
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering reversible cell–cell interactions using enzymatically lipidated chemically self-assembled nanorings†
Yiao
Wang
 a,
Ozgun
Kilic
b,
Clifford M.
Csizmar
b,
Sudhat
Ashok
c,
James L.
Hougland
c,
Mark D.
Distefano
*ab and
Carston R.
Wagner
*ab
a,
Ozgun
Kilic
b,
Clifford M.
Csizmar
b,
Sudhat
Ashok
c,
James L.
Hougland
c,
Mark D.
Distefano
*ab and
Carston R.
Wagner
*ab
aDepartment of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: diste001@umn.edu; wagne003@umn.edu
bDepartment of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA
cDepartment of Chemistry, Syracuse University, Syracuse, New York 13244, USA
First published on 26th October 2020
Abstract
Multicellular biology is dependent on the control of cell–cell interactions. These concepts have begun to be exploited for engineering of cell-based therapies. Herein, we detail the use of a multivalent lipidated scaffold for the rapid and reversible manipulation of cell–cell interactions. Chemically self-assembled nanorings (CSANs) are formed via the oligomerization of bivalent dihydrofolate reductase (DHFR2) fusion proteins using a chemical dimerizer, bis-methotrexate. With targeting proteins fused onto the DHFR2 monomers, the CSANs can target specific cellular antigens. Here, anti-EGFR or anti-EpCAM fibronectin-DHFR2 monomers incorporating a CAAX-box sequence were enzymatically prenylated, then assembled into the corresponding CSANs. Both farnesylated and geranylgeranylated CSANs efficiently modified the cell surface of lymphocytes and remained bound to the cell surface with a half-life of >3 days. Co-localization studies revealed a preference for the prenylated nanorings to associate with lipid rafts. The presence of antigen targeting elements in these bifunctional constructs enabled them to specifically interact with target cells while treatment with trimethoprim resulted in rapid CSAN disassembly and termination of the cell–cell interactions. Hence, we were able to determine that activated PBMCs modified with the prenylated CSANs caused irreversible selective cytotoxicity toward EGFR-expressing cells within 2 hours without direct engagement of CD3. The ability to disassemble these nanostructures in a temporally controlled manner provides a unique platform for studying cell–cell interactions and T cell-mediated cytotoxicity. Overall, antigen-targeted prenylated CSANs provide a general approach for the regulation of specific cell–cell interactions and will be valuable for a plethora of fundamental and therapeutic applications.
Introduction
Cell-based therapies have demonstrated notable therapeutic success when applied to tissue engineering, regenerative medicine, and cancer immunotherapy.1–3 These recent advances have highlighted the significance of controlling the fate and function of therapeutic cells. Typically, the efficacy of therapeutic cells is dependent on their ability to interact directly with other cells. Therefore, techniques that can efficiently regulate cell–cell interactions are regarded as valuable tools for cell-based therapies.4–6While the engineering of cell–cell interactions has been dominated by genetic methodologies such as chimeric antigen receptors (CARs), non-genetic cell surface modification is an attractive alternative since the cell surface can be directly modified without the need for genomic alterations or optimization of transfection efficiencies.7 Existing cell surface antigens are commonly utilized as handles for subsequent surface modification. For instance, bispecific T cell engagers are well-studied scaffolds that generally decorate the cancer cell surface with antigen-targeting scFvs that can also bind the pan-T cell antigen, CD3.8–10 While highly successful, the induced cell–cell interactions by bispecific antibodies are restricted to the binding specificity of the incorporated antibodies. While metabolic labeling can universally incorporate novel functional groups into the plasma membrane, the requisite cell culture steps and bio-orthogonal reactions are time-consuming.11 Finally, some one-step lipid insertion methods can directly anchor targeting molecules on the cell surface through hydrophobic interactions with membrane lipids; however, the modifications are usually stable for mere hours12,13 and, in some cases, both cell types must be modified.14
To address these issues, our lab has developed a multivalent protein scaffold referred to as chemically self-assembled nanorings (CSANs).15 We have previously demonstrated that CSANs can be engineered to stably associate with cell membranes and mediate reversible cell–cell interactions.16,17 CSANs are prepared by the oligomerization of bivalent dihydrofolate reductase (DHFR2) fusion proteins using a chemical dimerizer, bis-methotrexate (bisMTX). This results in the formation of a predominantly octameric nanoring structure with uniform size distribution.15,16 Importantly, the nanorings can be disassembled by treatment with the FDA-approved antibiotic, trimethoprim, thereby rendering ring formation reversible.17
Antibody-derived ligands are widely used scaffolds for cell targeting and cell–cell interaction studies. However, their biophysical properties make them less than optimal for many fundamental and clinical applications.18 As an alternative, our lab has evolved human tenth type III fibronectin (Fn3) domains capable of binding to epidermal growth factor receptor (EGFR) or epithelial cell adhesion molecule (EpCAM).19 When these Fn3 domains were recombinantly tethered to the DHFR2 fusion proteins, the resultant monomers were assembled into CSANs capable of binding to cells that overexpress these carcinoma antigens.20
Previously, we demonstrated that bifunctional CSANs can be installed on cell membranes via hydrophobic insertion using either of two approaches: (1) employing a phospholipid-bisMTX conjugate to form lipid-modified CSANs; or (2) first modifying the cell surface with biotinylated or clickable phospholipids, and then treating these cells with CSANs fused to monovalent streptavidin (mSA) or CSANs formed with trivalent azide-bisMTX, respectively.20,21 Nevertheless, these methods have significant limitations, as the first approach poses the synthetic challenge of preparing pure phospholipid-bisMTX, while the second approach requires a multi-step process and leaves residual conjugates on the cell surface. Therefore, the development of targeted CSANs that can be enzymatically lipidated would significantly streamline their preparation and hence dramatically expand their utility in engineering cell–cell interactions.
