
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking the barrier: an osmium photosensitizer with unprecedented hypoxic phototoxicity for real world photodynamic therapy†
John A.
Roque
III
 ab,
Patrick C.
Barrett‡
a,
Houston D.
Cole
b,
Liubov M.
Lifshits
b,
Ge
Shi§
c,
Susan
Monro
c,
David
von Dohlen
a,
Susy
Kim
d,
Nino
Russo
e,
Gagan
Deep
d,
Colin G.
Cameron
*ab,
Marta E.
Alberto
*e and
Sherri A.
McFarland
*abc
ab,
Patrick C.
Barrett‡
a,
Houston D.
Cole
b,
Liubov M.
Lifshits
b,
Ge
Shi§
c,
Susan
Monro
c,
David
von Dohlen
a,
Susy
Kim
d,
Nino
Russo
e,
Gagan
Deep
d,
Colin G.
Cameron
*ab,
Marta E.
Alberto
*e and
Sherri A.
McFarland
*abc
aDepartment of Chemistry and Biochemistry, The University of North Carolina at Greensboro, Greensboro, North Carolina, 27402 USA
bDepartment of Chemistry and Biochemistry, The University of Texas at Arlington, Arlington, Texas, 76019 USA. E-mail: colin.cameron@uta.edu; sherri.mcfarland@uta.edu
cDepartment of Chemistry, Acadia University, Wolfville, Nova Scotia, B4P 2R6 Canada
dDepartment of Cancer Biology, Wake Forest School of Medicine, Winston Salem, NC, 27157, USA
eDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Arcavacata di Rende, 87036 Italy. E-mail: marta.alberto@unical.it
First published on 3rd August 2020
Abstract
Hypoxia presents a two-fold challenge in the treatment of cancer, as low oxygen conditions induce biological changes that make malignant tissues simultaneously more aggressive and less susceptible to standard chemotherapy. This paper reports the first metal-based photosensitizer that approaches the ideal properties for a phototherapy agent. The Os(phen)2-based scaffold was combined with a series of IP-nT ligands, where phen = 1,10-phenanthroline and IP-nT = imidazo[4,5-f][1,10]phenanthroline tethered to n = 0–4 thiophene rings. Os-4T (n = 4) emerged as the most promising complex in the series, with picomolar activity and a phototherapeutic index (PI) exceeding 106 in normoxia. The photosensitizer exhibited an unprecedented PI > 90 (EC50 = 0.651 μM) in hypoxia (1% O2) with visible and green light, and a PI > 70 with red light. Os-4T was also active with 733 nm near-infrared light (EC50 = 0.803 μM, PI = 77) under normoxia. Both computation and spectroscopic studies confirmed a switch in the nature of the lowest-lying triplet excited state from triplet metal-to-ligand charge transfer (3MLCT) to intraligand charge transfer (3ILCT) at n = 3, with a lower energy and longer lifetime for n = 4. All compounds in the series were relatively nontoxic in the dark but became increasingly phototoxic with additional thiophenes. These normoxic and hypoxic activities are the largest reported to date, demonstrating the utility of osmium for phototherapy applications. Moreover, Os-4T had a maximum tolerated dose (MTD) in mice that was >200 mg kg−1, which positions this photosensitizer as an excellent candidate for in vivo applications.
1. Introduction
Cancer remains a leading cause of death in many countries.1 There are ongoing efforts to develop adjuvant or alternative approaches that complement conventional treatment modalities, e.g., surgery, radiotherapy, chemotherapy, and more recently immunotherapy.2–4 A persistent challenge has been resistance, either intrinsic or acquired, which causes subpopulations of cancerous cells to evade treatment and potentially metastasize.5–7 There are multiple mechanisms for resistance, but specific physiological conditions, particularly hypoxia, are known to drive the mutator phenotype, and increase the ability for cancer cells to survive and persist despite the utilization of different therapeutic approaches.7–10Light-based therapies, such as photodynamic therapy (PDT) and the relatively new field of photochemotherapy (PCT), are examples of locally administered treatments where light-responsive prodrugs are used to selectively target tumors while sparing healthy tissue. PDT, which has been known for over a hundred years and is clinically approved,11–23 employs an otherwise nontoxic photosensitizer (PS) that can be activated by therapeutic wavelengths of light to form cytotoxic singlet oxygen (1O2) and other reactive oxygen species (ROS). Due to this oxygen dependence, PDT can be less effective in treating hypoxic tumors. However, the very fact that ROS do not rely on a single biological mechanism of action (in contrast to kinase inhibitors, for example) makes it more difficult for cells to achieve resistance to PDT.24 This motivates new approaches to circumvent the limitations of low oxygen tension.
Alternatively, PCT has been proposed as a strategy that exploits oxygen-independent reaction pathways (e.g., photocaging or photocisplatin agents) with the potential advantage of maintaining activity toward hypoxic malignancies. Ru(II)-based metal complexes have emerged as PCT agents with modular architectures that can be tuned to be relatively inert in the absence of light, to photodecompose via ligand loss with visible light in the absence of oxygen, and to be less susceptible than other metallodrugs like cisplatin25 to deactivation by cellular thiols.26–41 Many of these Ru(II) coordination complexes are relatively simple to synthesize in high purity, exhibit aqueous solubility as their chloride salts, and have photophysical properties that can be tuned by rational design according to the well-developed principles from the Ru(II) polypyridyl literature.23,42–47
In contrast to PDT, PCT agents rely on engaging a specific cellular target or utilizing a specific mechanism of action, which cancer cells may already be able to evade, particularly under hypoxic conditions. In addition to the potential issues associated with utilizing specific mechanisms of action, PCT agents possess some technical limitations. Phototriggered ligand dissociation is a stoichiometric process that would require higher dosing than a catalytic drug to achieve efficacy. In addition, a mixture of potentially new (and difficult to identify) decomposition products are produced in situ. The impact of a hypoxic environment (with different pH, potential quenching mechanisms, and protein expression levels) on the photochemical process is challenging to ascertain.
Indeed, a careful review of the literature reveals that very few systems – that are believed to be active under hypoxia – have actually been tested against cells under low oxygen conditions.41,48–54 Likewise, relatively few systems have been tested for photodissociation in cell-free conditions under low oxygen conditions (i.e., 0–3%).37,49,50,52,55 Performing experiments under hypoxic conditions is challenging due to a variety of factors such as the initial establishment of low oxygen tension or differences in cellular growth and metabolism – if the cells can even grow at the desired oxygen level. A hypoxia chamber for cell incubation and processing is required to establish and maintain a low oxygen atmosphere, but many light devices do not fit inside the chamber and/or result in excessive heating in the chamber. Maintaining a low oxygen atmosphere during the time of irradiation outside the chamber is challenging but critical since the PCT agents are designed to release their phototoxic payloads immediately upon light exposure. We have applied qPCR (high transparency) films over well plates for this purpose (up to 384 wells). While not perfectly gas impermeable, they do mitigate gas exchange over the period of the light treatment (∼20 °C, ≤1.5 h). Interassay reproducibility (in terms of maintaining hypoxia during illumination) is excellent so long as oxygen-sensitive controls for leaky films or poor gas exchange are included.
We are committed to developing hypoxia active metal complexes and have established a protocol for effective testing. Using this rigorous experimental design for evaluation, we developed one compound with Glazer's group, [Ru(6,6′-dmb)2(1-NIP)]Cl2, that exhibited the largest known hypoxia phototherapeutic index (PI) at the time of its publication (PI = 15 at 1% O2). This is marginal compared to good normoxic values for PDT agents (PI > 1000), and unfortunately, it did not work with red light (630 nm), the clinically approved wavelength for PDT.41 Similar PIs for PCT agents that release cytotoxic ligands and/or metal complexes have been reported.27,29,50,56
These findings inspired us to consider other ways of instilling hypoxic activity in Ru(II) complexes that are photostable and exploit photocatalytic pathways/mechanisms for cytotoxic effects. Our approach is to create metal complex PSs that combine powerful 1O2 sensitization, such that some activity can be maintained even in hypoxia, with the possibility of light-induced electron transfer reactions via triplet intraligand charge transfer (3ILCT) excited states. Our own TLD1433, now in phase II human clinical trials (NCT03945162) for treating bladder cancer with PDT, was born out of this work.23
In the present study we demonstrate the feasibility of this approach using Os(II) as the metal combined with the same class of oligothienyl-appended ligands that feature in TLD1433.23,57 The Os(II) complexes provide longer wavelength absorption windows compared to their Ru(II) analogues, due to the more pronounced heavy atom effect.58 Larger spin–orbit coupling (SOC) associated with the Os(II) center also leads to faster intersystem crossing (ISC), producing triplet states in high yield.59,60 The prolonged excited state lifetimes characteristic of 3ILCT states result in sensitivity to trace oxygen.61 Finally, we have previously shown that certain Os(II) complexes are tolerated in vivo, can be panchromatically activated with visible and near-infrared (NIR) wavelengths of light, and give rise to phototoxic effects both in vitro and in vivo.62,63
Here we introduce an Os(II)-based PS (Os-4T) that is extremely potent, can be activated with NIR light (733 nm), and exhibits photocytotoxic effects in hypoxia (1% O2). To our knowledge, this complex is the first hypoxia-active Os(II)-based PS and it exhibits one of the largest PIs that we have encountered to date. This PS was part of a systematic study of a family of related [Os(phen)2(IP-nT)]Cl2 complexes, where phen = 1,10-phenanthroline, IP = imidazo[4,5-f][1,10]phenanthroline, and nT = thiophenes of varying chain lengths n (n = 0–4) (Chart 1). This allowed for a rigorous investigation of the effects of n on computed ground and excited state properties, time-resolved photophysics, and photobiology. In this report, we discuss these studies and provide a rationale for the observed photobiology, along with a road map for the theory-driven design of next-generation agents.
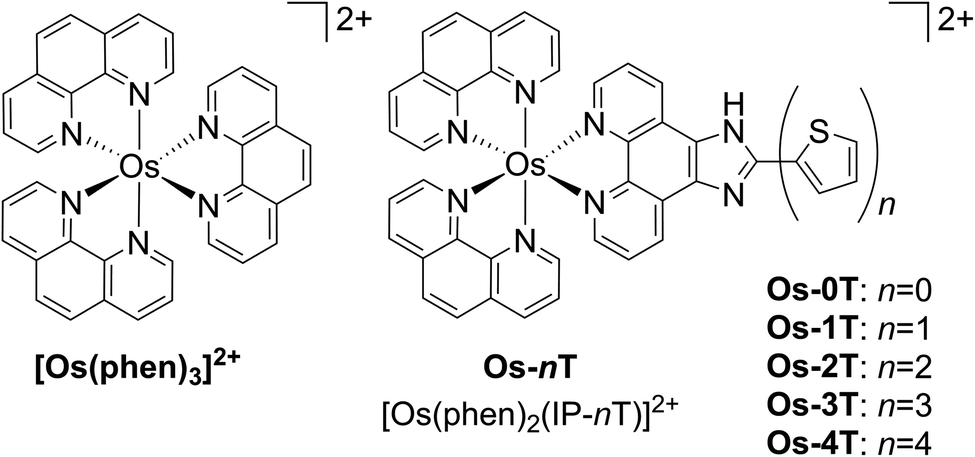 | ||
| Chart 1 Molecular structures of reference compound [Os(phen)3]2+ and Os-0T–Os-4T. The chloride salts were used unless otherwise specified. The compounds were racemic mixtures of the Δ/Λ isomers. | ||
2. Materials and methods
2.1 Materials
Unless otherwise specified, all reagents and solvents were purchased from commercial sources and used without further purification. Water used for all biological experiments was deionized to a resistivity ≥ 18.2 MΩ using either a Barnstead or Milli-Q® filtration system. Saturated solvents for log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D experiments were prepared in-house using 10 mM phosphate buffer solution (saline-free PBS) and 1-octanol (99.9%). Buffers were checked for an accurate pH against a two-point calibrated VWR® B10P pH meter (pHref. = 4.00, 7.00; Fisher Science Education, S25849A/B).
D experiments were prepared in-house using 10 mM phosphate buffer solution (saline-free PBS) and 1-octanol (99.9%). Buffers were checked for an accurate pH against a two-point calibrated VWR® B10P pH meter (pHref. = 4.00, 7.00; Fisher Science Education, S25849A/B).
2.2 Instrumentation
Microwave reactions were performed in a CEM Discover microwave reactor. Flash chromatography relied on the Teledyne Isco CombiFlash® EZ Prep system with Silicycle SiliaSep silica flash cartridges (FLH-R10030B-ISO25). Size-exclusion chromatography was performed on a gravity column packed with Sephadex® LH-20. NMR spectra were collected using a JEOL ECA 500 NMR spectrometer (1H, 1H–1H COSY) at the NMR facility at the University of North Carolina at Greensboro (UNCG) and Agilent 700 MHz NMR spectrometer (13C, 13C–1H HSQC, 13C–1H HMBC) at the Joint School of Nanoscience and Nanoengineering at Greensboro (JSNN). The chemical shifts are reported in parts per million (ppm) and were referenced to the residual solvent peaks. ESI mass spectra were obtained using a Thermo Fisher LTQ Orbitrap XL coupled to a Water's Acquity Ultra Performance Liquid Chromatography (UPLC) stack using a BEH C18 column at UNCG's Triad Mass Spectrometry facility. HPLC analyses were carried out on an Agilent/Hewlett Packard 1100 series instrument (ChemStation Rev. A. 10.02 software) using a Hypersil GOLD C18 column (Thermo 25005-254630, guard 25003-014001) with an A–B gradient (40 min run; 98% → 5% A; A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile (MeCN)). Reported retention times are accurate to within ±0.1 min.2.3 Synthesis
The Os(phen)2Cl2 intermediate58 and imidazo[4,5-f][1,10]phenanthroline (IP) based ligands64 were prepared according to adapted literature procedures. The synthesis of IP-based ligands follows the synthesis of IP-4T that is described below. [2,2′:5′,2′′:5′′,2′′′-Quaterthiophene]-5-carbaldehyde (4T-CHO) was prepared as previously described65via the coupling of 5-bromo-5′′-formyl-2,2′:5′,2′′-terthiophene with 2-(tributylstannyl)thiophene, which were purchased from Alfa Aesar and Fisher Scientific, respectively.Complexes Os-0T–Os-4T have not previously been reported and were isolated as their PF6− salts. Cl− salts were obtained from corresponding PF6− salts via anion metathesis on HCl-treated Amberlite IRA-410 resin (Alfa-Aesar, A1773436) with methanol as the eluent. Final complexes were a racemic mixture of Δ/Λ isomers. [Os(phen)3]2+ and Os-0T–Os-4T were characterized by TLC, 1H NMR and 1H–1H COSY NMR (Fig. S-1–S-6†), high-resolution ESI+-MS (Fig. S-10–S-15†), and HPLC (Fig. S-16–S-21†). COSY was not collected for [Os(phen)3]2+. NMR assignments were made in consultation with a literature source.66 Compound Os-4T required additional 13C, 13C–1H HSQC, and 13C–1H HMBC NMR data for the full assignment of the 1H NMR signals of the quaterthiophene group (Fig. S-7–S-9†). Biological studies were carried out on Cl− salts and photophysical measurements were carried out on PF6− salts.
2.4 Lipophilicity
The relative lipophilicities (or distribution coefficient, D, at pH = 7.4) of the complexes were determined using a modified “shake flask” method as described in previous work.65 A saturated solution of 1-octanol with water was prepared by mixing 16 mL of 1-octanol (99.9%) with 4 mL of 10 mM PBS. A saturated solution of PBS with 1-octanol was prepared by mixing 16 mL of PBS with 4 mL of 1-octanol. The saturated solutions were shaken for 24 h at ambient temperature (≈22 °C) using a New Brunswick Classic C25KC Incubator Shaker set at 230 rpm before further use. Starting with 500 μL of 50 μM complex (chloride salt) prepared in saturated 1-octanol, an equal volume of saturated water was added to give a total volume of 1 mL. The mixtures were shaken 200 times, centrifuged at 11000 rpm (≈10000 × g) for 2 minutes using a BioRad Model 16K Microcentrifuge; the two resultant layers were then separated with a syringe. The concentrations of the complexes in both octanol and water layers were calculated from absorption measurements using a microplate reader (SpectraMax M2e) and standard curves in each solvent.
