
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormal oxo- and aza-[3 + 2] reactions of α-enaminones and quinones: a double divergent process and the roles of chiral phosphoric acid and molecular sieves†
Weiwei
Luo
,
Zhicheng
Sun
 ,
E. H. Nisala
Fernando
,
Vladimir N.
Nesterov
,
Thomas R.
Cundari
* and
Hong
Wang
*
,
E. H. Nisala
Fernando
,
Vladimir N.
Nesterov
,
Thomas R.
Cundari
* and
Hong
Wang
*
Department of Chemistry, University of North Texas, Denton, TX 76203, USA. E-mail: thomas.cundari@unt.edu; hong.wang@unt.edu
First published on 9th July 2020
Abstract
A double divergent process has been developed for the reaction of α-enaminones with quinones through facile manipulation of catalyst and additive, leading to structurally completely different products. The two divergent processes, which involve formal aza- and oxo-[3 + 2] cycloaddition reactions, are mediated by chiral phosphoric acid and molecular sieves, respectively. While inclusion of phosphoric acid in the reaction switched the reaction pathway to favor the efficient formation of a wide range of N-substituted indoles, addition of 4 Å molecular sieves to the reaction switched the reaction pathway again, leading to enantioselective synthesis of 2,3-dihydrobenzofurans in excellent yields and enantioselectivities under mild conditions. Studies in this work suggest that the chiral phosphoric acid acts to lower the transition state energy and promote the formation of amide intermediate for the formal aza-[3 + 2] cycloaddition and the molecular sieves serve to facilitate proton transfer for oxo-[3 + 2] cycloaddition. The reactivity of α-enaminones is also disclosed in this work.
Introduction
Divergent reactivity is an effective tool to control product distribution, and has remained an intense research interest in chemical catalysis, synthetic organic chemistry and pharmaceutical sciences.1,2 Despite the challenges in developing divergent reactions, a growing number of switchable organic transformations have been achieved through divergent reactions in the past decade. The predominant strategy to access divergent reactions is to develop different catalysts to attain product selectivity via different catalytic intermediates (catalyst-controlled selectivity) (Scheme 1a). On the other hand, using the same catalyst to access structurally different compounds from the same reactants (additive-controlled) is extremely challenging,2a,b,p as the divergent reactions are expected to share the same catalytic intermediates. In an early work, Jørgensen and co-workers designed an additive-controlled divergent synthesis of tetrahydrothiophenes (Scheme 1b).2a This divergent reaction utilized the same proline catalyst and shared the same key intermediate (X). Treatment of the key intermediate (X) with different additives, i.e., PhCOOH and NaHCO3, respectively, leads to tetrahydrothiophenes with different substitution patterns through competitive aldol pathways. Jørgensen's work suggested the possibility for additive-controlled divergent reactions. | ||
| Scheme 1 Catalytic divergent transformations. | ||
In the present work, we reveal a rare double divergent process, which was mediated by phosphoric acid (catalyst-controlled) and molecular sieves (additive-controlled), respectively. α-Enaminone reacting with quinone in the absence of a catalyst or an additive afforded 2,3-dihydrobenzofurans (Scheme 1c, RX I). Upon addition of chiral phosphoric acid, the reaction pathway switched to forming N-substituted indoles (Scheme 1c, RX II). When molecular sieves were included in the reaction, the reaction pathway switched back to form enantioenriched tricyclic 2,3-dihydrobenzofurans (RX III). Thus, two divergent processes were realized. The first divergent process was controlled by phosphoric acid, and the second divergent process was controlled by molecular sieves (M.S.). This work represents, to our knowledge, the first example of a double divergent process. It is also the first example of molecular sieve-controlled divergent reaction. The functions of the phosphoric acid and the molecular sieves were investigated both experimentally and theoretically, and a new role of molecular sieves has been unveiled. This work provides concise new approaches to access important 2,3-dihydrobenzofurans and indoles, both of which are widespread structural skeletons found in biomolecules, natural products, pharmaceuticals, and have attracted significant research endeavors.3