Protein prenyltransferases catalyze the addition of C15 (farnesyl) and C20 (geranylgeranyl) lipids to proteins containing C-terminal CAAX-motifs.22–26 Following lipid modification, these prenylated proteins translocate to cellular membranes.27 Since the CAAX-box sequence is the only requirement for prenyltransferase recognition, essentially any protein can be lipidated by genetic fusion of a CAAX-box onto the C-terminus of the protein of interest.28,29 Hence, we envisioned that the fusion of a functionalized DHFR2 with such a sequence (CVIA) would facilitate the preparation of lipidated proteins suitable for CSAN assembly. This enzymatic process allows lipidation to be accomplished in a single step and is much more efficient than chemical conjugation reactions.28,30 Therefore, the resulting prenylated CSANs could directly modify any mammalian cell membrane with multivalent targeting elements through hydrophobic interactions between the isoprenoids and membrane phospholipids (Fig. 1). Moreover, the multivalency of these prenylated CSANs would make them more stable on the cell membrane than other monomeric lipid-modified constructs.31,32
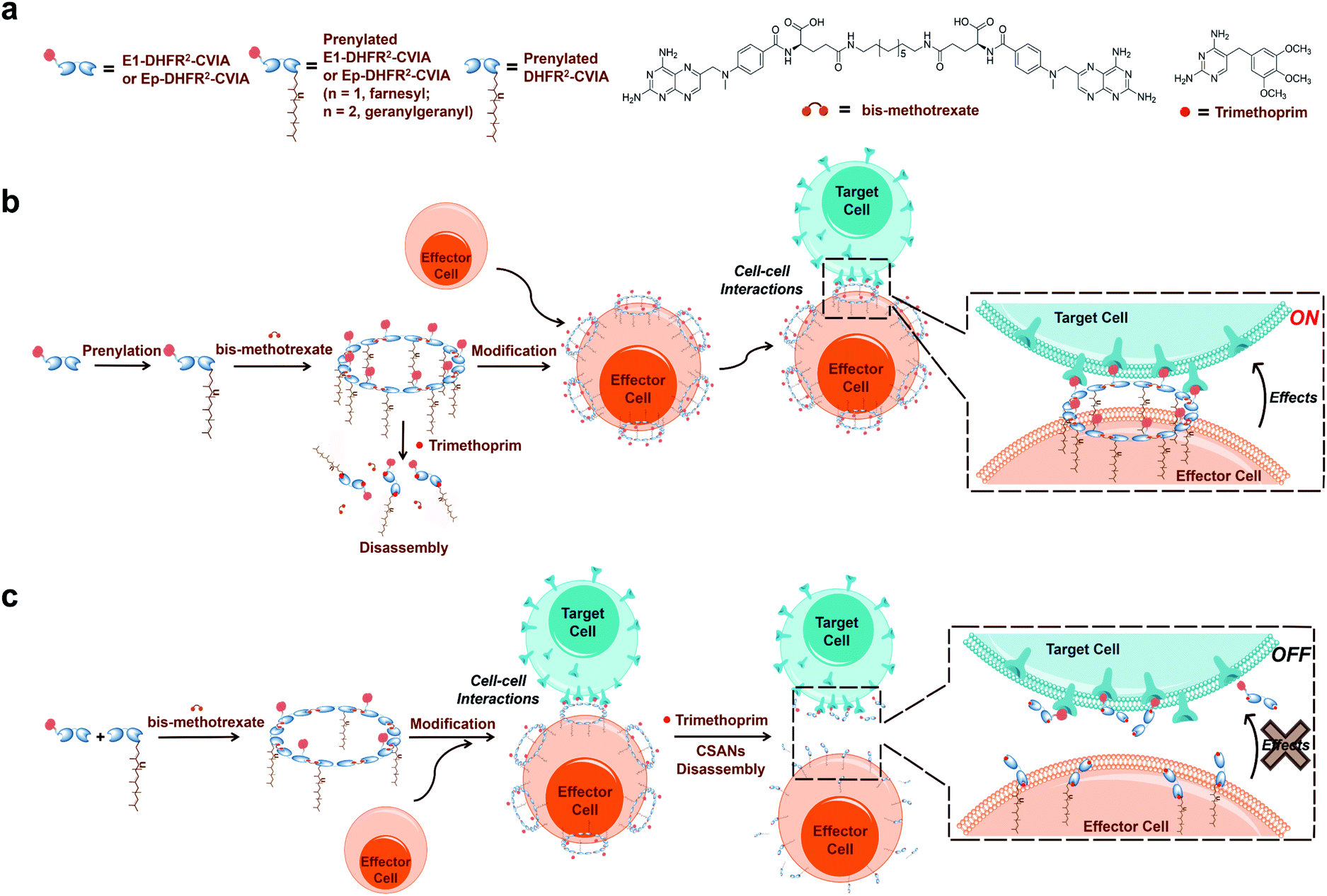 | ||
| Fig. 1 Approach to directing cell–cell interactions with prenylated CSANs. (a) Components of prenylated CSANs. (b) Anti-EGFR-DHFR2-CVIA (E1-DHFR2-CVIA) or anti-EpCAM-DHFR2-CVIA (Ep-DHFR2-CVIA) proteins can be farnesylated or geranylgeranylated, and these prenylated proteins can form multivalent CSANs in the presence of bisMTX. The prenylated nanorings can modify cell surfaces through hydrophobic interactions with the plasma membrane and then target the modified cells to EGFR+ or EpCAM+ cells to form cell–cell interactions. (c) Similarly, hybrid prenylated CSANs can be constructed from E1-DHFR2-CVIA (or Ep-DHFR2-CVIA) protein that is not prenylated and pre-prenylated but non-targeted DHFR2-CVIA protein. The hybrid CSANs can mediate cell–cell interactions and can be disassembled on the cell surface using clinically relevant concentrations of trimethoprim, thereby terminating the CSAN-mediated cell–cell interactions. | ||
In this study, we prepared three distinct DHFR2 fusion proteins, all of which include the CAAX-box sequence CVIA appended at their C-terminus. Two of these proteins also incorporate an Fn3-based targeting domain at their N-terminus, while the third lacks a targeting ligand and thereby remains agnostic (DHFR2-CVIA). One of the targeted entities utilizes an EGFR-targeting Fn3 (E1-DHFR2-CVIA) and the other uses an EpCAM-targeting Fn3 (Ep-DHFR2-CVIA). We demonstrate that both farnesyl transferase and geranylgeranyl transferase are able to efficiently and quantitatively generate lipid-modified E1-DHFR2-CVIA and Ep-DHFR2-CVIA proteins, and that these monomers easily assemble into nanorings that can modify cell surface membranes in a single step. The modification is stable for days and can direct specific, antigen-targeted cell–cell interactions that can be reversed using trimethoprim. This system was also used to demonstrate that activated T cells modified with prenylated anti-EGFR CSANs will fully initiate cell killing within two hours. Those results highlight the unique ensemble of properties including simplicity, tunability and reversibility that make prenylated CSANs particularly suitable for studying cell–cell interactions.
Results
Anti-EGFR and anti-EpCAM Fn3 proteins were previously generated based on the human tenth type III fibronectin domain.19,33 Here, the Fn3 proteins were genetically fused to the N-terminus of DHFR2 fusion proteins, while a CVIA sequence was appended to the C-terminus to generate the E1-DHFR2-CVIA and Ep-DHFR2-CVIA fusion proteins (ESI Note 1†). The proteins were expressed in soluble form in E. coli and characterized by gel electrophoresis (Fig. 2a). The CVIA-tagged proteins were shown to be substrates for both farnesyl transferase and geranylgeranyl transferase, which modify the C-terminal cysteine with either a farnesyl group or a geranylgeranyl group, respectively. In this manner, the farnesylated DHFR2 proteins (E1-DHFR2-Far, Ep-DHFR2-Far, and DHFR2-Far) and geranylgeranylated DHFR2 proteins (E1-DHFR2-GG, Ep-DHFR2-GG, and DHFR2-GG) were generated, with modifications confirmed via LC-MS (Fig. 2b and S1†).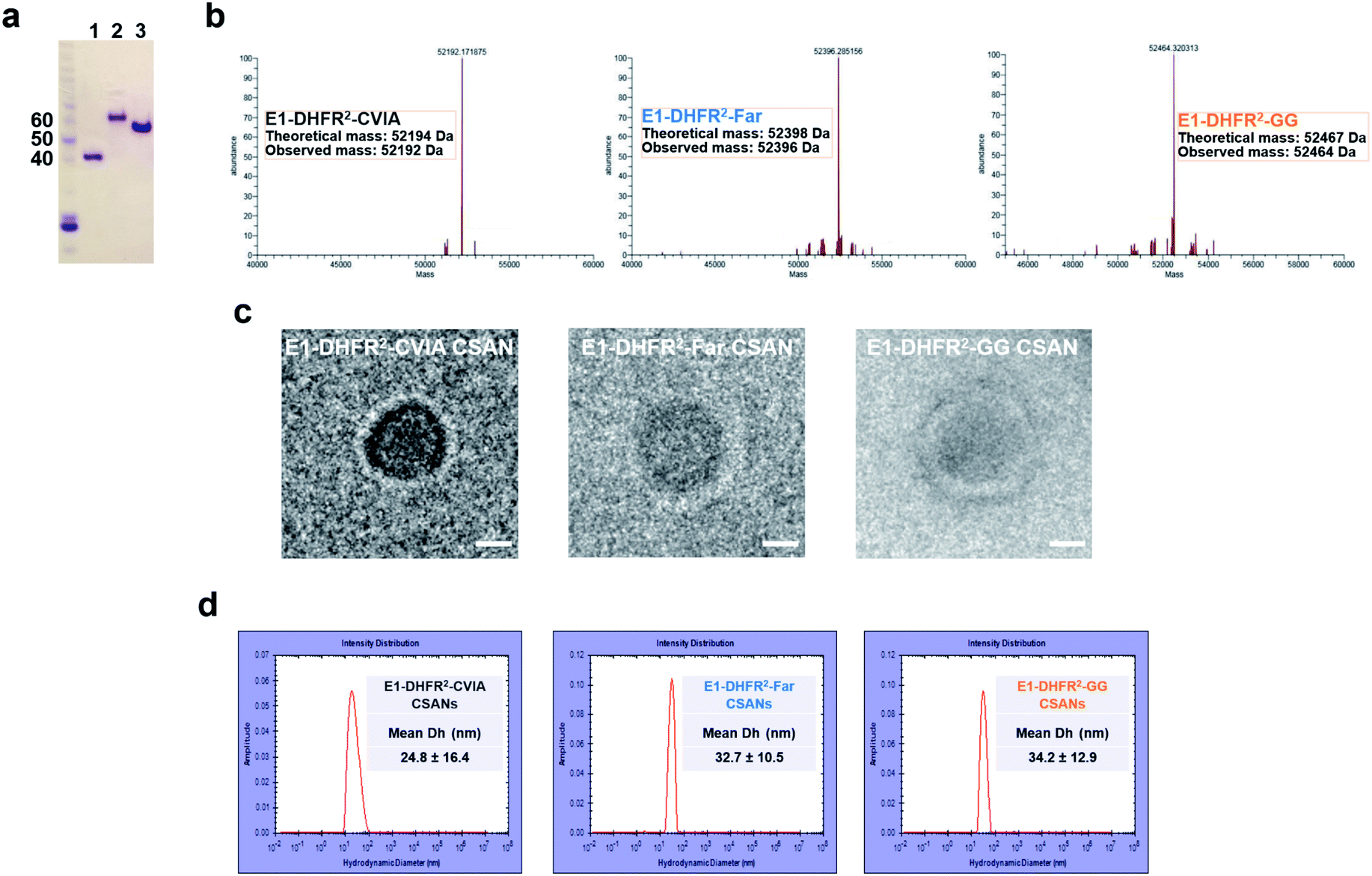 | ||
| Fig. 2 Characterization of prenylated CSANs. (a) SDS-PAGE gel of DHFR2-CVIA (1), Ep-DHFR2-CVIA (2), and E1-DHFR2-CVIA (3) proteins. (b) LC-MS of E1-DHFR2-CVIA, farnesylated E1-DHFR2-CVIA (E1-DHFR2-Far) and geranylgeranylated E1-DHFR2-CVIA (E1-DHFR2-GG) proteins. Ring formation was confirmed by (c) cryo-TEM imaging of anti-EGFR nanorings (scale bar, 10 nm). (d) Hydrodynamic diameter analysis of anti-EGFR nanorings using dynamic light scattering. The E1-DHFR2-CVIA nanorings have a DH of 25 ± 16 nm and prenylated nanorings have DH values of 33 ± 11 nm and 34 ± 13 nm, as shown. | ||
Upon the addition of a molar excess of the chemical dimerizer, bisMTX, the monomeric DHFR2 fusion proteins rapidly self-assembled into ring structures that were similar to previously reported CSANs,34 which was demonstrated by size exclusion chromatography (SEC; Fig. S2†). The hydrodynamic diameter of the nanorings was measured by dynamic light scattering (DLS) (Fig. 2d and S3†). The sizes of both the E1-DHFR2-CVIA CSANs and Ep-DHFR2-CVIA CSANs were found to be similar (24–25 nm) and somewhat larger than the simple DHFR2-CVIA rings (18 nm, Fig. S3†), which lack the added Fn3 targeting domain. Both farnesylated and geranylgeranylated CSANs were found to be modestly larger (33–35 nm) than their non-prenylated counterparts. Cryo-transmission electron microscopy (cryo-TEM) imaging analysis revealed that the morphology and the size of the anti-EGFR and anti-EpCAM CSANs were consistent with the results obtained via DLS studies (Fig. 2c and S4†).
