
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion binding to ubiquitin and its relevance to the Hofmeister effects†
Wei
Yao
 a,
Kaiyu
Wang
a,
Aide
Wu
b,
Wayne F.
Reed
b and
Bruce C.
Gibb
*a
a,
Kaiyu
Wang
a,
Aide
Wu
b,
Wayne F.
Reed
b and
Bruce C.
Gibb
*a
aDepartment of Chemistry, Tulane University, New Orleans, LA 70118, USA. E-mail: bgibb@tulane.edu
bDepartment of Physics and Engineering Physics, Tulane University, New Orleans, LA 70118, USA
First published on 4th November 2020
Abstract
Although the non-covalent interactions between proteins and salts contributing to the Hofmeister effects have been generally mapped, there are many questions regarding the specifics of these interactions. We report here studies involving the small protein ubiquitin and salts of polarizable anions. These studies reveal a complex interplay between the reverse Hofmeister effect at low pH, the salting-in Hofmeister effect at higher pH, and six anion binding sites in ubiquitin at the root of these phenomena. These sites are all located at protuberances of preorganized secondary structure, and although stronger at low pH, are still apparent when ubiquitin possesses no net charge. These results demonstrate the traceability of these Hofmeister phenomena and suggest new strategies for understanding the supramolecular properties of proteins.
Introduction
The supramolecular properties of proteins – how they interact non-covalently with other species in solution – are not fully understood. In particular, although the non-covalent interactions of salts with proteins that likely contribute to the Hofmeister effects1,2 have been mapped at a general level, there are countless open questions regarding the nature and specifics of all the interactions involved.3–9 Consider first, the charges on a protein. These can form coulombic interactions with co-solute ions, but the asymmetry in anion and cation solvation5,10 leads to stronger hydration free energies for negatively-charged groups on proteins than their cationic counterparts.11 This asymmetry means that negatively-charged proteins have greater solubility,12,13 which may explain why negatively-charged clusters of residues are common but positively-charged ones rare,14 and why the pI values of proteomes across all kingdoms of life are biased below 7.15 Nature prefers negativity.This asymmetry is exacerbated when salts are added to the milieu. Common laboratory salts frequently include weakly solvated anions (e.g., I−, SCN−, ClO4−) which can closely interact with weakly solvated cationic residues. In contrast, commonly utilized salt cations (Li+, Na+, K+) are strongly solvated,5 and therefore cannot interact strongly with the strongly solvated anionic residues. At least coulombically then, anions manifest Hofmeister effects in proteins more so than cations.8 Hence the reverse Hofmeister effect – the phenomenon whereby polarizable anions precipitate proteins – is most frequently investigated with basic proteins at pH values below their isoelectric point (pI),16–23 particularly lysozyme8,19,24–31 and amyloidogenic peptides and proteins;32–38 studying proteins at pH values below their pI value maximizes charge–charge interactions, the resulting attenuation of charge, and the salting-out phenomena this can induce. (See ref. 1 for a discussion of Hofmeister and reverse Hofmeister effects.)
Salts can of course interact with proteins through other non-covalent interactions.6,8,39–42 Amides are good hydrogen bond donors,43 and it is understood that in the solid-state, arrays of amide NH and CαH donors on protein surfaces can bind anions.44,45 Correspondingly, in the solution-state anion binding to amide groups in polyamide models has been observed (Ka = 20 M−1), and found to be stronger than cation binding to amide carbonyls in small models.46–48 However, whether selective anion binding to amide groups in proteins occurs in solution, and the degree to which this might be responsible for salting-in and reverse Hofmeister effects, is not clear. Further to hydrogen bonds, there is also evidence that polarizable anions bind to non-polar surfaces, and such non-coulombic interactions have also been proposed to be a component of salting-in Hofmeister effects.6,8,39,40,49–52
There are other potential non-covalent interactions to consider. For example cation–π interactions play a significant role in protein chemistry,53,54 but the general Hofmeister effects induced by the cations of typically explored salts is much weaker than that of anions. All of these possibilities noted, despite anion binding to proteins being first proposed in 1949,55 and models attempting to consider more than just coulombic forces between proteins and ions,7–9 there are still major difficulties parsing out all the non-covalent contributions to the Hofmeister effects writ large. Part of the difficulty here is simply the innate complexity of proteins. Additionally though, it has also been more common to examine general Hofmeister effects using indirect approaches, such as changes to catalytic rates, displacement assays with fluorescent dyes, or using macro-scale dependent variables such as surface tension, aggregation rates, solubility, or chromatographic retention.56 These strategies are not geared towards identifying the individual non-covalent interactions between water, salt, and protein contributing to all of the Hofmeister effects. Identifying these should however bring a wealth of information as to how salts affect amyloidogenesis,32–38 how buffers interact with proteins,56–59 how protein crystallization strategies can be improved,60–62 and more generally, how salts in the biological milieu affect proteins.
To examine for specific non-covalent interactions between proteins and ions from an added salt, we have examined the small protein ubiquitin (Ub, Mw = 8565 Da) in the presence of the sodium salts of a series of anions; from chloride at the middle of the Hofmeister series to weakly solvated anions such as hexafluorophosphate (PF6−). Specifically, we focus here primarily on the salt effects upon Ub in acidic to neutral media using Static Light Scattering (SLS), Differential Scanning Calorimetry (DSC), and 1H–15N HSQC Nuclear Magnetic Resonance spectroscopy (HSQC NMR). Ub was itself selected because its small size was expected to help identify specific non-covalent interactions between proteins and the ions, and because it is exceptionally well characterized by X-ray crystallography63 and NMR spectroscopy.64,65 Thus, Ub comprises one α-helix, two short 310 helices, a mixed five-strand β-sheet, and nine reverse turns (vide infra, Fig. 4). Moreover, although consisting of only 76 residues, Ub is extremely stable and highly resistant to chemical and thermal denaturation (melting temperature, Tm > 100 °C at pH = 7).66 As a host (receptor) therefore, Ub can be considered to be relatively preorganized and stable. Finally, Ub was also selected because it has no known metal binding sites, its normal role in biology67–70 is not as an anion binder, and because it is acidic rather than basic (isoelectric point, pI ∼ 6.5 to 6.9).71 We also extended the traditional anions used in Hofmeister studies to include anions such as ReO4− and PF6− because these have been shown to interact strongly not only with positively charged models, but negatively charged ones as well.52,72 The results describe here underscore three points. First, at low pH, interactions between weakly solvated anions of salts and the weakly solvated cationic groups of a protein correlate with the reverse Hofmeister effect, i.e., the ability of these anions to induce the precipitation of a protein. Second, at pH 5 where the net charge of Ub is close to zero these same charge-diffuse ions induce a weak salting-in Hofmeister effect. Third, in the case of Ub these ion–protein interactions involve specific anion binding sites. In combination our data suggest the importance of such specific interactions in contributing to these two Hofmeister phenomena.
