
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePanchromatic dirhodium photocatalysts for dihydrogen generation with red light†
Jie
Huang
,
Judith C.
Gallucci
and
Claudia
Turro
 *
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210, USA. E-mail: turro.1@osu.edu
First published on 5th August 2020
Abstract
A series of three dirhodium complexes cis-[Rh2(DPhB)2(bncn)2](BF4)2 (1, DPhB = diphenylbenzamidine; bncn = benzocinnoline), cis-[Rh2(DPhTA)2(bncn)2](BF4)2 (2, DPhTA = diphenyltriazenide), and cis-[Rh2(DPhF)2(bncn)2](BF4)2 (3, DPhF = N,N′-diphenylformamidinate) shown to act as single-molecule photocatalysts for H2 production was evaluated. Complexes 1–3 are able to generate H2 in the absence of any other catalyst in homogenous acidic solution upon irradiation with red light in the presence of the sacrificial electron donor BNAH (1-benzyl-1,4-dihydronicotinamide). The excited state of each complex is reductively quenched by BNAH, producing the corresponding one-electron reduced complex. The latter is also able to absorb a photon and oxidize another BNAH molecule, producing the doubly-reduced, activated form of the catalyst that is able to generate H2. The present work shows the effect of substitution on the bridging ligands on the driving force for reductive quenching and hydricity of the proposed active intermediate, both of which affect the efficiency of hydrogen production. Complexes 1–3 operate following a double reductive quenching mechanism and, importantly, are active with red light. This work lays the foundation for the design of single-molecule photocatalysts that operate from the ultraviolet to the near-infrared, such that solar photons throughout this entire range are harnessed and utilized for solar energy conversion.
Introduction
Photons from the sun represent an abundant and free source of energy, making it critical to develop systems that are able to harness and transform sunlight into chemical bonds, or fuels, for long-term storage.1–8 Carbon-free energy sources obtained from water and sunlight, such as H2, represent a desirable clean fuel generated from abundant sources.1–12 Traditional photocatalytic H2 evolution systems include a number of components, including a light-absorbing chromophore, an electron relay, and a catalyst, and these molecular systems have been coupled to semiconductors and/or electrodes in dye-sensitized photoelectrosynthesis cells.9–15 For optimal performance, the chromophore should efficiently absorb incident photons and its spectral profile should be well-matched with that of the solar irradiance spectrum, such that as many wavelengths of sunlight as possible are utilized to electronically excite the chromophore.5,13–17 Following excitation, the excited state of the light absorber must be able undergo desirable charge transfer reactions, such as reducing the relay molecule so that it can shuttle redox equivalents to a catalytic center or semiconductor.1–17 However, such multi-component systems suffer from energy losses during each of these charge transfer processes. Multi-component systems featuring covalently linked chromophores