The B-cell leukemia cell line, Raji, epidermoid carcinoma cell line, A431, and the breast cancer cell line, MCF-7, were used as models to assess the ability of the prenylated CSANs to modify mammalian cell membranes. The CVIA-tagged fusion proteins were farnesylated or geranylgeranylated by farnesyl transferase or geranylgeranyl transferase, respectively and oligomerized into CSANs using bisMTX (Fig. 2c and S4†). Accordingly, Raji cells, A431 cells, and MCF-7 cells were each incubated with prenylated CSANs at room temperature for 1 h and after washing, the membrane-bound CSANs were quantified using the anti-FLAG-PE antibody via flow cytometry. As shown, both farnesylated and geranylgeranylated CSANs effectively modified each of the cell surfaces (Fig. 3a).
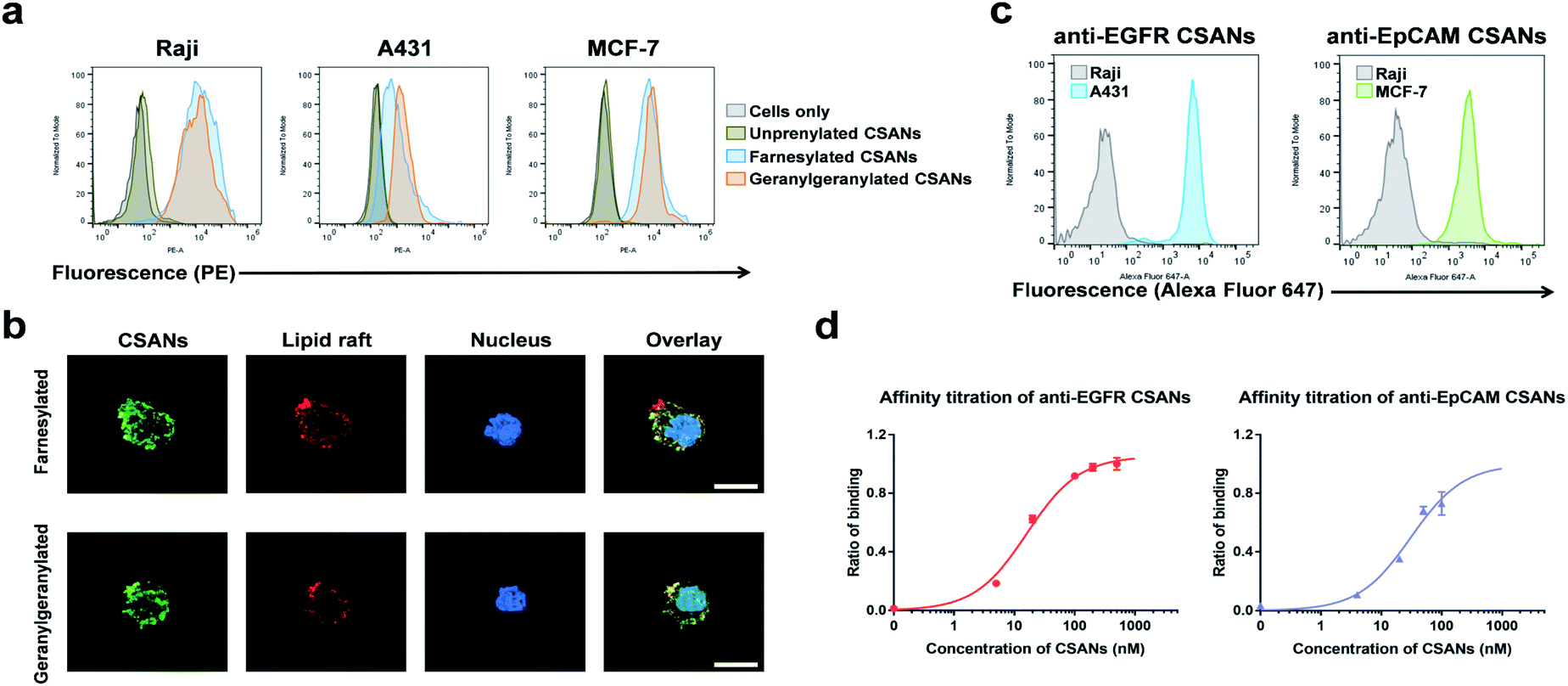 | ||
| Fig. 3 Prenylated CSANs insert into cell membranes and bind to their target antigens. (a) Flow cytometry demonstrates that prenylated CSANs hydrophobically insert into the cell membrane of Raji, A431, and MCF-7 cells. (b) Fluorescent microscopy shows the prenylated CSANs localized on the plasma membrane of Raji cells. Co-staining demonstrated that the prenylated CSANs localize to membrane lipid rafts, as shown (scale bar, 10 μm). (c) EGFR- and EpCAM-targeted CSANs bind selectively to EGFR+ A431 cells and EpCAM+ MCF-7 cells, respectively, via flow cytometry. Raji cells were used as the EGFR−/EpCAM− cell line for both assays. (d) The apparent affinity of anti-EGFR (16.3 ± 2.4 nM) and anti-EpCAM CSANs (31.1 ± 11 nM) was assessed by flow cytometry using EGFR+ MDA-MB-231 cells and EpCAM+ MCF-7 cells, respectively. | ||
To gain further insight into the nature of the interaction between prenylated CSANs and cell membranes, confocal microscopy studies were carried out with FITC labeled CSANs (Fig. 3b). Prenylated CSANs (2.5 μM) were shown to modify the surface of Raji cells in semi-discrete areas. Staining with the lipid raft specific agent, Alexa Fluor 594-conjugated cholera toxin subunit B, revealed co-localization of CSANs and lipid rafts. Consistent with these findings, previous studies have shown that farnesylation can target H-Ras to lipid rafts on the cytoplasmic membrane.35 Importantly, neither of the prenylated CSAN species were found to be toxic to Raji or MCF-7 cells at concentrations as high as 6 μM (Fig. S5†).
