
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural deformation and host–guest properties of doubly-reduced cycloparaphenylenes, [n]CPPs2− (n = 6, 8, 10, and 12)†
Zheng
Zhou
 a,
Zheng
Wei
a,
Tobias A.
Schaub
b,
Ramesh
Jasti
*b and
Marina A.
Petrukhina
*a
a,
Zheng
Wei
a,
Tobias A.
Schaub
b,
Ramesh
Jasti
*b and
Marina A.
Petrukhina
*a
aDepartment of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA. E-mail: mpetrukhina@albany.edu
bDepartment of Chemistry & Biochemistry, Materials Science Institute, Knight Campus for Accelerating Scientific Impact, University of Oregon, Eugene, OR 97403, USA. E-mail: rjasti@uoregon.edu
First published on 10th August 2020
Abstract
Chemical reduction of several cycloparaphenylenes (CPPs) ranging in size from [8]CPP to [12]CPP has been investigated with potassium metal in THF. The X-ray diffraction characterization of the resulting doubly-reduced [n]CPPs provided a unique series of carbon nanohoops with increasing dimensions and core flexibility for the first comprehensive structural analysis. The consequences of electron acquisition by a [n]CPP core have been analyzed in comparison with the neutral parents. The addition of two electrons to the cyclic carbon framework of [n]CPPs leads to the characteristic elliptic core distortion and facilitates the internal encapsulation of sizable cationic guests. Molecular and solid-state structure changes, alkali metal binding and unique size-dependent host abilities of the [n]CPP2− series with n = 6–12 are discussed. This in-depth analysis opens new perspectives in supramolecular chemistry of [n]CPPs and promotes their applications in size-selective guest encapsulation and chemical separation.
Introduction
[n]Cycloparaphenylenes ([n]CPPs) are polycyclic aromatic hydrocarbons (PAHs) that consist of a certain number (n) of fused para-connected benzene rings. CPPs have burst onto the scene as a fascinating new class of PAHs with radially oriented π-systems that can be considered as the shortest fragments of [n,n] armchair carbon nanotubes (CNTs) (Scheme 1). These cyclic molecules had long been sought since their early conceptualization in 1934 by Parekh and Guha,1 and Vögtle's pioneering synthetic efforts 60 years later.2 The first [n]CPPs were successfully synthesized by Jasti and Bertozzi in 2008.3 In their work, 3,6-syn-dimethoxy-cyclohexa-1,4-diene was used to provide enough curvature and rigidity for macrocyclization under standard Suzuki reaction conditions, and the intermediates were then reduced by lithium naphthalenide through a reductive aromatization reaction to afford the corresponding products, [9]CPP, [12]CPP, and [18]CPP.3 For the next decade, Jasti's group continued to report the selective synthesis of cycloparaphenylenes ranging from [5]CPP to [12]CPP, using a similar approach.4–8 Particularly, [8]CPP and [10]CPP can now be prepared on gram-scale, utilizing synthetic methodologies developed in their group.5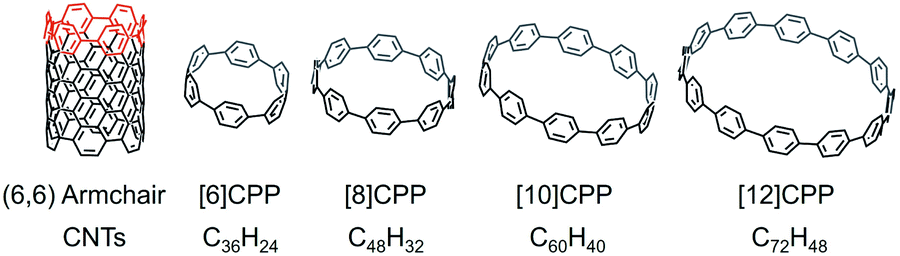 | ||
| Scheme 1 Depictions of (6,6) armchair carbon nanotube, [6]CPP, [8]CPP, [10]CPP, and [12]CPP. | ||
In conjunction with the work of the Jasti group, the selective syntheses of numerous [n]CPPs have also been successfully accomplished by the groups of Itami,9–12 Yamago,13–19 and others.20 Notably, the largest cycloparaphenylene to date, [36]CPP, has been prepared very recently by Müllen's group.20 Some latest works have been focused on the preparation of new CPP-based nanocarbons with different framework topologies, such as heteroatom-doped CPPs,21–23 π-surface extended CPPs,24–26 polymeric CPPs,27,28 catenanes and trefoil knots.29–31 Meanwhile, more extensive studies of [n]CPPs aimed at their structure–property relationships revealed their unique size-dependent optoelectronic properties and interesting supramolecular behavior.32–37
While the syntheses of [n]CPPs have been well developed,5,6 their solid-state structures remained unknown until the first X-ray diffraction characterization of [12]CPP was accomplished by Itami and coworkers in 2011.9 Single crystals of [12]CPP were grown from CHCl3/cyclohexane as [12]CPP·2(C6H12) and exhibited a herringbone packing in the solid state.9 Over the next year, [6]CPP,6 [8]CPP,5 and [10]CPP5 were structurally characterized by Jasti's group. While large cycloparaphenylenes, [8]CPP and [10]CPP, preserve a herringbone packing pattern,5 the smaller [6]CPP was shown to exhibit a linear column alignment in the crystalline state. It should be noted that none of these crystal structures are solvent-free; the interstitial and often disordered solvent molecules have been removed during the structure refinement and not accounted in the above solid-state structures. Interestingly, Itami's group in 2017 has found that [6]CPP can also form a herringbone packing under elevated temperature (70 °C) in the presence of CH2Cl2 molecules.38 In 2018, the only solvent-free crystal structure of [6]CPP obtained by Petrukhina's group was shown to exhibit unusual and very tight packing that minimizes the internal space of the nanohoop.22
One of the unique features of [n]CPPs is their large central void that may provide selective host–guest binding interactions for different encapsulated moieties, such as smaller π-systems. Thus, [n]CPPs can be potentially used as molecular π-hosts to trap or separate specific molecules based on size and shape complementarity. The host–guest complexation of CPPs was initially investigated by Yamago's group with UV-Vis and NMR spectroscopic techniques to reveal high selectivity of [10]CPP towards C60.39 In 2012, Jasti et al. reported the first crystal structure of the host–guest complex, C60⊂[10]CPP,5 demonstrating the perfect π–π concave–convex interaction between the host and guest. A few years later, Itami's group prepared an interesting ionic complex Li+@C60⊂[10]CPP·X−,40 where [10]CPP was capable of holding a charged {Li+@C60} moiety. In 2017, according to the NMR spectroscopy study, Yamago's group found that [n]CPPs can selectively interact with [n + 5]CPPs to afford the [n]CPP⊂[n + 5]CPP inclusion complexes, that can be viewed as the shortest double-walled carbon nanotubes.41 In the same year, Itami et al. prepared I2⊂[n]CPP, which exhibits turn-on electronic conductivity and white light luminescence upon voltage bias application.42 All these seminal works illustrate the outstanding host abilities of macrocyclic CPPs that can find unique applications in fundamental chemistry and materials science.