and catalysts aim to circumvent these losses, however, their synthesis and characterization is often challenging.18–20 Moreover, these systems often suffer from fast charge recombination, thus reducing their ability to effectively relay redox equivalents to the catalyst.21,22 Nearly half of the solar spectrum falls into the red to near-IR light, a spectral region that has been underutilized by most light absorbers developed to date.23,24 Single-molecule photocatalysts are important to circumvent the shortcomings of multi-component systems,25 however, it is critical for the photocatalyst is activated by light from the ultraviolet to the red/near-IR light to better harness the solar photons that reach the earth.In the field of single-chromophore photocatalysts, those for the reduction of CO2 and for the evolution of H2 have been reported. Re(I)(bpy)(CO)3Br (bpy = 2,2′-bipyridine) is able to reduce CO2 to CO with a turnover number (TON) of 27 after 4 h (λirr > 400 nm).26 Two-electron mixed-valence dirhodium complexes have been shown to reduce halogen acids, HX, to produce H2 through the activation of the RhII–X bond and the formation of RhII–H species, with the highest TON = 7 after 72 h of irradiation with ultraviolet/blue light.25,27 In addition, the unsymmetrically substituted diiron hydride [(μ-H)Fe2(pdt)−(CO)4(dppv)]+ (dppv = cis-1,2-C2H2(PPh2)2) can generate H2 with TON = 4 from a triflic acid solution with >400 nm irradiation.28 A mononuclear tungsten photocatalyst, W(pyNHC)(CO)4 (pyNHC = (pyridyl)-N heterocyclic carbene), was recently reported to absorb with maximum at 400 nm and to produce H2 with TON of 17 after 3 h of irradiation.29 In general, mechanistic studies of H2-producing catalysts have revealed that terminal metal-hydrides act as important intermediates in product formation.30,31
Dirhodium complexes of the type cis-[Rh(form)2(L)2](BF4)2 (form = formamidinate, L = chelating/bridging diimine ligands) feature an intense metal/ligand to ligand charge transfer (ML-LCT) transition in the visible to near-IR range, making them good candidates as panchromatic photosensitizers.32–35 The present work focuses on the comparison of the photocatalytic H2 production by cis-[Rh2(DPhB)2(bncn)2](BF4)2 (1, DPhB = diphenylbenzamidine; bncn = benzocinnoline) and cis-[Rh2(DPhTA)2(bncn)2](BF4)2 (2, DPhTA = diphenyltriazenide), shown in Fig. 1, and a comparison of their activity to that of cis-[Rh2(DPhF)2(bncn)2](BF4)2 (3; DPhF = N,N′-diphenylformamidinate). Complex 3 was recently shown by our group to act as a single-chromophore photocatalyst for H2 generation with TON of 170 over 24 h under red light irradiation (λirr = 670 nm), also operative with λirr = 735 nm as well as with higher energy light.36 Herein, the electron donating ability of the bridging formamidine ligands is tuned in 1 and 2 to improve the excited state lifetime and photocatalytic H2 evolution activity of this bimetallic paddlewheel architecture. These Rh2(II,II) complexes represent a new class of single-molecule, single-chromophore photocatalysts that are able to act as both the light absorber and the catalytic center with absorption profiles that make them operative from the ultraviolet to the near-IR.