The ability of targeted CSANs to bind to antigen-expressing cell lines was assessed by flow cytometry. The anti-EGFR CSANs bound selectively to the EGFR+ cell line, A431, and the anti-EpCAM CSANs bound selectively to the EpCAM+ cell line, MCF-7 (Fig. 3c). The binding affinity of the anti-EpCAM and anti-EGFR CSANs towards EpCAM+ MCF-7 cells or EGFR+ MDA-MB-231 cells, respectively, was also assessed by flow cytometry (Fig. 3d and S6†). The CSANs were shown to bind to their target cells with high affinity (anti-EpCAM CSANs: apparent Kd = 31 ± 11 nM; anti-EGFR CSANs: apparent Kd = 16 ± 2.4 nM). These results are comparable to our previous studies of these fibronectin-based targeting scaffolds.19,20,34
We have previously demonstrated that the apparent affinity of a targeted CSAN can be modulated by mixing different ratios of a targeting monomer with a non-targeting monomer, thereby tuning the valency of the scaffold.36 Accordingly, we chose to explore the effect of various prenylation valencies on farnesylated and geranylgeranylated CSANs' ability to insert into Raji cell membranes (Fig. 4a). Hybrid CSANs were prepared by oligomerizing prenylated DHFR2-CVIA protein (DHFR2-Far or DHFR2-GG) with unprenylated DHFR2-CVIA at different molar ratios, including 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 4:4 and 2:6. As such, the resultant CSANs contain different numbers of isoprenoids groups in each nanoring, with overall valencies ranging from 8–2. Cells were treated with the hybrid prenylated CSANs and incubated for 1 h at room temperature. The membrane insertion of the prenylated CSANs was then characterized by flow cytometry (Fig. S7†), and the Kd for each type of CSANs determined (Fig. 4b, c and Table S1†). In each case, CSANs composed of 100% prenylated monomers exhibited the highest membrane binding affinity, with geranylgeranylated CSANs exhibiting a higher membrane affinity (48 ± 22 nM) than farnesylated CSANs (150 ± 6.2 nM). As can be seen from Table S1,† a reduction in valency directly corresponded to a proportional reduction in the apparent affinity. Thus, although geranylgeranylated CSANs exhibited greater binding affinity, the cumulative cost per lipid loss on membrane binding was similar. It is noteworthy that the relationship between the apparent Kd values of prenylated CSANs and the prenylation valency (N) follows the equation:
:0, 4:4 and 2:6. As such, the resultant CSANs contain different numbers of isoprenoids groups in each nanoring, with overall valencies ranging from 8–2. Cells were treated with the hybrid prenylated CSANs and incubated for 1 h at room temperature. The membrane insertion of the prenylated CSANs was then characterized by flow cytometry (Fig. S7†), and the Kd for each type of CSANs determined (Fig. 4b, c and Table S1†). In each case, CSANs composed of 100% prenylated monomers exhibited the highest membrane binding affinity, with geranylgeranylated CSANs exhibiting a higher membrane affinity (48 ± 22 nM) than farnesylated CSANs (150 ± 6.2 nM). As can be seen from Table S1,† a reduction in valency directly corresponded to a proportional reduction in the apparent affinity. Thus, although geranylgeranylated CSANs exhibited greater binding affinity, the cumulative cost per lipid loss on membrane binding was similar. It is noteworthy that the relationship between the apparent Kd values of prenylated CSANs and the prenylation valency (N) follows the equation:

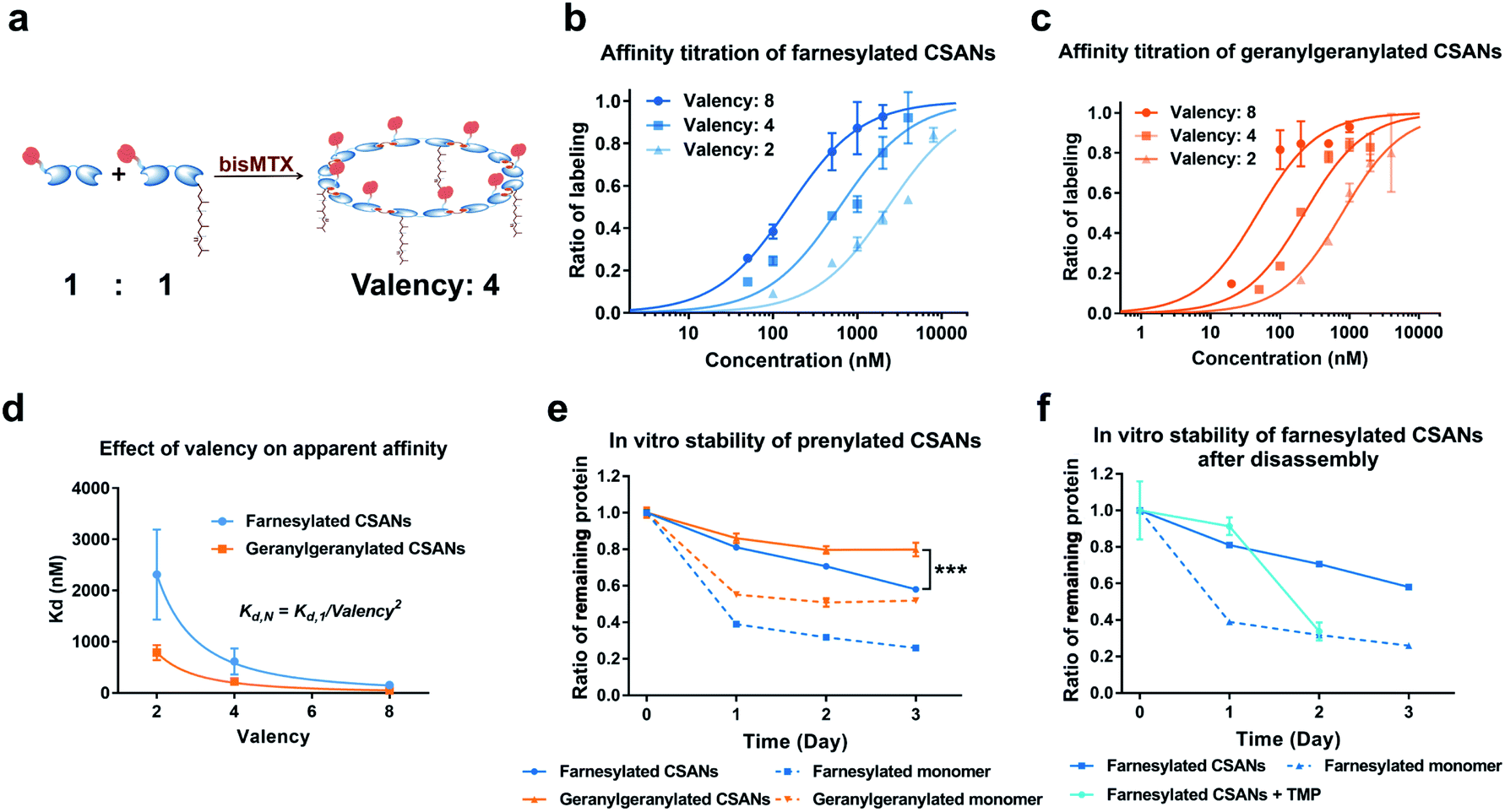 | ||
| Fig. 4 Affinity and stability study of prenylated CSANs. (a) The number (valency) of isoprenoids groups in the hybrid CSANs can be tuned by assembling different molar ratios of monomeric subunits. The apparent affinity of (b) farnesylated and (c) geranylgeranylated CSANs for the plasma membrane varies directly with isoprenoid valency, as demonstrated by flow cytometry. (d) The apparent Kd values of the prenylated CSANs for membrane binding exhibited an inverse relationship with the square of the valency (Kd,N = Kd,1/valency2, R2 > 0.997 for both prenylated CSANs). (e) Multivalent prenylated CSANs exhibit a longer cell surface half-life (>72 hours) compared to the monomeric prenylated proteins in vitro. (f) The membrane residence half-life of the farnesylated CSANs decreases significantly (<20 hours) upon scaffold disassembly with trimethoprim. Significance in (e) (data on Day 3) was tested using a two-tailed, unpaired t-test. And is indicated as ***P < 0.001. Data in panels (b), (c), (e), and (f), are represented as the mean ± standard deviation from n = 3 independent trials. For panel (d), data are shown as the mean Kd ± standard error (from n = 3 independent samples). In some instances, small error bars are obscured by the symbols denoting the mean value. | ||
Previously, we have demonstrated that octameric phospholipid-based CSANs are stable on the cell membrane for >72 h.20,21 To assess the cell surface stability of prenylated CSANs, we stained Raji cells with CellTrace Violet (CTV) and then treated them with farnesylated or geranylgeranylated CSANs prepared from prenylated DHFR2-CVIA monomers. The CSAN-modified cells were then returned to culture for 0–72 h. Every 24 h, an aliquot of cells was taken for analysis, and the remaining culture media was refreshed to partially stimulate physiologic clearance. Samples were stained using an anti-FLAG-PE antibody to label remaining cell surface CSANs before analysis by flow cytometry. The quantification of surface-bound CSANs was corrected for the dilution effect of cell division over this time period, as dictated by the CTV labeling. The results revealed that, similar to the studies utilizing phospholipid attachment, the half-life for the association of the farnesylated and geranylgeranylated CSANs was >72 h (Fig. 4e). Within this extended time frame, farnesylated CSANs appear to dissociate from the cell membrane more rapidly than the geranylgeranylated counterparts. Indeed, this difference in membrane longevity was more pronounced when comparing the farnesylated and geranylgeranylated DHFR2-CVIA monomers (Fig. 4e).