Enaminones are important synthetic intermediates in organic synthesis.4,5 Enaminones including α-enaminones and β-enaminones (Fig. 1) often display multi-functionality, as they possess both nucleophilic (enamine) and electrophilic (enone) characteristics. Unlike β-enaminones, which have been frequently used in the synthesis of natural products and other biological active compounds,4 little attention has been paid to α-enaminones due to the lack of appropriate synthetic routes of their synthesis.5 We became interested in α-enaminones during our investigation in cooperative enamine-metal Lewis acid catalysis.6 It was found that α-enaminones could be readily formed from arylamine and cyclohexanone in the presence of a metal Lewis acid under ambient conditions.5e This discovery prompted us to investigate the reactivities of α-enaminones as they remain largely unexplored. It is expected that α-enaminones will display different set of reactivities from β-enaminones, as β-enaminones are linearly conjugated molecules and α-enaminones are cross-conjugated molecules. In this work, we demonstrate that α-enaminones display multifunctionality, acting as both an enamine and a nitrogen nucleophile, as well as an imine electrophile to initiate two novel phosphoric acid-catalyzed [3 + 2] cycloaddition reactions through 1,4-ketone/imine intermediate (Paal–Knorr type intermediates).7
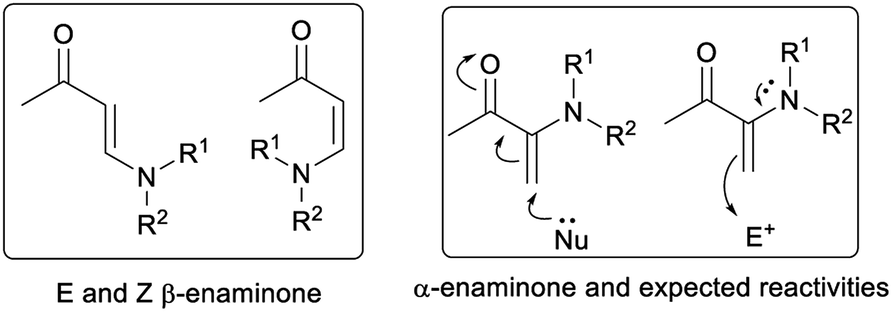 | ||
| Fig. 1 α- and β-enaminones. | ||
Results and discussion
Quinone ester was selected as the reaction partner for α-enaminone due to its high activity and multifunctionality.8 Quinone ester 1a reacted smoothly with enaminone 2a without any catalyst, affording a formal oxo-[3 + 2] product 3a in 85% yield (Table 1, entry 1). While it was exciting that an interesting new reaction was discovered, this strong background reaction makes it challenging to develop an asymmetric version of this reaction.|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Yieldb (%) | ee of 3ac (%) | |
| 3a | 4a | |||
| a Unless otherwise noted, all reactions were carried out with 1a (0.1 mmol), 2a (0.11 mmol), CPA (10 mol%) in 1.0 mL DCM at −78 °C for 16 h. b Isolated yield. c Determined by chiral HPLC analysis. d 5.0 mg of 4 Å M.S. e 1.0 mol% of catalyst was used. f Toluene (1.0 mL) as solvent. g 0.1 mmol of 2a was used. h 5.0 mol% of catalyst was used. | ||||
| 1 | — | 85 | — | 0 |
| 2 | CPA1 | 10 | 90 | 18 |
| 3 | CPA2 | 14 | 77 | 20 |
| 4 | CPA3 | 34 | 63 | 90 |
| 5 | CPA4 | 78 | 17 | 72 |
| 6 | CPA5 | 16 | 78 | 99 |
| 7d | CPA5 | 97 | — | 99 |
| 8d,e | CPA5 | 98 | — | 99 |
| 9f,g | CPA3 | — | 96 | — |
| 10f,g,h | CPA3 | — | 99 | — |
| 11g | PA | 8 | 84 | — |
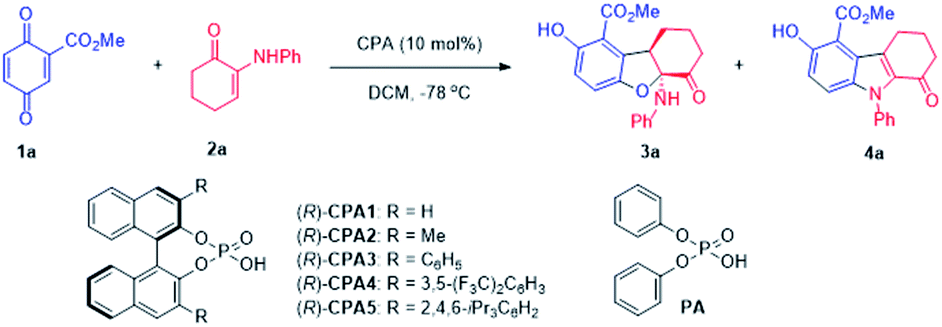
Considering the possible mechanism and multiple functional groups capable of H-bonding present in the substrates, a chiral phosphoric acid (CPA1) was chosen to mediate this reaction. Surprisingly, a new product was obtained in 90% yield along with 3a in 10% yield and 18% ee (entry 2). This new product turned out to be a N-substituted indole (4a) formed via a formal aza-[3 + 2] cycloaddition. These data suggest: (1) phosphoric acid can catalyze this reaction, initiating a divergent process; (2) chiral phosphoric acid could induce ee of 3a. A series of chiral phosphoric acid catalysts were then tested (Table 1, entries 3–6). Increasing the steric bulk (from H, Me, to Ph) at the 3,3′-position of the CPA resulted in increased yield and ee of 3a at the cost of 4a (entries 2–4). Based on these data, it appeared that increasing the steric bulkiness of CPA slightly favors the formation of 3a and results in higher ee of 3a. When CPA4 containing bulky 3,5-bis(trifluoromethyl)phenyl groups was used, the yield of 3a increased to 78% but the ee decreased to 72% (entry 5). When a highly bulky group, i.e., 2,4,6-tri(isopropyl)phenyl, was attached at the 3,3′-positions of CPA (entry 6), although the yield of 3a decreased significantly with increased yield of 4a, the ee of 3a increased to 99%. These data suggest that a stronger acid is likely to facilitate the formation of 3a, however, at the cost of enantioselectivity; on the other hand, increased steric bulkiness of CPA enhances the enantioselectivity of 3a.