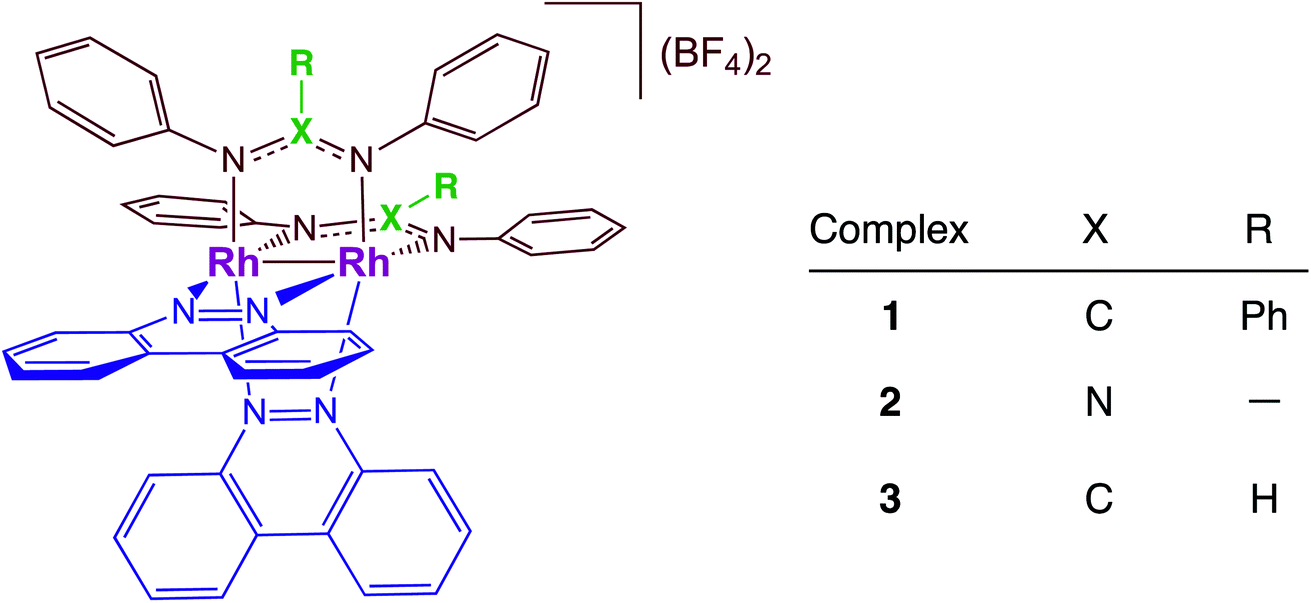 | ||
| Fig. 1 Schematic representation of the structures of 1–3 with the bncn ligands colored blue. | ||
Experimental
Materials
The solvents acetonitrile, N,N-dimethylformamide (DMF), dichloromethane, diethyl ether, and chlorobenzene were purchased from Fisher Scientific or Sigma Aldrich. Prior to use, the solvents were distilled over calcium hydride or filtered over dried alumina and stored over molecular sieves. Benzo[c]cinnoline (bncn) and p-toluenesulfonic acid (TsOH) were purchased from Sigma-Aldrich, Rh2(OAc)4 (OAc = acetate) and 1-benzyl-1,4-dihydronicotinamide (BNAH) were obtained from TCI. Cobaltocene was stored in a glove box preventing it from exposure to moisture and oxygen. Synthetic procedures were performed under a N2 atmosphere and the purification processes were opened to air; the general procedure is shown in Scheme S1† and 1H NMR spectra of the starting materials and products are shown in Fig. S1–S6 (ESI†). The ligands diphenylbenzamidine (DPhB) and diphenyltriazenide (DPhTA), as well as the precursor cis-[Rh2(DPhTA)2(CH3CN)6][BF4]2 were synthesized according to literature procedures.37,38Rh2(DPhB)3(OAc)
Rh2(OAc)4 (50 mg, 0.11 mmol) and 3 eq. of diphenylbenzamidine ligand were dissolved in 8 mL chlorobenzene from a sealed bottle under N2 and refluxed for 2 days. The solution turned dark-green. The reaction mixture was then cooled to room temperature and concentrated to dryness. The residue was washed with methanol to give a green solid. Yield: 80 mg (0.074 mmol, 66%).cis-[Rh2(DPhB)2(CH3CN)6][BF4]2
Rh2(DPhB)3(OAc) (100 mg, 0.093 mmol) was dissolved in 30 mL CH2Cl2/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), was heated to 45 °C, and 0.75 mL of 0.1 M (C2H5)3O(BF4) was added to the solution and allowed to stir at this temperature for 6 h, during which time it changed color from the initial green to brown/red. The reaction mixture was then cooled to room temperature and concentrated to dryness. The remaining solid was redissolved in ∼2 mL CH3CN, was added dropwise to 150 mL of rapidly stirring diethyl ether, was allowed to stir 1 h, and was then filtered through a medium frit under vacuum. The filtrate was pink and was confirmed to be Rh2(DPhB)4. The solid was washed with excess diethyl ether, resulting in a light green solid. Yield: 83% (90 mg, 0.077 mmol).
:1, v/v), was heated to 45 °C, and 0.75 mL of 0.1 M (C2H5)3O(BF4) was added to the solution and allowed to stir at this temperature for 6 h, during which time it changed color from the initial green to brown/red. The reaction mixture was then cooled to room temperature and concentrated to dryness. The remaining solid was redissolved in ∼2 mL CH3CN, was added dropwise to 150 mL of rapidly stirring diethyl ether, was allowed to stir 1 h, and was then filtered through a medium frit under vacuum. The filtrate was pink and was confirmed to be Rh2(DPhB)4. The solid was washed with excess diethyl ether, resulting in a light green solid. Yield: 83% (90 mg, 0.077 mmol).