As previously reported, the treatment of CSANs with the FDA-approved antibiotic and DHFR antagonist, trimethoprim, results in rapid CSANs disassembly and a corresponding decrease in their cell membrane stability.20,21 To demonstrate this, Raji cells modified with CSANs were treated with a 20-fold molar excess of trimethoprim. Within 24 h, greater than 60% of the farnesylated CSANs had been lost from the cell surface, with the presumed monomeric species exhibiting similar stability on the cell membrane as the parental monomeric farnesylated DHFR2-CVIA (Fig. 4f). Thus, these observations confirm that the stability of the CSANs and their multivalent nature can be pharmacologically altered on the cell membrane by treatment with trimethoprim.
To study the cell–cell interactions mediated by prenylated CSANs, CFSE-stained Raji cells were modified with prenylated anti-EGFR CSANs or incubated with control solutions and then co-cultured with CTV-stained A431 cells that were plated on glass coverslips. After washing off unbound Raji cells, the cells were fixed and imaged via fluorescence microscopy. Both farnesylated and geranylgeranylated anti-EGFR CSANs were able to efficiently mediate cell–cell interactions, as shown by the adherence of CFSE-labeled Raji cells to the CTV-labeled A431 cells (Fig. 5a, b and S8a†). In contrast, the prenylated DHFR2-CVIA CSANs or unprenylated E1-DHFR2-CVIA CSANs that lack the targeting moiety or the prenyl group, respectively, were unable to induce cell–cell interactions and did not significantly differ from the PBS control group (Fig. 5a). As illustrated in Fig. 1c, the disassembly of the hybrid prenylated anti-EGFR CSANs was expected to completely terminate the CSANs-mediated cell–cell interactions. Treatment of the co-cultures with trimethoprim (TMP) for 1 h was shown to significantly reduce the amount of cell–cell interactions induced by the hybrid anti-EGFR CSANs constructed from prenylated DHFR2-CVIA and unprenylated E1-DHFR2-CVIA protein (Fig. 5c and S8b†), consistent with the disassembly of the CSANs by TMP.
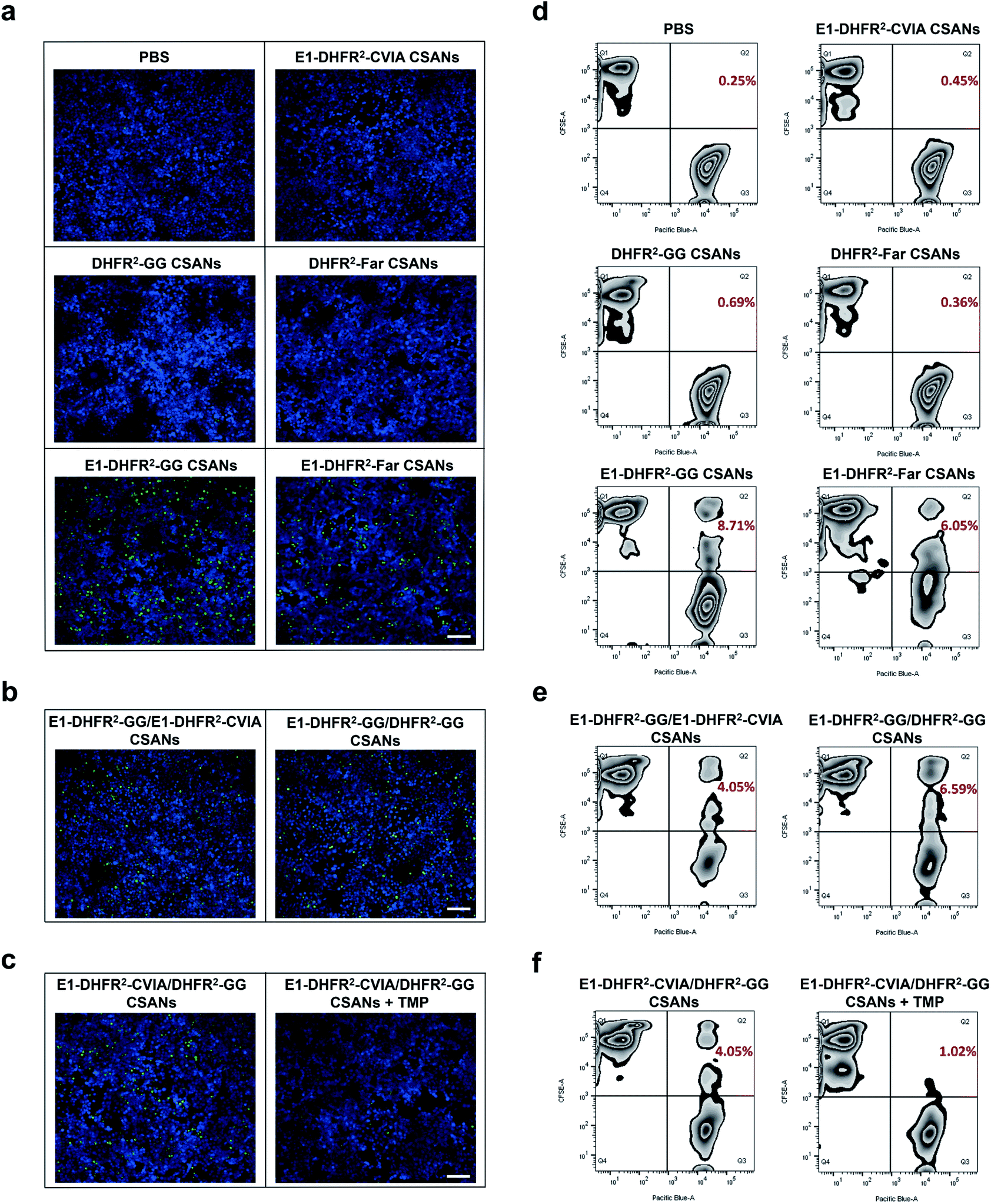 | ||
| Fig. 5 Prenylated CSANs mediate reversible cell–cell interactions. (a) Fluorescent microscopy demonstrates that prenylated anti-EGFR CSANs can mediate reversible cell–cell interactions in the fluorescent microscopy imaging experiment (scale bar, 100 μm). Raji cells were stained by CFSE, while EGFR+ A431 cells were plated on the coverslips and stained by CellTrace Violet (CTV). The E1-DHFR2-GG CSANs and E1-DHFR2-Far CSANs maintained the cell–cell interactions, which were shown by the remaining green Raji cells on the coverslip after PBS wash, while the CSANs without targeting elements or isoprenoids groups (E1-DHFR2-CVIA, DHFR2-GG and DHFR2-Far CSANs) failed to mediate cell–cell interactions. (b) The hybrid CSANs with 50% molar isoprenoids groups (E1-DHFR2-GG/E1-DHFR2-CVIA CSANs) or 50% molar anti-EGFR domains (E1-DHFR2-GG/DHFR2-GG CSANs) were able to mediate cell–cell interactions (scale bar, 100 μm). (c) The cell–cell interactions were shown to be dissociated by the disassembly of the hybrid prenylated anti-EGFR CSANs (E1-DHFR2-CVIA/DHFR2-GG CSANs) through TMP treatment (scale bar, 100 μm). (d) The cell–cell interactions were also investigated using flow cytometry, where the CFSE-stained Raji cells were modified with CSANs and incubated with CTV-stained A431 cells. The resulting CFSE+/CTV+ cell pairs were then quantified by flow cytometry. Both prenylated and geranylgeranylated CSANs induced cell–cell interactions, though the latter was more efficient. (e) Reduction of isoprenoids groups or EGFR-targeting domains by 50% in the CSANs diminished cell–cell interactions. (f) TMP was shown to significantly interrupt the cell–cell interactions mediated by E1-DHFR2-CVIA/DHFR2-GG CSANs. | ||
To quantitatively characterize the cell–cell interactions mediated by the prenylated anti-EGFR CSANs, CFSE-stained Raji cells were labeled with CSANs and incubated with EGFR+ A431 cells in PBS, followed by flow cytometry analysis. The CFSE+/CTV+ double-positive cell population, which represents induced Raji/A431 cell clusters, was quantified as the indicator of cell–cell interactions. Specifically, both prenylated anti-EGFR CSANs induced cell–cell interactions, though the geranylgeranylated anti-EGFR CSANs were shown to induce 44% more cell–cell interactions than comparable farnesylated CSANs (Fig. 5d). When hybrid CSANs formed from a 1:1 molar mixture of E1-DHFR2-CVIA and prenylated E1-DHFR2-CVIA were used, a 50% decrease in the percentage cell–cell interactions was observed (Fig. 5e and S9a†). Consistent with these observations, the hybrid CSANs prepared from a 1:1 molar mixture of prenylated E1-DHFR2-CVIA and prenylated DHFR2-CVIA also exhibited a reduction in the percentage of cell–cell interactions (Fig. 5e and S9a†). Moreover, the cell clusters induced by the hybrid prenylated anti-EGFR CSANs, whether geranylgeranylated or farnesylated, were greatly reduced in number after treatment with trimethoprim (Fig. 5f and S9b†). Thus, this data indicates that these strong and reversible cell–cell interactions are mediated by prenylated anti-EGFR CSANs.