Results and discussion
Static light scattering
Using a model host, we previously showed that polarizable anions could induce hallmarks of both the salting-in Hofmeister and reverse Hofmeister effects.72 The former came about because polarizable anions have an affinity for the non-polar pocket of the host, an affinity that weakened the apparent association constant of non-polar guests binding to the pocket. The latter was engendered by concomitant association with the positively charged groups on the exterior of the host, and in particular a second, charge-rich, binding site. As a result, simultaneous to the weakening of guest binding to the non-polar pocket, anion ‘accumulation’ to the outside of the host led to charge attenuation, neutralization, and precipitation.To explore the reverse Hofmeister effect in Ub, we employed SLS to probe the irreversible, thermally-induced denaturation and aggregation of Ub with the sodium salts of PF6−, ReO4−, ClO4−, TfO−, NO3−, Br−, Cl− and MeSO3− across the pH range from 2.3–5.73 The results at three pH values are shown in Fig. 1a–c (see also ESI, Fig. S9–S27†). At pH = 2.3 Ub (pI ≈ 6.8) has a net charge of approximately +13,71 and underwent no aggregation in the absence of salt (as was the case at all pH values examined). However, in the presence of 1 M salt anion binding and the resulting charge-attenuation and aggregation was extensive.73 Thus, ReO4− caused the instant precipitation of Ub, whilst the other anions gave (condition specific) Tagg values ranging from 29 °C for PF6− to 81 °C for MeSO3−. The power of ReO4− to induce precipitation was evident by decreasing its concentration one order of magnitude; a low Tagg value of 40 °C was recorded at 100 mM ReO4−. Overall there was considerable similarity between the order of precipitation of Ub and previously studied, positively charged models.72
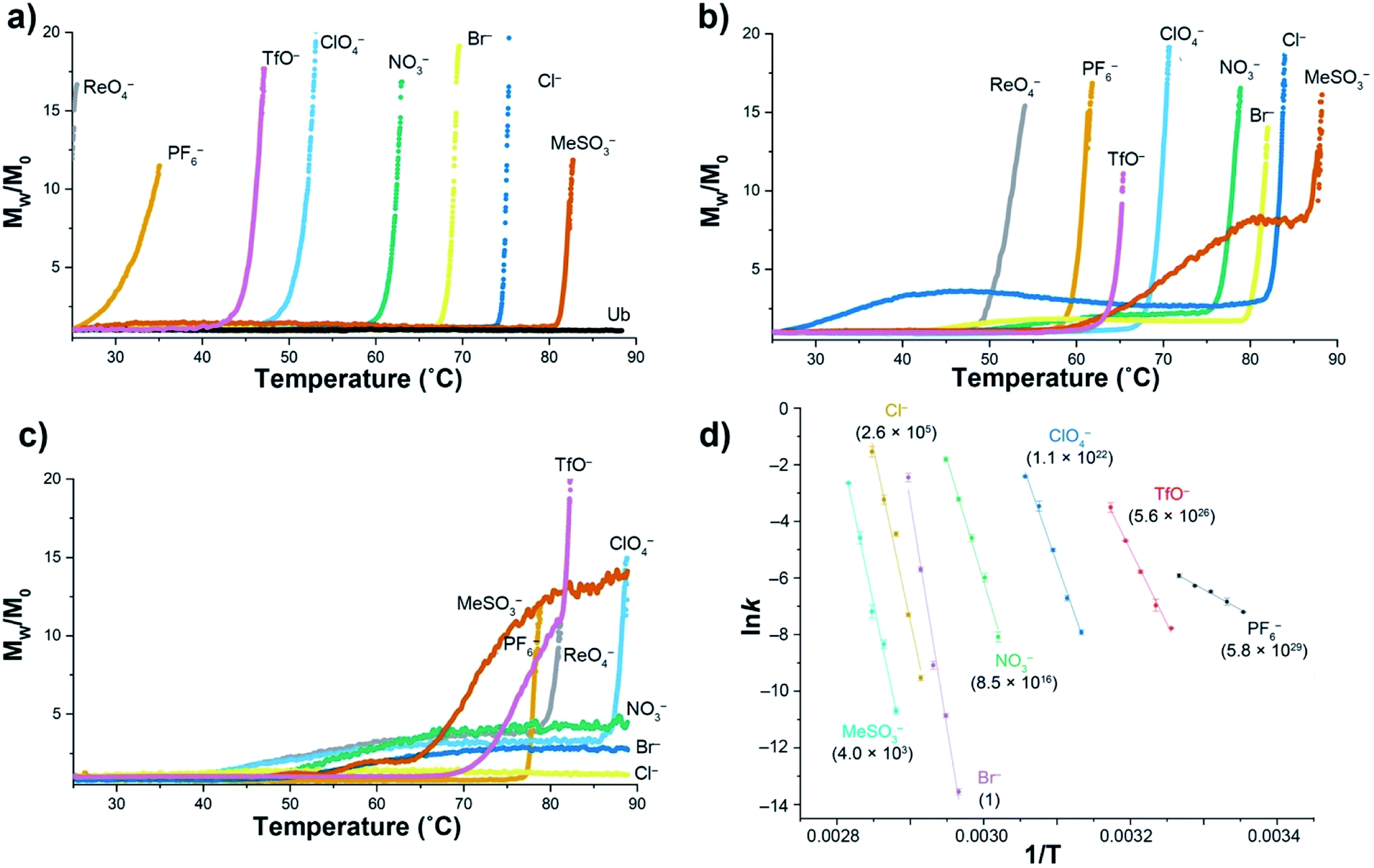 | ||
| Fig. 1 SLS data for the thermally induced aggregation of Ub (1 mg mL−1, 117 μM) in the presence of 10 mM phosphate buffer and 1 M sodium salts in H2O. Figures (a), (b) and (c) show temperature ramp data (25 to 90 °C, over 7 h) for three pH values: 2.3, 3.7, and 5.0 respectively, and express aggregation as the normalized, average scattering intensity Mw/M0. As no precipitation of Ub was observed in the absence of added salt, data is only shown at pH 2.3 (Ub). Figure (d) shows the corresponding Arrhenius plots for the aggregation of Ub in the presence of seven salts at pH = 2.3, with data collected at constant temperature between 25 and 82 °C depending on the salt. Relative aggregation rate constants at 298 K (krel) are shown in parenthesis. Data collection and handling is fully described in the ESI.† | ||
At pH 3.7 (charge ≈ +10), the ability of each anion to bind to Ub and induce aggregation decreased. Thus, the Tagg values ranged from 49 °C for ReO4− to 85 °C for MeSO3−. Interestingly, at this pH the Mw/M0 value of MeSO3− (and to a lesser extent Cl−) plateaus, suggesting the formation of one or more metastable aggregation states, but overall, the order of Tagg values followed that of pH 2.3. In contrast, at pH 5 (charge ≈ +3), the order of (attenuated) precipitating power changed, suggesting that ion selective sites on Ub are selectively switched off as the pH is raised. Focusing only on the well-behaved data (no plateau), the observed Tagg, values were: PF6− (77 °C), TfO− and ReO4− (78 °C), and ClO4− (87 °C). In contrast, little if any aggregation was observed for NO3−, Br−, or Cl−.
Confirming the conclusions from Fig. 1a–c, Fig. 1d shows Arrhenius plots for salt-induced aggregation at constant temperature (see ESI, Fig. S1–S8†). We found that relative to Br−, PF6− reduced the Ea for aggregation by an order of magnitude (Table S1†). Moreover, the relative aggregation rate constants at 298 K (krel) vary by nearly 30 orders of magnitude, with PF6− the fastest, and Br− the slowest at inducing precipitation.
The combined SLS data reveals how weakly solvated anions can induce aggregation of Ub. In general terms there are two possibilities at play here: changes on long-range charge screening, or anion binding via a mix of non-covalent interactions that lead to short range salt-bridges between positively charged groups, and protein charge attenuation. Charge screening can be well modeled classically (anions as point charges), so the fact that aggregation is anion specific strongly suggests specific binding to the surface of Ub. However, the extent to which the native fold of the protein is altered by this anion binding is unclear from this SLS data.
Differential scanning calorimetry
As measured by melting temperature (Tm), Ub is an exceedingly stable protein (Tm > 100 °C, pH 7) and unfolds reversibly and cooperatively.74 However, reducing the pH and protonating all the ionizable residues75 leads to considerable (coulombic) destabilization. Thus, in phosphate buffer DSC revealed Tm values of 99.4 °C at pH 5, but only 57.4 °C at pH 2.3 (errors < ±0.1). As previously observed, at low pH weak Cl− binding slightly increases Ub stability via salt-bridging.76To probe anion stabilization further, we examined the influence of the eight aforementioned anions at pH 2.3. Fig. 2a shows that all anions stabilized Ub, from between 5.2 (MeSO3−) and 11.8 °C (PF6−) at a low 50 mM concentration (see also ESI, Fig. S29–S37†). The plateauing of data in the case of, for example, ReO4− is evidence of anion binding to both the folded and unfolded state,77 whereas the absence of plateauing in much of the data indicates binding mostly to the folded state; the continued rise in Tm reflective of the additional free energy required to remove the ligand from the protein prior to its thermally induced unfolding, which is itself based largely on the entropy of mixing of the dissociated ligand.77 This data reveals that at pH = 2.3 the strongest precipitators as determined by SLS, for example ReO4− and PF6−, are the stronger binders to the unfolded state.