Overall, the size-dependent physical and chemical properties of [n]CPPs attract significant attention to this intriguing class of strained and cyclic PAHs.8,43–46 Surprisingly, the HOMO–LUMO gaps of CPPs become narrow with decreasing size, which contrasts the open-chain linear paraphenylenes and most other PAHs that show narrowing HOMO–LUMO gaps with an increased number of aromatic rings.47,48 Thus, the redox chemistry of [n]CPPs should be of special interest for a variety of applications, including new organic electroconductive or energy storage materials.36,45,48,49 However, these investigations have been mainly limited to oxidation of several [n]CPPs reported by Yamago and coworkers.48 While various spectroscopic and computational tools have been used for investigation of the oxidized cationic species,35,47–49 multi-electron reduction properties of CPPs remained largely unexplored.50 The X-ray crystallographic characterization of the only highly-reduced [8]CPP tetraanion dates back to 2013.51 More recently, the accessibility of the first and second reduction steps for a small and rigid [6]CPP has been demonstrated, along with the successful crystallographic characterization of both carbanions.52 This has opened a broad investigation of chemical reduction properties and host abilities of larger and more flexible cycloparaphenylenes.
Herein, we report the controlled chemical reduction study of [8]CPP, [10]CPP, and [12]CPP (Scheme 1) that allowed the first successful isolation and X-ray diffraction characterization of the doubly-reduced states of these carbon nanohoops. Structural analysis of the resulting products and comparison with a smaller [6]CPP dianion revealed systematic structural deformation trends upon two-electron charging, as well as the unique size-dependent ability of the doubly-reduced hoop-shaped hosts, [n]CPPs2− (n = 6–12), for selective guest binding.
Results and discussion
Chemical reduction of [n]CPPs
The chemical reduction of [n]CPPs with K metal has been carried out at room temperature in THF and monitored by UV-Vis spectroscopy (Fig. S1–S3†), following the procedures reported for [6]CPP.52 These reactions, passing through a deep brown color initially, generate dark-purple solutions associated with the second reduction step for all CPPs. Crystallization of the resulting doubly-reduced [n]CPP2− anions (n = 8–12) with potassium counterions has been facilitated by the addition of 18-crown-6 or benzo-15-crown-5 ethers (Scheme 2), producing single-crystals of good quality (see ESI† for more details). The X-ray diffraction analysis of these crystals confirmed the formation of [{K+(18-crown-6)(THF)}2([8]CPP2−)] (1); [{K+(18-crown-6)}2([10]CPP2−)] (2), which was crystallized with two interstitial THF molecules as 2·2THF; as well as [K+(benzo-15-crown-5)2]2([12]CPP2−) (3), which was crystallized with seven interstitial THF molecules as 3·7THF.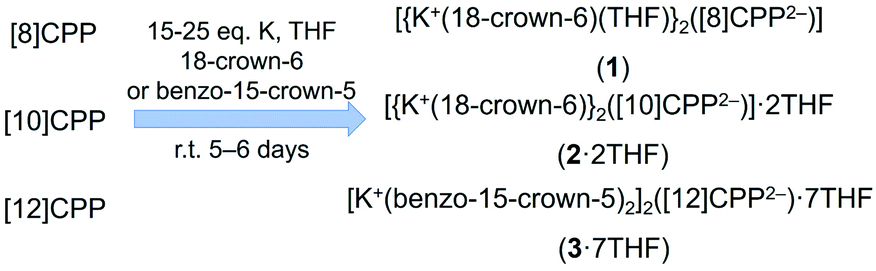 | ||
| Scheme 2 Chemical reduction of [8]CPP, [10]CPP, and [12]CPP. | ||
Crystal structures and supramolecular packing