Finally, as an alternative antigen, the cell–cell interactions mediated by prenylated anti-EpCAM CSANs were also investigated through analogous imaging and flow cytometry experiments using EpCAM+ MCF-7 cells as the target cells. Consistent with our observations described above for the prenylated anti-EGFR CSANs, both types of prenylated anti-EpCAM CSANs efficiently mediated the cell–cell interactions, with the geranylgeranylated CSANs again providing more robust interactions (Fig. S10 and S11†).
We next assessed the ability of prenylated CSANs to direct PBMC-mediated lysis of target cells using a lactate-dehydrogenase (LDH) release assay. Activated PBMCs were modified with prenylated anti-EGFR CSANs (E1-DHFR2-Far or E1-DHFR2-GG CSANs) and incubated with EGFR+ A431 cells. The CSAN-functionalized PBMCs exhibited dose-dependent lysis of the target cells that was significantly higher than the cytotoxicity observed for unmodified control PBMCs (Fig. 6a and b). In addition, the prenylated anti-EGFR CSANs failed to enhance the cell lysis of EGFR-MCF-7 cells, by CSAN-modified PBMCs, further demonstrating the specificity of the prenylated anti-cancer CSANs (Fig. S12†). Consistent with the cell–cell interactions and membrane stability studies, geranylgeranylated anti-EGFR CSANs induced a significantly greater amount of cellular toxicity than farnesylated anti-EGFR CSANs, even at effector/target cell ratios (E/T) as high as 20:1 (Fig. 6c).
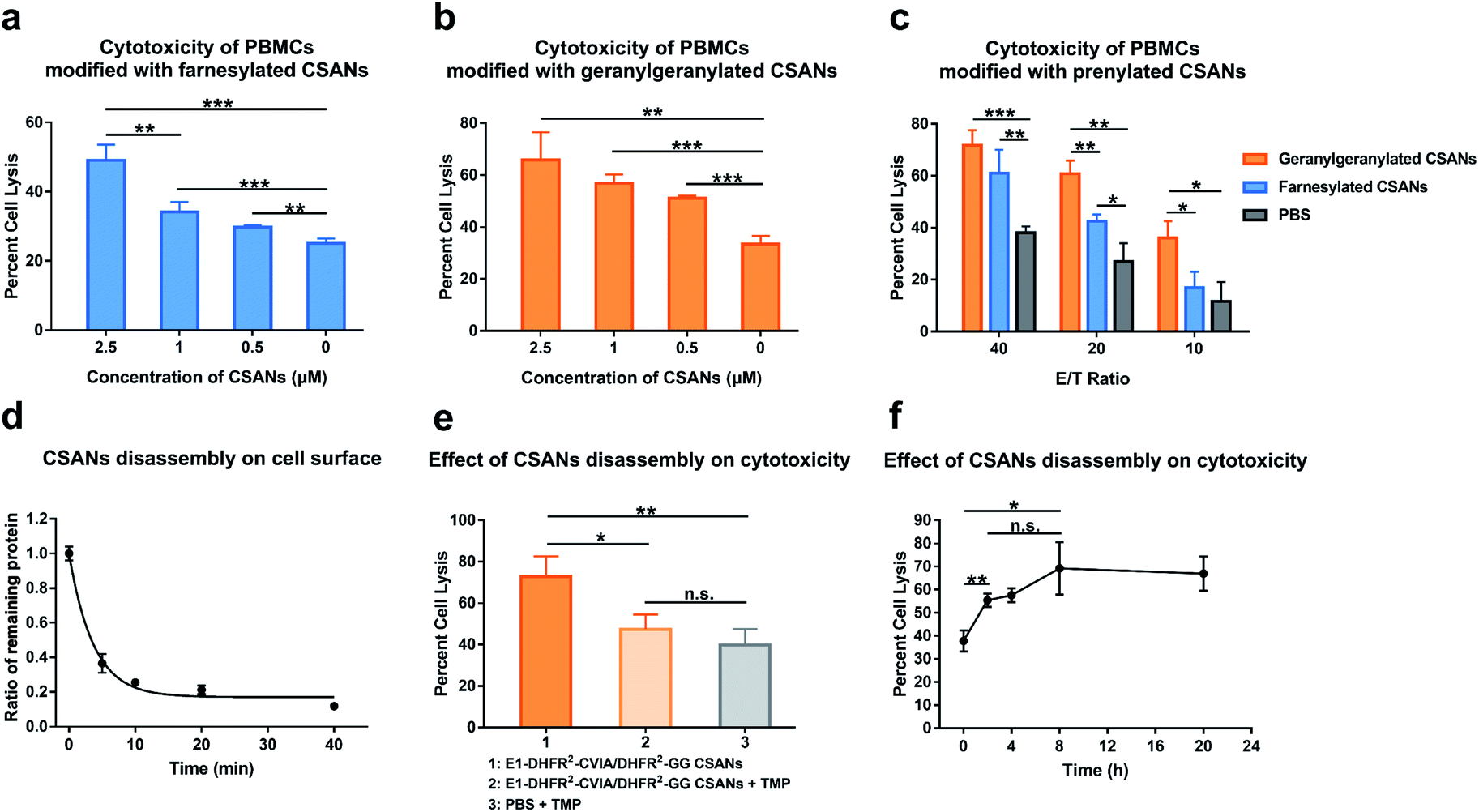 | ||
| Fig. 6 Prenylated CSANs enhance cytotoxicity towards EGFR+ target cells. The cytotoxicity of PBMCs against EGFR+ A431 cells after modification with (a) farnesylated anti-EGFR CSANs (E1-DHFR2-Far CSANs) or (b) geranylgeranylated anti-EGFR CSANs (E1-DHFR2-GG CSANs) was studied using an LDH release assay. Both prenylated CSANs enhanced the cytotoxicity of PBMCs in a concentration-dependent manner. (c) The impact of different effector:target (E/T) ratios on the cytotoxicity of CSAN-modified PBMCs was also assessed using an LDH release assay. (d) Kinetic study of CSAN disassembly on cell surface. Raji cells were modified with the hybrid E1-DHFR2-CVIA/DHFR2-GG CSANs, and the E1-DHFR2-CVIA protein was disassociated from the cell surface using trimethoprim. The remaining E1-DHFR2-CVIA protein was then quantified via flow cytometry. The half-life of CSAN disassembly is 3.5 min. (e) CSAN disassembly was shown to significantly reduce the cytotoxicity of PBMCs modified with the hybrid E1-DHFR2-CVIA/DHFR2-GG CSANs, as expected. (f) The E1-DHFR2-CVIA/DHFR2-GG CSANs were disassembled at different time points (0 h, 2 h, 4 h, 8 h, 20 h) during the co-culture of CSAN-modified PBMCs and A431 cells, and the cytotoxicity of the PBMCs was studied after the CSANs disassembly. Significance in (a), (b), (c), (e) and (f) was tested using a two-tailed, unpaired t-test. And is indicated as **P < 0.01 or *P < 0.05. For panel (d), data are shown as the mean ± standard deviation from n = 3 independent samples. Error bars in (a), (b), (c), (e) and (f) denote the standard error calculated from n = 3 independent trials and each trial has n = 3 independent samples. | ||
As was demonstrated above, treatment with trimethoprim can disassemble prenylated CSANs, resulting in their loss from the cell surface. When Raji cells modified with geranylgeranylated anti-EGFR CSANs were treated with a 20-fold excess of trimethoprim, approximately 80% of the nanorings were disassembled on the cellular membrane within 10 min, yielding a half-life of 3.5 min (Fig. 6d). To facilitate complete dissociation of the lipidated subunit from the targeting ligand, thereby ensuring that no cross-links between the cells would remain upon scaffold disassembly, we prepared geranylgeranylated anti-EGFR CSANs from E1-DHFR2-CVIA (unprenylated) and non-targeted geranylgeranylated DHFR2-CVIA. Treatment with trimethoprim at time zero (right after mixing between CSAN-modified PBMCs and A431 cells) was shown to abolish the ability of the modified PBMCs to induce target cell cytotoxicity (Fig. 6e).