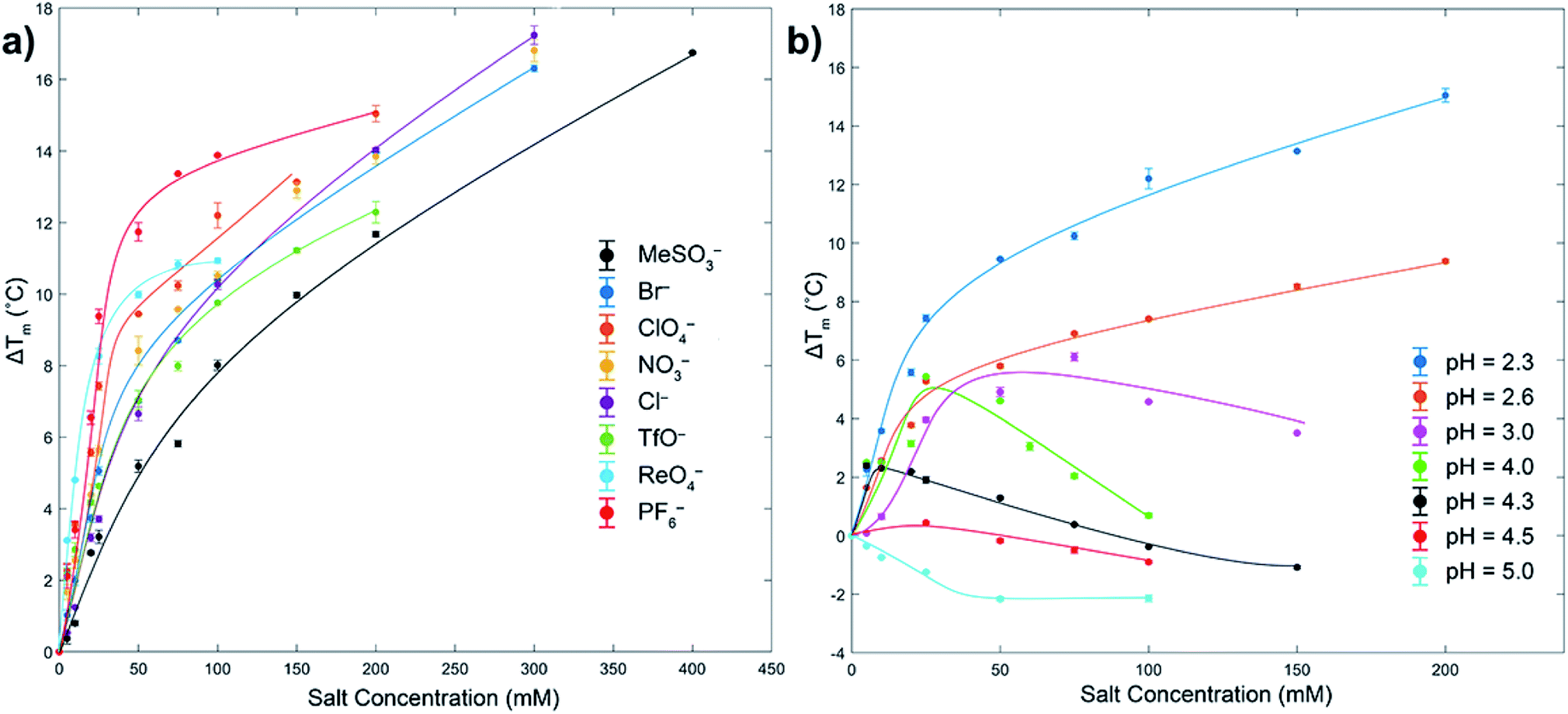 | ||
| Fig. 2 DSC melting temperature (Tm) data for Ub showing how anions stabilize or destabilize the protein (ΔTm positive or negative respectively). Figure (a) shows ΔTm at pH 2.3 for eight anions up to 400 mM, with ΔTm relative to the Tm of Ub in the absence of salt (57.4 °C). Figure (b) shows the effects of pH and ClO4− concentration on ΔTm. In both figures the shown error bars represent the variance in duplication or triplication of each run, whist the lines shown are only for guiding the eye. In figure (a), aggregation prevented data collection above the maximum salt concentration indicated. Similarly, for figure (b) aggregation prevented data collection above pH 5. All samples were 1 mg mL−1 or 117 μM, 10 mM phosphate buffer in H2O. See ESI.† | ||
We attribute binding to the unfolded state to the fact that polarizable anions can form multiple (non-coulombic) interactions with normally buried residues in the folded protein,6,8,39,40,49–52 including amide–anion hydrogen bonding and nonpolar surface-anion interactions arising from anion–dipole, anion-induced dipole, and van der Waals interactions. These can be surprisingly strong, allowing charge diffuse anions (with potentially some assistance from attendant, counter cations) to even bind to hosts possessing strongly negative electrostatic potential fields.50–52
To get some insight to the power of non-coulombic interactions to affect Ub, we carried out DSC studies with ClO4− at different pH values. As Fig. 2b reveals (see also ESI, Fig. S38–S44†), as the pH was increased from 2.3 to 5 (charge ≈ +13 to +3), so there is a transition from ClO4− stabilization of Ub and preferential anion binding to the folded state (no plateau in data), to a destabilization or salting-in of Ub arising from ClO4− binding to both the folded and unfolded state (plateauing of data). This salting-in Hofmeister effect – how charge diffuse anions can destabilize the fold of a protein – is even evident at pH 12. Thus, whereas the addition of NaCl or NaClO4 to Ub at this pH causes a slight stabilization of the protein attributed to Na+ ions non-specifically binding to the surface and shielding the negative charges of the protein, addition of NaReO4 actually causes a slight decrease in stability (Fig. S45†); even with a net charge of −11, ReO4− (or arguably ReO4− and an associating Na+ counter ion) can weakly associate with Ub. Returning to Fig. 2b, the counterposing interactions at play here are most evident with the non-monotonic trends at intermediate pH. Thus, increasing ΔTm values at low anion concentrations correspond to large stabilizing contributions from coulombic interactions (salt-bridges), but at higher concentrations weaker and counteracting non-coulombic forces involving the anions and nonpolar portions of the protein become more prominent and ΔTm decreases. These DSC results demonstrate that although polarizable anions have only a limited capacity to induce aggregation at pH = 5 (vide supra, SLS data), they do nevertheless still bind to the protein. With Ub only possessing a charge of +3 at this pH, this suggests anion binding does not (necessarily) involve salt-bridging, but that binding may be quite remote from the few positively charged groups present.
The combined SLS and DSC data suggest that for the stronger binding anions, aggregation of Ub is a combination of anion binding to the folded state and the attendant charge attenuation, and binding to the unfolded state which both attenuates charge and exposes non-polar residues to the aqueous medium. The balance between these two phenomena, as well as the degree to which these phenomena are present in weakly binding anions, is however unclear.
1H–15N HSQC NMR spectroscopy studies
Although there is a plethora of anion-binding proteins with highly specific binding sites, there are many open questions regarding the degree of specificity of anion binding responsible for general Hofmeister effects. Do solubilized proteins possess anion binding sites akin to Nests44 and CαNN motifs45 that can control salting-in (or reverse) Hofmeister phenomena? To address this question, we carried out titrations utilizing 1H–15N HSQC NMR spectroscopy. As per previous studies,78,79 seventy N–H signals were apparent from these 2D NMR experiments. Absent were signals from: M1 (no amide group), the proline residues P19, P37 and P37 (no amide N–H), and residues E24 and G53 (typically not observed in the 1H–15N HSQC NMR experiment). In each titration Ub (1 mM) was dissolved in 50 mM phosphate buffer of the required pH, and titrated from 0 mM salt up to between 300–800 mM. Regardless of the nature of the anion, most N–H signals underwent small linear shifts, whereas some residues underwent more significant, non-linear shifts. In these cases, we defined an anion binding site from a cluster of local residues whose N–H signal underwent the largest (top 20%) shift, i.e., a Δδ value greater than 0.18 ppm (Fig. S69†). In such cases we used the Δδ shifts as a function of the protein–salt ratios to fit to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model using non-linear regression analysis. Reproducibility of affinity data was confirmed by comparison between similar pH values rather than repetition at individual pH values.80 These studies used the sodium salts of Cl−, NO3−, TfO−, ClO4−, ReO4−, and PF6−, examining affinity first at pH = 2.8 (Ub charge ≈ +12). Full details are given in the ESI (Fig. S49–S101†). For brevity our discussion is focused on the binding of ClO4− at pH = 2.8.