cis-[Rh2(DPhB)2(bncn)6][BF4]2 (1)
Complex 1 was prepared from the reaction of cis-[Rh2(DPhB)2(CH3CN)6][BF4]2 (50 mg, 0.043 mmol) and benzo[c]cinnoline (15.4 mg, 0.086 mmol) in 20 mL dry CH3CN and refluxed overnight under N2 to give a green-blue solution. The reaction mixture was then cooled to room temperature and concentrated to dryness. The remaining solid was dissolved in ∼2 mL of CH3CN and was added dropwise to 150 mL of rapidly stirring diethyl ether, and allowed to stir for 1 h. The solution was then filtered through a medium frit under vacuum the solid product was washed with excess diethyl ether on the filter frit. Yield: 73% (42 mg, 0.031 mmol). 1H NMR (CD3CN, 400 MHz, 298 K) δ/ppm (int., mult., coupl., assignment): 9.57 (4H, d, J = 8.6, bncn), 8.85 (4H, d, J = 7.8, bncn), 8.19 (8H, m, bncn), 7.16 (2H, d, J = 7.6, Phenyl), 6.91 (13H, m, DPhB phenyl), 6.73 (3H, t, J = 7.6, Phenyl), 6.53(10H, t, J = 7.8 Phenyl), 6.09 (2H, d, J = 7.7, Phenyl). ESI-MS: m/z = 554.4, z = 2+.cis-[Rh2(DPhTA)2(bncn)6](BF4)2 (2)
Complex 2 was prepared by refluxing a solution of 20 mL of dry CH3CN containing cis-[Rh2(DPhTA)2(CH3CN)6](BF4)2 (50 mg, 0.05 mmol) and bncn (18 mg, 0.10 mmol) in 20 mL overnight under N2, resulting in a purple-blue reaction mixture. The solution was then cooled to room temperature and dried under vacuum. The solid was redissolved in ∼2 mL CH3CN and this concentrated solution was added dropwise to 150 mL of rapidly stirring diethyl ether. The mixture was stirred for 1 h and was then filtered through a medium frit under vacuum. The residue was washed with excess diethyl ether to obtain dry product. Yield: 59% (35 mg, 0.029 mmol). 1H NMR (CD3CN, 400 MHz, 298 K) δ/ppm (int., mult., coupl., assignment): 9.70 (4H, d, J = 8.4, bncn), 8.83 (4H, d, J = 7.7, bncn), 8.33 (4H, t, J = 7.9, bncn), 8.24 (4H, t, J = 7.2, bncn), 7.34 (12H, m, J = 7.3, DPhTA phenyl), 7.05 (8H, dd, J = 7.6, J = 1.9 Phenyl). ESI-MS: m/z = 479.3, z = 2+.Instrumentation and methods
1H NMR spectra were recorded on a Bruker 400 MHz or 250 MHz spectrometer and the peaks were referenced to the residue CD3CN solvent peak at 1.94 ppm. Electronic spectra were collected on a Hewlett–Packard 8454 diode array spectrometer in 1 cm × 1 cm quartz cuvettes. Electrospray ionization mass spectroscopy (ESI-MS) was performed on a Bruker MicroTOF spectrometer in positive low ion mode with samples dissolved in CH3CN.A single crystal of 1 was obtained by slowly diffusing diethyl ether over a CH3CN solution containing the complex. The diffraction pattern was measured on a Bruker D8 Venture diffractometer with a Photon II detector, which indicated a monoclinic crystal system conducted at 150 K using an Oxford Cryosystems Cryostream Cooler. The data collection strategy was set up to measure a quadrant of reciprocal space with a redundancy factor of 6, such that 90% of the reflections were measured at least 6 times. Omega and phi scans with a frame width of 0.5° and a frame time of 20 seconds were used and additional details are listed in Table S1 (ESI†). The data frames were collected using the program APEX3 and processed with the SAINT program within APEX3.39 Absorption and beam corrections were made with the multiscan technique in SADABS.40
Cyclic voltammetry was conducted using a BASi CV-50 W potentiostat with glassy carbon disc as the working electrode, Ag/AgCl as the reference electrode, and Pt wire as the counter electrode under a N2 atmosphere. The sweep rate was constant at 200 mV s−1, the potential was referenced to ferrocene (E1/2 = +0.55 V vs. Ag/AgCl in DMF or +0.44 V vs. Ag/AgCl in CH3CN), and 0.1 M nBu4NPF6 (TBAPF6) was used as the supporting electrolyte. For electrocatalysis experiments, aliquots of 3 μL of 2 M TsOH, salicylic acid, or acetic acid in CH3CN or DMF solution were titrated into the solution containing the photocatalyst (3 mL total volume). Before each scan, the working electrode was polished to eliminate the possibility of the accumulation of decomposition products or aggregation on the surface. Spectroelectrochemistry experiments were recorded using a homemade H-shape two-compartment cell. The glassy carbon rod working electrode and Ag/AgCl reference electrode were placed in the working compartment equipped with a quartz cuvette, and the carbon mesh counter electrode was positioned in the auxiliary compartment. Spectroelectrochemistry solutions containing 0.1 M TBABF4 as the electrolyte were held ∼200 mV more negative potential than the one- and two-electron couples to generate the respective species until the electronic absorption spectrum no longer changed.