To further study the effects of CSANs disassembly timing on induced cytotoxicity, PBMCs were modified with the hybrid geranylgeranylated anti-EGFR CSANs (E1-DHFR2-CVIA/DHFR2-GG CSANs), mixed with target A431 cells, and then treated with trimethoprim at various time points. The level of targeted cytotoxicity was then determined at the end of 20 h for each time point. As can be seen from Fig. 6f, cytotoxicity is initiated within the first few minutes with maximum cytotoxicity observed after at least 2 h. Thus, the initiation of target tumor cell apoptosis and likely necrosis can be abolished if interrupted early but is irreversible past this time point.
Discussion
Cell–cell interactions play vital roles in many aspects of physiological and pathological processes; therefore, the ability to manipulate such interactions is valuable for fundamental studies of cellular biology and for potential therapeutic applications. To streamline the preparation of CSANs designed to bind to membranes via non-covalent interactions, we explored here the use of enzymatic methods to append naturally occurring lipids onto the desired nanostructured targets. An important advantage of such an approach is that it capitalizes on the catalytic efficiency afforded by enzymatic conjugation. Such methods allow protein attachment to occur with high conversion even at low concentrations driven by the formation of an energetically favorable enzyme–substrate complex. Moreover, the use of natural substrates obviates the need for complex synthetic methods, thus enhancing the ease of implementation.Accordingly, we explored the potential of enzymatically labeling the monomeric protein component of the CSANs to directly install a natural C15 or C20 lipid, which upon multivalent display, would result in high-affinity membrane binding. We designed DHFR2-CVIA monomers that would be substrates for either farnesyl transferase or geranylgeranyl transferase, thus allowing enzymatic lipidation with either farnesyl or geranylgeranyl moieties, respectively. The DHFR2-CVIA monomers were easily expressed in E. coli and quantitatively enzymatically labeled with either farnesyl or geranylgeranyl groups. Despite being prenylated, the rings did not aggregate and were found to be stable for days in solution. Moreover, the prenylated CSANs quickly and stably (T1/2 > 72 h) labeled the cell membrane, with longer geranylgeranyl modifications exhibiting significantly more longevity than the shorter farnesyl groups. Since the number of displayed isoprenoids groups on the rings can be tuned by varying the ratio of prenylated to non-prenylated protein components prior to oligomerization, we were able to demonstrate that both fully farnesylated and geranylgeranylated CSANs yielded the highest membrane binding affinity. The appending of an anti-EGFR or anti-EpCAM fibronectin targeting domain did not affect membrane binding and allowed labeled cells to form direct contact with EGFR-expressing or EpCAM-expressing cells respectively in vitro.
In addition, significant differences were observed between the ability of fully farnesylated or geranylgeranylated CSANs to promote targeted cell–cell interactions, with geranylgeranylated CSANs forming significantly more cell–cell interactions than the farnesylated counterparts. When the valency of either isoprenoid groups or targeting ligands incorporated into the CSAN was reduced, a corresponding reduction of cell–cell interactions was observed. Consistent with previously reported CSANs, each construct was readily disassembled by treatment with a non-toxic excess of trimethoprim. Given the lower affinity of the farnesylated DHFR2 monomers, there may be an advantage to employing the farnesylated CSANs over geranylgeranylated CSANs, since dissociation of the resulting farnesylated monomers from the cell surface is likely to be faster than geranylgeranylated monomers due to the lower affinity of the shorter lipid for the membrane. Interestingly, similar to observations for prenylated proteins on the cytoplasmic membrane, the prenylated CSANs were shown to preferentially concentrate in lipid rafts. The differences in the ability of, for example, A431 and MCF-7 cells to bind to the prenylated CSANs may reflect differences in their lipid raft composition. Further studies with a range of cell lines and well characterized level of lipid rafts should shed light on the selectivity of prenylated CSANs to associate with lipid rafts over other regions of the cell membrane.
The T cell receptor in complexation with CD3, CD4, and CD8 has been found to be highly associated with and dependent on lipid rafts for the propagation of Lck based signaling.37,38 Consequently, we evaluated the ability of prenylated anti-EGFR CSANs, to direct T cell-targeted killing of an EGFR-overexpressing epidermoid carcinoma line, A431 cells. Activated PBMCs, which are 90% T cells, were treated with geranylgeranylated anti-EGFR CSANs. The modified T cells were shown to induce targeted killing of A431 cells. Given that we were employing activated T cells and that trimethoprim treatment can lead to rapid disassembly of the CSANs, we chose to investigate the time frame within which T cell killing is initiated once the targeted tumor cells have been engaged by dosing with excess trimethoprim at various time points and measuring the amount of induced cytotoxicity that occurred prior to CSAN abrogation. Our results indicate that similar to CAR-T cells at a similar E:T ratio, cytotoxicity is induced within the first two hours, after which removal of the CSANs has no effect on the targeted cytotoxicity.39 Whether the structure of the cytotoxic immune synapse is similar to CAR-T cells or BiTes remains to be determined, since there is no direct binding or engagement of CD3.40,41 Given the complex order and processes engaged by TCRs through the immune synapse, it is surprising that simply associating the T cell surface with the target cell through the prenylated CSANs results in T cell induced cytotoxicity. On-going studies of the engineering of interactions between the T cells and the target cells by prenylated CSANs should expand our understanding of the constraints or lack of constraints governing T cell-mediated cytotoxicity. The ability to study the time-dependence of T cell target engagement highlights the utility of the lipidated CSANs described here. The unique reversibility and tunability of the CSAN-based constructs described here make them particularly versatile tools for the study of cell–cell interactions in general. Given the complex order and processes engaged by TCRs through the immune synapse, it is interesting that the prenylated anti-EGFR CSANs are able to induce T cell-targeted cell killing.
Conclusions
In summary, we developed the multivalent prenylated chemically self-assembled nanorings (CSANs) for mediating reversible cell–cell interactions. Prenylated CSANs offer a facile one-step approach for the stable membrane labeling of virtually any cell type, providing accessible and pharmacologically reversible cell–cell interactions for a range of fundamental scientific and therapeutic applications.Author contributions
C. R. W., M. D. D., and Y. W. designed the project. O. K. designed the E1-DHFR2-CVIA (initial version) protein, and C. M. C. designed the Ep-DHFR2 proteins. Y. W. designed and generated the Ep-DHFR2-CVIA, E1-DHFR2-CVIA and DHFR2-CVIA proteins based on the protein scaffolds from O. K. and C. M. C. Y. W. prepared the farnesyl transferase. S. A. and J. L. H. prepared the geranylgeranyl transferase. Y. W. performed all the experiments and analyses presented in the paper. Y. W. wrote the manuscript and all authors provided edits and comments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by GM084152 (M. D. D.), CA185627 (C. R. W.), CA247681 (CRW) and NSF Grant ECCS-2025124 to the Minnesota Nano Center. We thank Dr Robert Hafner for his help with the cryo-TEM experiments, and these experiments were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program. We thank Dr Yingchun Zhao for his help with the LC-MS experiments that were conducted in the Masonic Cancer Center's Analytical Biochemistry Shared Resource. We thank Dr Mark Sanders for his help with the fluorescent imaging experiments, which were carried out in the University Imaging Center, University of Minnesota. We thank Dr Barry Finzel and William McCue for their assistance with the DLS analysis. We thank Dr Lakmal Rozumalski for the help with the manuscript.Notes and references
- T. N. Yamamoto, R. J. Kishton and N. P. Restifo, Nat. Med., 2019, 25, 1488–1499 CrossRef CAS.