Inspired by Rueping's9a and Wang's reviews9b on phosphoric acids and additive effects, in which M.S. could promote some organic transformations catalyzed by phosphoric acid, M.S. were then attempted to mediate the 3a forming reaction. It was a surprise that addition of 4 Å M.S. completely switched the reaction pathway of the CPA5 catalyzed reaction (entry 7) to favor 3a formation. While the yield of 3a increased to 97%, the enantioselectivity of 3a remained at 99%. It is notable that decreasing CPA5 loading to 1 mol% led to comparable results (entry 8, 98% yield, 99% ee). These data suggest that molecule sieves initiated another divergent process.
To optimize the reaction condition for 4a, solvent was surveyed. In the presence of CPA without M.S., reactions in most of the solvents favored the formation of 4a. Toluene appeared to be the best to promote 4a (entries 9–11, 92–96% yields). Decreasing CPA3 loading from 10% to 5 mol% has no negative effect (entry 10, 99% yield). Considering that the employment of a chiral phosphoric acid to produce achiral products is not cost-effective, we examined an achiral phosphoric acid as the catalyst. Diphenyl phosphate (PA) turned out to be suitable for such reaction, and the desired product 4a was obtained in 84% yield (entry 11).
Having a controllable new synthesis in hand, the substrate scope for both reactions were investigated. The substrate scope of 2,3-dihydrobenzofuran synthesis is broad (Table 2). Various quinones and α-enaminones reacted smoothly to afford tricyclic 2,3-dihydrobenzofurans (3a–3x) in good yields (39–99%) with excellent diastereoselectivities (91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 → 95:5 d.r.) and enantioselectivities (95–99% ee). Changing the ester (R1) group of quinone from methyl to ethyl and benzyl didn't affect the reactivity and enantioselectivity (3a–c). Regarding the ortho substituents on the phenyl ring of α-enaminones, a weak electron-donating group such as methyl (3d) gave slightly better yield (96%) than slightly electron-withdrawing groups such as chlorine and bromine (3e & 3f, 85% and 81% respectively), while the stereoselectivity of these substrates were similar. Substituents at the meta and para position are equally well tolerated. Both electron-donating and electron-withdrawing groups provided the corresponding products in high yields and enantioselectivities (3g–s). Multisubstituted and naphthalenyl-substituted α-enaminones also proceeded smoothly to give the corresponding products in up to 99% yield with excellent stereoselectivity (3t–v). α-Enaminone derived from cyclopentanone was also investigated. While the enantioselectivity remained high (3w, 99% ee), the yield decreased to 39% likely due to ring strain. Enaminone derived from substituted cyclohexanone generated product 3x in good yield of the major diastereomer with excellent enantioselectivity. The absolute configuration of 3a was established unambiguously to be (5aS,9aR) through X-ray crystallographic analysis (Table 2, CCDC 1919282).10
:9 → 95:5 d.r.) and enantioselectivities (95–99% ee). Changing the ester (R1) group of quinone from methyl to ethyl and benzyl didn't affect the reactivity and enantioselectivity (3a–c). Regarding the ortho substituents on the phenyl ring of α-enaminones, a weak electron-donating group such as methyl (3d) gave slightly better yield (96%) than slightly electron-withdrawing groups such as chlorine and bromine (3e & 3f, 85% and 81% respectively), while the stereoselectivity of these substrates were similar. Substituents at the meta and para position are equally well tolerated. Both electron-donating and electron-withdrawing groups provided the corresponding products in high yields and enantioselectivities (3g–s). Multisubstituted and naphthalenyl-substituted α-enaminones also proceeded smoothly to give the corresponding products in up to 99% yield with excellent stereoselectivity (3t–v). α-Enaminone derived from cyclopentanone was also investigated. While the enantioselectivity remained high (3w, 99% ee), the yield decreased to 39% likely due to ring strain. Enaminone derived from substituted cyclohexanone generated product 3x in good yield of the major diastereomer with excellent enantioselectivity. The absolute configuration of 3a was established unambiguously to be (5aS,9aR) through X-ray crystallographic analysis (Table 2, CCDC 1919282).10
| a All reactions were carried out with 1 (0.1 mmol), 2 (0.11 mmol), 4 Å M.S. (5.0 mg) and (R)-CPA5 (1.0 mol%) in CH2Cl2 (1.0 mL) at −78 °C for 16 h. b Isolated yield. c The d.r. and ee values of the products were determined by HPLC analysis on a chiral stationary phase. d 20.0 mg of 4 Å M.S. e 5.0 mol% of (R)-CPA5. f The reaction was conducted on 0.05 mmol scale with 10 mol% of (R)-CPA5 and 10.0 mg of 4 Å M.S. at −60 °C. g 2w (0.22 mmol) and (R)-CPA5 (10.0 mol%) at −60 °C, isolated yield of the major diastereomer. |
|---|

|
α-Enaminones derived from an aliphatic amine, i.e., benzylamine, did not give the expected 2,3-dihydrobenzofuran derivative (3y). Instead, 2,3-dihydrobenzofuran bearing a hydroxy group (3z) was obtained in 92% yield and 92% ee (Scheme 2, eqn (1)). The structure and the absolute configuration of 3z was assigned to be (5aS,9aR) with X-ray crystallographic analysis (CCDC 1921254).10 Theoretically, product 3z could result from the hydrolysis of 3y (Scheme 2, eqn (1)). It is also possible that 2u first decomposes to 1,2-cyclohexanedione 2x, then react with 1a producing 3z. Reaction of 1a with 2x generated 3z in high yield (95%), however, the ee was low (11%) (Scheme 2, eqn (2)). These data suggest that 3z obtained from enaminone 2u was through hydrolysis of intermediate 3y, and that the activity of aliphatic amine derived enaminones is different from arylamine derived enaminones.