2.5 Computational details
The photophysical properties and the feasibility of Type I and Type II photoreactions of Os(II)-compounds were explored at DFT and TDDFT levels of theory,68 as implemented in Gaussian09 code.69 Ground singlet and triplet state geometry optimizations were performed in water without constraints by using PBE0 exchange–correlation functional (XC),70 and adopting the IEFPCM model (integral equation formalism polarizable continuum model)71,72 to simulate the solvent environment (ε = 80). The Os(II) metal ion was described by using the quasi-relativistic Stuttgart–Dresden pseudopotential73 while the 6-31+G(d,p) basis set was adopted for all the other atoms.Absorption spectra were obtained in water on the ground state equilibrium structures, using the same basis set as for the optimizations by employing the M06 XC-functional,74 which has been widely tested and verified in terms of its reliable prediction of the photophysical properties of metallic compounds.75 The selected protocol allows a direct comparison with results previously obtained for analogous Ru(II)-compounds.76
2.6 Spectroscopy
Spectroscopic measurements were obtained from dilute (5–20 μM) solutions of the PF6− salts of the complex in spectroscopic grade MeCN that had been further distilled over CaH2 under argon. Deaerated solutions were prepared by five freeze–pump–thaw repetitions in custom Schlenk-style quartz cuvettes (10 mm path length) for transient absorption measurements and by sparging with argon in septum-capped quartz cuvettes (10 mm path length) for emission. Ambient air was bubbled through solutions to be used for singlet oxygen measurements.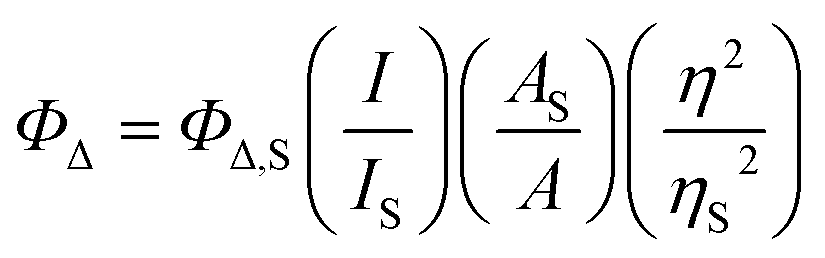 | (1) |
The ΦΔ experiments were performed in triplicate for MeCN solutions of the PF6− salts. MeCN was used because water quenches the 1O2 emission, and PF6− salts in MeCN are commonly used in the literature for comparison. The excitation wavelength was the longest wavelength in the excitation spectrum that maximized emission at 1268 nm. The emission was collected between 1200–1350 nm using a 1000 nm long-pass filter. Values were generally reproducible within ±5%.
2.7 Cellular assays
2.7.2.1 Normoxia. Cell viability experiments in normoxia were performed and data analyzed as previously described.23,41 The light treatments used for full dose–response included cool white visible (400–700 nm; maximum ≈450 nm), green (523 nm), and red (633 nm) light at an irradiance of 20 mW cm−2 for a total fluence of 100 J cm−2. External validation experiments were specific to Acadia University and used a broadband BenQ projector visible light source and red (630 nm) LED panel at an irradiance of 33–40 mW cm−2 for a total fluence of 100 J cm−2. Plates were read with a Molecular Devices M2e (λexc = 530 nm, long-pass 570 nm, λem = 620 nm) at UNCG/UTA or a Cytofluor 4000 (λexc/em 530 ± 25/620 ± 40 nm) reader at Acadia University.
Specialized dosimetry experiments on the Modulight ML8500 platform included well-by-well illumination with lasers centered at 525, 630, and 753 nm at various fluences (0–400 J cm−2) and constant irradiance of 300 mW cm−2. Additional experiments with Os-4T included 384-well plates using (a) illumination with a CivilLaser 733 nm laser (∼5 mW cm−2) in tandem with a frontal diffuser by Medlight (model FD Frontal Light Distributor) and (b) monitoring Os-4T performance over a month with varying conditions (pipette tip, cell parent seed stock, plate map). The assay procedure for a 384-well plate is outlined in SI method SM-1. The spectral profiles of the light sources are shown in Fig. S-22 and S-23.† Spectral output was monitored using the Luzchem SPR fiber optic detector in tandem with an Ocean Optics USB4000 spectrophotometer and an Ocean Optics UV-Vis XSR fiber optic of 230 μm diameter. Irradiance was measured using a Thorlabs Optical Power Meter PM100D and their corresponding thermal power sensor S310C. Irradiance was generally within 5% across entire plate area for full-plate illumination.
2.7.2.2 Hypoxia. Cell culture and compound evaluation in hypoxia were performed as previously reported for SK-MEL-28 cells.41 Only SK-MEL-28 cells and full dose–response experiments (EC50 determination) were applied in hypoxia for comparison to normoxic data. As in our previous work, resazurin-based cell viability endpoints had strong interference on hypoxia-treated well plates. An additional sulforhodamine B (SRB) endpoint was included for any hypoxic assay comparisons due to lack of compound interference at lower concentrations. Comparisons of results with resazurin versus SRB and the required corrections are shown for both oxygen treatments in Fig. S-24.† Following resazurin reads, the plates were treated in accordance with the precedent literature.78–80 Well plates were briefly centrifuged in a prechilled (4 °C) Thermo Scientific™ Sorvall™ Legend™ RT centrifuge with swinging bucket rotor (7500 6445, 7500 6449) at 180 × g for 5 min. Protein was precipitated by incubation for 45 min to 1 h with final 10% w/v trichloroacetic acid (TCA, pre-chilled) per well at 4 °C. Supernatant was removed via simple plate inversion and flick. Precipitate was washed five times with 150–200 μL per well DI water at ambient temperature. Afterwards, protein was stained with 50 μL per well of 1 mM SRB in 1% v/v acetic acid, briefly incubated for 15–30 min at ambient temperature (∼20 °C), and washed five times with 200 μL per well of 1% v/v acetic acid.
Protein was finally dissolved in 200 μL per well of 10 mM Trizma base. Well solutions were read after 1 h incubation at 565 nm. Note that shorter incubation times can be used with a plate shaker. Alternative endpoints (510 nm abs. or fluorescence) were also explored but found to be less selective for SRB due to spectral overlap with the metal complexes. Day 0 reference plates were prepared, and values subtracted from final readings to test treatment effect on relative changes in protein. Rather than use as a true cytotoxicity assay as sometimes ascribed, SRB results were interpreted similarly to the NCI60 panel,81 as a reference to the cell death response.80 Interpolated endpoints are as follows:
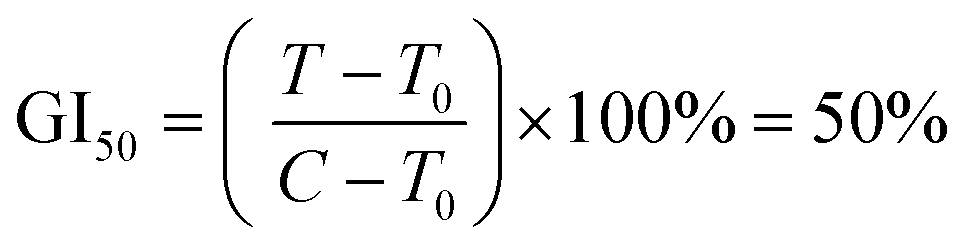 | (2) |
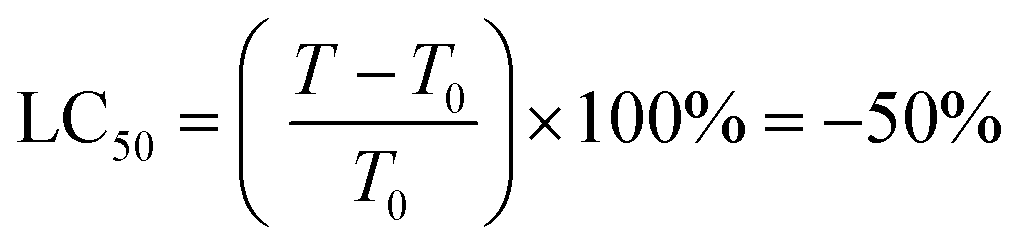 | (3) |
 | (4) |
For the longitudinal study or repeats of (photo)cytotoxicity experiments (normoxic and hypoxic) with Os-4T in SK-MEL-28, resazurin was used as the viability indicator. Results were plotted with original run (repeat #0) using log scale EC50 and PI values to monitor change over time. Repeats 1–5 were conducted within a month of each other in 384 well-plates (see SM-1); the study began several months after the original run (repeat 0) in 96-well-plates. Variables were adjusted as follows to simulate differences common in assay comparisons across long periods of time. Repeats used different plate maps (all), different tips (Sartorius 790352 repeats #0–1, VWR 83007-352 repeats #2–3, low retention Sartorius LH-L790352 repeats #4–5), changed cell parent seed stock for repeats 4–5, and overhead lights were off in #5. Serum and consumable lots were identical for repeats 1–5. Cell passage numbers were equal and between 10–15 passages from seed stock receipt.
2.8 Maximum tolerated dose in mice
An 8 week old litter of female C57BL/6J mice, averaging 20 g per mouse, were treated by intraperitoneal injection of Os-4T in accordance with protocol A20-006 (approved by WFU Animal Care and Use Committee). Mice were dosed from 25–200 mg kg−1 with 200 μL injections and 10% DMSO in 0.9% saline as the vehicle. Solutions of Os-4T were prepared immediately with sonication before injection. Female mice were dosed by slow intraperitoneal injection (the lower right abdominal quadrant) only after visible confirmation of complete compound dissolution. Animals were continuously monitored for 2 hours, frequently over the next 6, and periodically for up to 2 weeks before being sacrificed. Mice were accordingly euthanized if (a) a combination of moderate severity signs appeared, (b) a single severe sign appeared, or (c) the study period was complete, 2 weeks post-injection. The maximum tolerated dose (MTD) was defined as the dose that produces moderate signs of clinical toxicity in the final tested animal.3. Results and discussion
3.1 Synthesis and characterization
Complexes [Os(phen)2(IP-nT)]2+Os-0T–Os-4T were synthesized following adapted literature procedures.82–84 The complexes were isolated as their PF6− salts and purified with flash chromatography on silica, affording final products in 16–60% yields. The PF6− salts were converted to their corresponding Cl− salts in quantitative yields via anion metathesis using Amberlite IRA-410 and were further purified using size-exclusion chromatography on Sephadex LH-20, if needed. HPLC analysis confirmed the purities of complexes Os-0T–Os-4T to be 95% or higher (Fig. S-17–S-21†). The structures of complexes Os-0T–Os-4T were confirmed using high resolution ESI+ mass spectrometry (Fig. S-11–S-15†) and a detailed analysis of their 1D 1H NMR (Chart 2 and Fig. 1) and 2D 1H–1H COSY NMR spectra (Fig. S-2–S-6†). Assignments were made based on the connectivities observed in 1H–1H COSY NMR and in consultation with literature sources.65,66 Compound Os-4T required additional 13C, 13C–1H HSQC, and 13C–1H HMBC NMR experiments for full assignment of the 1H NMR signals of the quaterthiophene group (Fig. S-7–S-9†). The chemical shifts and resolution of signals from the thiophene ring hydrogens exhibited a concentration dependence, especially for complexes with longer thiophene chains. The best resolution of these NMR signals was obtained at concentrations near 1.6 mM.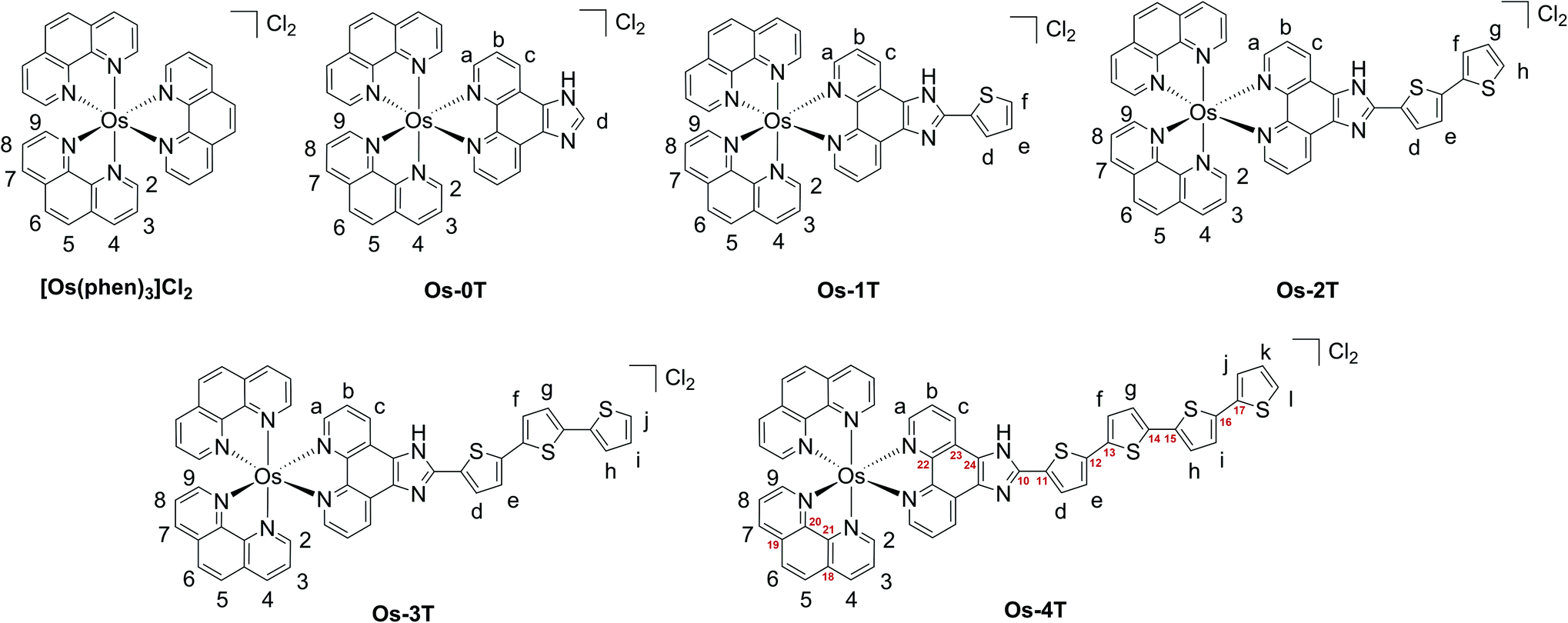 | ||
| Chart 2 Hydrogen labels (black) and selected carbon labels (red) used in 1H peak assignments of reference compound [Os(phen)3]2+ and Os-0T–Os-4T. | ||
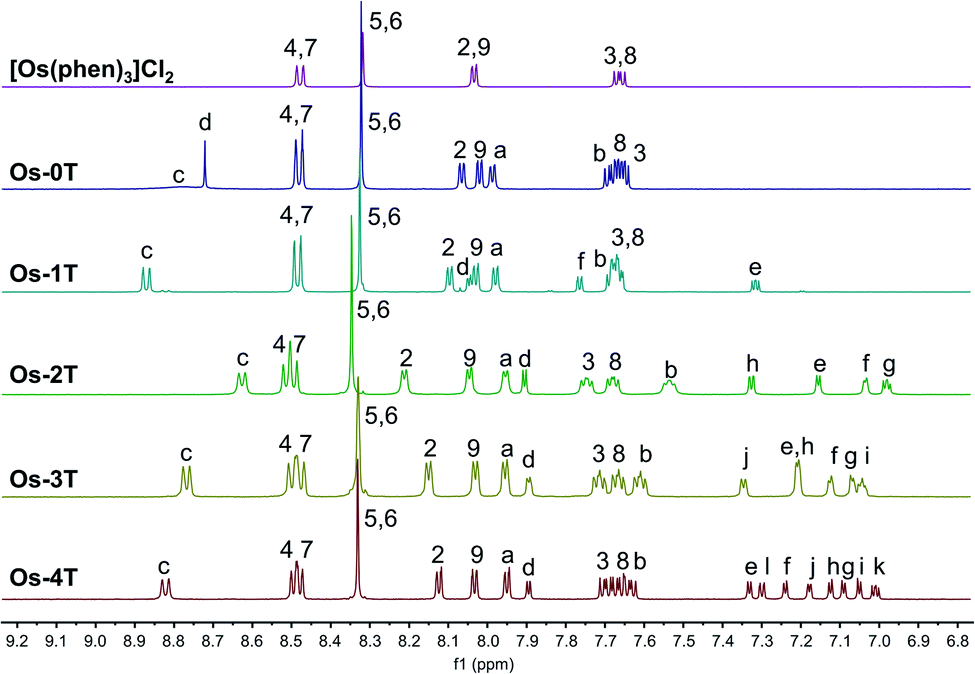 | ||
| Fig. 1 500 MHz 1H NMR spectra of reference compound [Os(phen)3]Cl2 and Os-0T–Os-4T (Cl− salts) in MeOD-d3 at 298 K, aromatic region. | ||
The a–b–c spin system was assigned next. It appeared as c > a > b, repeating the pattern observed for spin systems 2–3–4 and 7–8–9, where the signal for the hydrogen in the meta-position relative to the Os-coordinated nitrogen was shifted the farthest upfield and that for the hydrogen in the para-position appeared the most downfield of the three. Due to similarities between these spin systems, a appeared close to 2,9, and b appeared close to 3,8, and c appeared relatively close to 4,7. The signal for the imidazole-NH hydrogen was not observed, in agreement with the literature,85,86,89 where quick exchange between the two nitrogens of the imidazole ring occurs in solution.