:1 binding model using non-linear regression analysis. Reproducibility of affinity data was confirmed by comparison between similar pH values rather than repetition at individual pH values.80 These studies used the sodium salts of Cl−, NO3−, TfO−, ClO4−, ReO4−, and PF6−, examining affinity first at pH = 2.8 (Ub charge ≈ +12). Full details are given in the ESI (Fig. S49–S101†). For brevity our discussion is focused on the binding of ClO4− at pH = 2.8.
The X-ray structure reveals that one third of Ub is comprised of a five-strand, mixed β-sheet (vide infra, Fig. 4).63 We identified two binding sites, Sites 1 and 4, at protruding β-turns (Fig. 3a and d and 4). The former is centered on the NH of L8 and includes a β-bulge (T7, G10, and K11), whilst the latter involves residues F45 to K48. Both turns possess free and/or weakly hydrogen-bonded NH groups and constitute polydentate NH and CαH arrays capable of forming Nest/CαNN hybrid sites.44,45 ΔG° for ClO4− binding was in the region of 7.5–8.3 kJ mol−1 (Table 1).
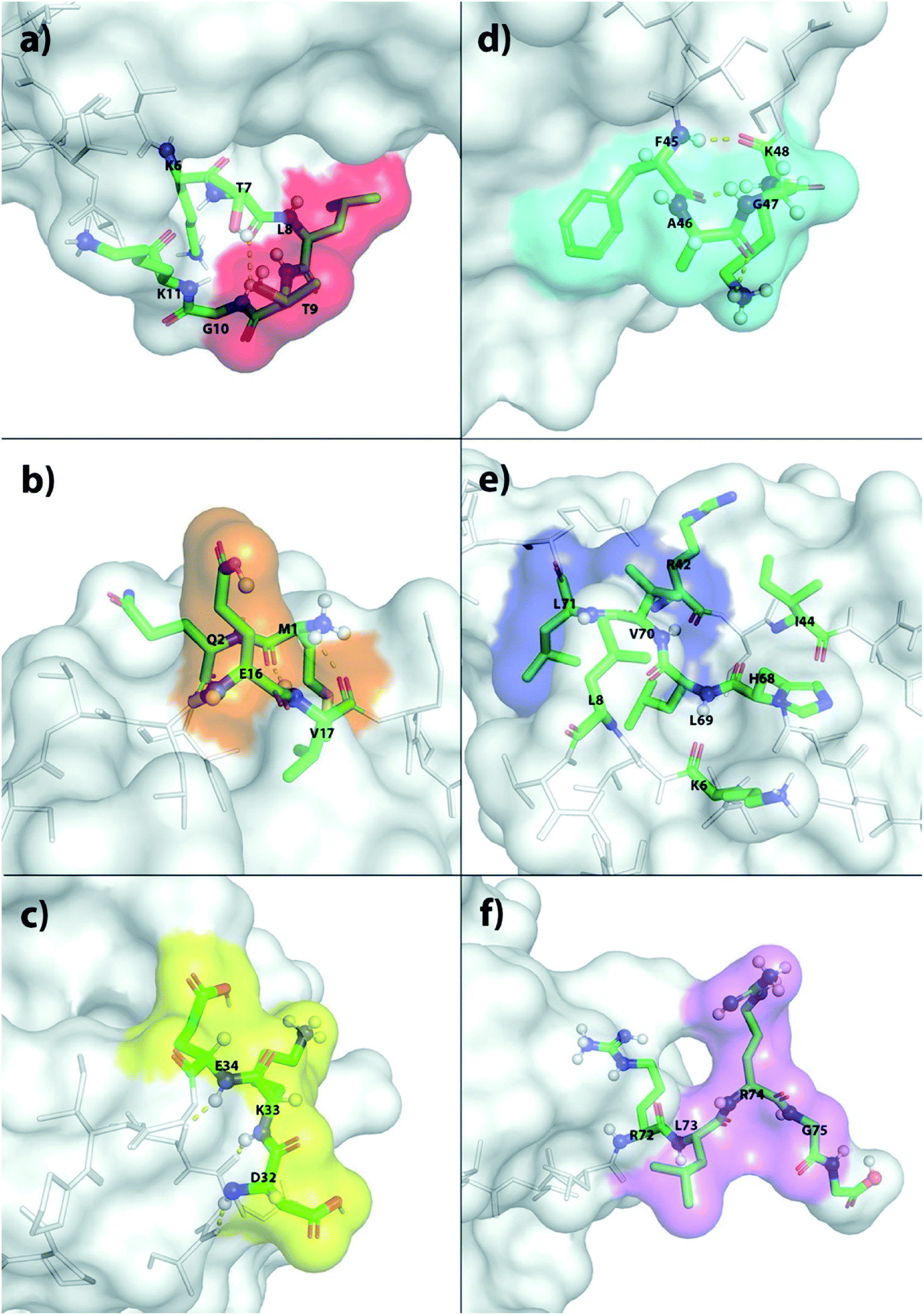 | ||
| Fig. 3 The six primary anion binding sites in Ub as determined by 1H–15N HSQC NMR. (a) Sites 1–6 are shown in (a)–(f) respectively. In each case, the primary binding residues are highlighted using color surface plots. Additionally, the atoms of the primary residues, and the surrounding residues presumed key to anion affinity, are highlighted using colored tubes. Protons undergoing large Δδ shifts, as well as those presumed involved in key non-covalent interactions, are depicted as spheres. | ||
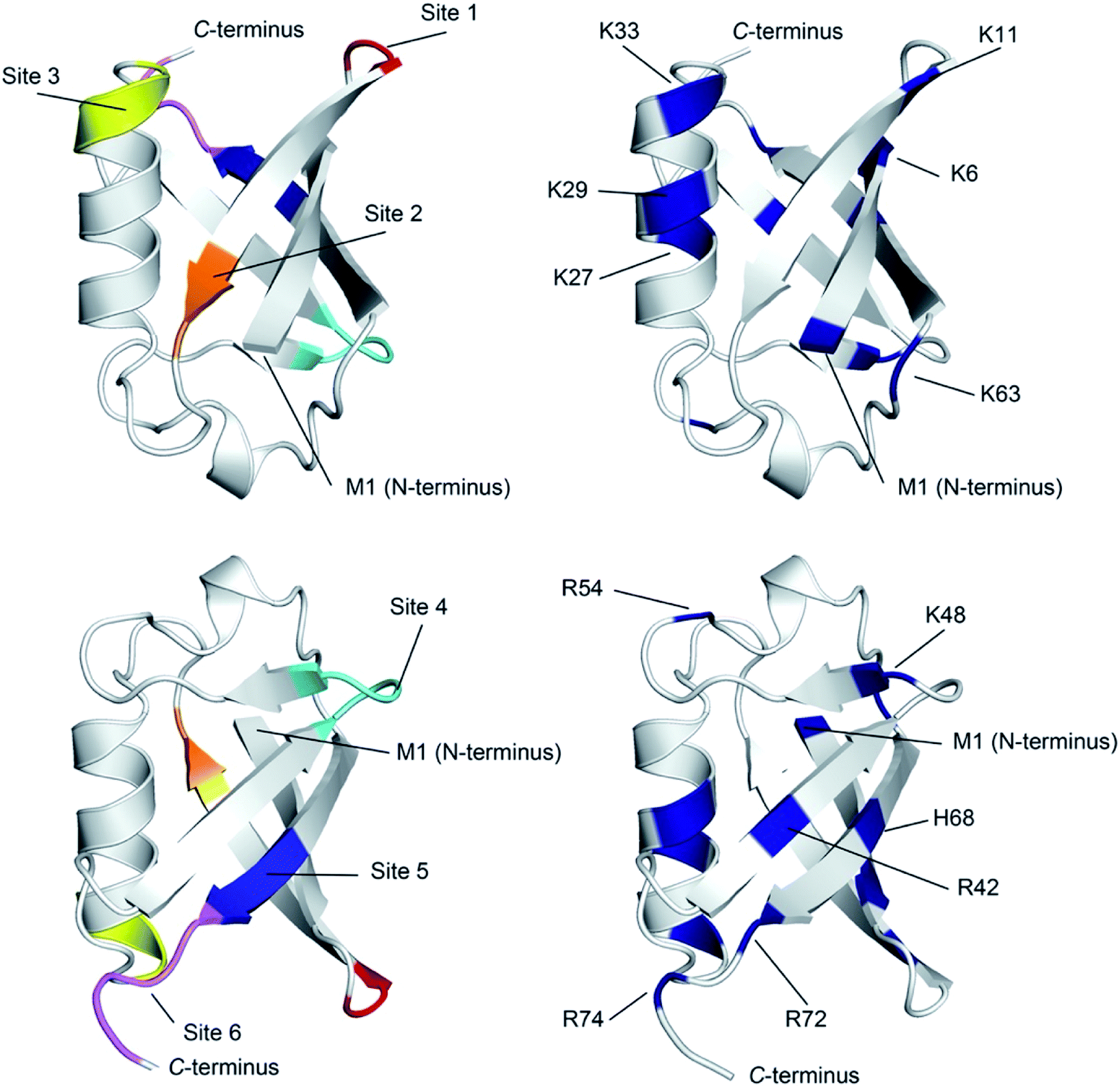 | ||
| Fig. 4 Ribbon diagram representations of (left) the six primary anion binding sites in Ub, and (right) the locations of the positive charged groups in Ub (N-terminus, H, K and R residues). In both cases, the lower figure differs from the upper one by a 180° flip around a horizontal axis in the plane of the media. For the structures showing the binding sites, each site is color coded as Fig. 3. The less structured mainchain of Ub between E18–I23 and Q49–H68 is represented by the lower 1/3 of structure in the top pair of ribbon diagrams. | ||
| Site 1 | Site 2 | Site 3 | Site 4 | Site 5 | Site 6 | |
|---|---|---|---|---|---|---|
| a Average of all the individual Ka determinations from the NH donors in each binding site. For precise NH signals used and attendant errors, see ESI. | ||||||
| Cl− | 7 | 6 | 10 | 9 | 7 | 13 |
| NO3− | 10 | 4 | 7 | 13 | 17 | 11 |
| TfO− | 24 | 4 | 9 | 11 | 10 | 12 |
| ClO4− | 29 | 9 | 14 | 21 | 20 | 31 |
| PF6− | 26 | 3 | 14 | 18 | 12 | 17 |
| ReO4− | 54 | 19 | 21 | 36 | 61 | 36 |
Sites 2 and 5 were identified on the frayed edges of β-sheet (Fig. 3b, e and 4). Located at the C-terminal end of strand 2, Site 2 involves E16 and V17, whereas Site 5 is centered around L69. Again, free and/or weakly hydrogen bonded NH groups63 appear key. It may be the case that the weaker affinity at Site 2 (5.4 kJ mol−1) arises because of the singular proximal positive charge; Site 5 is surrounded by three positively charged residues (K6, R42, and H68) and has a higher affinity (7.5 kJ mol−1).
Being located at the C-terminal of the α-helix, Site 3 (Fig. 3c) is unique. Here the NH signals of residues D32–E34 undergo large, non-linear shifts, but pointing towards the center of the protein, are hydrogen-bonded to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of A28–I30. We therefore attribute the observed shifts to an anomeric effect arising from anion binding to the three CαH methines of D32–E34 on the surface of the protein. Perhaps not surprisingly, like Site 2, anion affinity was relatively weak.
O groups of A28–I30. We therefore attribute the observed shifts to an anomeric effect arising from anion binding to the three CαH methines of D32–E34 on the surface of the protein. Perhaps not surprisingly, like Site 2, anion affinity was relatively weak.
Finally, Site 6 is located at the C-terminus (Fig. 3f) and involves residues L73, R74 and G75. Residue L73 is sandwiched (Fig. 4) between positively charged R72 and R74, and the determined affinity the strongest for a single NH group; Ka for ClO4− = 66 M−1. However, weaker affinity determinations from R74 and G75 attenuated the overall affinity, and suggest that the flexibility of the C-terminus may limit its ability to contribute to binding.
This overall pattern of six binding sites is seen for all anions examined, but binding is more pronounced and stronger for the largest, charged-diffuse ions (Table 1). Although the strongest precipitators ReO4− and PF6− are also the strongest binders, there is little evidence they bind strongly to any additional site (ESI Section 5.4†).