In the crystal structure of 1 (Fig. 1a), the K+ ions are bound to the opposite six-membered rings of [8]CPP2− in a η2-fashion, with the corresponding K⋯C distances of 3.184(2) Å and 3.239(2) Å. The K⋯Ccentroid distance of 3.291(2) Å is notably longer than the value of 3.059(6) Å in the highly-reduced product with [8]CPP4−.51 The coordination of K+ ion is completed by an axially bound 18-crown-6 ether molecule (K⋯Ocrown: 2.751(2)–2.867(2) Å) and a capping THF molecule (K⋯OTHF: 2.728(2) Å), with all K⋯Ocrown and K⋯OTHF distances being close to those previously reported.51–55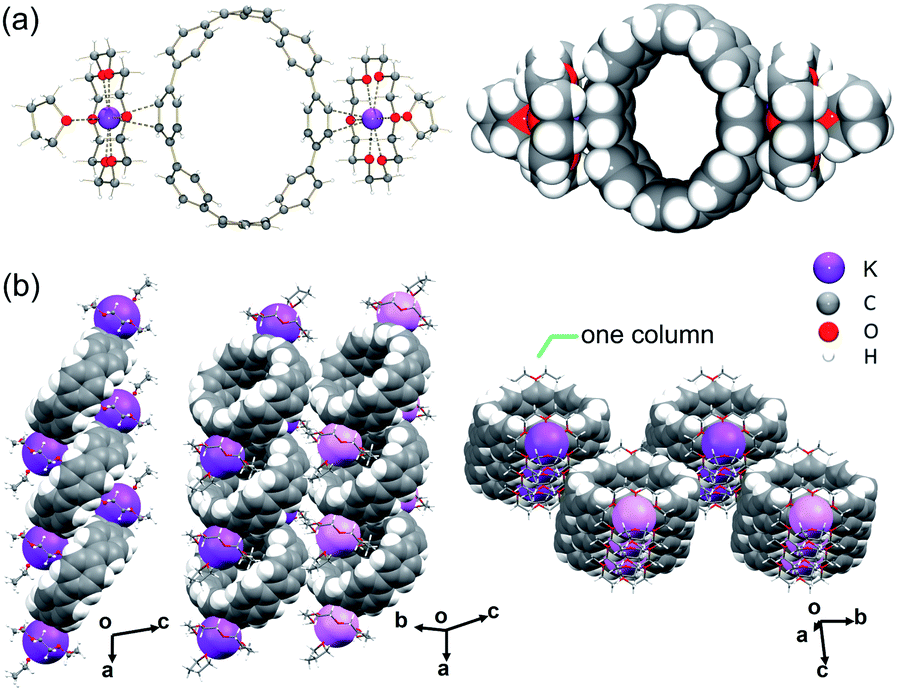 | ||
| Fig. 1 (a) Crystal structure of [{K+(18-crown-6)(THF)}2([8]CPP2−)] (1), ball-and-stick and space-filling models, (b) 1D column, two adjacent columns, and a fragment of solid-state packing of 1, mixed models. K+ ions from different columns are shown in different shades of purple. | ||
In the solid-state structure of 1, the {K+(18-crown-6)(THF)} moieties from adjacent units are interacting with the internal surface of the neighboring [8]CPP2− dianions (Fig. S8†) with the C–H⋯π contacts ranging over 2.679(4)–2.785(4) Å. Specifically, two cationic {K+(18-crown-6)} moieties embed in the internal cavity of one [8]CPP2− host from opposite directions, forming the stepped 1D columns with a slip distance of 7.97 Å (Fig. 1b). In contrast to the highly-reduced [8]CPP4−,51 only partial insertions of the crown ether groups are observed in this case. No strong interactions are found between the adjacent 1D columns.
In the crystal structure of 2 (Fig. 2a), the K+ ions bind to the six-membered rings of the [10]CPP2− exterior at the opposite positions in a η6-mode, with the K⋯C distances ranging over 3.227(3)–3.422(3) Å. The K⋯Ccentroid distance of 3.013(3) Å is shorter than that in 1 and in [{K+(18-crown-6)(THF)}2([6]CPP2−)].52 These short K⋯C contacts in 2 result in asymmetric coordination of 18-crown-6 ether that could prevent additional THF binding to the K+ ion observed in the above cases. The K⋯Ocrown distances of 2.788(2)–2.832(2) Å are comparable to those previously reported.51–55 Two interstitial THF molecules were found in the crystal structure of 2·2THF, filling the structural voids with no close contacts.
 | ||
| Fig. 2 (a) Crystal structure of [{K+(18-crown-6)}2([10]CPP2−)] (2), ball-and-stick and space-filling models, (b) 2D layer and solid-state packing of 2, mixed models. K+ ions from different layers are shown in different shades of purple. | ||
In the solid-state structure of 2, the internal surface of the [10]CPP2− anions accommodates two {K+(18-crown-6)} moieties from the adjacent units to form a 2D layer (Fig. 2b and S9†), with the shortest C–H⋯π contacts of 2.632(5)–2.755(5) Å. No significant interactions are found between the adjacent layers.
In the crystal structure of 3 (Fig. 3a), there are two external {K+(benzo-15-crown-5)2} moieties that are solvent-separated from the [12]CPP2− core. Each K+ ion is fully wrapped by two benzo-15-crown-5 ether molecules with the K⋯Ocrown distances (2.752(2)–2.945(2) Å) being comparable to those previously reported.51–53,55–58 Seven interstitial THF molecules are found to fill the structural voids with no close contacts.