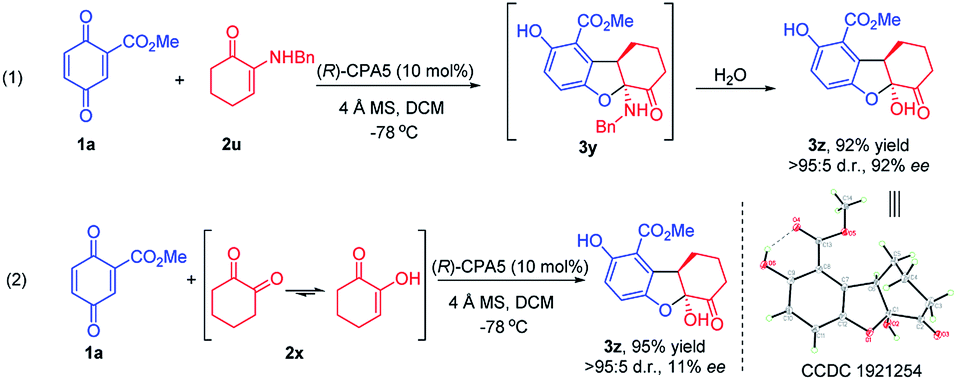 | ||
| Scheme 2 Catalytic formation of 2,3-dihydrobenzofuran 3w. | ||
To probe the interactions between CPA with α-enaminone 2u and 1,2-cyclohexanedione 2x, control 1H NMR experiments were carried out. Analysis of these 1H NMR spectra (Fig. S5 and S6, see ESI†) suggests that the interaction between CPA5 and α-enaminone 2u is significantly stronger than that of CPA and 2x, supporting the reaction pathway hypothesized above. These data also further support our earlier hypothesis that chiral phosphoric acid could mediate this reaction due to the existence of multifunctional groups capable of H-bonding in the starting materials.
Next, the substrate scope for N-substituted indoles was screened (Table 3). Quinone 1a reacted with α-enaminone 2a to afford 4a in 99% yield. When the ester group of quinone 1 changed from methyl to ethyl and benzyl, the yields were somewhat decreased (4b & 4c). α-Enaminones, which bear both electron-donating and electron-withdrawing substituents at the para-position of the phenyl ring gave high yields of indoles (4i–4p). α-Enaminones carrying electron-donating substitutes at the meta-position of the phenyl ring resulted in higher yields than α-enaminones carrying electron-withdrawing substitutes at the meta-position. 3,4-Disubstitution on the phenyl ring of α-enaminones also led to the formation of indoles in good yields (4q & 4r). This reaction turned out to be challenging for α-enaminones with ortho substituents on the phenyl ring, producing the desired indoles in only 10% yield (Table S1, see ESI†). Nevertheless, this method could potentially provide an easy access to indoles with axial chirality, which is a hot topic currently,11 and is worthy of further investigation. N-Benzyl protected indoles (4s) was obtained in low yield (31%), once again demonstrating different activity of arylamine-based α-enaminones from aliphatic amine-based α-enaminones. Notably, α-enaminones derived from cyclopentanone and 4-methyl cyclohexanone generated adducts 4t (40% yield) and 4u (48% yield), respectively. It was noteworthy that condition B using an achiral phosphoric acid as the catalyst exhibited good yields with selected substrates. The structure of 4a was confirmed with X-ray crystal analysis (Table 3, CCDC 1919286).10
| a Condition A: 1 (0.1 mmol), 2 (0.1 mmol) and (R)-CPA3 (5.0 mol%) in toluene (1.0 mL) at −78 °C for 16 h; condition B: 1 (0.1 mmol), 2 (0.1 mmol) and PA (10 mol%) in DCM (1.0 mL) at −78 °C for 16 h. b Isolated yield of condition A, the results in parentheses were obtained with condition B. c 10 mol% of (R)-CPA3. d The reaction was conducted on 0.05 mmol scale with 10 mol% of (R)-CPA5 at −60 °C. e At −60 °C for 18 h. |
|---|

|
To show the prospect of the current methodologies in synthesis, the divergent reaction of quinone ester 1a with α-enaminone 2a was expanded to gram scale (3.0 mmol), and the desired 2,3-dihydrobenzofuran product 3a was obtained in 94% yield with 98% ee (Scheme 3a). Reduction of the carbonyl group in 3a using NaBH4 afforded β-amino alcohol 5 in excellent yield with the retention of the enantioselectivity (Scheme 3b).