In thiophene-containing complexes Os-1T–Os-4T, the signals from the thiophenes integrated as one hydrogen each and were assigned last. Among those, three hydrogen signals were most diagnostic. The signal for hydrogen d was the most downfield compared to the signals for the rest of the thienyl hydrogens due to the strong deshielding effect of the nitrogens of the neighboring imidazole. Another strongly deshielded hydrogen was the hydrogen positioned closest to the sulfur (f in Os-1T, h in Os-2T, j in Os-3T, and l in Os-4T). The signal for the most shielded hydrogen appeared as a distinct doublet of doublets and corresponded to the middle hydrogen of the most distal thiophene ring (e in Os-1T, g in Os-2T, i in Os-3T, and k in Os-4T). Using these characteristic signals as starting points, the rest of the thiophene signals from the proximal and the most distal thiophene ring were assigned via the observed 1H–1H COSY correlations.
Assigning the signals from the internal thiophenes was less straightforward and was accomplished for Os-4T with the aid of 13C–1H HSQC and 13C–1H HMBC NMR experiments (Fig. S-8 and S-9†). The two internal spin systems (f–g and h–i) were first established using 1H–1H COSY correlations. Then, 13C–1H HSQC was used to identify which 13C peaks corresponded to the thiophene hydrogens d–l. Then, 13C–1H HMBC was used to establish diagnostic correlations, starting with hydrogens d and e. Hydrogens d and e both correlated to two of the same 13C peaks (132.20 ppm and 141.39 ppm), which were assumed to be carbons 11 and 12. Hydrogen d correlated more strongly to the peak at 132.20 ppm, and hydrogen e correlated more strongly to the peak at 141.39 ppm, which allowed for carbon 11 and carbon 12 to be assigned as signals at 132.20 ppm and 141.39 ppm, respectively. In addition to correlations with d and e, carbon 12 exhibited correlations with one more hydrogen, which led to the assignment of this hydrogen as f. Hydrogen g was then readily identified via its correlation to f in the 1H–1H COSY NMR spectrum. Next, a diagnostic 13C signal at 137.79 ppm exhibited correlations to hydrogens l, j, and one more 1H signal that was assigned as hydrogen i. This 13C signal was assigned as carbon 17 due to the greatest possibility of both l and j showing correlation to it. Lastly, hydrogen h was then readily identified via its correlation to i observed in the 1H–1H COSY NMR spectrum.
3.2 Lipophilicity
The lipophilicities of the chloride salts of the compounds, including the reference [Os(phen)3]Cl2, were evaluated by determining their logDo/w values (Fig. 2 and Table S-1†). This parameter reflects the solubility preference of a given compound for either octanol or aqueous PBS prepared at neutral pH, where positive values indicate higher lipophilicity (octanol solubility) and negative values indicate lower lipophilicity (aqueous solubility).90 Among the series, the logDo/w values increased systematically with the number of thienyl rings appended to the IP ligand. The compounds were essentially hydrophilic from n = 0 to 2 (Os-0T–Os-2T), with an abrupt switch to lipophilic at n = 3 (Os-3T). Os-0T was the only compound that was more hydrophilic than [Os(phen)3]Cl2, reflecting the influence of the imidazo group of the IP ligand (relative to phen) on aqueous solubility. The difference in lipophilicities for Os-0T, lacking thienyl groups, to three in Os-3T was approximately 103. The logDo/w value for Os-4T could not be determined owing to its precipitation from the solvent mixture under all conditions, suggesting an even greater lipophilicity that favors self-aggregation through favorable π–π interactions.
 | ||
| Fig. 2 Octanol–water distribution coefficients (logDo/w ± SD) for reference compound [Os(phen)3]2+ and Os-0T–Os-3T (Cl− salts); SD = standard deviation. The value of log Do/w for Os-4T could not be determined due to its precipitation from the solvent mixture. | ||
3.3 Computational studies
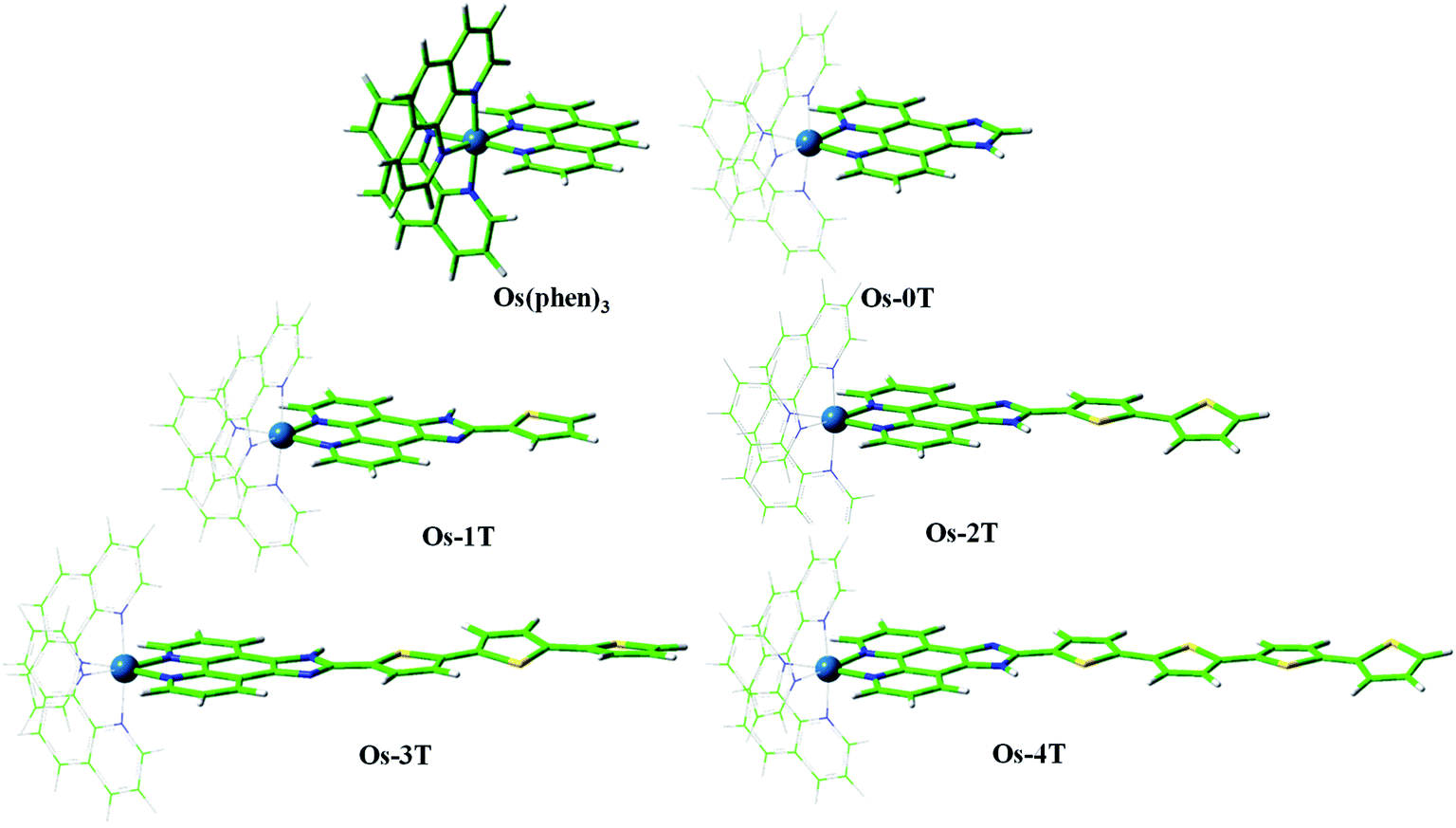 | ||
| Fig. 3 Optimized geometries of reference compound [Os(phen)3]2+ and Os-0T–Os-4T in a water environment at the PBE0/6-31+G**/SDD/level of theory. | ||
The ground state electronic structures of these complexes can be described by the frontier molecular orbitals (HOMO−1, HOMO, LUMO, LUMO+1) shown in Fig. S-27–S-31.† The calculated percent contribution of the Os d-orbitals, phen, IP and thienyl chain to the frontier orbitals of each complex in the singlet ground state are reported in Table 1. The nature of the HOMO (Fig. 4) changed with the number of thienyl groups in the chain. The considerable amount of Os d character (∼35%) in the HOMO of [Os(phen)3]2+, Os-0T and Os-1T nearly vanished (<5%) when two or more thiophenes were incorporated. The increasing contribution of the distal thiophene rings to the HOMO is illustrated by the energy trend plot in Fig. 5a. For Os-2T through Os-4T, the HOMO orbital becomes progressively more ligand-based and reaches a maximum value for thienyl contribution in Os-4T. The incorporation of a polythiophene chain (with n ≥ 2) thus, produces the installation of a HOMO orbital of organic character lying at higher energies, reflected by the HOMO–LUMO gaps shown in Fig. 5a and the HOMO orbital plots in Fig. 4. By sharp contrast, the composition of the HOMO−1 orbital was relatively unchanged throughout the series (Fig. S-28†), being largely comprised of Os d character (30–39%) mixed with phen and IP (61–68%) but without any contribution from the thiophene chains.
| % | HOMO−1 | HOMO | LUMO | LUMO+1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Os | Phen | IP | Thienyl | Os | Phen | IP | Thienyl | Os | Phen | IP | Thienyl | Os | Phen | IP | Thienyl | |
| [Os(phen)3]2+ | 36 | 64 | — | — | 35 | 65 | — | — | 3 | 97 | — | — | 2 | 98 | — | — |
| Os-0T | 39 | 42 | 19 | 0 | 34 | 46 | 19 | 0 | 3 | 59 | 39 | 0 | 4 | 69 | 28 | 0 |
| Os-1T | 30 | 34 | 34 | 2 | 34 | 40 | 25 | 0 | 3 | 59 | 39 | 0 | 3 | 72 | 25 | 0 |
| Os-2T | 34 | 35 | 31 | 0 | 4 | 47 | 24 | 25 | 3 | 57 | 38 | 3 | 4 | 57 | 24 | 15 |
| Os-3T | 34 | 40 | 25 | 0 | 1 | 38 | 17 | 45 | 3 | 57 | 39 | 1 | 7 | 42 | 25 | 26 |
| Os-4T | 35 | 42 | 23 | 0 | 1 | 32 | 13 | 55 | 3 | 54 | 40 | 3 | 8 | 42 | 28 | 22 |
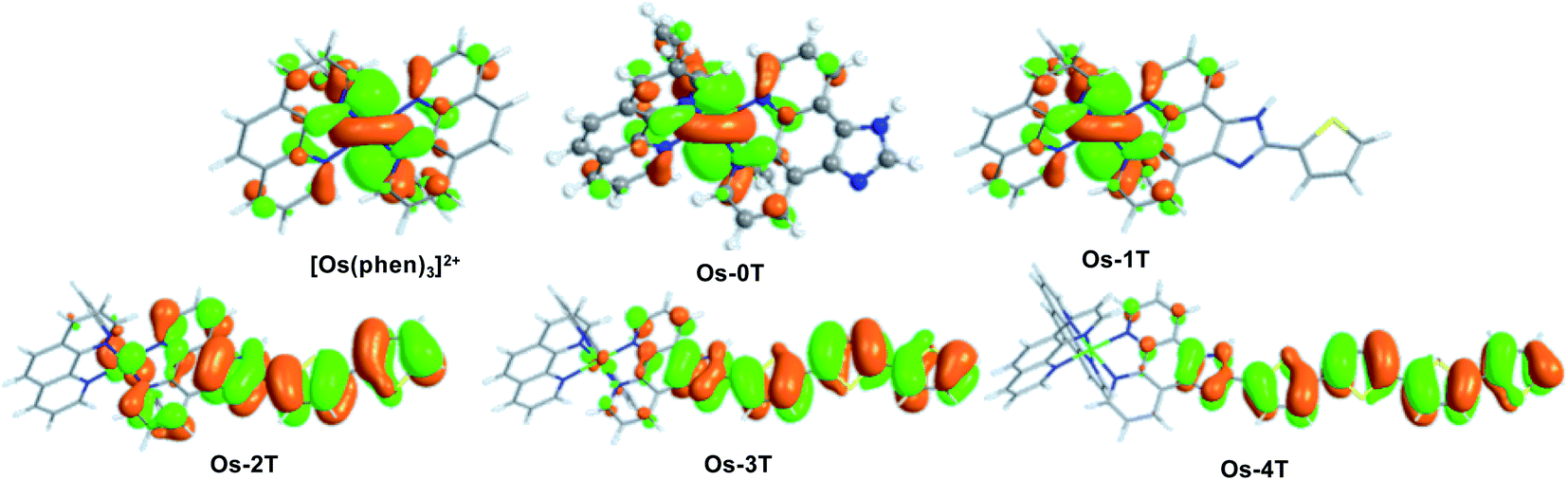 | ||
| Fig. 4 HOMO orbital plots for reference compound [Os(phen)3]2+ and Os-0T–Os-4T, calculated in water at M06/6-31+G(d,p)/SDD/level of theory. | ||
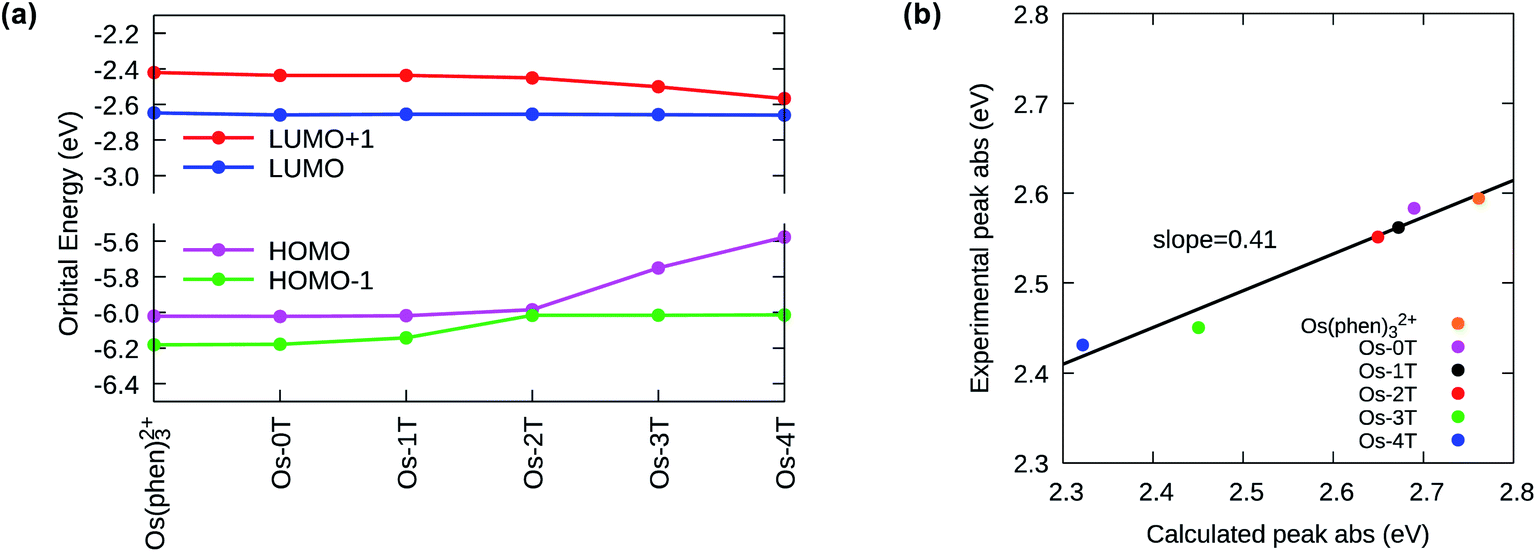 | ||
| Fig. 5 (a) Calculated frontier orbital energies for reference compound [Os(phen)3]2+ and Os-0T–Os-4T. (b) Correlation of calculated and experimental longest wavelength absorption maxima for reference compound [Os(phen)3]2+ and Os-0T–Os-4T measured in water. | ||
The lowest unoccupied molecular orbitals (LUMOs and LUMO+1s, Fig. S-29 and S-30†) were almost exclusively localized to the phen and IP (phen portion) ligands. The contribution of the thienyl chain produced opposite effects on the HOMO versus LUMO+1 orbitals and had almost no effect on the LUMOs (Fig. 5a). While the energy of the LUMO orbital remains constant with the same % composition regardless of the number of thienyl groups, the LUMO+1 drops slightly in energy with ≥2 thiophene rings, consistent with an increased contribution from the thienyl ligand observed in the orbital composition (15, 26 and 22% in Os-2T, Os-3T and Os-4T, respectively).