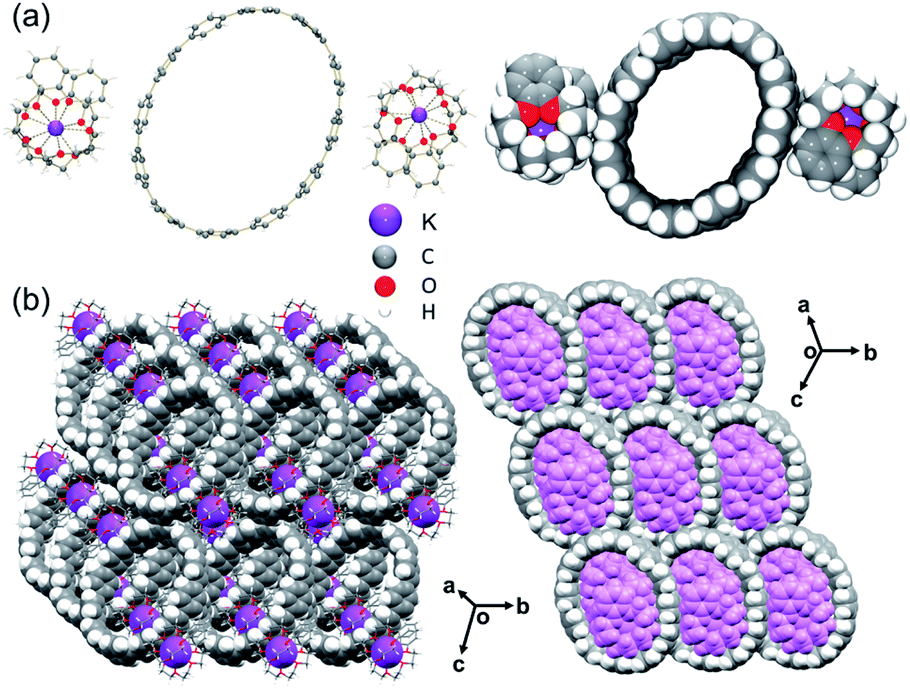 | ||
| Fig. 3 (a) Crystal structure of [K+(benzo-15-crown-5)2]2([12]CPP2−) (3), ball-and-stick and space-filling models, (b) solid-state packing of 3, mixed models. | ||
In the solid-state structure of 3, the internal cavities of the [12]CPP2− anions are occupied by {K+(benzo-15-crown-5)2} cations from the adjacent molecules, with C–H⋯π interactions ranging over 2.621(8)–2.735(8) Å. Notably, the C–H⋯π contacts as short as 2.559(8) Å are also found between the two encapsulated {K+(benzo-15-crown-5)2} moieties (Fig. S10†). In addition, the C–H⋯π contacts of 2.655(8)–2.740(8) Å can be identified between the external surface of [12]CPP2− and four adjacent {K+(benzo-15-crown-5)2} moieties, resulting in the formation of an extended 3D network in the solid state (Fig. 3b).
Deformation of [n]CPPs upon two-step reduction
In the course of these investigations, four dianions ranging in size from [6]CPP2− to [12]CPP2− have been crystallographically characterized, providing a unique series for comprehensive structural analysis and comparison (Fig. 4). The addition of two electrons and direct metal binding lead to the deformation of the [n]CPP core, which is clearly reflected by the change of C–C bond distances of the series. In [8]CPP2−, the C–C bond (d) connecting the six-membered rings is significantly shortened from 1.486(2) Å in the neutral parent to 1.443(4) Å upon reduction (Δ = 0.043 Å) (see Tables S2–S4† for more details). The corresponding C–C bond in [10]CPP2− has shrunk by 0.034 Å. In [12]CPP2−, this C–C bond length has only reduced by 0.008 Å in comparison to the neutral state. The largest C–C bond length shortage of this type has been observed upon a two-fold reduction of the smallest [6]CPP with Δ = 0.050 Å.52 The revealed C–C bridging bond changes of the [n]CPP2− anions point to an increasing quinoidal character of their core structures,59 which is the most pronounced in the smallest members of the series.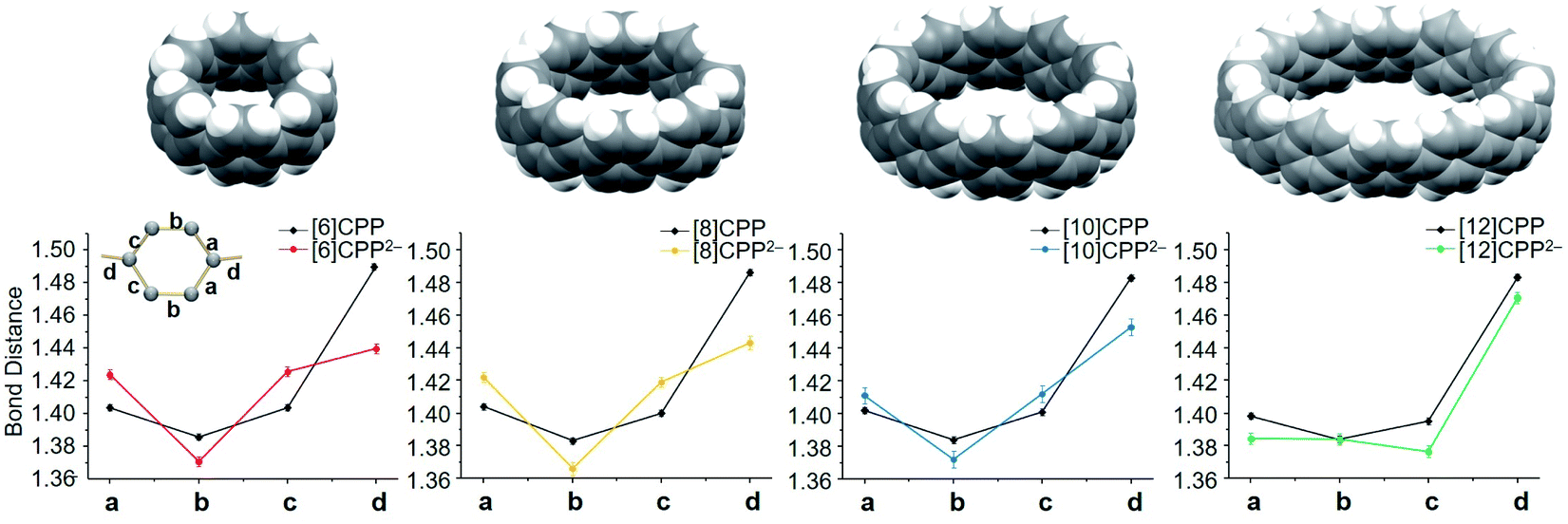 | ||
| Fig. 4 Average C–C bond distances (Å) in [6]CPP2−,52 [8]CPP2−, [10]CPP2−, and [12]CPP2− in comparison to neutral parents, along with their space-filling models. | ||
The structural changes of [n]CPPs upon reduction can also be analyzed by comparison of the inner diameters and selected dihedral angles of the dianions vs. their neutral parents. In the doubly-reduced CPPs (n = 6–10), the D.P. (deformation parameter) values are increased by 0.06–0.10 (Table 1), indicating that the cyclic core becomes only slightly elongated along the dmax axis (the longest inner diameter, Fig. S12†) upon two-electron acquisition. Interestingly, the largest deformation is observed in the [12]CPP2− macrocycle (D.P. = 1.22) that shows an increased elliptical distortion by 18% along the dmax direction upon two-electron addition. Notably, the D.P. of [8]CPP2− (1.19) could be further increased to 1.45 upon further reduction, as observed in its tetrareduced form, [8]CPP4−.51