There are several conclusions that can be made about the six sites (Fig. 4). First, all possess multiple NH and/or CαH donors and are located at protuberances on the protein surface. Second, all are part of well-defined secondary structure rather than disordered loops. Third, sites 1, 3, 4 and 6 all have G residues in (or directly adjacent to) the binding site, providing an extra CαH donor and greater access to other donors in the site. Fourth, all binding sites are proximal to 1–3 positively charged residues.
Where is binding not observed? Binding is not observed to the less structured mainchain of Ub between E18–I23 and Q49–H68 (lower 1/3 of structure in top two ribbon diagrams in Fig. 4). This suggest that some level of preorganization is needed for anion binding. Coincidently however, this section of Ub also possesses little in the way of positively charged residues (R54 and K63). Thus, in Ub it is not clear if this region fails to bind anions because of the level of preorganization, or because this part of the protein possesses a relatively weak electrostatic potential field. Regardless, this area where binding is not observed forms a ∼270° belt that runs from Site 1 to Site 6 along and around the long axis of the protein (Fig. S76†).
We also examined ClO4− binding as a function of pH, examining affinity at pH 3.8, 4.8, 5.8 and 7.3. As anticipated, affinity decreased as the net positive charge decreased. Thus, as a function of increasing pH both the number of residues that underwent a significant Δδ shift and the calculated Ka values decreased. Unfortunately it was not possible to observe site-specific pH dependencies (Fig. S97†), but rather collectively Ka values decreased sharply between pH 3.8 and 4.8, corresponding to the range where half or more of the aspartic and glutamic acid residues become deprotonated.75 Importantly though, despite little net charge at pH 7.3, and despite the titrations only extending to 400 mM NaClO4, all of the NH signal movements at the six sites still possessed significant curvature that fitted a 1:1 binding model (Fig. S98–S101†). Hence, association constants for ClO4− of up to 15 M−1 (3.4 kJ mol−1) could still be reliably calculated for the different binding sites of neutral Ub.
Conclusions
Despite working within a relatively narrow window of salt concentrations to avoid protein aggregation and precipitation, the 2D NMR spectroscopy titration studies have revealed six anion-binding sites on major protuberances of Ub. Binding constants for sizable polarizable anions such as ReO4− or ClO4− range from 61 M−1 (5.1 kJ mol−1) to 15 M−1 (3.4 kJ mol−1) where the net charge on Ub is approximately +13 and 0 respectively. No significant binding is observed on the disordered loops of the protein, only at exposed, preorganized secondary structure.What is the effect of binding polarizable, charge-diffuse anions at these sites? At low pH, DSC reveals that anion binding enhances the stability of Ub by partial screening of the positive charges on the protein. In this regard, the more strongly binding anions are, as might be expected, much better at stabilizing the protein fold at a given concentration. At higher concentrations though (but as low as 100 mM in the case of ReO4−), SLS reveals that anion binding induces sufficient charge attenuation for aggregation and precipitation (the reverse Hofmeister effect).
Importantly, DSC also reveals that at pH = 3 there is a counterposing effect at play that leads to the destabilization (salting-in) of Ub at higher salt concentrations. Moreover, as the coulombic attraction between anion and protein is switched off by raising the pH further, so this salting-in Hofmeister effect comes to the fore. Indeed, even at pH = 12, where Ub has a formal charge of −11, polarizable anions (potentially paired with their counter cations) still bind and destabilize the protein. NMR spectroscopy and DSC data both suggest that this salting-in phenomenon is tied to anion binding to the folded and the unfolded state. Thus, we envision that this destabilization is a combination of amide–anion hydrogen bonding, anion–diploe, anion-induced dipole, and van der Waals interactions that are independent of pH and can compete with the normal intramolecular forces holding the protein together. There is no reason to suppose that these interactions are not present at low pH. However, under these conditions the stronger coulombic forces responsible for the fold stabilization and the observed reverse Hofmeister effect are too dominant for them to be apparent.