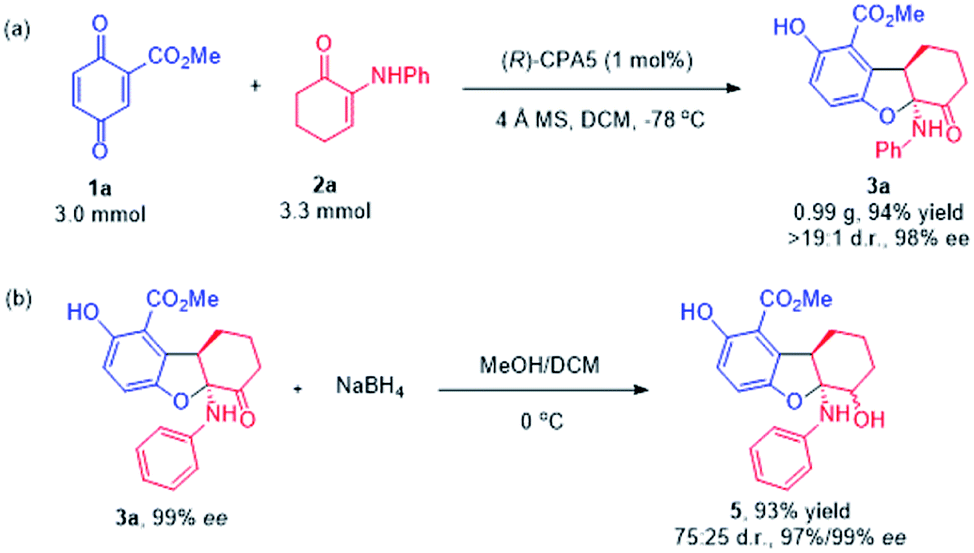 | ||
| Scheme 3 (a) Gram-scale version of the reactions. (b) Synthetic utility. | ||
Control experiments were carried out to probe the roles of phosphoric acid and M.S. playing in this reaction (Table 4). The general function of M.S. is to take out water from a reaction. Molecule sieves are also known to mediate the interaction of water with substrates/intermediates in a reaction.9b,12 Reactions with nonactivated 4 Å M.S. or freshly activated 4 Å M.S. (entries 1 and 2) gave similar results, both generating benzofuran 3a as the major product in high enantioselectivity (99% ee). However, when dry MgSO4 was used instead of M.S. (entry 9), the reaction pathway switched to favor the formation of 4a, similar to the reaction without inclusion of M.S. (entry 3). When wet 4 Å M.S. was used (entries 6 & 8), the formation of 3a slowed down, at the same time the formation of 4a sped up. These results suggest that, (1) M.S. is not serving a drying reagent for this reaction; (2) extra water added to this reaction influences the reaction pathways to slightly favor indole 4a formation. It was noticed that the size and format of M.S. also affected the results of this reaction (entries 1, 4, 5, & 7). We suspect that the M.S. acts to facilitate proton (H+) transfer.9b1H NMR and 31P NMR spectroscopy of chiral phosphoric acid ((R)-CPA5) with and without M.S. were then conducted in benzene-d6 (Fig. 2). In the absence of M.S., a broad peak was found for the phosphoric acid proton centered at 1.80 ppm; addition of M.S. broadened and shifted this peak significantly to the downfield centered at 5.19 ppm. Similar changes were observed on 31P NMR spectra, with a phosphine peak at 3.23 ppm without M.S., and 3.94 ppm in the presence M.S. It is evident from these experiments that M.S. can potentially affect the acidity of phosphoric acid and mediate the interaction between a substrate and a catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | Variation from the “standard conditions” | Conv.b (%) |
3a:4ab |
ee of 3ac (%) |
| a All reactions were carried out with 1a (0.1 mmol), 2a (0.11 mmol), 4 Å M.S. (5.0 mg) and (R)-CPA5 (10 mol%) in CH2Cl2 (1.0 mL) at −78 °C for 16 h. b Determined by 1H NMR. c Determined by HPLC analysis on a chiral stationary phase. d Freshly activated 4 Å M.S. and H2O (2 μL). e M.S. and phosphoric acid were not present. | ||||
| 1 | None | 97 | >20:1 |
99 |
| 2 | Freshly activated 4 Å M.S. | 86 | 20:1 |
99 |
| 3 | No M.S. | 94 | 1:4.9 |
99 |
| 4 | 3 Å M.S. instead of 4 Å M.S. | 95 | >20:1 |
99 |
| 5 | 5 Å M.S. instead of 4 Å M.S. | 94 | 11:1 |
99 |