| Distance | [6]CPPa | [6]CPP2−a | [8]CPPb | [8]CPP2− | [10]CPPb | [10]CPP2− | [12]CPPc | [12]CPP2− | |
|---|---|---|---|---|---|---|---|---|---|
| a Ref. 52. b Ref. 5. c Ref. 9. d D.P. = dmax/dmin. | |||||||||
| d A | 8.049 | 7.848 | 10.364 | 9.958 | 13.742 | 14.197 | 16.557 | 18.155 |

|
| d B | 8.050 | 8.156 | 10.376 | 10.678 | 13.471 | 13.830 | 16.554 | 17.413 | |
| d C | 8.117 | 8.374 | 11.281 | 11.856 | 13.436 | 13.148 | 16.331 | 15.511 | |
| d D | — | — | 11.351 | 10.098 | 13.720 | 13.279 | 16.353 | 14.285 | |
| d E | — | — | — | — | 13.702 | 13.943 | 16.349 | 14.929 | |
| d F | — | — | — | — | — | — | 16.078 | 16.910 | |
| D.P.d | 1.01 | 1.07 | 1.09 | 1.19 | 1.02 | 1.08 | 1.03 | 1.22 | |
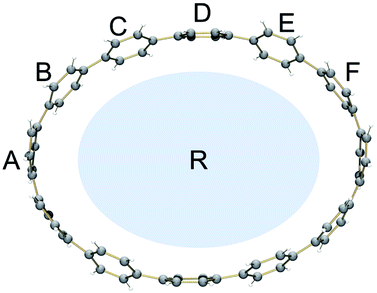
Compared to the neutral CPPs, the average dihedral angles (Fig. S11†) of the six-membered rings in [6]CPP2−, [8]CPP2−, [10]CPP2−, and [12]CPP2− are notably increased by about 8–15° (Table 2), indicating that the rings become more vertically oriented in respect to the central plane R (See scheme in Table 2). This structural change is consistent with the coplanarization of the phenyl rings expected for the increased quinoidal character of the doubly-reduced CPPs. The observed ring re-orientation also increases an internal open space of the [n]CPP2− macrocycles, thus enhancing their host–guest binding abilities.
| Angle | [6]CPPa | [6]CPP2−a | [8]CPPb | [8]CPP2− | [10]CPPb | [10]CPP2− | [12]CPPc | [12]CPP2− | |
|---|---|---|---|---|---|---|---|---|---|
| a Ref. 52. b Ref. 5. c Ref. 9. | |||||||||
| a A/R | 73.6 | 89.4 | 64.6 | 84.1 | 88.3 | 89.0 | 89.1 | 86.3 |
|
| a B/R | 71.1 | 88.7 | 70.1 | 85.0 | 61.6 | 89.9 | 61.1 | 85.1 | |
| a C/R | 75.6 | 87.5 | 83.0 | 88.7 | 65.0 | 82.2 | 83.1 | 78.9 | |
| a D/R | — | — | 81.5 | 83.4 | 88.6 | 81.2 | 74.0 | 71.8 | |
| a E/R | — | — | — | — | 68.0 | 86.0 | 85.8 | 87.8 | |
| a F/R | — | — | — | — | — | — | 57.3 | 89.1 | |
| a avg. | 73.4 | 88.5 | 74.8 | 85.3 | 74.5 | 85.7 | 75.1 | 83.2 | |
Host–guest properties of doubly-charged [n]CPPs
The unique size selectivity of [n]CPPs able to encapsulate various guests in their neutral forms5 prompted us to look into the host–guest binding abilities of the doubly-negatively charged [n]CPPs with increasing size and core flexibility. To simplify the comparison, we used the shortest inner diameter to estimate the potential spherical volume of the void space and compared it with the volume of the encapsulated moieties found in the X-ray crystal structures (Fig. 5 and Table 3).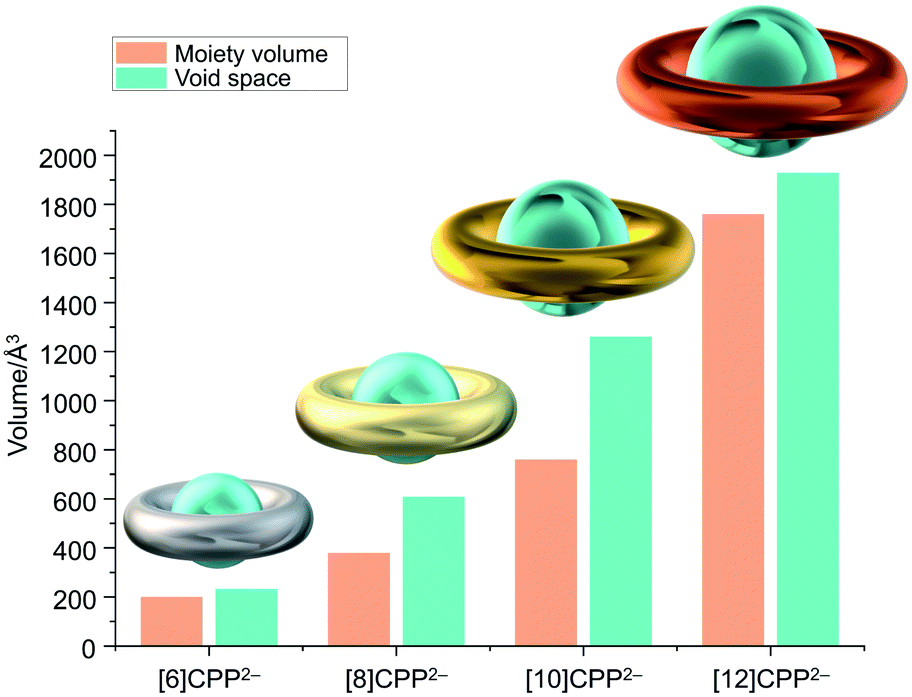 | ||
| Fig. 5 Depictions of [6]CPP2− (silvery), [8]CPP2− (yellow), [10]CPP2− (golden), and [12]CPP2− (orange), along with a chart of volume comparison between the encapsulated moieties and void space in the solid-state structures. The estimated internal space is shown as a blue-green sphere. | ||
| [6]CPP2−a | [8]CPP2− | [10]CPP2− | [12]CPP2− | |
|---|---|---|---|---|
a
Ref. 52.