Based on these findings and our work with model compounds,50–52,72Table 2 summarizes our current viewpoint of the relationships between the different, classical Hofmeister effects and potential contributing non-covalent interactions involving those between ions and water, and those between ions and proteins. It is yet to be ascertained the extent to which the anion–protein interactions observed here are responsible for the salting-in and reverse Hofmeister effects; protein–water and ion–water interactions also likely play a role; especially with highly concentrated solutions of strongly solvated ions that induce the salting-out Hofmeister effect.
| Observation | Traditional Hofmeister designation | Principle non-covalent interactions involved |
|---|---|---|
| a Protein–water interactions also likely play a significant role in the classic Hofmeister effects. | ||
| High concentrations of highly solvated anions induce precipitation of a protein | Salting-out Hofmeister effect | Anion–water (ion–dipole interactions). Salts out-compete a protein for water |
| Charge-diffuse anions induce the destabilization and solubilization of a protein | Salting-in Hofmeister effect | Ion–amide NH hydrogen bonding (HB), ion–dipole, anion-induced-dipole, and van der Waals (vdW) interactions |
| Charge-diffuse anions induce the stabilization and precipitation of a protein | Reverse Hofmeister effect | Coulombic (plus HB, ion–dipole, anion-induced-dipole and vdW) interactions |
In conclusion, many open questions remain concerning the complex interplay between ion–water, ion–protein, and protein–water interactions that engender the Hofmeister effects. As we demonstrate here however, ‘stacking the deck’ by selecting a small protein and charge-diffuse ions allows specific anion–protein interactions to be pin-pointed. Our understanding is that mapping such interactions will be of considerable utility in addressing the aforementioned open questions, and towards that we will report on further studies of the supramolecular properties of ubiquitin in due course.
Contributions
WY was the lead researcher on the project. KW carried out substantial portions of the described DSC and NMR studies and led the generation of the protein images used in the paper. AW provided instrument guidance during the initial SLS studies, whilst WR provided guidance for the described SLS experiments. BCG conceived and directed the research and contributed to the data analysis and manuscript preparation.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors wish to express their sincere gratitude to the National Institutes of Health for financial support of this work (GM 125690). AW and WR acknowledge support from Tulane’s PolyRMC.References
- The term “Hofmeister effect” tends to be used deus ex machina, and therefore means different things to different people. Here, our definition of “Hofmeister effects” (plural) is as general as possible: namely water or aqueous solution phenomena that arise and are dependent on the nature and concentration of co-solute salts. One common demarcation within this general definition are the “traditional Hofmeister effect” and the “reverse (or inverse) Hofmeister effect”. The former is comprised of two facets: first, charge-dense ions can salt-out (make less soluble or precipitate) molecules and biomacromolecules from water. This is classically ascribed to competition for solvation between the ions of the salt and the protein. Second, charge-diffuse, polarizable ions salt-in (solubilize) co-solute species. The causes of this phenomenon may be manifold. In contrast, the “reverse Hofmeister effect” is only a partial reversal: this phenomenon pertains to charge-diffuse and polarizable ions inducing the salting-out (rather than salting-in) of co-solutes such as proteins. Again, the causes of this phenomenon may be manifold. Our view is that the etymology of this terminology is more based on history rather than on a rational attempt at clarification. Instead, as we describe here, we believe that a more illuminating approach to the whole problem of how salts affect the properties of co-solutes is to think in terms of the non-covalent interactions that can arise between water, salt and solute. This paper focuses on anion–protein interactions.
- For an English translation of the seminar papers of Franz Hofmeister see: W. Kunz, J. Henle and B. W. Ninham, Zur Lehre von der Wirkung der Salze (About the science of the effect of salts): Franz Hofmeister's historical papers, Curr. Opin. Colloid Interface Sci., 2004, 9(1–2), 19–37 CrossRef CAS.
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and Their Biological Functions, J. Phys. Chem. B, 2017, 121(9), 1997–2014 CrossRef CAS.
- P. Jungwirth and P. S. Cremer, Beyond Hofmeister, Nat. Chem., 2014, 6(4), 261–263 CrossRef CAS.
- K. D. Collins, Why continuum electrostatics theories cannot explain biological structure, polyelectrolytes or ionic strength effects in ion–protein interactions, Biophys. Chem., 2012, 167, 43–59 CrossRef.
- E. S. Courtenay, M. W. Capp and M. T. Record Jr, Thermodynamics of interactions of urea and guanidinium salts with protein surface: Relationship between solute effects on protein processes and changes in water-accessible surface area, Protein Sci., 2001, 10(12), 2485–2497 CrossRef CAS.
- A. Salis and B. W. Ninham, Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited, Chem. Soc. Rev., 2014, 43(21), 7358–7377 RSC.
- M. Boström, F. W. Tavares, S. Finet, F. Skouri-Panet, A. Tardieu and B. W. Ninham, Why forces between proteins follow different Hofmeister series for pH above and below pI, Biophys. Chem., 2005, 117(3), 217–224 CrossRef.
- D. F. Parsons, M. Bostrom, P. Lo Nostro and B. W. Ninham, Hofmeister effects: interplay of hydration, nonelectrostatic potentials, and ion size, Phys. Chem. Chem. Phys., 2011, 13(27), 12352–12367 RSC.
- G. Hummer, L. R. Pratt and A. E. García, Free Energy of Ionic Hydration, J. Phys. Chem., 1996, 100(4), 1206–1215 CrossRef CAS.
- Y. Marcus, Ion Properties, Marcel Dekker, New York, 1st edn, 1997 Search PubMed.
- R. M. Kramer, V. R. Shende, N. Motl, C. N. Pace and J. M. Scholtz, Toward a Molecular Understanding of Protein Solubility: Increased Negative Surface Charge Correlates with Increased Solubility, Biophys. J., 2012, 102(8), 1907–1915 CrossRef CAS.
- S. DasSarma and P. DasSarma, Halophiles and their enzymes: negativity put to good use, Curr. Opin. Microbiol., 2015, 25, 120–126 CrossRef CAS.
- Z. Y. Zhu and S. Karlin, Clusters of charged residues in protein three-dimensional structures, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(16), 8350–8355 CrossRef CAS.
- L. P. Kozlowski, Proteome-pI: proteome isoelectric point database, Nucleic Acids Res., 2016, 45(D1), D1112–D1116 CrossRef.
- O. A. Francisco, C. J. Clark, H. M. Glor and M. Khajehpour, Do soft anions promote protein denaturation through binding interactions? A case study using ribonuclease A, RSC Adv., 2019, 9(6), 3416–3428 RSC.
- C. H. I. Ramos and R. L. Baldwin, Sulfate anion stabilization of native ribonuclease A both by anion binding and by the Hofmeister effect, Protein Sci., 2002, 11(7), 1771–1778 CrossRef CAS.
- C. H. Henkels, J. C. Kurz, C. A. Fierke and T. G. Oas, Linked Folding and Anion Binding of the Bacillus subtilis Ribonuclease P Protein, Biochemistry, 2001, 40(9), 2777–2789 CrossRef CAS.
- G. I. Makhatadze and P. L. Privalov, Protein interactions with urea and guanidinium chloride: A calorimetric study, J. Mol. Biol., 1992, 226(2), 491–505 CrossRef CAS.
- J. W. Bye, N. J. Baxter, A. M. Hounslow, R. J. Falconer and M. P. Williamson, Molecular Mechanism for the Hofmeister Effect Derived from NMR and DSC Measurements on Barnase, ACS Omega, 2016, 1(4), 669–679 CrossRef CAS.
- E. Sedlák, L. Stagg and P. Wittung-Stafshede, Effect of Hofmeister ions on protein thermal stability: Roles of ion hydration and peptide groups?, Arch. Biochem. Biophys., 2008, 479(1), 69–73 CrossRef.
- G. Battistuzzi, M. Borsari, A. Ranieri and M. Sola, Effects of Specific Anion-Protein Binding on the Alkaline Transition of Cytochrome c, Arch. Biochem. Biophys., 2001, 386(1), 117–122 CrossRef CAS.