The increase of the HOMO and concomitant decrease of the LUMO+1 energies with n is reflected in the calculated UV-visible absorbance spectra of the Os-nT series (Fig. 5b, S-32, S-33 and Table S-3†). There is a clear and systematic increase in the absorption intensity throughout the visible region with increasing n on going from Os-0T–Os-4T, reaching singlet–singlet transition energies into the red.
Close inspection of the computed transitions and their orbital compositions allowed assignment of the orbital parentage and configuration of the experimental lowest-energy spin-allowed, singlet–singlet bands in the UV-vis absorption spectra (i.e., transitions occurring <550 nm). There was an abrupt change in the nature of these lowest-energy spin-allowed transitions with n > 2, which reflected the decreased contribution of the metal-based orbitals and increased contribution of the thienyl-based orbitals to the lowest-energy singlet–singlet electronic transitions as thiophene rings were added. The lowest-energy transitions had predominantly 1MLCT character for n ≤ 2 (Os-0T–Os-2T), while 1ILCT character made a significant contribution to those with n ≥ 3 (Os-3T, Os-4T) where the HOMO orbital was exclusively localized over the thienyl groups. This change in the nature of the lowest-energy spin-allowed transitions was also supported by a solvent dependence (water versus acetonitrile, Fig. S-35†) in the experimental absorption spectra for Os-3T and Os-4T but not for Os-0T–Os-2T.
Os-3T showed an intense band contributed mostly by a transition centered at 484 nm (versus an experimental value of 487 nm) characterized as ILCT (48%) mixed with MLCT (29%), and another intense band near 506 nm, which was also mixed ILCT/MLCT (Table 2 and Fig. S-33†). Os-4T had its most intense band bathochromically shifted to 534 nm and was unequivocally assigned to ILCT. The charge-transfer nature of these ligand-localized 1ππ* transitions in Os-3T and Os-4T was corroborated by the solvent dependence of these transitions in the experimental absorption spectra (Fig. S-34 and S-35†).
| Complex | λ max calc | λ max exp | Transitions | Assignment |
|---|---|---|---|---|
| Os-0T | 461 | 480 | H−1 → L+1 (45%) | MLCT |
| H−2 → L+2 (37%) | ||||
| Os-1T | 464 | 484 | H−1 → L+1 (49%) | MLCT |
| H−2 → L+2 (41%) | ||||
| Os-2T | 468 | 486 | H−2 → L+2 (44%) | MLCT |
| Os-3T | 506 | 506 | H−3 → L (48%) | MLCT + ILCT |
| H → L (38%) | ||||
| 484 | 487 | H → L+1 (48%) | ILCT | |
| Os-4T | 534 | 520 | H → L+1 (55%) | ILCT |
| H → L (34%) | ||||
| 451 | 436 | H−1 → L+5 (52%) | MLCT |
The nature of the lowest-lying triplet states of the Os-nT dyads was examined by Mulliken spin density (MSD) calculations and was found to vary among the series. The MSD on the Os(II) center for complexes with n ≤ 2 was one, indicative of one unpaired electron on the metal center as would be expected for the 3MLCT state. No involvement of the metal was detected for Os-3T and Os-4T, supporting the assignment of the lowest-energy triplet states in these two complexes as the 3ILCT state. The installation of low-lying 3ILCT states for Os-3T and Os-4T can be thus connected with greater π expansion, which was also responsible for a drop in the triplet state energy. The calculated T1 state energies of [Os(phen)3]2+, Os-0T, and Os-1T were nearly identical (≈2.09 eV), but decreased systematically with added thiophene rings, falling to 1.45 eV in Os-4T (Fig. 6). Earlier work has connected similar stabilization of the 3ILCT energy with longer lifetimes in Ru(II) complexes,57,93,94 and this could account for Os-4T having exceptional in vitro activity (vide infra).
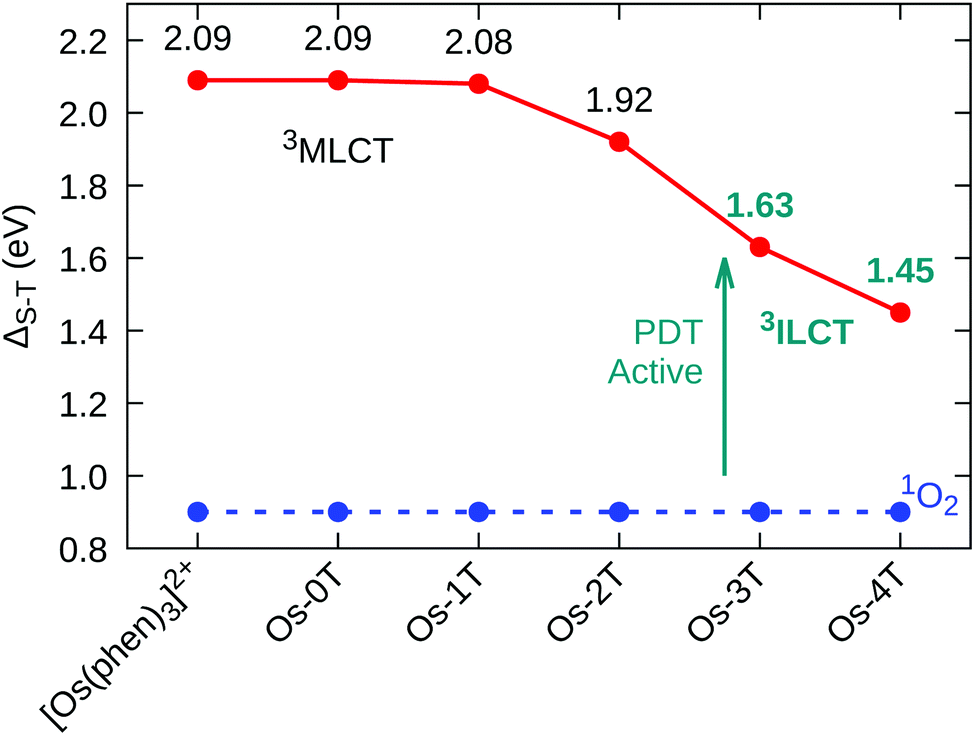 | ||
| Fig. 6 Singlet–triplet energy gap (ΔS–T) for reference compound [Os(phen)3]2+ and Os-0T–Os-4T, in water, at the M06/6-31+G(d,p)/SDD level of theory. | ||
The capacity of the triplet-state Os(II) complexes to promote Type I and Type II photoreactions was explored. To produce the cytotoxic singlet oxygen species, the singlet–triplet splitting energy of the complex must be larger than that of O2, thereby allowing energy transfer to promote the O23Σg− → 1Δg transition; the lowest oxygen singlet state energy was previously computed to be 0.90 eV,95 in good agreement with the experimental value of 0.98 eV. The calculated singlet–triplet energy gaps (ΔS–T) show that every compound in the series is sufficiently energetic to form 1O2 through a Type II pathway (Fig. 6).
There also exists the possibility that the triplet state could generate the superoxide anion O2˙− through Type I photoreactions. This is a potentially important process since O2˙− can participate in rapid bimolecular reactions that lead to other reactive oxygen species (ROS) such as H2O2 and OH˙, which in turn could damage biomolecules.95 The O2˙− species can be formed by direct electron transfer from the photosensitizer (PS) to O2(5), or by an electron transfer mechanism from a reduced form of the PS to O2(6). The reduced form of the PS could be generated by autoionization reactions (7) and (8), through the reduction of a PS T1 state by a neighboring PS in the S0 or T1 state. Such reactions were explored computationally through the vertical electron affinity (VEA) and ionization potentials (VIP) of each Os(II) compound and for molecular oxygen.76,96–98 The results are summarized in Table 3.
| PS(T1) + 3O2 → PS˙+ + O2˙− | (5) |
| PS˙− + 3O2 → PS + O2˙− | (6) |
| PS(T1) + PS(S0) → PS˙+ + PS˙− | (7) |
| PS(T1) + PS(T1) → PS˙+ + PS˙− | (8) |
| [Os(phen)3]2+ | Os-0T | Os-1T | Os-2T | Os-3T | Os-4T | |
|---|---|---|---|---|---|---|
| VEA | −2.92 | −2.94 | −2.94 | −2.94 | −2.94 | −2.94 |
| VIP | 5.52 | 5.51 | 5.51 | 5.68 | 5.43 | 5.26 |
| VEA(T1) | −5.01 | −5.03 | −5.02 | −4.86 | −4.57 | −4.39 |
| VIP(T1) | 3.43 | 3.42 | 3.43 | 3.76 | 3.80 | 3.81 |
The feasibility of direct electron transfer to molecular O2 through reaction (5) decreases as the thiophene chain grows, as was previously observed for the related Ru(II) complexes.76 The triplet states of the Os(II) complexes become weaker electron donors with increasing n, thus preventing the direct electron transfer process in water. This process could be possible for [Os(phen)3]2+, Os-0T and Os-1T but can be ruled out for Os-2T–Os-4T since the sum of VIP(T1) of the latter compounds and the electron affinity of 3O2 are positive.
However, the T1 state of all the Os(II) compounds can undergo indirect Type I photoreactions. The results demonstrate that compounds in the T1 state can be reduced through autoionization (8) with neighboring compounds in the T1 state given that the sum of VEA(T1) and VIP(T1) is negative. Importantly, the higher electron affinity of oxygen compared to the Os(II)-complexes suggests that any of complexes in the reduced state is able to generate the superoxide species (6). It should be also noted that O2˙− could behave as a further reducing agent for Os(II) T1 states that could also be implicated in biomolecule-damaging processes, but this was not examined.
To summarize, computational studies conclude that all of the Os(II) complexes have the requisite triplet state energies to sensitize 1O2 through the Type II energy transfer pathway but only [Os(phen)3]2+, Os-0T and Os-1T can directly form superoxide. However, all of the Os(II) complexes are capable of generating superoxide through Type I autoionization photoreactions, suggesting at least two potential modes of activity in this class of compounds. The relative contributions of these pathways in cells would likely depend on the local concentrations of these complexes and O2 as well as the redox environment. It is possible that other intracellular mediators of electron transfer could participate in any intracellular Type I photochemistry.
3.4 Photophysical properties
The photophysical properties of the complexes were measured as dilute solutions of the PF6− salts in MeCN. This solvent choice facilitates comparison with related compounds in the photophysical literature.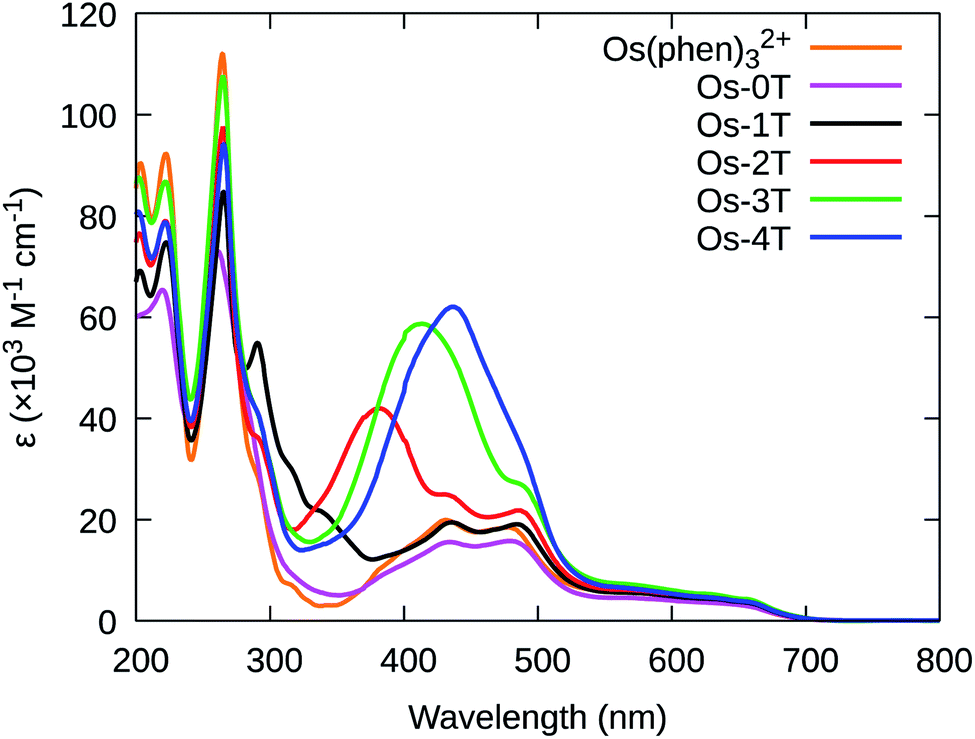 | ||
| Fig. 7 UV-vis spectra of reference compound [Os(phen)3]2+ and Os-0T–Os-4T (PF6− salts) in room temperature acetonitrile. | ||
| Complex |
λ
abs/nm (logε)a |
|---|---|
| a Absorption band maxima and extinction coefficients (M−1 cm−1) determined in acetonitrile at room temperature. | |
| [Os(phen)3]2+ | 222 (4.97), 265 (5.05), 431 (4.30), 478 (4.27), 653 (3.56) |
| Os-0T | 220 (4.82), 261 (4.87), 434 (4.20), 480 (4.20), 655 (3.49) |
| Os-1T | 222 (4.88), 265 (4.93), 291 (4.73), 338 (4.35), 435 (4.29), 484 (4.28), 658 (3.55) |
| Os-2T | 222 (4.89), 265 (4.98), 288 (4.56), 382 (4.62), 432 (4.40), 486 (4.33), 658 (3.58) |
| Os-3T | 222 (4.94), 265 (5.03), 413 (4.77), 487 (4.43), 658 (3.63) |
| Os-4T | 222 (4.89), 265 (4.96), 436 (4.79), 656 (3.57) |
Additional peaks between 330–440 nm were observed for Os(II) compounds containing thienyl groups. These absorption bands in Os-1T through Os-4T (338, 382, 413, and 436 nm, respectively) increased in intensity and decreased in energy, consistent with the increased conjugation of added thiophene rings, and are hence assigned to ILCT ππ* transitions involving nT of the IP-nT ligands.