b
V
void ≈ 4/3 × π × (dmin/2 × cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) amin)2 × (dmax/2 × cosamax).
c
V
moiety ≈ N × 20. N is the number of non-hydrogen atoms in the moiety. amin)2 × (dmax/2 × cosamax).
c
V
moiety ≈ N × 20. N is the number of non-hydrogen atoms in the moiety.
|
||||
| V void | 233 | 608 | 1261 | 1928 |
| Moiety | THF ×2 | ½{K+(18-crown-6)} ×2 | {K+(18-crown-6)} ×2 | {K+(benzo-15-crown-5)2} ×2 |
| V moiety | 200 | 380 | 760 | 1760 |
| Host–guest formula | {THF}2⊂[6]CPP2− | ½{K+(18-crown-6)}2⊂[8]CPP2− | {K+(18-crown-6)}2⊂[10]CPP2− | {K+(benzo-15-crown-5)2}2⊂[12]CPP2− |
For the series of [n]CPP (n = 6, 8, 10, 12) hosts, due to a significant internal volume increase (233 Å3,52 493 Å3,5 1021 Å3,5 and 1587 Å3,9 respectively) the encapsulation of larger guests can be expected. The host ability is further enhanced in the doubly-reduced [n]CPP species. For example, the measured internal volume of [6]CPP2− is about 233 Å3, thus matching the size of two THF molecules that filled the void, as found in the X-ray crystal structure of [{K+(18-crown-6)(THF)}2([6]CPP2−)] (Table 3 and Fig. 6a).52 Consistent with the size increase of the [n]CPP series, the estimated internal space of [8]CPP2− is expanded to 608 Å3, allowing the dianion to trap larger guests than THF (Fig. 6b). In the crystal structure of 1, two cationic {K+(18-crown-6)} moieties (ca. 380 Å3) from the neighboring units are partially inserted into the internal cavity of [8]CPP2−, thus forming the 1D stepped columns in the solid-state (Fig. 1b). Taking advantage of the larger size, the internal volume of [10]CPP2− is doubled (1261 Å3) compared to that of [8]CPP2−, with the void being capable of accommodating two cationic moieties from the adjacent units (Fig. 6c). Two {K+(18-crown-6)} cations show perfect parallel alignment inside the [10]CPP2− cavity with a slip distance of 2.82 Å. Due to some space restriction, the cationic guest moiety is tilted in respect to the central plane of the [10]CPP2− host with an approximate tilt angle of 25.0°. This head-to-head encapsulation of two {K+(18-crown-6)} moieties inside the [10]CPP2− host is responsible for the formation of a 2D layered structure in 2. For the largest [12]CPP2−, the internal volume is significantly increased to 1928 Å3, allowing the encapsulation of two {K+(benzo-15-crown-5)2} moieties from the adjacent units (Fig. 6d) in the solid-state structure of 3.
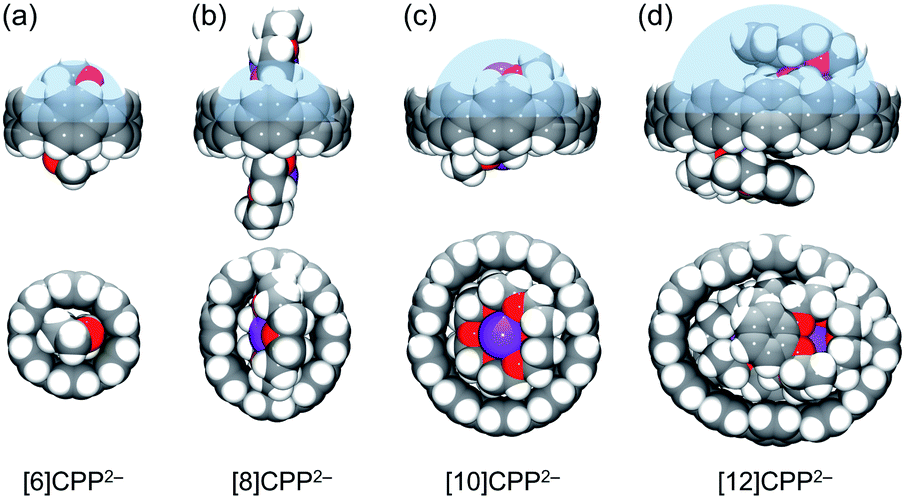 | ||
| Fig. 6 Host–guest assemblies of (a) [6]CPP2−, (b) [8]CPP2−, (c) [10]CPP2− and (d) [12]CPP2− dianions, side and face views in space-filling models. Half of the estimated internal space is shown in blue. | ||
Overall, the increased internal volume along the series of [n]CPP2− (n = 6, 8, 10, and 12) allows the accommodation of larger guests with enhanced host–guest interactions. This affects the intermolecular interactions between the neighboring cationic and anionic units and results in different solid-state packing arrangements, as revealed crystallographically.