- C. Jolivalt, A. Bockmann, M. Ries-Kautt, A. Ducruix and E. Guittet, Characterization of the interaction between bovine pancreatic trypsin inhibitor and thiocyanate by NMR, Biophys. Chem., 1998, 71(2–3), 221–234 CrossRef CAS.
- Y. Zhang and P. S. Cremer, The inverse and direct Hofmeister series for lysozyme, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15249–15253 CrossRef CAS.
- Y. R. Gokarn, R. M. Fesinmeyer, A. Saluja, V. Razinkov, S. F. Chase, T. M. Laue and D. N. Brems, Effective charge measurements reveal selective and preferential accumulation of anions, but not cations, at the protein surface in dilute salt solutions, Protein Sci., 2011, 20(3), 580–587 CrossRef CAS.
- J. W. Bye and R. J. Falconer, Thermal stability of lysozyme as a function of ion concentration: A reappraisal of the relationship between the Hofmeister series and protein stability, Protein Sci., 2013, 22(11), 1563–1570 CrossRef CAS.
- J. W. Bye and R. J. Falconer, Three stages of lysozyme thermal stabilization by high and medium charge density anions, J. Phys. Chem. B, 2014, 118(16), 4282–4286 CrossRef CAS.
- A. Salis, F. Cugia, D. F. Parsons, B. W. Ninham and M. Monduzzi, Hofmeister series reversal for lysozyme by change in pH and salt concentration: insights from electrophoretic mobility measurements, Phys. Chem. Chem. Phys., 2012, 14(13), 4343–4346 RSC.
- M. Boström, D. F. Parsons, A. Salis, B. W. Ninham and M. Monduzzi, Possible Origin of the Inverse and Direct Hofmeister Series for Lysozyme at Low and High Salt Concentrations, Langmuir, 2011, 27(15), 9504–9511 CrossRef.
- R. A. Curtis, J. M. Prausnitz and H. W. Blanch, Protein-protein and protein-salt interactions in aqueous protein solutions containing concentrated electrolytes, Biotechnol. Bioeng., 1998, 57(1), 11–21 CrossRef CAS.
- M. M. Ries-Kautt and A. F. Ducruix, Relative effectiveness of various ions on the solubility and crystal growth of lysozyme, J. Biol. Chem., 1989, 264(2), 745–748 CAS.
- K. L. Stewart and S. E. Radford, Amyloid plaques beyond Aβ: a survey of the diverse modulators of amyloid aggregation, Biophys. Rev., 2017, 9(4), 405–419 CrossRef CAS.
- S. Jain and J. B. Udgaonkar, Salt-Induced Modulation of the Pathway of Amyloid Fibril Formation by the Mouse Prion Protein, Biochemistry, 2010, 49(35), 7615–7624 CrossRef CAS.
- D. Thirumalai, G. Reddy and J. E. Straub, Role of Water in Protein Aggregation and Amyloid Polymorphism, Acc. Chem. Res., 2012, 45(1), 83–92 CrossRef CAS.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Self-assembling peptide and protein amyloids: from structure to tailored function in nanotechnology, Chem. Soc. Rev., 2017, 46(15), 4661–4708 RSC.
- D. P. Raleigh, Guilt by Association: The Physical Chemistry and Biology of Protein Aggregation, J. Phys. Chem. Lett., 2014, 5(11), 2012–2014 CrossRef CAS.
- S. Campioni, B. Mannini, J. P. Lopez-Alonso, I. N. Shalova, A. Penco, E. Mulvihill, D. V. Laurents, A. Relini and F. Chiti, Salt anions promote the conversion of HypF-N into amyloid-like oligomers and modulate the structure of the oligomers and the monomeric precursor state, J. Mol. Biol., 2012, 424(3–4), 132–149 CrossRef CAS.
- P. J. Marek, V. Patsalo, D. F. Green and D. P. Raleigh, Ionic Strength Effects on Amyloid Formation by Amylin Are a Complicated Interplay among Debye Screening, Ion Selectivity, and Hofmeister Effects, Biochemistry, 2012, 51(43), 8478–8490 CrossRef CAS.
- M. T. Record Jr, E. Guinn, L. Pegram and M. Capp, Introductory lecture: interpreting and predicting Hofmeister salt ion and solute effects on biopolymer and model processes using the solute partitioning model, Faraday Discuss., 2013, 160, 9–44 RSC ; discussion 103–120.
- L. M. Pegram, T. Wendorffa, R. Erdmanna, I. Shkela, D. Bellissimoa, D. J. Felitskya and M. T. J. Record, Why Hofmeister effects of many salts favor protein folding but not DNA helix formation, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7716–7721 CrossRef CAS.
- X. Chen, T. Yang, S. Kataoka and P. S. Cremer, Specific Ion Effects on Interfacial Water Structure near Macromolecules, J. Am. Chem. Soc., 2007, 129(40), 12272–12279 CrossRef CAS.
- R. L. Baldwin, How Hofmeister Ion Interactions Affect Protein Stability, Biophys. J., 1996, 71, 2056–2063 CrossRef CAS.
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- J. D. Watson and E. J. Milner-White, A Novel Main-chain Anion-binding Site in Proteins: The Nest. A Particular Combination of f,c Values in Successive Residues Gives Rise to Anion-binding Sites That Occur Commonly And Are Found Often at Functionally Important Regions, J. Mol. Biol., 2002, 315, 171–182 CrossRef CAS.
- K. A. Denessiouk, M. S. Johnson and A. I. Denesyuk, Novel CαNN structural motif for protein recognition of phosphate ions, J. Mol. Biol., 2005, 345(3), 611–629 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, Specific Ion Effects on the Water Solubility of Macromolecules: PNIPAM and the Hofmeister Series, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- K. B. Rembert, J. Paterova, J. Heyda, C. Hilty, P. Jungwirth and P. S. Cremer, Molecular mechanisms of ion-specific effects on proteins, J. Am. Chem. Soc., 2012, 134(24), 10039–10046 CrossRef CAS.
- H. I. Okur, J. Kherb and P. S. Cremer, Cations bind only weakly to amides in aqueous solutions, J. Am. Chem. Soc., 2013, 135(13), 5062–5067 CrossRef CAS.
- K. D. Collins, The behavior of ions in water is controlled by their water affinity, Q. Rev. Biophys., 2019, 52, e11 CrossRef CAS.
- C. L. D. Gibb and B. C. Gibb, Anion binding to hydrophobic concavity is central to the salting-in effects of Hofmeister chaotropes, J. Am. Chem. Soc., 2011, 133(19), 7344–7347 CrossRef CAS.
- R. Carnagie, C. L. D. Gibb and B. C. Gibb, Anion Complexation and The Hofmeister Effect, Angew. Chem., Int. Ed., 2014, 53(43), 11498–11500 CrossRef.
- P. Sokkalingam, J. Shraberg, S. W. Rick and B. C. Gibb, Binding Hydrated Anions with Hydrophobic Pockets, J. Am. Chem. Soc., 2016, 138(1), 48–51 CrossRef CAS.
- A. L. Koenig and M. Waters, Cation-π Interactions in Biomolecular Recognition, in Monographs in Supramolecular Chemistry: Aromatic Interactions: Frontiers in Knowledge and Application, ed. D. W. Johnson and F. Hof, Royal Society of Chemistry, 2016, pp. 214–237 Search PubMed.
- K. D. Daze and F. Hof, The cation-pi interaction at protein-protein interaction interfaces: developing and learning from synthetic mimics of proteins that bind methylated lysines, Acc. Chem. Res., 2013, 46(4), 937–945 CrossRef CAS.
- G. Scatchard and E. S. Black, The Effect of Salts on the Isoionic and Isoelectric Points of Proteins, J. Phys. Colloid Chem., 1949, 53(1), 88–100 CrossRef CAS.
- A. Salis and M. Monduzzi, Not only pH. Specific buffer effects in biological systems, Curr. Opin. Colloid Interface Sci., 2016, 23, 1–9 CrossRef CAS.
- F. Cugia, M. Monduzzi, B. W. Ninham and A. Salis, Interplay of ion specificity, pH and buffers: insights from electrophoretic mobility and pH measurements of lysozyme solutions, RSC Adv., 2013, 3(17), 5882–5888 RSC.