Emission spectra were recorded on a FluoroMax-4 spectrometer (Horiba Jobin Yvon) at 77 K in CH3CN using a homemade Dewar. An appropriate long-pass filter was used at the entrance slit of the detection monochromator to eliminate scattered excitation light. A PMT single wavelength detector was used to record λ < 800 nm signals and an iCCD was used for λ > 800 nm emission; the reported results are the average of five trials. Femtosecond transient absorption spectroscopy was performed on a homebuilt system previously described in detail.41 The excitation (2–3 μJ) and white light probe beams are focused onto a 1 mm pathlength flow cell that contained the sample. Nanosecond transient absorption data was collected using a basiScan tunable optical parametric oscillator (Spectra-Physics) pumped by a frequency-tripled Quanta-Ray INDI Nd:YAG laser (Spectra-Physics, ∼6 ns pulses at 10 Hz), and an LP980 spectrometer system (Edinburgh Instruments) equipped with a 150 W Xe arc lamp as the white light probe. Spectral measurements were collected using an iCCD camera (iStar, Andor Technology) and single wavelength kinetic traces were collected using a PMT and digital oscilloscope (Tektronix MDO3022, 200 MHz, 2.5 GS s−1). The instrument response function (IRF) in CH3CN was measured to be 12 ns (fwhm).42 The samples were purged with N2 for 15 min prior to each measurement and the electronic absorption spectrum were recorded before and after the experiment to ensure no sample degradation had taken place.
Photolysis experiments were performed using four light emitting diodes (LEDs, Luxeon Star) centered at 655 nm as the light source. The sum of the irradiation intensity was kept at 800 mW. Photolysis solutions contained 30 mM BNAH, 0.1 M TsOH (p-toluenesulfonic acid) and 64 μM 1 or 100 μM 2 in 3 mL DMF; the samples were prepared in the glove box using a cuvette with a side arm equipped with a Kontes stopcock. The amount of H2 produced in head space was quantified by a Shimadzu GC-2014 gas chromatography with He as the carrier gas. Absorption spectra were collected at time intervals until no further spectral changes were observed.
Results and discussion
X-ray crystal structure
The single crystal X-ray structure of 1 is depicted in Fig. 2 and additional details are provided in Table S1 (ESI†). The cis disposition of the two diphenylbenzamidine (DPhB) anions and the two neutral benzo[c]cinnoline (bncn) ligands that bridge the two Rh(II) centers is readily evident in Fig. 2, resulting a heteroleptic paddlewheel structure. Each axial site is coordinated by an acetonitrile solvent molecule, as is typical for this class of complexes,43 along with two BF4− counterions and a free acetonitrile molecule in the outer coordination sphere (not shown). The shorter Rh–Rh bond distance determined for 1, 2.3878(4) Å, as compared to that of 3, 2.4049(5) Å,36 is attributed to steric strain afforded by central phenyl ring on the bridging DPhB ligand, not present in diphenylformamidinate (DPhF) in 3. The torsion angle between the coordinating N atoms on the formamidinate ligand with the two rhodium atoms increases from 4.7° in 3 to 9.4° in 1, a measure that is also indicative of the additional steric strain in the latter. Although crystals of 2 were not suitable for X-ray diffraction, the corresponding homoleptic complex, Rh2(DPhTA)4, was reported to feature a Rh–Rh distance of 2.377(3) Å, shorter than that of Rh2(DPhB)4 as 2.389(1) Å.44,45 Based on the these results, it may be hypothesized that the Rh–Rh bond in 2 is similar to or shorter than that in 1.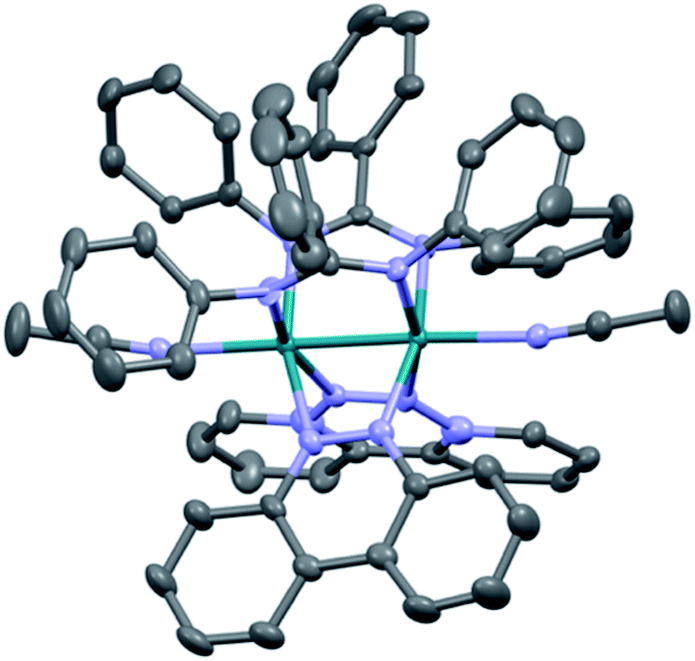 | ||