| 6d | Wet 4 Å M.S. | 99 | 1:1.1 |
99 |
| 7 | 4 Å M.S. (beads) | 99 | 1:1 |
99 |
| 8d | Wet 4 Å M.S. (beads) | 99 | 1:5.3 |
99 |
| 9 | Dry MgSO4 instead of 4 Å M.S. | 99 | 1:4.2 |
99 |
| 10 | Benzoic acid only | 85 | 5:1 |
0 |
| 11e | Et3N without 4 Å M.S. | 15 | >20:1 |
0 |
| 12 | Et3N with 4 Å M.S. | 13 | >20:1 |
0 |
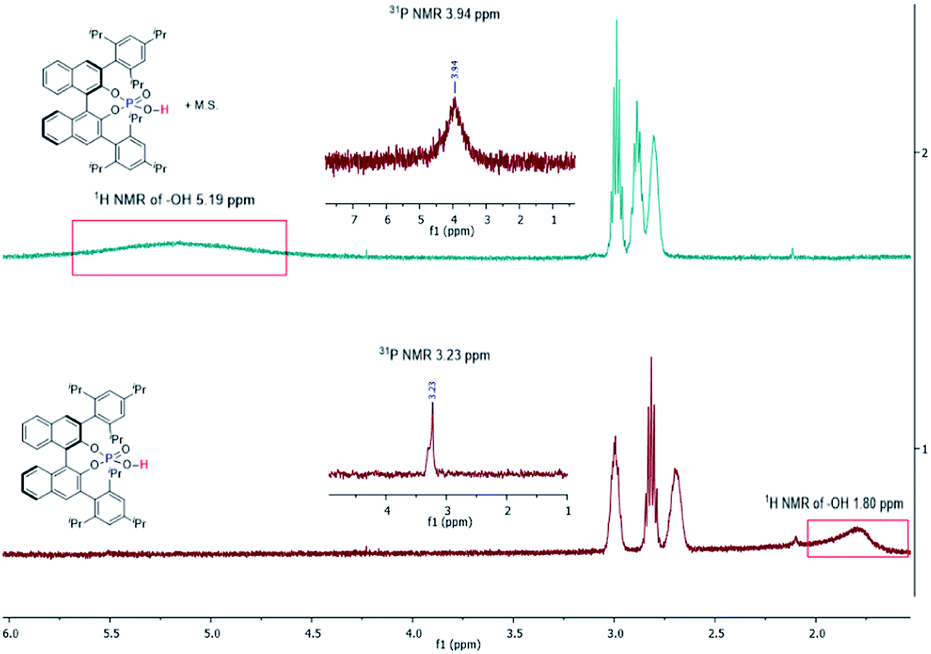 | ||
| Fig. 2 1H and 31P NMR spectra for the investigation of the interaction of phosphoric acid with M.S. in C6D6. | ||
In order to further illustrate the role of the phosphoric acid, an experiment with a different acid, i.e., benzoic acid, was conducted (entry 10), leading to similar results with the background reaction (Table 1, entry 1). This result suggests that the strength of the acid matters in this reaction. Experiments with inclusion of a tertiary amine in the presence and absence of M.S. were also conducted (entries 11 & 12). Both reactions generated 3a as the only product, however, with 0% ee, and in much decreased yields as compared with the background reaction. These data indicate: (1) the acid function of the phosphoric acid is important for this reaction, (2) M.S. is likely to mediate proton transfer in this reaction, as no effect of M.S. was observed after the phosphoric acid was neutralized with a tertiary amine.
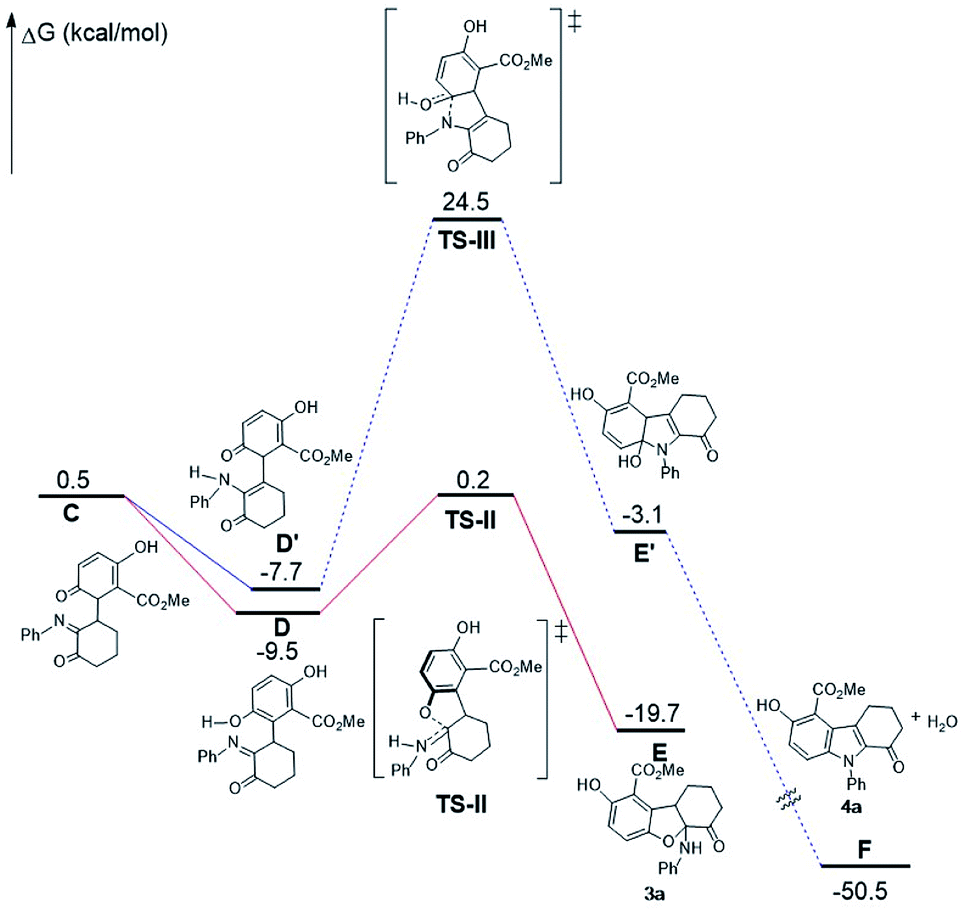
A mechanism is proposed to illustrate the reaction process and stereocontrol of the divergent reaction based on the experimental data and X-ray structures of 3a and 4a (Scheme 4). α-Enaminone acts as an enamine nucleophile to attack from the Si face at the Re face of the quinone (Michael addition) through TS-I to form intermediate (1R,1′R)-int A. TS-I′ is expected to be disfavored due to steric hindrance (Fig. S12, see ESI†). Intermediate (1R,1′R)-int A equilibrates to intermediate int B and int C through O-tautomerization (enol/phenol from quinone) and N-tautomerization (enamine from imine), respectively. Intermediate int B and int C then bind CPA as illustrated in transition states TS-II and TS-III. TS-II leads to the formation of 2,3-dihydrobenzofuran ((5aS,9aR)-3a) through hydroxy group attack at the Si face of the imine, and TS-III gives rise to intermediate int D though amine group attack at the Si face of the carbonyl group followed by dehydroxylation to afford indole (4a). TS-II is more polarized in nature than TS-III, as imine nitrogen is a better proton acceptor than carbonyl oxygen, and hydroxy group in phenol is a better acid than amines. As such, in a more polar solvent such as DCM, TS-II is better stabilized. The addition of 4 Å molecular sieves (M.S.) is likely to facilitate the proton transfer in TS-II through absorbing/releasing proton.9b In non-polar solvent such as toluene, TS-II is destabilized, and the reaction through TS-III becomes dominant.