- W. J. Ferguson, K. I. Braunschweiger, W. R. Braunschweiger, J. R. Smith, J. J. McCormick, C. C. Wasmann, N. P. Jarvis, D. H. Bell and N. E. Good, Hydrogen ion buffers for biological research, Anal. Biochem., 1980, 104(2), 300–310 CrossRef CAS.
- N. E. Good, G. D. Winget, W. Winter, T. N. Connolly, S. Izawa and R. M. M. Singh, Hydrogen Ion Buffers for Biological Research, Biochemistry, 1966, 5(2), 467–477 CrossRef CAS.
- A. McPherson and J. A. Gavira, Introduction to protein crystallization, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2014, 70(Pt 1), 2–20 CrossRef CAS.
- T. M. Bergfors, Protein Crystallization, in Protein Crystallization, ed. T. M. Bergfors, International University Line, 2nd edn, 2009, vol. 8, p. 504 Search PubMed.
- M. Riès-kautt and A. Ducruix, [3] Inferences drawn from physicochemical studies of crystallogenesis and precrystalline state, in Methods Enzymol., Academic Press, 1997, vol. 276, pp 23–59 Search PubMed.
- S. Vijay-Kumar, C. E. Bugg and W. J. Cook, Structure of Ubiquitin Refined at the 1.8 Å Resolution, Mol. Biol., 1987, 194, 531–544 CrossRef CAS.
- D. L. Di Stefano and A. J. Wand, Two-Dimensional 1H NMR Study of Human Ubiquitin: A Main Chain Directed Assignment and Structure Analysis, Biochemistry, 1987, 26, 7272–7281 CrossRef CAS.
- G. Cornilescu, J. L. Marquardt, M. Ottiger and A. Bax, Validation of Protein Structure from Anisotrpic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase, J. Am. Chem. Soc., 1998, 120, 6836–6837 CrossRef CAS.
- R. E. Lenkinski, D. M. Chen, J. D. Glickson and G. Goldstein, Nuclear Magnetic Resonance Studies of the Denaturation of Ubiquitin, Biochim. Biophys. Acta, 1977, 494, 126–130 CrossRef CAS.
- J.-M. Peters, J. R. Harris and D. Finley, Ubiquitin and the Biology of the Cell, Plenum Press, New York, 1998 Search PubMed.
- J. M. Argilés, F. J. López-Soriano and J. Pallarés-Trujillo, Ubiquitin and Disease, R. G. Landes Company, Austin, Texas, 1998 Search PubMed.
- J. Marx, Ubiquitin Lives Up To Its Name, Science, 2002, 297, 1792–1794 CrossRef.
- S. Jentsch and B. Haendler, The Ubiquitin System in Health and Disease, Springer-Verlag, 2009 Search PubMed.
- Note that measuring the isoelectric point of a protein requires the presence of a buffer which is assumed to not interact with the protein. Here, we use the Protein Calculator (http://protcalc.sourceforge.net/) which gave a pI value for Ub of 6.8, arising from: four arginine residues, seven lysine residues, one tyrosine, one histidine, six glutamic and five aspartic acids, as well as the N and C-termini. This value of 6.8 is however an approximation. As we show here, many anions interact specifically with Ub. Moreover, the phosphate buffers used also presumably have some affinity for the protein, particularly at low pH values where the major anionic form is the monovalent dihydrogen phosphate. Hence although for convenience we assume a pI of 6.8, we understand that the effective pI of Ub under most of the conditions studied, i.e., the ensemble average pI values of the different Ub-anion complexes, is lower than this. Hence the positive charge values of Ub quoted in the main text are all over estimations (for the SLS experiments, pH = 2.3 (charge = +12.8), 2.7 (+12.5), 3.0 (+12.1), 3.3 (+11.6), 4.0 (+9.0), 4.3 (+7.2), 4.6 (+5.3), 5.0 (+3.2); for the DSC experiments: 2.3 (+12.8), 2.6 (+12.6), 3.0 (+12.1), 4.0 (+9.0), 4.3 (+7.2), 4.5 (+5.9), 5.0 (+3.2), and; for the NMR experiments: 2.3 (+12.8), 2.8 (+12.4), 3.8 (+10.0), 4.8 (+4.1), 5.8 (+1.3), 7.3 (0.0)).
- J. H. Jordan, C. L. D. Gibb, A. Wishard, T. Pham and B. C. Gibb, Ion-Hydrocarbon and/or Ion-Ion Interactions: Direct and Reverse Hofmeister Effects in a Synthetic Host, J. Am. Chem. Soc., 2018, 140(11), 4092–4099 CrossRef CAS.
- It is understood that the extent of unfolding during aggregation is system dependent see: G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. Knowles, Nat. Protoc., 2016, 11(2), 252–272 CrossRef CAS; W. Wang and C. J. Roberts, Non-Arrhenius Protein Aggregation, AAPS J., 2013, 15(3), 840–851 CrossRef; W. Wang, S. Nema and D. Teagarden, Int. J. Pharm., 2010, 390, 89–99 CrossRef; A. M. Morris, M. A. Watzky and R. G. Finke, Biochim. Biophys. Acta, 2009, 1794, 375–397 CrossRef . For Ub at pH = 2.3 and 25 °C we saw no evidence of unfolding by Circular Dichroism (CD, Fig. S28†), but signal attenuation and precipitation occurred at high temperatures and salt concentrations. Thus, these CD studies only reveal that solubilized protein remains folded in the presence of salts. Relatedly, extrapolation of our DSC data (vide infra) suggests that the melting temperature (Tm) for Ub in the presence of 1 M ClO4− is >75 °C; a value greater than the Tagg from SLS. Thus, ClO4− may not cause significant unfolding ahead of aggregation, but the opposite may be true where the Tagg value of an anion is close to or greater than the corresponding Tm value of Ub.
- G. I. Makhatadze, M. M. Lopez, J. M. I. Richardson and S. T. Thomas, Anion binding to the ubiquitin molecule, Protein Sci., 1998, 7, 689–697 CrossRef CAS.
- M. Sundd, N. Iverson, B. Ibarra-Molero, J. M. Sanchez-Ruiz and A. D. Robertson, Electrostatic Interactions in Ubiquitin: Stabilization of Carboxylates by Lysine Amino Groups, Biochemistry, 2002, 41(24), 7586–7596 CrossRef CAS.
- M. F. M. Sciacca, D. Milardi, M. Pappalardo, C. LaRosa and D. M. Grasso, Role of electrostatics in the thermal stability of ubiquitin, J. Therm. Anal. Calorim., 2006, 86(2), 311–314 CrossRef CAS.
- A. Cooper, M. A. Nutley and A. Wadood, Differential scanning microcalorimetry, in Protein-Ligand Interactions: Hydrodynamics and Calorimetry, ed. S. E. Harding and B. Z. Chowdhry, Oxford University Press, Oxford, 2001 Search PubMed.
- P. L. Weber, S. C. Brown and L. Mueller, Sequential proton NMR assignments and secondary structure identification of human ubiquitin, Biochemistry, 1987, 26(23), 7282–7290 CrossRef CAS.
- L. J. Edwards, D. V. Savostyanov, Z. T. Welderufael, D. Lee and I. Kuprov, Quantum mechanical NMR simulation algorithm for protein-size spin systems, J. Magn. Reson., 2014, 243, 107–113 CrossRef CAS.
- It is important to note here the logistics of gathering the described affinity data. At an NMR spectroscopy frequency of 500 MHz, each point on a titration curve required approximately ∼1 hour of data acquisition. Thus, only 8–10 points per titration curve were recorded to keep one titration experiment to less than 10 hours of acquisition time. Moreover, the protein host had up 70 NMR signals to analyze which, due to NMR software restrictions, was performed manually. Consequently, contrary to the routine protocols in supramolecular chemistry, we did not perform titrations in duplicate. Rather we used the affinity determinations at pH 2.8 and 3.8 (vide infra) – which we presumed would yield the same binding constants – as a measure of repeatability. Based on this data the errors in affinity for the six identified binding sites are respectively (Sites 1–6): <5%, 30%, <5%, 17%, 23%, 6%. These errors in reproducibility, as well as the fitting errors to the 1:1 model are given in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04245e |
| This journal is © The Royal Society of Chemistry 2021 |