- M. De Luca, A. Aiuti, G. Cossu, M. Parmar, G. Pellegrini and P. G. Robey, Nat. Cell Biol., 2019, 21, 801–811 CrossRef CAS.
- A. Golchin and T. Z. Farahany, Stem Cell Rev. Rep., 2019, 15, 166–175 CrossRef.
- C. A. Custódio and J. F. Mano, ChemNanoMat, 2016, 2, 376–384 CrossRef.
- M. Rothbauer, H. Zirath and P. Ertl, Lab Chip, 2018, 18, 249–270 RSC.
- D. J. Irvine and E. L. Dane, Nat. Rev. Immunol., 2020, 20, 321–334 CrossRef CAS.
- C. M. Csizmar, J. R. Petersburg and C. R. Wagner, Cell Chem. Biol., 2018, 25, 931–940 CrossRef CAS.
- S. R. Frankel and P. A. Baeuerle, Curr. Opin. Chem. Biol., 2013, 17, 385–392 CrossRef CAS.
- M. Ciof, J. Dorado, P. A. Baeuerle and C. Heeschen, 2012, 465–475.
- J. Wu, J. Fu, M. Zhang and D. Liu, J. Hematol. Oncol., 2015, 1–7 Search PubMed.
- Y. L. Huang and A. Chen, Advances in Proteomics Research, 2012 Search PubMed.
- M. Takeo, C. Li, M. Matsuda, H. Nagai, W. Hatanaka, T. Yamamoto, A. Kishimura, T. Mori and Y. Katayama, J. Biomater. Sci., Polym. Ed., 2015, 26, 353–368 CrossRef CAS.
- Y. Bagheri, S. Chedid, F. Shafiei, B. Zhao and M. You, Chem. Sci., 2019, 10, 11030–11040 RSC.
- Y. Teramura, H. Chen, T. Kawamoto and H. Iwata, Biomaterials, 2010, 31, 2229–2235 CrossRef CAS.
- J. C. T. Carlson, S. S. Jena, M. Flenniken, T. F. Chou, R. A. Siegel and C. R. Wagner, J. Am. Chem. Soc., 2006, 128, 7630–7638 CrossRef CAS.
- Q. Li, C. R. So, A. Fegan, V. Cody, M. Sarikaya, D. A. Vallera and C. R. Wagner, J. Am. Chem. Soc., 2010, 132, 17247–17257 CrossRef CAS.
- J. Shen, D. A. Vallera and C. R. Wagner, J. Am. Chem. Soc., 2015, 137, 10108–10111 CrossRef CAS.
- M. Gebauer and A. Skerra, Curr. Opin. Chem. Biol., 2009, 13, 245–255 CrossRef CAS.
- L. A. Stern, C. M. Csizmar, D. R. Woldring, C. R. Wagner and B. J. Hackel, ACS Comb. Sci., 2017, 19, 315–323 CrossRef CAS.
- C. M. Csizmar, J. R. Petersburg, A. Hendricks, L. A. Stern, B. J. Hackel and C. R. Wagner, Bioconjugate Chem., 2018, 29, 1291–1301 CrossRef CAS.
- K. Gabrielse, A. Gangar, N. Kumar, J. C. Lee, A. Fegan, J. J. Shen, Q. Li, D. Vallera and C. R. Wagner, Angew. Chem., Int. Ed., 2014, 53, 5112–5116 CAS.
- K. T. Lane and L. S. Beese, J. Lipid Res., 2006, 47, 681–699 CrossRef CAS.
- T. Scott Reid, K. L. Terry, P. J. Casey and L. S. Beese, J. Mol. Biol., 2004, 343, 417–433 CrossRef.
- T. C. Turek-Etienne, C. L. Strickland and M. D. Distefano, Biochemistry, 2003, 42, 3716–3724 CrossRef CAS.
- Y. Wang, J. K. Dozier, L. S. Beese and M. D. Distefano, ACS Chem. Biol., 2014, 9(8), 1726–1735 CrossRef CAS.
- J. L. Hougland, K. A. Hicks, H. L. Hartman, R. A. Kelly, T. J. Watt and C. A. Fierke, J. Mol. Biol., 2010, 395, 176–190 CrossRef CAS.
- E. Choy, V. K. Chiu, J. Silletti, M. Feoktistov, T. Morimoto, D. Michaelson, I. E. Ivanov and M. R. Philips, Cell, 1999, 98, 69–80 CrossRef CAS.
- Y. Zhang, M. J. Blanden, C. Sudheer, S. A. Gangopadhyay, M. Rashidian, J. L. Hougland and M. D. Distefano, Bioconjugate Chem., 2015, 26, 2542–2553 CrossRef CAS.
- B. P. Duckworth, J. Xu, T. A. Taton, A. Guo and M. D. Distefano, Bioconjugate Chem., 2006, 3, 967–974 CrossRef.
- M. Rashidian, S. C. Kumarapperuma, K. Gabrielse, A. Fegan, C. R. Wagner and M. D. Distefano, J. Am. Chem. Soc., 2013, 135, 16388–16396 CrossRef CAS.
- T. J. Kean, L. Duesler, R. G. Young, A. Dadabayev, A. Olenyik, M. Penn, J. Wagner, D. J. Fink, A. I. Caplan and J. E. Dennis, J. Drug Targeting, 2012, 20, 23–32 CrossRef.
- J. De Kruif, M. Tijmensen, J. Goldsein and T. Logtenberg, Nat. Med., 2000, 6, 223–227 CrossRef CAS.
- S. L. Emanuel, L. J. Engle, G. Chao, R. R. Zhu, C. Cao, Z. Lin, A. Yamniuk, J. Hosbach, J. Brown, E. Fitzpatrick, J. Gokemeijer, P. Morin, B. Morse, I. M. Carvajal, D. Fabrizio, M. C. Wright, R. Das Gupta, M. Gosselin, D. Cataldo, R. P. Ryseck, M. L. Doyle, T. W. Wong, R. T. Camphausen, S. T. Cload, H. N. Marsh, M. M. Gottardis and E. S. Furfine, mAbs, 2011, 3, 38–48 CrossRef.
- O. Kilic, M. R. de Souza, A. A. Almotlak, Y. Wang, J. M. Siegfried, M. D. Distefano and C. R. Wagner, J. Med. Chem., 2020, 63, 10235–10245 CrossRef CAS.
- I. A. Prior, A. Harding, J. Yan, J. Sluimer, R. G. Parton and J. F. Hancock, Nat. Cell Biol., 2001, 3, 368–375 CrossRef CAS.
- C. M. Csizmar, J. R. Petersburg, T. J. Perry, L. Rozumalski, B. J. Hackel and C. R. Wagner, J. Am. Chem. Soc., 2019, 141, 251–261 CrossRef CAS.
- R. Fragoso, D. Ren, X. Zhang, M. W.-C. Su, S. J. Burakoff and Y.-J. Jin, J. Immunol., 2003, 170, 913–921 CrossRef CAS.
- A. Larbi, G. Dupuis, A. Khalil, N. Douziech, C. Fortin and T. Fülöp, Cell. Signalling, 2006, 18, 1017–1030 CrossRef CAS.
- K. Fousek, J. Watanabe, S. K. Joseph, A. George, X. An, T. T. Byrd, J. S. Morris, A. Luong, M. A. Martínez-Paniagua, K. Sanber, S. A. Navai, A. Z. Gad, V. S. Salsman, P. R. Mathew, H. N. Kim, D. L. Wagner, L. Brunetti, A. Jang, M. L. Baker, N. Varadarajan, M. Hegde, Y. M. Kim, N. Heisterkamp, H. Abdel-Azim and N. Ahmed, Leukemia, 2020 DOI:10.1038/s41375-020-0792-2.
- A. J. Davenport, R. S. Cross, K. A. Watson, Y. Liao, W. Shi, H. M. Prince, P. A. Beavis, J. A. Trapani, M. H. Kershaw, D. S. Ritchie, P. K. Darcy, P. J. Neeson and M. R. Jenkins, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2068–E2076 CrossRef CAS.
- S. Offner, R. Hofmeister, A. Romaniuk, P. Kufer and P. A. Baeuerle, Mol. Immunol., 2006, 43, 763–771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03194a |
| This journal is © The Royal Society of Chemistry 2021 |