Conclusions
In summary, the first in-depth structural comparison of the series of the doubly-reduced [n]CPPs (n = 6–12) has been accomplished, revealing the consequences of two-electron addition to a cycloparaphenylene core with increasing dimensions and core flexibility. Specifically, the bridging C–C bonds connecting the six-membered rings become significantly shortened in the doubly-reduced CPPs, consistent with a quinoidal structural transformation. The effect is enhanced in the smallest [6]CPP2− and it gradually weakens for larger [n]CPPs with n ranging from 8 to 12. Upon a two-fold reduction, a notable elliptical core distortion is observed in all CPPs with the D.P. values increased by 0.06–0.19 along the series. This effect is more pronounced for larger CPPs reflecting their enhanced core flexibility. Interestingly, the dianion of [10]CPP seems an outlier in the series in that it has a smaller D.P. than that of [8]CPP. This difference is likely due to solid state packing effects and weak intermolecular interactions, but further detailed computational studies are required to understand these subtle differences. Notably, all six-membered rings of the doubly-reduced macrocycles become more vertically oriented with respect to the central plane compared to their neutral parents. This change along with the systematic internal volume increase along the [n]CPP series allows gradual expansion of the void space available for guest accommodation in the doubly-negatively charged nanohoops. The above trend is well illustrated by the analysis of the encapsulated guests, ranging from THF molecules in the smallest [6]CPP2− host to larger cationic moieties in the [n]CPP2− anions with n = 8–12. The size-dependent host–guest interactions result in the different solid-state structural arrangements that range from 1D columns to 2D layers and 3D supramolecular networks. This comprehensive structural analysis of the [n]CPP series facilitates understanding of the systematic carbon core deformation upon electron acquisition. It also illustrates the unique host abilities of the doubly-reduced nanohoops of variable sizes, providing new perspectives in supramolecular chemistry and applications of cyclic carbon cages in chemical separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial and instrumentational support of this work from the U.S. National Science Foundation (CHE-1608628, CHE-2003411, and MRI-1726724) is gratefully acknowledged (M. A. P.). R. J. was supported by the National Science Foundation (CHE-1800586) and T. A. S. acknowledges the German Research Council (DFG, SCHA 2061/1-1) for a postdoctoral fellowship. NSF's ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1834750. The use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.Notes and references
- V. C. Parekh and P. C. Guha, J. Indian Chem. Soc., 1934, 11, 95–100 CAS.
- R. Friederich, M. Nieger and F. Vögtle, Chem. Ber., 1993, 126, 1723–1732 CrossRef CAS.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- T. J. Sisto, M. R. Golder, E. S. Hirst and R. Jasti, J. Am. Chem. Soc., 2011, 133, 15800–15802 CrossRef CAS PubMed.
- J. Xia, J. W. Bacon and R. Jasti, Chem. Sci., 2012, 3, 3018–3021 RSC.
- J. Xia and R. Jasti, Angew. Chem., Int. Ed., 2012, 51, 2474–2476 CrossRef CAS PubMed.
- P. J. Evans, E. R. Darzi and R. Jasti, Nat. Chem., 2014, 6, 404–408 CrossRef CAS PubMed.
- E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- Y. Segawa, S. Miyamoto, H. Omachi, S. Matsuura, P. Šenel, T. Sasamori, N. Tokitoh and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 3244–3248 CrossRef CAS PubMed.
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572–576 CrossRef CAS PubMed.
- F. Sibbel, K. Matsui, Y. Segawa, A. Studer and K. Itami, Chem. Commun., 2014, 50, 954–956 RSC.
- Y. Segawa, T. Kuwabara, K. Matsui, S. Kawai and K. Itami, Tetrahedron, 2015, 71, 4500–4503 CrossRef CAS.
- E. Kayahara, Y. Sakamoto, T. Suzuki and S. Yamago, Org. Lett., 2012, 14, 3284–3287 CrossRef CAS PubMed.
- E. Kayahara, T. Iwamoto, T. Suzuki and S. Yamago, Chem. Lett., 2013, 42, 621–623 CrossRef CAS.
- E. Kayahara, V. K. Patel and S. Yamago, J. Am. Chem. Soc., 2014, 136, 2284–2287 CrossRef CAS PubMed.
- S. Yamago, E. Kayahara, V. Patel, J. Xia and R. Jasti, Synlett, 2015, 26, 1615–1619 CrossRef.
- E. Kayahara, L. Sun, H. Onishi, K. Suzuki, T. Fukushima, A. Sawada, H. Kaji and S. Yamago, J. Am. Chem. Soc., 2017, 139, 18480–18483 CrossRef CAS PubMed.
- E. Kayahara, Y. Cheng and S. Yamago, Chem. Lett., 2018, 47, 1108–1111 CrossRef CAS.
- T. Kawanishi, K. Ishida, E. Kayahara and S. Yamago, J. Org. Chem., 2020, 85, 2082–2091 CrossRef CAS PubMed.
- W. Zhang, A. Abdulkarim, F. E. Golling, H. J. Räder and K. Müllen, Angew. Chem., Int. Ed., 2017, 56, 2645–2648 CrossRef CAS PubMed.
- K. Matsui, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 1888–1891 CrossRef CAS PubMed.
- J. M. Van Raden, S. Louie, L. N. Zakharov and R. Jasti, J. Am. Chem. Soc., 2017, 139, 2936–2939 CrossRef CAS PubMed.
- S. Hashimoto, E. Kayahara, Y. Mizuhata, N. Tokitoh, K. Takeuchi, F. Ozawa and S. Yamago, Org. Lett., 2018, 20, 5973–5976 CrossRef CAS PubMed.
- A. Yagi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2012, 134, 2962–2965 CrossRef CAS PubMed.
- B. Farajidizaji, C. Huang, H. Thakellapalli, S. Li, N. G. Akhmedov, B. V. Popp, J. L. Petersen and K. K. Wang, J. Org. Chem., 2017, 82, 4458–4464 CrossRef CAS PubMed.
- Q. Huang, G. Zhuang, H. Jia, M. Qian, S. Cui, S. Yang and P. Du, Angew. Chem., Int. Ed., 2019, 58, 6244–6249 CrossRef CAS PubMed.