 | ||
| Scheme 4 Proposed mechanism. | ||
DFT calculations (B3LYP/6-311+G(d,p)/CPCM-DCM//B3LYP/6-31G(d)) are performed to gain more mechanistic insight into this novel double divergent process. The calculated free energy profile is shown in Fig. 3 to illustrate the reaction process and stereocontrol of the divergent reaction based on the experimental and computational data and X-ray structures of 3a and 4a. α-Enaminone and quinone ester coordination to the phosphoric acid forming the heterotrimer is used as the reference point for the free energy profile. Formation of the adduct (B) is shown to be 2 kcal mol−1 more favored in free energy than the separated reactants (A). To form intermediate int A (D), α-enaminone acts as an enamine nucleophile to attack the quinone (Michael addition) through TS-I with a barrier of 10.9 kcal mol−1. The formation of int A intermediate coordination complex (C) is close to thermoneutral, which is 0.7 kcal mol−1 more stable than the reactant adducts (B). The dissociation of int A and the rotation of C–C bond formed in TS-I is calculated to be 3.2 kcal mol−1 uphill from C to D. Intermediate int A equilibrates to intermediate int B and int C through O-tautomerization (enol/phenol from quinone) and N-tautomerization (enamine from imine), respectively. Calculations show that O-tautomerization and forming new hydrogen bonds with CPA (E′) is 15.2 kcal mol−1 downhill and that of N-tautomerization (E) is exergonic by 16.9 kcal mol−1 relative to reactant adduct B.
 | ||
| Fig. 3 Free-energy profiles calculated for the pathways catalyzed by phosphoric acid. The relative free-energies are in kcal mol−1. | ||
TS-II leads to the formation of 2,3-dihydrobenzofuran (3a) through hydroxy group attack of the imine. TS-III gives rise to intermediate int D though amide group attack the carbonyl group followed by dehydroxylation to afford indole (4a). The calculated barriers for TS-II and TS-III are 8.8 kcal mol−1 and 9.1 kcal mol−1, respectively. We calculated different initial geometries of hydrogen-bonding modes for TS-III between the amide and the phosphoric acid. The optimized structure of TS-III suggests the proton transfer between the catalyst and int C and the amide dissociation from the catalyst happen prior to the amide attack. TS-II is more polarized in nature than TS-III, as amide nitrogen is a better proton acceptor than carbonyl oxygen, and hydroxy group in phenol is a better acid than amines.