| Complex | MLCT λem [λex] (nm) | τ em(ns) | Φ Δ [λex] (nm) |
|---|---|---|---|
| [Os(phen)3]2+ | 718 [471] | 2.1 × 102 | 0.53 [472] |
| Os-0T | 732 [472] | 1.8 × 102 | 0.43 [473] |
| Os-1T | 737 [471] | 1.7 × 102 | 0.42 [473] |
| Os-2T | 740 [470] | 1.8 × 102 | 0.49 [472] |
| Os-3T | 743 [468] | τ 1 1.4–2.9 × 102 | 0.51 [525] |
| τ 2 3.8–7.1 × 102 | |||
| Os-4T | 746 [470] | τ 1 1.8–2.2 × 102 | 0.95 [525] |
| τ 2 1.4–1.6 × 104 |
Emission lifetimes were measured with a 355 nm laser pulse (pulse width ≈ 5 ns) excitation. The 2.1 × 102 ns lifetime of the series archetype [Os(phen)3]2+ in MeCN compared well to the literature lifetime in CH2Cl2 (315 ns)67 and is shorter than that of the analogous [Ru(phen)3]2+ complex (τ ≈ 500 ns).101,102 The similarity of the lifetimes (≈200 ns) across the series further supports the conclusion that the 3MLCT state involves the phen-based LUMO as the π* acceptor in all the complexes. Complexes Os-3T and Os-4T also exhibited delayed emission (τ2 = 3.8–7.1 × 102 ns and 14–16 μs, respectively), which indicates that the 3MLCT state can be populated from a state different than the initially-formed 1MLCT state that leads to prompt 3MLCT emission (τ2 ≈ 200 ns). This proposed model is illustrated in the Jablonski diagram in Scheme 1. Given that the estimated 3MLCT energy from emission measurements for Os-4T was about 1.66 eV and the computed value for the 3ILCT state was 1.45 eV, we hypothesized that this energetic proximity could enable the longer-lived 3ILCT state to act as an excited state energy reservoir for delayed 3MLCT emission. This 3MLCT–3ILCT gap was estimated to be even smaller for Os-3T.
 | ||
| Scheme 1 Jablonski diagrams depicting the proposed decay pathways in (a) Os-1T, (b) Os-3T, and (c) Os-4T. | ||
The ESA spectra of [Os(phen)3](PF6)2 and Os-0T through Os-2T (Fig. 8a) were qualitatively similar, featuring three regions. A strong bleach with peaks at around 440 and 480 nm corresponding to the loss of the ground state 1A1 → MLCT transition. A broad, weaker bleach spanned ≈525–680 nm, also corresponding to loss of the MLCT transition. The absorption below 400 nm arises from a new excited state transition on the 3MLCT radical anion ligand. The lifetimes were monoexponential and essentially the same (≈150–200 ns) at all wavelengths for each compound (Table 6), indicating that the phen-based LUMO is involved in all these cases. The TA lifetimes also corresponded well with the emission lifetimes (Table 5), indicating that phosphorescent decay of the phen-based 3MLCT state is the dominant relaxation pathway in these four compounds.
 | ||
| Fig. 8 Transient absorption profiles for (a) [Os(phen)3](PF6)2, Os-0T–Os-2T, and (b) Os-3T and Os-4T, showing the indicated integration slices. ΔOD = 0 is indicated by the dashed line of the corresponding color. | ||
| Complex | Wavelength (nm) | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) |
|---|---|---|---|---|
| [Os(phen)3]2+ | 360 | 2.2 × 102 | ||
| 440 | 2.1 × 102 | |||
| 480 | 2.1 × 102 | |||
| 600 | 2.0 × 102 | |||
| Os-0T | 350 | 1.7 × 102 | ||
| 440 | 1.9 × 102 | |||
| 480 | 1.8 × 102 | |||
| 600 | 1.8 × 102 | |||
| Os-1T | 360 | 1.6 × 102 | ||
| 440 | 1.6 × 102 | |||
| 480 | 1.7 × 102 | |||
| 600 | 1.8 × 102 | |||
| Os-2T | 390 | 1.6 × 102 | ||
| 440 | 1.5 × 102 | 4.0 × 102 | ||
| 490 | 1.7 × 102 | 3.9 × 102 | ||
| 600 | 1.5–1.7 × 102 | 2.9–5.5 × 102 | ||
| Os-3T | 430 | 2.8–4.5 × 102 | 7.4 × 102 | |
| 610 | 3.6–4.8 × 102 | 7.1 × 102 | ||
| Os-4T | 440 | 2.6 × 102 | 3–4 × 103 | 12 × 103 |
| 660 | 2.4 × 102 | 3–4 × 103 | 12–13 × 103 |
In contrast, Os-3T and Os-4T exhibited ESA spectra consistent with long-lived ligand-based triplet states (Fig. 8b) and which qualitatively resembled the spectra of the IP-nT ligands alone (Fig. S-44 and S-45†). The bleach in the 400–450 nm region corresponds to the loss of the strong ππ* transition, which peaks at around 413 nm and 436 nm in the ground state UV-vis absorbance spectra of the Os-3T and Os-4T complexes, respectively, and at 408 nm and 424 nm for the free IP-3T and IP-4T ligands. The broad and intense positive absorption in the ESA spectra of Os-3T and Os-4T was characteristic of transitions associated with one or more ligand-localized 3ππ* states, which computational studies indicated were 3ILCT in nature. These bands resembled those observed in the ESA spectra of the uncomplexed IP-3T and IP-4T ligands, indicating that the initially excited 1ILCT and/or 1MLCT states populate 3ILCT states centered on the thiophene-containing ligand. The lack of this signature in the complexes with n ≤ 2 suggests that this state is not energetically accessible and/or is not a dominant relaxation pathway in systems with less π-conjugated chains.
Os-0T and Os-1T displayed similar TA kinetics to [Os(phen)3](PF6)2, with only one lifetime (τ1 = 200 ns) assigned as the emitting 3MLCT state. This state was also observed for the other Os(II) complexes. The decay of the TA signal for Os-2T had an additional component (τ2) that was slightly longer than τ1 and not observed in the emission measurements. While this state could have 3ILCT character, its contribution was weak (3–5% relative to τ1) and the TA spectral signature for Os-2T was almost identical to the 3MLCT signature of Os-0T and Os-1T (Fig. 8a).
The TA lifetimes of Os-3T and Os-4T were in agreement with their emission lifetimes and corroborated the model proposing delayed 3MLCT emission through a second channel (Scheme 1). The TA kinetics of Os-3T were described by time constants of τ1 < 500 ns and τ2 ≈ 700 ns, corresponding to the prompt and delayed kinetics observed in the emission measurements. The shorter component was assigned to prompt emission from the 3MLCT state, while the longer component was assigned to the decay of an 3ILCT state of similar energy via population of the emitting 3MLCT state. The involvement of the 3ILCT was invoked based on its computed energy being very similar to that of the 3MLCT state and the characteristic signature of the 3ILCT state observed in the TA spectrum that was absent from the complexes with fewer thiophene rings.
A similar model was invoked for Os-4T, which displayed three lifetimes in the TA measurements. The prompt decay (τ1 ≈ 250 ns) was assigned to the emitting 3MLCT state and agreed with the shorter component (τ1) in the emission measurements. The longest TA lifetime (τ3 ≈ 12 μs) agreed with the lifetime for delayed emission (τ2 = 14–16 μs) and was ascribed to the emitting 3MLCT state populated from a lower-energy 3ILCT state. The intermediate component (τ2 ≈ 3–4 μs) was not observed in the emission measurements and was assumed to be nonradiative decay of the dark 3ILCT state back to the ground state. In this model, the 3ILCT state has two discrete decay pathways whereby the longer lifetime could reflect the uphill process for 3MLCT population due to the larger 3MLCT-3ILCT energy gap for Os-4T relative to Os-3T (Scheme 1).
3.5 Biological results
3.5.1.1 Normoxic cellular assays. The dark and light cytotoxicities for [Os(phen)3]Cl2 through Os-4T were measured in SK-MEL-28 human skin melanoma cells growing as 2D adherent monolayers. Briefly, cells growing in log phase were seeded, dosed with compound (1 nM to 300 μM), and then incubated for 48 h before cell viability was determined using the resazurin viability assay for cytotoxicity. To determine photocytotoxicity, an analogous set of plates were prepared but were irradiated 15–22 h after compound addition. Dose–response curves were prepared from both dark and light conditions and analyzed to provide dark and light EC50 values, the effective concentration to reduce relative viability by 50%. PIs, or the light-induced amplification of cytotoxicity, were calculated as the ratios of the dark EC50 values to the light EC50 values. For highly potent compounds, the assay was adjusted to include a wider concentration range (10−12 to 300 μM). Any deviations from this method (hypoxia, different cell lines, etc.) are described as they are discussed. All of the complexes exhibited excellent solubility in high ionic strength solvents (DPBS, media) up to 50 μM, with [Os(phen)3]Cl2 as most soluble (>300 μM).
3.5.1.2 Dark cytotoxicity. The Os(II) oligothienyl complexes as well as reference compounds [Os(phen)3]Cl2 and Os-0T were relatively nontoxic toward SK-MEL-28 cells in the absence of a light treatment (Fig. 9a and Table S-4†), with dark EC50 values ranging from 158 to 35.7 μM. The least dark cytotoxic compound of the series was the reference compound [Os(phen)3]Cl2 (EC50 = 158 μM), and the most dark cytotoxic compound was Os-2T (EC50 = 35.7 μM). The dark cytotoxicities of Os-1T, Os-3T, and Os-4T (EC50 = 65.1 μM) were intermediate at 65–75 μM. These dark cytotoxicities did not correlate with the lipophilicities of the complexes, which increased with number of thienyl units. The reason behind the almost two-fold greater dark toxicity for Os-2T, with two thienyl groups, is unclear but agrees with our observations for other Os(II)- and Ru(II)-oligothiophene complexes (unpublished results).
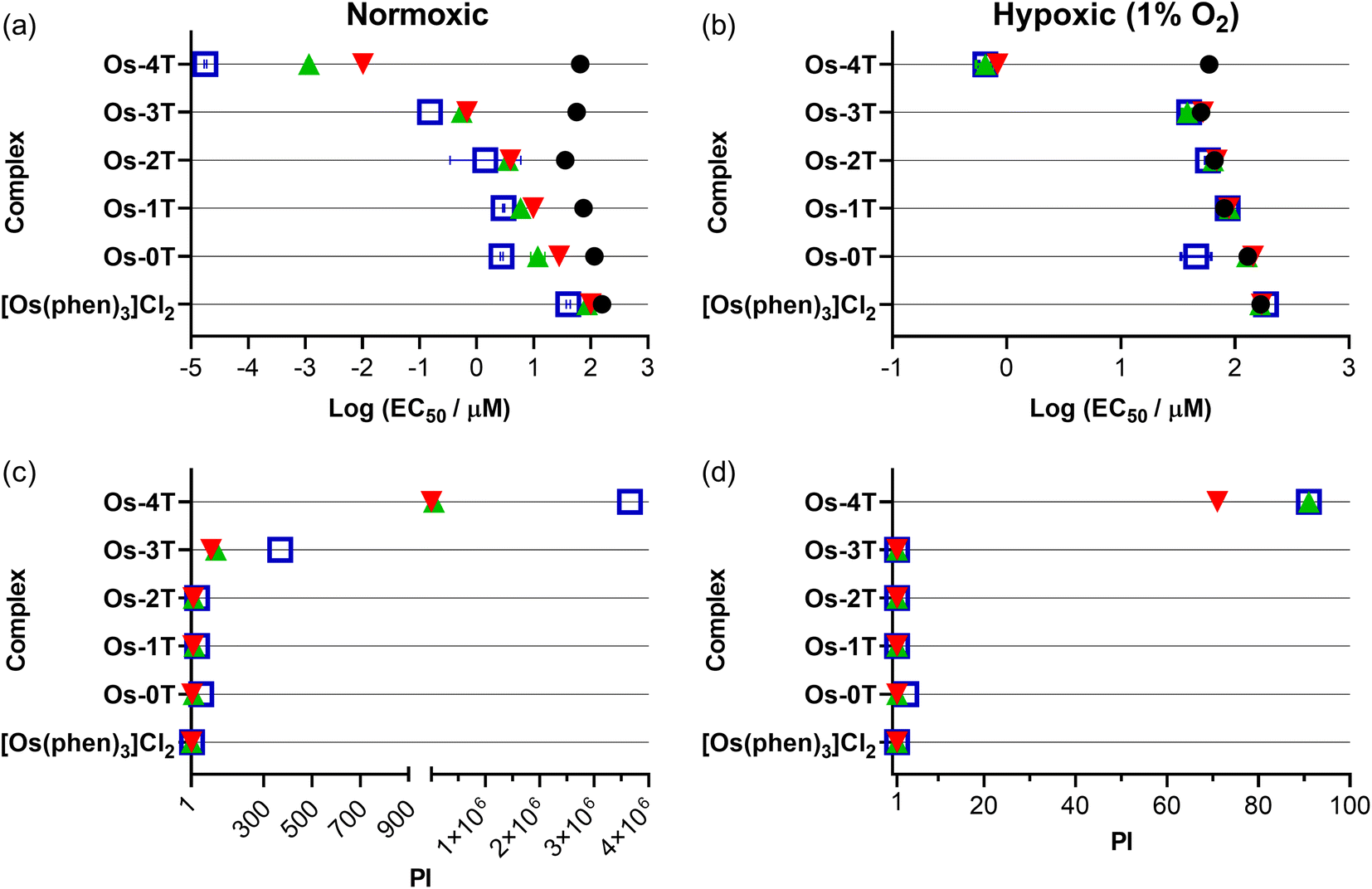 | ||
| Fig. 9 Summary resazurin-based (photo)cytotoxicity of Os-0T–Os-4T compared to reference compound [Os(phen)3]Cl2 against SK-MEL-28 in normoxia (18.5–21% O2;106 left-column (a and c)) and hypoxia (1% O2; right-column (b and d)). The phototherapeutic index (PI) as the ratio of light to dark cytotoxicity is plotted in (c) and (d). Figures are labelled as dark (no light; black circles) and 100 J cm−2 treatments at ∼20 mW cm−2 as cool white visible (blue, open square), 523 nm (green, triangle) and 633 nm (red, inverted triangle). Results are tabulated in Tables S-4–S-7.† | ||
3.5.1.3 Photocytotoxicity. The photocytotoxicities of the compounds in the series were assessed using a light fluence of 100 J cm−2 with broadband visible (400–700 nm, 21 mW cm−2), green (523 nm, 18 mW cm−2), and red (633 nm, 18 mW cm−2) light. The spectral output of the light sources is shown in Fig. S-22 and S-23,† and the comparisons of the photocytotoxicity as EC50 values are shown in Fig. 9a.
The measured light EC50 values, which reflect contributions from both the dark and light cytotoxicity of each complex, generally increased with n in Fig. 9a and Table S-4.† The reference compound [Os(phen)3]Cl2 was considered relatively inactive, with visible EC50 values of 40–100 μM regardless of the light condition. The visible-light photocytotoxicities of the other compounds spanned almost six orders of magnitude. The values for compounds with n = 0–2 were in the 1–3 μM range, while that for Os-3T was nanomolar (EC50 = 153 nM). Os-4T was notable in its activity, with its visible EC50 value of 18 pM. These photocytotoxicities were attenuated slightly (but still within an order of magnitude) with the longer green (523 nm) and red (633 nm) wavelengths of light. However, the photocytotoxicity exerted by Os-4T exhibited a strong wavelength-dependence. Its 18 pM EC50 value fell to 1 nM with green light and 10 nM with red light. These EC50 values are still considered to be quite potent, but the wavelength dependence suggests a possible change in the mechanism with one mechanism clearly being more efficient than another.
