J. L. Greenfield and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 1707–1715 CrossRef CAS.
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. J. Deeth, A. D. Faulkner, D. H. Simpson and P. Scott, Dalton Trans., 2011, 40, 10416–10433 RSC.
- D. Zhang, T. K. Ronson, J. L. Greenfield, T. Brotin, P. Berthault, E. Léonce, J.-L. Zhu, L. Xu and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 8339–8345 CAS.
- J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958–7962 CrossRef CAS.
- K. Wu, K. Li, Y.-J. Hou, M. Pan, L.-Y. Zhang, L. Chen and C.-Y. Su, Nat. Commun., 2016, 7, 10487 CrossRef CAS.
- P. Howlader, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 9070–9078 CrossRef CAS.
- T. Hong, Z. Zhang, Y. Sun, J. J. Tao, J. D. Tang, C. Xie, M. Wang, F. Chen, S. S. Xie, S. Li and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 10244–10249 CrossRef CAS.
- V. Martínez-Agramunt, T. Eder, H. Darmandeh, G. Guisado-Barrios and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 5682–5686 CrossRef.
- A. M. Castilla, M. A. Miller, J. R. Nitschke and M. M. J. Smulders, Angew. Chem., Int. Ed., 2016, 55, 10616–10620 CrossRef CAS.
- A. J. Metherell and M. D. Ward, Dalton Trans., 2016, 45, 16096–16111 RSC.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406–4422 RSC.
- R. A. Bilbeisi, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 9027–9030 CrossRef CAS.
- M. Kieffer, B. S. Pilgrim, T. K. Ronson, D. A. Roberts, M. Aleksanyan and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 6813–6821 CrossRef CAS.
- S. Zarra, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 4837–4840 CrossRef CAS.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 751–756 CrossRef CAS.
- F. J. Rizzuto, J. P. Carpenter and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 9087–9095 CrossRef CAS.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS.
- M. Käseborn, J. J. Holstein, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2018, 57, 12171–12175 CrossRef.
- W. Wang, Z. Zhou, J. Zhou, B. Shi, B. Song, X. Li, F. Huang and P. J. Stang, Inorg. Chem., 2019, 58, 7141–7145 CrossRef CAS.
- P. N. W. Baxter, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Chem.–Eur. J., 1999, 5, 113–120 CAS.
- R. Kramer, J. M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 CrossRef CAS.
- F. Durola, V. Heitz, F. Reviriego, C. Roche, J.-P. Sauvage, A. Sour and Y. Trolez, Acc. Chem. Res., 2014, 47, 633–645 CrossRef CAS.
- B. Champin, P. Mobian and J.-P. Sauvage, Chem. Soc. Rev., 2007, 36, 358–366 CAS.
- C. O. Dietrich-Buchecker and J. P. Sauvage, Chem. Rev., 1987, 87, 795–810 CrossRef CAS.
- T. Hashimoto, T. Nishimura, J. M. Lim, D. Kim and H. Maeda, Chem.–Eur. J., 2010, 16, 11653–11661 CrossRef CAS.
- S. Evariste, A. Moustafa Khalil, M. Elsayed Moussa, A. Kwun-Wa Chan, E. Yau-Hin Hong, H.-L. Wong, B. Le Guennic, G. Calvez, K. Costuas, V. Wing-Wah Yam and C. Lescop, J. Am. Chem. Soc., 2018, 140, 12521–12526 CAS.
- R. A. S. Vasdev, J. A. Findlay, A. L. Garden and J. D. Crowley, Chem. Commun., 2019, 55, 7506–7509 RSC.
- J. S. Mugridge, D. Fiedler and K. N. Raymond, J. Coord. Chem., 2010, 63, 2779–2789 CrossRef CAS.
- B. Quinodoz, G. Labat, H. Stoeckli-Evans and A. Von Zelewsky, Inorg. Chem., 2004, 43, 7994–8004 CrossRef CAS.
- B. Quinodoz, H. Stoeckli-Evans and A. von Zelewsky, Mendeleev Commun., 2003, 13, 146–147 CrossRef.