Calculations on enantio-determining TSs with full CPA5 catalyst shows that TS-I′ is disfavored by 13.7 kcal mol−1 compared to TS-I (Fig. 4). No significant steric clashes were observed in the TS structures. But the structure of TS-I shows that the quinone ester is not coordinated to the catalyst, while CPA5, 1a and 2a form heterotrimer via hydrogen bonding with the catalyst in TS-I′. This result suggests that locking the two substrates is not necessary for lowering the activation barrier, especially if larger and more sterically shielding groups are used on the catalyst. Computational results also show that the formation of (1R,1′R)-int A is favored by 5.6 kcal mol−1 in free energy compared to (1R,1′S)-int A. The background reaction without catalyst is also calculated with the same level of theory (Fig. S15, see ESI†). TS-I has a much higher barrier without the catalyst (18.9 kcal mol−1) than with catalyst (10.9 kcal mol−1). Calculations show that TS-II is slightly stabilized (∼1 kcal mol−1) when catalyst is present. The reaction did not proceed viaTS-III without catalyst is likely due to the inability to form amide for the attack without activation by phosphoric acid (Fig. 5).
 | ||
| Fig. 4 Optimized geometries and the key bond lengths of TS-I and TS-I′ with CPA5. | ||
 | ||
| Fig. 5 Free-energy profiles calculated for the background reaction without catalyst. | ||
Conclusions
The first example of a double divergent process has been developed for the reaction of α-enaminone with quinone ester. This double divergent process involves a catalyst (phosphoric acid)-controlled and an additive (molecular sieves)-controlled process. Facile manipulation of catalyst and additive leads to structurally completely different products. The roles of phosphoric acid and the molecular sieves were investigated through experiments and theoretical calculations. Our studies suggest that the phosphoric acid serves to lower the transition state energy and to promote amide formation leading to the formal aza-[3 + 2] cycloaddition to give the thermodynamic product. Our studies also show that M.S. do not play its traditional role as drying agent and/or H2O mediator in the reactions. It was found that M.S. could affect the acidity of the phosphoric acid and serve to facilitate electron transfer in the process, disclosing a new role of molecular sieves. It is proposed that M.S. initiate a kinetic pathway to favor the formal oxo-[3 + 2] cycloaddition in the presence of phosphoric acid catalyst.This work has introduced new reaction patterns, providing two novel formal oxo- and aza-[3 + 2] cycloaddition reactions. Both reactions are highly efficient, producing the corresponding 2,3-dihydrobenzofurans and N-substituted indoles in good yields with wide substrate scopes. The formal oxo-[3 + 2] cycloaddition reaction is highly stereoselectivity with up to >95:5 d.r. and 99% ee. Theoretical calculations conducted for enantio-determining TSs support the experimental data.
This work has also revealed a new set of reactivity of rarely explored α-enaminones. The α-enaminones displayed multifunctionality serving as both an enamine and a nitrogen nucleophile, and an imine electrophile in the divergent reaction. This divergent reaction offers new concise routes to access enantioenriched 2,3-dihydrobenzofurans and N-substituted indoles, both of which are of biological and synthetic importance. Investigation of new reactions of α-enaminones are underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Science Foundation MRI Program (CHE-1726652) and the University of North Texas for supporting the acquisition of the Rigaku XtaLAB Synergy-S X-ray diffractometer. Z. S. and T. R. C. thank the NSF for their support of this work via grants CHE-1464943 and CHE-1531468, the latter funding the purchase of the UNT Dept. of Chemistry high-performance computing cluster. We thank Dr Guido Verbeck and the Laboratory for Imaging Mass Spectrometry at the University of North Texas for Mass Spectrometry data. W. L. is grateful for University of North Texas for providing financial support.Notes and references
- For selected reviews, see:
(a) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954 CrossRef CAS PubMed
; (b) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC
- For selected examples, see:
(a) S. Brandau, E. Maerten and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 14986 CrossRef CAS PubMed
-
(a) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303 CrossRef PubMed
-
(a) J. V. Greenhill, Chem. Soc. Rev., 1977, 6, 277 RSC
-
(a) M. Rueping and A. Parra, Org. Lett., 2010, 12, 5281 CrossRef CAS PubMed
-
(a) Z. Xu, L. Liu, K. Wheeler and H. Wang, Angew. Chem., Int. Ed., 2011, 50, 3484 CrossRef CAS PubMed
-
(a) C. Paal, Ber. Dtsch. Chem. Ges., 1884, 17, 2756 CrossRef
-
(a) D. A. Evans and J. Wu, J. Am. Chem. Soc., 2003, 125, 10162 CrossRef CAS PubMed
-
(a) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed
- CCDC 1919282 (3a), 1921254 (3w) and 1919286 (4a) contain the supplementary crystallographic data for this paper.
-
(a) S. L. Li, C. Yang, Q. Wu, H. L. Zheng, X. Li and J. P. Cheng, J. Am. Chem. Soc., 2018, 140, 12836 CrossRef CAS PubMed
-
(a) L. Chang, Y. L. Kuang, B. Qin, X. Zhou, X. H. Liu, L. L. Lin and X. M. Feng, Org. Lett., 2010, 12, 2214 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1919282, 1921254 and 1919286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02078h |
| This journal is © The Royal Society of Chemistry 2020 |