3.5.1.4 Phototherapeutic index (PI). Because the photocytotoxicity, quantified as an EC50 value, contains contributions from any inherent dark cytotoxicity, the PI is the more appropriate benchmark for light-induced toxicity. The trends for the light EC50 values were reflected in the PIs that are compared in Fig. 9 and Table S-4.† Light had very little effect on the cytotoxicity of reference compound [Os(phen)3]Cl2 (PI = 2–4, regardless of wavelength). The PIs for compounds with n = 0–2 were larger than the reference compound, but still relatively low overall: <50 with visible light and near 10 or less for both green and red light. Os-3T had PI values that were generally ten-fold greater (PIvis = 369, PIgreen = 103, PIred = 83) despite having a 1O2 quantum yield (ΦΔ = 51%) that was similar to that of [Os(phen)3]Cl2 and Os-0T−Os-2T. The spike in phototoxic effects for Os-3T is most likely due to the low-energy 3ILCT state that becomes accessible at n ≥ 3 as supported by computational studies and TA measurements.
Os-4T stood out among the series, not only with its near unity 1O2 quantum yield and long triplet excited state lifetimes (3–4 μs and 14–16 μs), but also its exceptionally large PIvis that exceeded 106. Even with attenuation at the lower-energy wavelengths, PIs were still >56000 and >6500 with green and red light, respectively. McKenzie et al. have reported that a ratio of PI to light dose > 100 is rare.107 In this respect, Os-4T's visible and green PIs are among the largest in the literature and very intriguing.
Os-4T did have a much higher 1O2 quantum yield (ΦΔ = 95%) and the longest triplet excited state lifetime relative to the other compounds in the family. However, Os-4T is not unique in terms of its triplet state lifetime or its near unity 1O2 quantum yield. Other complexes have such characteristics and even longer lifetimes.23 It is also not particularly unusual in terms of its ground state absorption properties and ILCT triplet configuration; other complexes have similar profiles and exhibit less potency (i.e., Os-3T as one example).23 There were also no clear differences between compounds of the present family in terms of their computed excited state reactivities with oxygen or autoionization capacity, except that the complexes with n ≤ 2 could directly form superoxide. The lower-energy 3ILCT states in Os-4T, including the additional τ2 ≈ 3–4 μs process that was observed in the TA kinetics of Os-4T but absent for Os-3T, could be a starting point for better understanding this unique compound. The pronounced wavelength-dependence for the activity of Os-4T may also support that initially populated 1ILCT states are more efficient at producing the reactive triplets important for phototoxic effects.
3.5.1.5 Hypoxic cellular assays. The compounds were also assessed for their abilities to maintain a phototoxic response toward SK-MEL-28 cells under low oxygen conditions (hypoxia, 1% O2). We have been investigating photobiological agents under hypoxic conditions, and have developed an optimized protocol for these difficult experiments.41 Multiple parameters required optimization, including: incubation time in hypoxia prior to light treatment (DLI in hypoxia), the method for maintaining hypoxia during the light treatment (e.g., low gas permeable films versus irradiating inside the hypoxia chamber), the post-PDT incubation period, and the cell viability determination protocol since metabolic viability indicators such as resazurin are strongly affected by hypoxic media. The reproducibility of this new protocol was evaluated over the past few years and found fairly robust with routine screens. The use of internal control(s) known to be strongly oxygen-dependent for their activity proved to be invaluable for experiment validation. Furthermore, passive gas exchange (surface-level) is only feasible to qualify hypoxic conditions if an appropriate time is allotted. We suggest that effective experiments to evaluate biological systems under hypoxia require either confirmation of (low) dissolved oxygen levels or sufficient measures to both control and maintain low oxygen at the point of light treatment. Without rigorous experimental design, studies may give misleading or irreproducible results. Unfortunately, this biases compounds to appear more promising than they truly are, distracting researchers from better agents or scaffolds remaining to be discovered.
These assays were run in parallel with normoxic assays (Fig. 9b and d) using our optimized hypoxia method41 and included [Ru(bpy)2(dppn)]Cl2 as an O2-dependent reference photosensitizer. Briefly, cells were equilibrated in hypoxia 2–3 h before treatment with the compounds and kept in hypoxia throughout the experiment (≥16 h). To mitigate any interference with the resazurin cell viability dye in hypoxia as we previously reported,41 the samples were allowed to recover in normoxia after the light treatment. We have found post-irradiation recovery in normoxia to be a robust strategy for preserving the reliability of the resazurin indicator, confirmed by the absence of phototoxic effects for compounds that rely on oxygen for photocytotoxicity. The results are shown in Fig. 9 and tabulated in Table S-5.†
Almost all of compounds had dark EC50 values in hypoxia that were within about 10% of those in normoxia, with some compounds being slightly less dark toxic in hypoxia and others being slightly more. The exception was Os-2T, which showed a two-fold reduction in dark cytotoxicity in hypoxia. The photocytotoxicities of [Os(phen)3]Cl2 and Os-0T–Os-3T were very similar to their dark cytotoxicities, giving rise to PI values that were less than 2 in most cases and thus considered inactive as hypoxic phototoxic agents. Os-4T, again, was a notable exception. While its phototoxic effects were attenuated by several orders of magnitude, EC50 values were still submicromolar (651–835 nM) with PIvis,green = 91 and PIred = 71 (Table S-5†). A striking observation was that the wavelength-dependence that was observed in normoxia (Fig. 9) was almost absent in hypoxia, suggesting that the photocytotoxicity in the two different oxygen conditions may derive from different excited states and/or pathways.
Since Os-4T stood out for its remarkable activity and because its activity in hypoxia was much larger than any we had previously published,41 we used sulforhodamine B (SRB) as a secondary endpoint to confirm the activity determined with resazurin as the cell viability indicator. This additional endpoint confirmed the activity of Os-4T in hypoxia with total growth inhibition (TGI) values of 140–170 nM for light-treated samples (Tables S-6 and S-7†). The SRB quantification also revealed that the resazurin-interference observed previously for hypoxic experiments was not an issue for more potent compounds (defined as compounds with subnanomolar EC50 in the normoxic resazurin assay) such as Os-4T.
Clearly, there remains much work to be done in order to understand the potency observed for Os-4T. Before attempting a mechanistic understanding, we first wanted to validate and test the robustness of our findings (e.g., reproducibility within our own lab and across different laboratories and researchers) and to explore the scope of this activity (e.g., hypoxia, NIR, other cell lines).
3.5.1.6 In-house validation. To validate the photocytotoxicity we observed for Os-4T, we tested the reproducibility of this response in normoxia and hypoxia several months after the initial screen was conducted. The results are compiled in Fig. 10, Tables S-8 and S-9,† where 0 represents the initial data obtained for Os-4T from Fig. 9, and 1–5 are repeats of that experiment performed over a one-month period where certain factors were purposely varied: location of the various Os-4T concentrations on the microplate (by row), pipette tips used for serial dilution of the Os-4T stock solution, SK-MEL-28 seed stock batch, and room lighting. Consumables were of the same lot number and all cells were between 10–15 passages. Each repeat consisted of three replicates. In Fig. 10, hypoxic data points (1% O2) are connected by dashed lines with open shapes and normoxic data points are connected by solid lines and solid shapes.
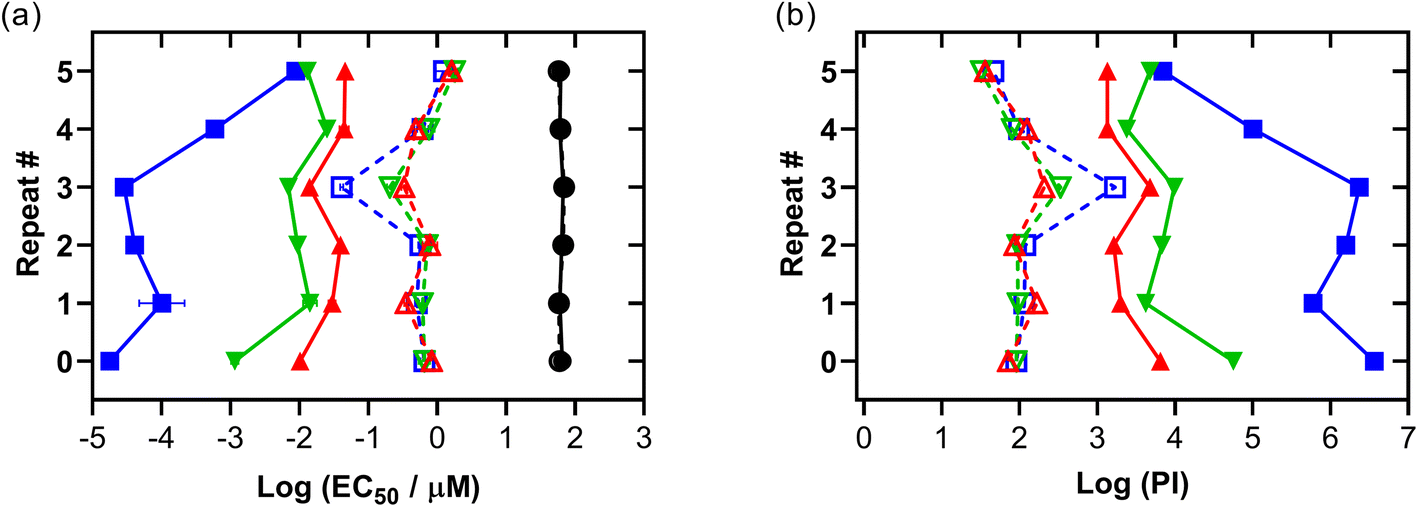 | ||
| Fig. 10 Summary of Os-4T interassay performance in SK-MEL-28 with various factors changed across each repeat as described in the experimental. Normoxic points are connected by solid lines and labelled as dark (no light; black circles) and 100 J cm−2 treatments at ∼20 mW cm−2 as cool white visible (blue, square), 523 nm (green, inverted triangle) and 633 nm (red, triangle) ± log(SEM). SEM = standard error of the mean. Hypoxic treated plates (1% O2) are connected by dashed lines and open shapes for the same color scheme. | ||
The dark cytotoxicity associated with Os-4T did not change substantially between the repeated experiments in either oxygen condition, with the average dark EC50 value being 62.8 μM with less than 7% variation. The photocytotoxicities and PIs for Os-4T showed variation across the repeated experiments compared to the dark cytotoxicity, but the variation was notably less than that for the normoxic condition. In hypoxia, the variation in phototoxic effects was relatively consistent across the three light conditions, with average EC50 values and PIs ranging from 351 nM to 1.80 μM and 32 to 167, respectively. The average hypoxic EC50 value and PI across the three light conditions were 0.816 μM and 90, respectively, with the exception of the repeat 3 (which was not included in the average because 1% O2 was not maintained for that specific experiment).
The larger variation in normoxic photocytotoxicities and PIs also exhibited a wavelength dependence, with the most variation in the visible light condition (222%). The EC50 value ranges were greatest for the visible light treatment (0.0178 to 8.91 nM) and least for red (10.0 to 46.2 nM), with green being intermediate (1.16 to 25.4 nM). The values for PIvis varied from 103 to 106, while values for PIred were consistently >103 and PIgreen ranged between 103 and 104. The average normoxic EC50 values for the visible, green, and red light treatments were 1.62 nM, 11.7 nM, and 30.8 nM, respectively. The corresponding average values for PI were on the order of 106, 104, and 103, respectively.
Together, the data indicates that the phototoxic mechanism that is operative in normoxia with shorter wavelengths of light is subject to a much greater degree of variability (three orders of magnitude) and different from the hypoxic phototoxic mechanism. Despite the variation, Os-4T remained extremely potent in normoxia even for the least potent repeat. Importantly, the hypoxic activity that we observed in the original set of experiments was confirmed across repeated screening and even the least potent repeats were still considered potent for hypoxia.
3.5.1.7 External validation. The family, excluding [Os(phen)3]Cl2, was further evaluated for their (photo)cytotoxicities in two cell lines in another laboratory by different researchers, whereby SK-MEL-28 was the human melanoma cell line used in-house (but not from the same seed stock) and the mouse B16-F10 melanoma cell line was an additional line. The assay was similar, including the light wavelengths and fluences employed, but the light sources and irradiances were different than those used to generate the data in Fig. 9.
While the Os(II) complexes were already relatively nontoxic to SK-MEL-28 cells in the absence of a light trigger, they were two-fold less dark toxic in the external validation experiments despite using similarly low cell passage numbers and had EC50 values that ranged from 123 to 238 μM (Fig. 11a and Table S-10†). The overall trend in photocytotoxicities followed the in-house trend, with potency increasing with the number of thiophenes rings in the chain. The PIvis of Os-4T (Fig. 11b) was at the lower end of the range measured in-house at approximately 5 × 104 (visible EC50 = 2.91 nM), but its PIred was ten-fold larger than in-house at 1.5 × 104 (red EC50 = 9.44 nM). In the case of shorter-wavelength visible light, the attenuated PIvis externally measured could arise from the inherent variability already noted at the shorter wavelengths (Fig. 10) in combination with the different light source and higher irradiance used. For the larger PIred measured externally, the discrepancy could be due to different light sources and irradiances having been used and that the external SK-MEL-28 cells were more dark-resistant compared to those that were used in-house.
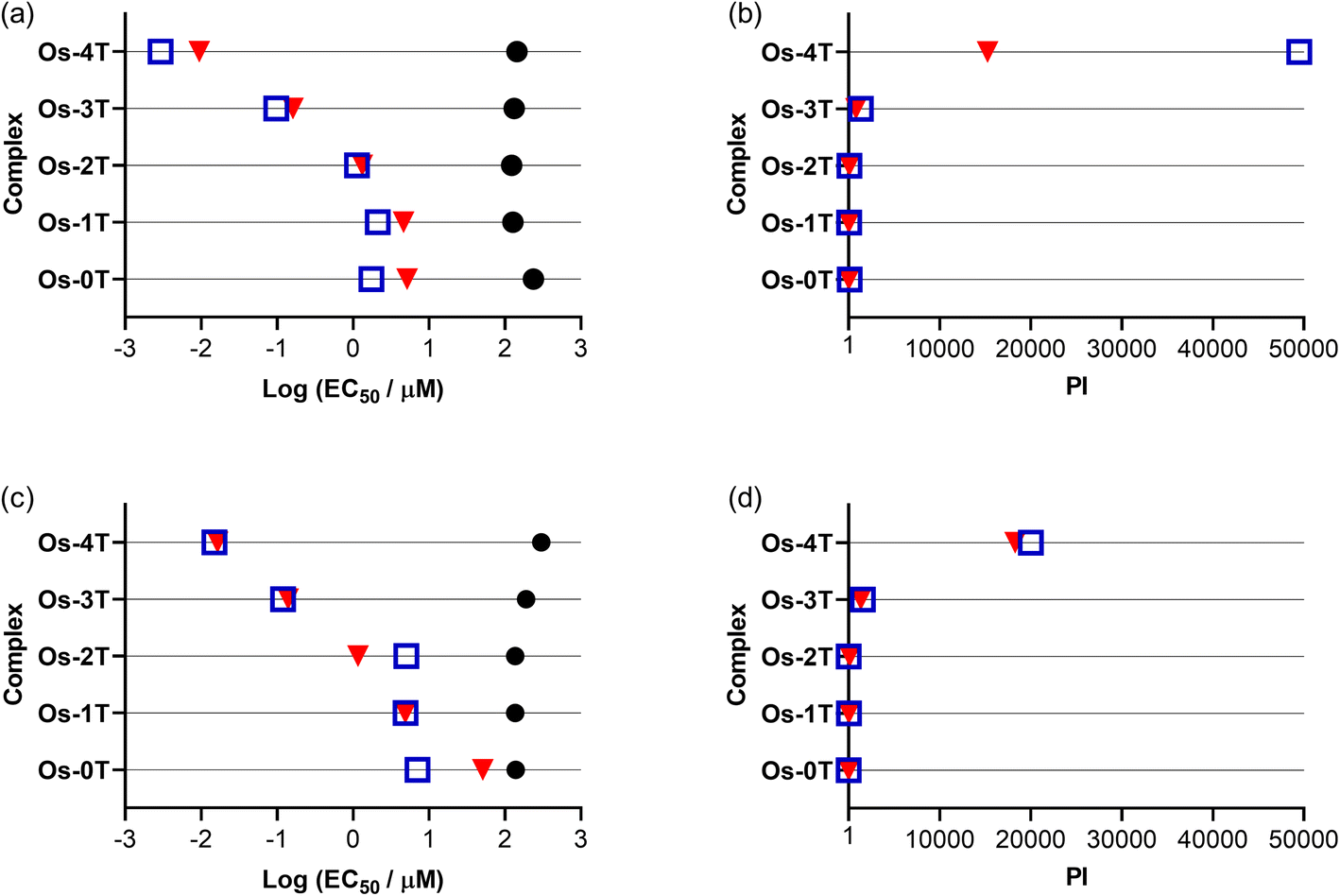 | ||
| Fig. 11 Summary resazurin-based (photo)cytotoxicity of Os-0T–Os-4T in normoxia against human amelanotic SK-MEL-28 (a and b) and murine melanotic B16-F10 (c and d) cell lines. Figures are labelled as dark (no light; black circles) and 100 J cm−2 treatments using 33 mW cm−2 visible (blue, open square) or 40 mW cm−2 633 nm (red, inverted triangle). | ||
To test the robustness of this response toward melanoma cells, the external validation study also compared the activities of the compounds toward a highly pigmented murine B16-F10 cell line since melanoma cells can vary widely in pigmentation108 and pigmentation can reduce phototoxic effects.109,110 The compounds were also relatively nontoxic to the highly pigmented murine B16-F10 cell line in the dark (Fig. 11c and Table S-10†). Their dark EC50 values were 140–190 μM and 300 μM for Os-0T–Os-3T and Os-4T, respectively. Os-3T and Os-4T were less dark toxic toward the B16-F10 cell line relative to the SK-MEL-28 line.