- Y. Ishii, S. Matsuura, Y. Segawa and K. Itami, Org. Lett., 2014, 16, 2174–2176 CrossRef CAS PubMed.
- Q. Huang, G. Zhuang, M. Zhang, J. Wang, S. Wang, Y. Wu, S. Yang and P. Du, J. Am. Chem. Soc., 2019, 141, 18938–18943 CrossRef CAS PubMed.
- K. Matsui, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2014, 136, 16452–16458 CrossRef CAS PubMed.
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota and K. Itami, Science, 2019, 365, 272–276 CrossRef CAS PubMed.
- Y. Segawa, M. Kuwayama and K. Itami, Org. Lett., 2020, 22, 1067–1070 CrossRef CAS PubMed.
- T. Nishihara, Y. Segawa, K. Itami and Y. Kanemitsu, Chem. Sci., 2014, 5, 2293–2296 RSC.
- N. Kubota, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 1356–1361 CrossRef CAS PubMed.
- E. Kayahara, V. K. Patel, A. Mercier, E. P. Kündig and S. Yamago, Angew. Chem., Int. Ed., 2016, 55, 302–306 CrossRef CAS PubMed.
- M. P. Alvarez, M. C. Ruiz Delgado, M. Taravillo, V. G. Baonza, J. T. Lopez Navarrete, P. Evans, R. Jasti, S. Yamago, M. Kertesz and J. Casado, Chem. Sci., 2016, 7, 3494–3499 RSC.
- N. Ozaki, H. Sakamoto, T. Nishihara, T. Fujimori, Y. Hijikata, R. Kimura, S. Irle and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11196–11202 CrossRef CAS PubMed.
- Y. Xu, R. Kaur, B. Wang, M. B. Minameyer, S. Gsänger, B. Meyer, T. Drewello, D. M. Guldi and M. von Delius, J. Am. Chem. Soc., 2018, 140, 13413–13420 CrossRef CAS PubMed.
- T. Fukushima, H. Sakamoto, K. Tanaka, Y. Hijikata, S. Irle and K. Itami, Chem. Lett., 2017, 46, 855–857 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed. Engl., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- H. Ueno, T. Nishihara, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 3707–3711 CrossRef CAS PubMed.
- S. Hashimoto, T. Iwamoto, D. Kurachi, E. Kayahara and S. Yamago, ChemPlusChem, 2017, 82, 1015–1020 CrossRef CAS PubMed.
- S. Sato, A. Unemoto, T. Ikeda, S.-i. Orimo and H. Isobe, Small, 2016, 12, 3381–3387 CrossRef CAS PubMed.
- H. Omachi, Y. Segawa and K. Itami, Acc. Chem. Res., 2012, 45, 1378–1389 CrossRef CAS PubMed.
- M. R. Golder and R. Jasti, Acc. Chem. Res., 2015, 48, 557–566 CrossRef CAS PubMed.
- D. Wu, W. Cheng, X. Ban and J. Xia, Asian J. Org. Chem., 2018, 7, 2161–2181 CrossRef CAS.
- J. B. Lin, E. R. Darzi, R. Jasti, I. Yavuz and K. N. Houk, J. Am. Chem. Soc., 2019, 141, 952–960 CrossRef CAS PubMed.
- M. Fujitsuka, S. Tojo, T. Iwamoto, E. Kayahara, S. Yamago and T. Majima, J. Phys. Chem. A, 2015, 119, 4136–4141 CrossRef CAS PubMed.
- E. Kayahara, T. Kouyama, T. Kato and S. Yamago, J. Am. Chem. Soc., 2016, 138, 338–344 CrossRef CAS PubMed.
- Y. Masumoto, N. Toriumi, A. Muranaka, E. Kayahara, S. Yamago and M. Uchiyama, J. Phys. Chem. A, 2018, 122, 5162–5167 CrossRef CAS PubMed.
- M. Fujitsuka, S. Tojo, T. Iwamoto, E. Kayahara, S. Yamago and T. Majima, J. Phys. Chem. Lett., 2014, 5, 2302–2305 CrossRef CAS PubMed.
- A. V. Zabula, A. S. Filatov, J. Xia, R. Jasti and M. A. Petrukhina, Angew. Chem., Int. Ed., 2013, 52, 5033–5036 CrossRef CAS PubMed.
- S. N. Spisak, Z. Wei, E. Darzi, R. Jasti and M. A. Petrukhina, Chem. Commun., 2018, 54, 7818–7821 RSC.
- G. E. Rudebusch, G. L. Espejo, J. L. Zafra, M. Pena-Alvarez, S. N. Spisak, K. Fukuda, Z. Wei, M. Nakano, M. A. Petrukhina, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 12648–12654 CrossRef CAS PubMed.
- T. Wombacher, R. Goddard, C. W. Lehmann and J. J. Schneider, Dalton Trans., 2018, 47, 10874–10883 RSC.
- Z. Zhou, R. K. Kawade, Z. Wei, F. Kuriakose, O. Ungor, M. Jo, M. Shatruk, R. Gershoni-Poranne, M. A. Petrukhina and I. V. Alabugin, Angew. Chem., Int. Ed., 2020, 59, 1256–1262 CrossRef CAS PubMed.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, V. M. Grigoryants and M. A. Petrukhina, Chem.–Eur. J., 2012, 18, 6476–6484 CrossRef CAS PubMed.
- Z. Zhou, X.-Y. Wang, Z. Wei, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 14969–14973 CrossRef CAS PubMed.
- S. N. Spisak, M. U. Bühringer, Z. Wei, Z. Zhou, R. R. Tykwinski and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 2023–2028 CrossRef CAS PubMed.
- M. Fujitsuka, T. Iwamoto, E. Kayahara, S. Yamago and T. Majima, ChemPhysChem, 2013, 14, 1570–1572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of preparation and X-ray diffraction study. CCDC 2005328–2005330. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03072d |
| This journal is © The Royal Society of Chemistry 2020 |