- F. Chen-jie, D. Chun-ying, M. Hong, H. Cheng, M. Qing-jin, L. Yong-jiang, M. Yu-hua and W. Zhe-ming, Organometallics, 2001, 20, 2525–2532 CrossRef.
- K.-J. Wei, J. Ni, Y.-S. Xie, Y. Liu and Q.-L. Liu, Dalton Trans., 2007, 3390–3397 RSC.
- M. S. Inkpen, S. Scheerer, M. Linseis, A. J. P. White, R. F. Winter, T. Albrecht and N. J. Long, Nat. Chem., 2016, 8, 825–830 CrossRef CAS.
- S. Ø. Scottwell, J. E. Barnsley, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Commun., 2017, 53, 7628–7631 RSC.
- V. Martínez-Agramunt, S. Ruiz-Botella and E. Peris, Chem.–Eur. J., 2017, 23, 6675–6681 CrossRef.
- V. Martínez-Agramunt, D. G. Gusev and E. Peris, Chem.–Eur. J., 2018, 24, 14802–14807 CrossRef.
- G. Szalóki, V. Croué, V. Carré, F. Aubriet, O. Alévêque, E. Levillain, M. Allain, J. Aragó, E. Ortí, S. Goeb and M. Sallé, Angew. Chem., Int. Ed., 2017, 56, 16272–16276 CrossRef.
- C. Colomban, G. Szalóki, M. Allain, L. Gómez, S. Goeb, M. Sallé, M. Costas and X. Ribas, Chem.–Eur. J., 2017, 23, 3016–3022 CAS.
- V. Croué, S. Goeb, G. Szalóki, M. Allain and M. Sallé, Angew. Chem., Int. Ed., 2016, 55, 1746–1750 Search PubMed.
- K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CAS.
- K. Yazaki, S. Noda, Y. Tanaka, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 15031–15034 CAS.
- F. Jia, H. V. Schröder, L.-P. Yang, C. von Essen, S. Sobottka, B. Sarkar, K. Rissanen, W. Jiang and C. A. Schalley, J. Am. Chem. Soc., 2020, 142, 3306–3310 CAS.
- H. V Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 Search PubMed.
- Z. Lu, T. K. Ronson and J. R. Nitschke, Chem. Sci., 2020, 11, 1097–1101 RSC.
- J. Jung, W. Liu, S. Kim and D. Lee, J. Org. Chem., 2019, 84, 6258–6269 CrossRef CAS.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 CAS.
- S. Krykun, M. Dekhtiarenko, D. Canevet, V. Carré, F. Aubriet, E. Levillain, M. Allain, Z. Voitenko, M. Sallé and S. Goeb, Angew. Chem., Int. Ed., 2020, 59, 716–720 CAS.
- T. Y. Kim, R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Eur. J., 2018, 24, 14878–14890 CrossRef CAS.
- D. D. Swanson, K. M. Conner and S. N. Brown, Dalton Trans., 2017, 46, 9049–9057 RSC.
- D. A. Roberts, B. S. Pilgrim, G. Sirvinskaite, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 9616–9623 CAS.
- C. J. E. Haynes, J. Zhu, C. Chimerel, S. Hernández-Ainsa, I. A. Riddell, T. K. Ronson, U. F. Keyser and J. R. Nitschke, Angew. Chem., Int. Ed., 2017, 56, 15388–15392 CrossRef CAS.
- I. A. Riddell, T. K. Ronson, J. K. Clegg, C. S. Wood, R. A. Bilbeisi and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 9491–9498 CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CAS.
- F. J. Rizzuto, D. M. Wood, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 11008–11011 CAS.
- S. Sundaresan, J. A. Kitchen and S. Brooker, Inorg. Chem. Front., 2020, 7, 2050–2059 CAS.
- A. J. Plajer, E. G. Percástegui, M. Santella, F. J. Rizzuto, Q. Gan, B. W. Laursen and J. R. Nitschke, Angew. Chem., Int. Ed., 2019, 58, 4200–4204 CAS.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2006764. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04352d |
| ‡ These authors contributed equally. |
| § Present address: Kekulé-Institut für Organische Chemie und Biochemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Str. 1, 53121, Bonn, Germany. |
| This journal is © The Royal Society of Chemistry 2020 |