Their photocytotoxicity trends with increasing n paralleled those in SK-MEL-28 (external validation), with nanomolar activity for Os-3T and Os-4T (Fig. 11c and d). However, their visible EC50 values were larger across the series compared to SK-MEL-28 and indicated greater resistance to visible light-induced toxicity in the B16-F10 cell line, where EC50 values ranged from 15 nM for Os-4T to 7.04 μM for Os-0T. Only Os-0T and Os-4T were attenuated with red light in B16-F10 with respect to red EC50 values in the SK-MEL-28 cell line: ten-fold for Os-0T and two-fold for Os-4T. Despite this attenuation, Os-3T and Os-4T had similar PI values as those measured in the SK-MEL-28 cell line for both light conditions due to their lower dark cytotoxicity toward the murine B16-F10 cell line. For Os-4T, the visible and red PIs were still >20000 and >18000, respectively and were comparable to those measured externally for the SK-MEL-28 cell line. The absence of a difference in photocytotoxicity between the visible and red light treatments in the B16-F10 cell line for Os-4T, which was observed for SK-MEL-28 (both in-house and externally), may reflect the ability of increased melanin (from more advanced melanosome stages)108 in the highly pigmented B16-F10 cells to absorb the shorter wavelengths.
3.5.1.8 Fluence and irradiance. The scope of photoactivity toward the SK-MEL-28 cell line was explored further for Os-4T using monochromatic laser light with a smaller bandwidth rather than LEDs, with the opportunity to precisely deliver various fluence and irradiance combinations well-by-well (37 °C, 5% CO2) using the Modulight ML8500 platform. The response as a function of light fluence was explored at two different concentrations (0.5 and 1.0 μM) and wavelengths (525 and 630 nm; Fig. 12a and b). Higher concentrations combined with larger fluences yielded greater potency following what would be expected for PDT dosing (μM J cm−2). Green light was more effective than red light at lower fluences for the two concentrations tested, but the two wavelengths both yielded maximal and indistinguishable potency around 100 J cm−2 and higher. The irradiance dependence in Fig. 12c and d was explored using a fluence of 100 J cm−2 and 525 nm light (100 nM) or 630 nm light (250 nM), which were conditions where the Modulight illumination system gave sub-maximal potency. In this region, we were able to observe that lower irradiances yielded higher photocytotoxicity and that the higher concentration used for the red-light condition was more susceptible. In other words, the phototoxicity with red light can be improved at a defined fluence by lowering the irradiance and increasing the concentration of Os-4T. This flexibility is advantageous when progressing to in vivo models.
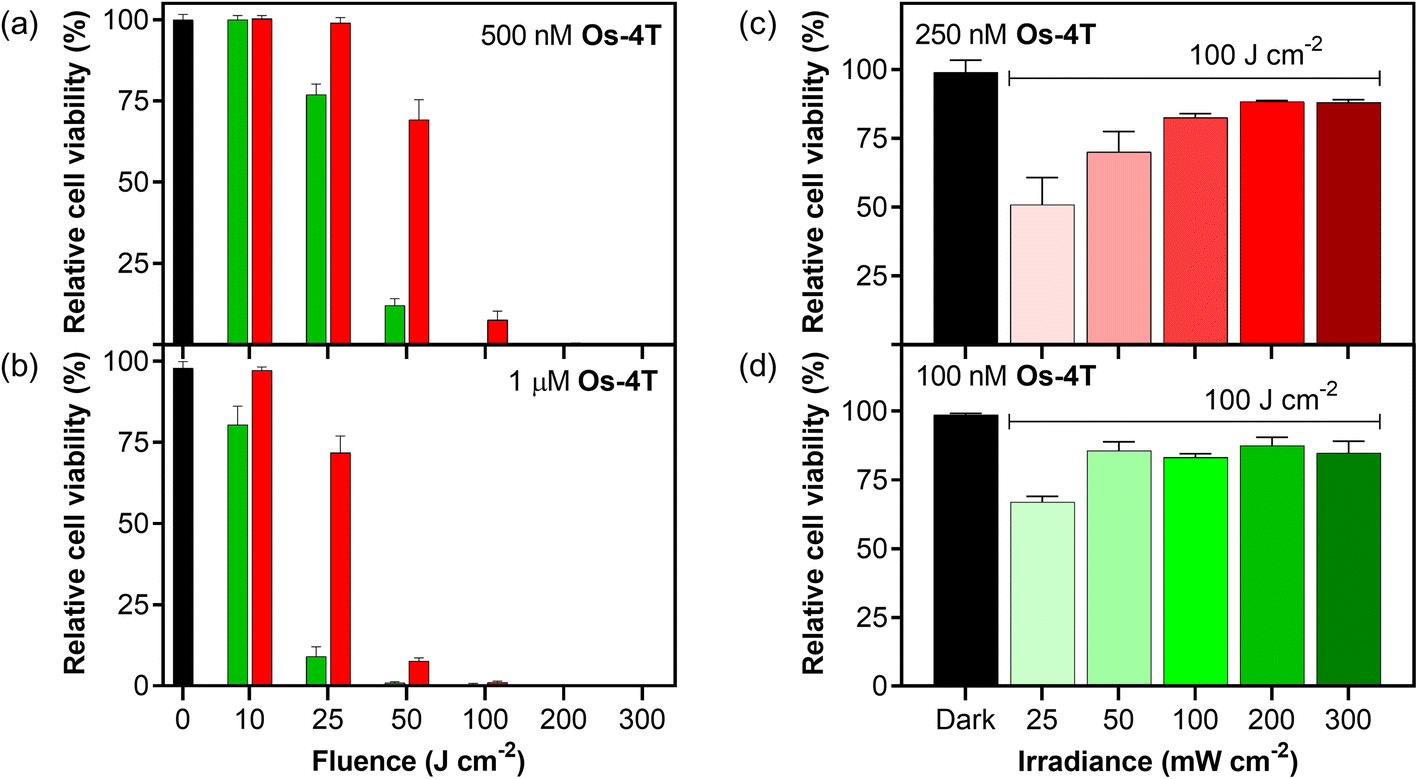 | ||
| Fig. 12 Dosimetry experiments on the Modulight ML8500 (37 °C, 5% CO2) in triplicate (±SD) with viability based on resazurin. Fluence dependence of (a) 500 nM or (b) 1 μM Os-4T against SK-MEL-28 at 300 mW cm−2. The left-most column is a dark (no-light; black bar) reference of compound cytotoxicity. Light treatments included 10–300 J cm−2 of either 525 nm (left bar, green) or 630 nm (right bar, red). Irradiance dependence of Os-4T with either no light (left, dark, black bar), (c) 630 nm and 250 nM, or (d) 525 nm and 100 nM treatments at 100 J cm−2 and 25–300 mW cm−2. | ||
3.5.1.9 Near-infrared (NIR) activation. We also investigated the photocytotoxicity of Os-4T toward SK-MEL-28 cells using wavelengths in the so-called PDT window (700–900 nm) despite minimal absorption in this region. Only high concentrations (≥10 μM) combined with very high fluences (≥400 J cm−2) yielded a marginal response (14% viability reduction) with 753 nm delivered at an irradiance of 300 mW cm−2 (Fig. S-46†). However, shortening the excitation by only 20 nm produced a substantial change in the NIR response of Os-4T. When 733 nm light was delivered at low irradiance (5 mW cm−2), we observed submicromolar response with EC50 = 0.803 μM and a PI733 nm of 77 with 100 J cm−2 fluence (Fig. 13). This was the same fluence (albeit at lower irradiance) used in the dose–response data plotted in Fig. 9, where the molar extinction coefficient at 733 nm for Os-4T was less than 300 M−1 cm−1. This NIR photocytotoxicity was sensitive to oxygen tension and disappeared in hypoxia (1% O2, PI733 nm = 1).
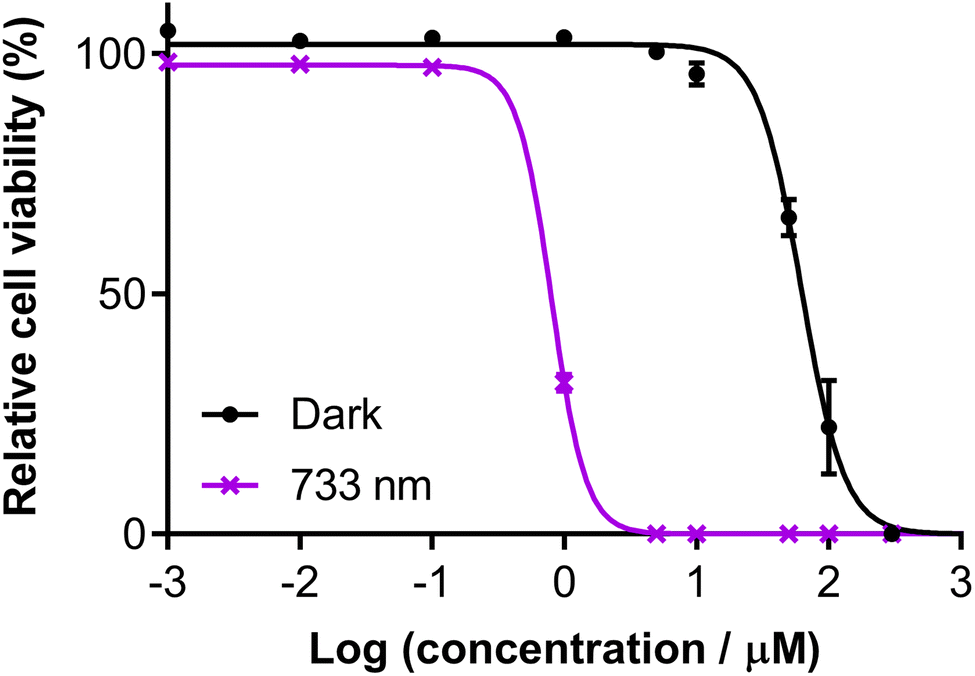 | ||
| Fig. 13 Dose–response (±SD) of Os-4T in SK-MEL-28 cells with 733 nm light at 100 J cm−2 and 5 mW cm−2. Dark (sham) treatments are black filled circles and 733 nm violet crosses. | ||
3.5.1.10 Maximum tolerated dose (MTD) in mice. Since our in vitro models indicated low dark toxicity (and even lower in the murine line), maximum tolerated dose (MTD) studies were performed on female C57BL/6J mice to gauge whether in vivo phototoxicity experiments were a logical next step. The mice treated by intraperitoneal injection of Os-4T were observed to experience mild toxicity at doses above 100 mg kg−1. At the three tested doses of 100, 125, and 200 mg kg−1—all animals displayed signs of stress (intermittent hunching, ears laid back), were subdued but responsive, and experienced weight loss. Their irregular behavior, however, resolved after 2 h. Since no signs of moderate toxicity were detected over the 2 week study, the intraperitoneal MTD of Os-4T was determined to be ≥200 mg kg−1. Since the complex Os-4T is highly phototoxic in normoxia, active in hypoxia, active in the photodynamic window at 733 nm, and nontoxic in mice, we plan to investigate this compound for its in vivo light-triggered antitumor properties.
4. Conclusions
This study highlights the structure–activity relationships for a series of [Os(phen)2(IP-nT)]Cl2 complexes from n = 0 to 4 with respect to ground and excited state geometries and configurations, 1O2 quantum yields, excited state lifetimes, and dark and light cytotoxicities toward melanoma cells in normoxia and hypoxia using various light parameters. Our hypothesis was that an abrupt switch in photophysical behavior would occur at some critical n, where a lowest energy 3ILCT state would dominate the excited state dynamics and lead to increased sensitivity to oxygen that might be maintained even in hypoxia. This strategy was applied successfully for the analogous Ru(II) series and resulted in TLD1433, which is the first non-tetrapyrrole-based metal complex to enter human clinical trials for treating cancer with PDT. Replacement of Ru(II) with the heavier Os(II) center serves to shift the optical window for activation to longer wavelengths with higher quantum yields for triplet state formation for all of the complexes, while keeping the highly reactive 3ILCT state at the requisite energy for potent phototoxicity only for certain values of n.The computational studies corroborated the critical point (n ≥ 3) for switching to a lowest lying 3ILCT state as determined by TA spectroscopy and did suggest that the 3ILCT state for Os-4T would be better positioned energetically to dominate the photophysics, but did not explain the unusual reactivity of Os-4T. While the high 1O2 quantum yield and prolonged lifetime for Os-4T could explain why this compound stood out among the series, these factors cannot be solely responsible for the picomolar potency and PI > 106 with visible light. An interesting facet to its activity was that the photocytotoxic response with visible light (enriched in the shorter, bluer wavelengths) varied by over three orders of magnitude with repeated experiments in normoxia, but even at its least potent was nanomolar with PIs >104. Even at its least potent, it still has one of the largest PIs reported. This potency was attenuated with the longer green and red wavelengths of light, but was not subject to the large variation in response observed with the shorter wavelengths. These results seem to point toward a different mechanism of action for the phototoxic effect with visible light. Interestingly, there was no drastic variation in the visible light response in hypoxia, and there was also almost no difference in the potencies between the shorter and longer wavelengths in hypoxia. This observation points toward a different phototoxic mechanism in hypoxia (at least with visible light) compared to normoxia.
While we are currently investigating the mechanism, we felt it more urgent to confirm our potency in an additional melanoma cell line and by other researchers given that results can be highly variable between laboratories running cytotoxic assays, especially with the added complexity of the light treatment (different light sources, wavelengths, fluences, irradiances, etc.). While the PI of Os-4T with visible light in normoxia measured externally was at the lower end of the range we measured in-house (but still low nanomolar with a PI > 104), the red PI was ten-fold higher. In the highly pigmented mouse melanoma cells, these PIs were attenuated but still >104.
The photocytotoxicity of Os-4T demonstrated an irradiance dependence, where larger PIs were obtained with lower irradiance. This afforded the opportunity to optimize the light parameters to achieve a response with NIR light at 733 nm, where the EC50 value was still submicromolar with a PI of almost 102. Given this versatility and robustness in response combined with its high aqueous solubility, we examined the MTD in mice and found that Os-4T was tolerated very well (MTD > 200 mg kg−1 by intraperitoneal injection). As we demonstrated for our previous Os(II) panchromatic absorbers in an in vivo investigation, Os-4T is yet another example that dispels the myth that Os (regardless of oxidation state) is an inherently toxic metal that cannot be used in medicine
The unprecedented photoactivities observed for Os-4T will certainly be explored using more sophisticated models in future work. In addition, next steps are aimed at delineating the mechanism of action across the different wavelengths and oxygen concentrations, which would lay the foundation for increasing activities in hypoxia and with NIR wavelengths of light while developing our understanding of the photophysical relationship to photocytotoxicity in compounds of this class.
Conflicts of interest
S. A. M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.Acknowledgements
S. A. M., C. G. C., and G. D. thank the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) for partial support of this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S. A. M. also acknowledges the Canadian Institutes of Health Research (CIHR), the Natural Sciences and Engineering Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), and the Nova Scotia Research and Innovation Trust (NSRIT) for support. S. A. M. would also like to thank Prof. Edith (Phoebe) Glazer for insightful discussions. N. R. and M. E. A. are grateful to the Università della Calabria. M. E. A. would also like to thank Dr Antonio Francés Monerris for the valuable and constructive discussion. S. A. M. also thanks Dr Daniel Todd as UNCG's Triad Mass Spectrometry Facility manager and his assistants Jennifer Simpson and Diane Wallace. We thank Dr Franklin Moy for his experimental support and instrument maintenance as UNCG's NMR facility manager.References
- World Health Organization, World health statistics 2019: monitoring health for the SDGs, sustainable development goals., World Health Organization, S.l., 2019 Search PubMed.
- Prodrugs: challenges and rewards, ed. V. J. Stella, Springer: AAPS Press, New York, NY, 2007 Search PubMed.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef PubMed.
- I. Giang, E. L. Boland and G. M. K. Poon, AAPS J., 2014, 16, 899–913 CrossRef CAS PubMed.
- R. S. Kerbel, H. Kobayashi and C. H. Graham, J. Cell. Biochem., 1994, 56, 37–47 CrossRef CAS PubMed.
- A. Sharma, Nanomed, 2017, 12, 2137–2148 CrossRef CAS PubMed.
- E. C. Fiedler and M. T. Hemann, Annu. Rev. Cancer Biol., 2019, 3, 409–428 CrossRef.
- A. S. Azmi, B. Bao and F. H. Sarkar, Cancer Metastasis Rev., 2013, 32, 623–642 CrossRef CAS PubMed.
- E. B. Rankin and A. J. Giaccia, Science, 2016, 352, 175–180 CrossRef CAS PubMed.
- L. Vera-Ramirez and K. W. Hunter, F1000Research, 2017, 6, 2134 Search PubMed.
- Photodynamic therapy: basic principles and clinical applications, ed. B. W. Henderson and T. J. Dougherty, Dekker, New York, 1992 Search PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- R. Bonnett, Chemical aspects of photodynamic therapy, Gordon and Breach Science Publishers, Amsterdam, The Netherlands, 2000 Search PubMed.
- Advances in photodynamic therapy: basic, translational, and clinical, ed. M. R. Hamblin and P. Mróz, Artech House, Boston, Mass, 2008 Search PubMed.
- K. Plaetzer, B. Krammer, J. Berlanda, F. Berr and T. Kiesslich, Lasers Med. Sci., 2009, 24, 259–268 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- Handbook of photomedicine, ed. M. R. Hamblin and Y.-Y. Huang, CRC Press, Boca Raton, FL, 2014 Search PubMed.
- R. R. Allison, Future Oncol., 2014, 10, 123–124 CrossRef CAS PubMed.
- L. Benov, Med. Princ. Pract., 2015, 24, 14–28 CrossRef PubMed.
- Photodynamic medicine: from bench to clinic, ed. H. Kostron, T. Hasan and Royal Society of Chemistry (Great Britain), Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- D. van Straten, V. Mashayekhi, H. de Bruijn, S. Oliveira and D. Robinson, Cancers, 2017, 9, 19 CrossRef PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- B. Q. Spring, I. Rizvi, N. Xu and T. Hasan, Photochem. Photobiol. Sci., 2015, 14, 1476–1491 RSC.
- T. Minervini, B. Cardey, S. Foley, C. Ramseyer and M. Enescu, Metallomics, 2019, 11, 833–844 RSC.
- T. Respondek, R. N. Garner, M. K. Herroon, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2011, 133, 17164–17167 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- E. C. Glazer, Isr. J. Chem., 2013, 53, 391–400 CrossRef CAS.
- E. Wachter and E. C. Glazer, J. Phys. Chem. A, 2014, 118, 10474–10486 CrossRef CAS.
- A. Bahreman, J.-A. Cuello-Garibo and S. Bonnet, Dalton Trans., 2014, 43, 4494–4505 RSC.
- J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS.
- T. Sainuddin, M. Pinto, H. Yin, M. Hetu, J. Colpitts and S. A. McFarland, J. Inorg. Biochem., 2016, 158, 45–54 CrossRef CAS.
- J.-A. Cuello-Garibo, M. S. Meijer and S. Bonnet, Chem. Commun., 2017, 53, 6768–6771 RSC.
- J.-A. Cuello-Garibo, E. Pérez-Gallent, L. van der Boon, M. A. Siegler and S. Bonnet, Inorg. Chem., 2017, 56, 4818–4828 CrossRef CAS PubMed.
- J. K. White, R. H. Schmehl and C. Turro, Inorg. Chim. Acta, 2017, 454, 7–20 CrossRef CAS.
- S. Bonnet, Dalton Trans., 2018, 47, 10330–10343 RSC.
- M. H. Al-Afyouni, T. N. Rohrabaugh, K. F. Al-Afyouni and C. Turro, Chem. Sci., 2018, 9, 6711–6720 RSC.
- A. Li, C. Turro and J. J. Kodanko, Chem. Commun., 2018, 54, 1280–1290 RSC.
- D. Havrylyuk, M. Deshpande, S. Parkin and E. C. Glazer, Chem. Commun., 2018, 54, 12487–12490 RSC.
- D. Havrylyuk, K. Stevens, S. Parkin and E. C. Glazer, Inorg. Chem., 2020, 59, 1006–1013 CrossRef PubMed.
- J. Roque, D. Havrylyuk, P. C. Barrett, T. Sainuddin, J. McCain, K. Colón, W. T. Sparks, E. Bradner, S. Monro, D. Heidary, C. G. Cameron, E. C. Glazer and S. A. McFarland, Photochem. Photobiol., 2020, 96, 327–339 CrossRef PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef.
- K. Kalyanasundaram, Photochemistry of polypyridine and porphyrin complexes, Acad. Pr., London, 1992 Search PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–834 CrossRef.
- V. Balzani, A. Credi and M. Venturi, Coord. Chem. Rev., 1998, 171, 3–16 CrossRef CAS.
- V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, vol. 280, pp. 117–214 Search PubMed.
- C. L. Evans, A. O. Abu-Yousif, Y. J. Park, O. J. Klein, J. P. Celli, I. Rizvi, X. Zheng and T. Hasan, PLoS One, 2011, 6, e23434 CrossRef CAS PubMed.
- W. Feng, C. Gao, W. Liu, H. Ren, C. Wang, K. Ge, S. Li, G. Zhou, H. Li, S. Wang, G. Jia, Z. Li and J. Zhang, Chem. Commun., 2016, 52, 9434–9437 RSC.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. Le Dévédec and S. Bonnet, Angew. Chem., Int. Ed., 2017, 56, 11549–11553 CrossRef CAS PubMed.
- Z. Lv, H. Wei, Q. Li, X. Su, S. Liu, K. Y. Zhang, W. Lv, Q. Zhao, X. Li and W. Huang, Chem. Sci., 2018, 9, 502–512 RSC.
- V. H. S. van Rixel, V. Ramu, A. B. Auyeung, N. Beztsinna, D. Y. Leger, L. N. Lameijer, S. T. Hilt, S. E. Le Dévédec, T. Yildiz, T. Betancourt, M. B. Gildner, T. W. Hudnall, V. Sol, B. Liagre, A. Kornienko and S. Bonnet, J. Am. Chem. Soc., 2019, 141, 18444–18454 CrossRef PubMed.
- Q. Yu, T. Huang, C. Liu, M. Zhao, M. Xie, G. Li, S. Liu, W. Huang and Q. Zhao, Chem. Sci., 2019, 10, 9091–9098 RSC.
- V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Angew. Chem., Int. Ed., 2019, 58, 6311–6315 CrossRef.
- M.-D. Li, N.-K. Wong, J. Xiao, R. Zhu, L. Wu, S.-Y. Dai, F. Chen, G. Huang, L. Xu, X. Bai, M. R. Geraskina, A. H. Winter, X. Chen, Y. Liu, W. Fang, D. Yang and D. L. Phillips, J. Am. Chem. Soc., 2018, 140, 15957–15968 CrossRef.
- A. Li, R. Yadav, J. K. White, M. K. Herroon, B. P. Callahan, I. Podgorski, C. Turro, E. E. Scott and J. J. Kodanko, Chem. Commun., 2017, 53, 3673–3676 RSC.
- G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2015, 282–283, 127–138 CrossRef CAS.
- E. M. Kober, J. V. Caspar, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4587–4598 CrossRef CAS.
- Photochemistry and photophysics of coordination compounds. 1, ed. G. Accorsi and V. Balzani, Springer, Berlin, 2007 Search PubMed.
- Photodynamic therapy: From Theory to Application, ed. M. Abdel-Kadel, Springer, New York, 2013 Search PubMed.
- G. E. Shillito, C. B. Larsen, J. R. W. McLay, N. T. Lucas and K. C. Gordon, Inorg. Chem., 2016, 55, 11170–11184 CrossRef CAS PubMed.
- S. Lazic, P. Kaspler, G. Shi, S. Monro, T. Sainuddin, S. Forward, K. Kasimova, R. Hennigar, A. Mandel, S. McFarland and L. Lilge, Photochem. Photobiol., 2017, 93, 1248–1258 CrossRef CAS PubMed.
- E. C. Glazer, Photochem. Photobiol., 2017, 93, 1326–1328 CrossRef CAS.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagentsang, 2010, vol. 3 Search PubMed.
- G. Ghosh, K. L. Colón, A. Fuller, T. Sainuddin, E. Bradner, J. McCain, S. M. A. Monro, H. Yin, M. W. Hetu, C. G. Cameron and S. A. McFarland, Inorg. Chem., 2018, 57, 7694–7712 CrossRef CAS.
- L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szłyk, Magn. Reson. Chem., 2010, 48, 450–457 CrossRef CAS PubMed.
- W. J. Vining, J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1985, 89, 1095–1099 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. E. Casida, in Recent developments and applications of modern density functional theory, ed. J. M. Seminario, Elsevier, Amsterdam, Netherlands, 1996, pp. 155–192 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2000, 112, 2427–2435 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) C. Latouche, D. Skouteris, F. Palazzetti and V. Barone, J. Chem. Theory Comput., 2015, 11, 3281–3289 CrossRef CAS; (b) M. E. Alberto and C. Adamo, Chem.–Eur. J., 2017, 23, 15124–15132 CrossRef CAS.
- M. E. Alberto, J. Pirillo, N. Russo and C. Adamo, Inorg. Chem., 2016, 55, 11185–11192 CrossRef CAS PubMed.
- M. DeRosa, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- J. Blois, A. Smith and L. Josephson, Cancer Chemother. Pharmacol., 2011, 68, 795–803 CrossRef CAS.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS.
- Y. Arenas, S. Monro, G. Shi, A. Mandel, S. McFarland and L. Lilge, Photodiagn. Photodyn. Ther., 2013, 10, 615–625 CrossRef CAS.
- S. A. McFarland, Metal-based Thiophene Photodynamic Compounds and Their Use, US Pat., number 9,345,769, issued May 24, 2016.
- S. A. McFarland, Metal-based Thiophene Photodynamic Compounds and Their Use, US Pat., number 9,676,806 B2, issued June 13, 2017.
- L.-F. Tan, F. Wang, H. Chao, Y.-F. Zhou and C. Weng, J. Inorg. Biochem., 2007, 101, 700–708 CrossRef CAS PubMed.
- H. Xu, K.-C. Zheng, L.-J. Lin, H. Li, Y. Gao and L.-N. Ji, J. Inorg. Biochem., 2004, 98, 87–97 CrossRef CAS PubMed.
- Q.-X. Zhen, B.-H. Ye, J.-G. Liu, Q.-L. Zhang, L.-N. Ji and L. Wang, Inorg. Chim. Acta, 2000, 303, 141–147 CrossRef CAS.
- J.-Z. Wu and L.-N. Ji, Transition Met. Chem., 1999, 24, 299–303 CrossRef CAS.
- C.-W. Jiang, H. Chao, R.-H. Li, H. Li and L.-N. Ji, Polyhedron, 2001, 20, 2187–2193 CrossRef CAS.
- R. A. Scherrer and S. M. Howard, J. Med. Chem., 1977, 20, 53–58 CrossRef CAS PubMed.
- G. Lanzani, M. Nisoli, S. De Silvestri and R. Tubino, Chem. Phys. Lett., 1996, 251, 339–345 CrossRef CAS.
- J.-P. Yang, W. Paa and S. Rentsch, Chem. Phys. Lett., 2000, 320, 665–672 CrossRef CAS.
- Y. Arenas, S. Monro, G. Shi, A. Mandel, S. McFarland and L. Lilge, Photodiagn. Photodyn. Ther., 2013, 10, 615–625 CrossRef CAS.
- P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2016, 15, 481–495 RSC.
- M. E. Alberto, B. C. De Simone, G. Mazzone, E. Sicilia and N. Russo, Phys. Chem. Chem. Phys., 2015, 17, 23595–23601 RSC.
- J. Llano, J. Raber and L. A. Eriksson, J. Photochem. Photobiol., A, 2003, 154, 235–243 CrossRef CAS.
- M. E. Alberto, N. Russo and C. Adamo, Chem.–Eur. J., 2016, 22, 9162–9168 CrossRef CAS.
- M. E. Alberto, C. Iuga, A. D. Quartarolo and N. Russo, J. Chem. Inf. Model., 2013, 53, 2334–2340 CrossRef CAS.
- B. J. Pankuch, D. E. Lacky and G. A. Crosby, J. Phys. Chem., 1980, 84, 2061–2067 CrossRef CAS.
- S. Decurtins, F. Felix, J. Ferguson, H. U. Guedel and A. Ludi, J. Am. Chem. Soc., 1980, 102, 4102–4106 CrossRef CAS.
- V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS.
- R. C. Young, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1976, 98, 286–287 CrossRef CAS.
- M. B. Majewski, N. R. de Tacconi, F. M. MacDonnell and M. O. Wolf, Inorg. Chem., 2011, 50, 9939–9941 CrossRef CAS.
- B. Maubert, N. D. McClenaghan, M. T. Indelli and S. Campagna, J. Phys. Chem. A, 2003, 107, 447–455 CrossRef CAS.
- N. D. McClenaghan, Y. Leydet, B. Maubert, M. T. Indelli and S. Campagna, Coord. Chem. Rev., 2005, 249, 1336–1350 CrossRef.
- R. Wenger, V. Kurtcuoglu, C. Scholz, H. Marti and D. Hoogewijs, Hypoxia, 2015, 35 CrossRef PubMed.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef.
- K. G. Chen, R. D. Leapman, G. Zhang, B. Lai, J. C. Valencia, C. O. Cardarelli, W. D. Vieira, V. J. Hearing and M. M. Gottesman, J. Natl. Cancer Inst., 2009, 101, 1259–1271 CrossRef CAS.
- K. V. Sharma and L. M. Davids, Photodiagn. Photodyn. Ther., 2012, 9, 156–163 CrossRef CAS.
- A. Sparsa, S. Bellaton, T. Naves, M.-O. Jauberteau, J.-M. Bonnetblanc, V. Sol, M. Verdier and M.-H. Ratinaud, Oncol. Rep., 2013, 29, 1196–1200 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental methodology, synthetic characterization (1D and 2D NMR, HPLC, HRMS), spectroscopic characterization (emission, TA), computational predictions (ground and excited states), (photo)biological evaluations, and the various acronyms used in-text. See DOI: 10.1039/d0sc03008b |
| ‡ Present address: Carilion School of Medicine, Virginia Polytechnic Institute and State University, Roanoke, Virginia, 24016, USA. |
| § Present address: Department of Pathology, Dalhousie University, Halifax, Nova Scotia, B3H 4R2 Canada. |
| This journal is © The Royal Society of Chemistry 2020 |