| Fig. 2 Crystal structure of 1; hydrogen atoms, solvent molecules, and BF4− counterions have been omitted for clarity (ellipsoids drawn at 50% probability). | ||
Electronic absorption spectroscopy and electrochemistry
The lowest energy absorption exhibit maxima at 645 nm (ε = 8900 M−1 cm−1) for 1 and 575 nm (ε = 7660 M−1 cm−1) for 2 (Fig. S7†). This band is observed at 624 nm (ε = 8600 M−1 cm−1) in 3 and has been previously attributed to a metal/ligand-to-ligand charge transfer (ML-LCT) from the Rh2(δ*)/DPhF(π*) HOMO (highest occupied molecular orbital) to the bncn LUMO (lowest unoccupied molecular orbital).36 Given the similarity in structure and absorption profiles of 1 and 2 to that of 3, the lowest energy transition is also ascribed to the HOMO, Rh2(δ*)/DPhB(π*) in 1 and Rh2(δ*)/DPhTA(π*) in 2, to the bncn(π*) LUMO in each complex.33,34 The blue shift of the charge-transfer band in 2 relative to that in 3 is attributed to the better electron withdrawing ability of the DPhTA ligand which lowers the energy of the HOMO. Complexes 1–3 also exhibit absorption in the 330–344 nm range attributed to a bncn-centered ππ* transition and peaks in the 401–433 nm range believed to arise from 1ML-LCT transitions, as previously reported for 3, as well as other dirhodium complexes with similar structures.33,34,36The redox potentials obtained from cyclic voltammetry for 1 and 2 are summarized in Table 1 and shown in Fig. S8,† and are compared to those previously reported for 3. The first reversible oxidation is observed at +1.33 V for 1 and +1.57 V for 2 in CH3CN vs. Ag/AgCl, which compare well to that measured for 3 in the same solvent, +1.28 V vs. Ag/AgCl.36 In 3, this oxidation event is assigned to the removal of an electron from the Rh2(δ*)/form(π*) HOMO.36 A similar assignment of this couple can be made for 1 and 2, with HOMOs of mixed Rh2(δ*)/DPhB(π*) and Rh2(δ*)/DPhTA(π*) character, respectively. Both DPhB and DPhTA ligands are expected to be more difficult to oxidize than DPhF in 3, attributed to the more electron withdrawing phenyl substituent on the bridgehead carbon atom in DPhB in 1 and the presence of a nitrogen atom in DPhTA in 2. Two reversible reduction couples are observed for 1 and 2, the first at −0.44 V and −0.35 V vs. Ag/AgCl in CH3CN, at potentials slightly more positive than that of 3 at −0.46 V under similar conditions (Table 1).36 A similar trend is observed for the second reversible reduction waves in 1 and 2, which are comparable to that of 3 (Table 1). These two reduction events in 1–3 are ascribed to placement of two consecutive electrons on each of the bncn ligands.36 It should be noted that HOMO–LUMO gap calculated from the difference between the first oxidation and first reduction waves in 2, 1.92 V, are greater than those of 1 (1.77 V) and 3 (1.74 V), by 0.15 V and 0.18 V, respectively (Table 1). This difference is consistent with the higher energy LM-LCT transition observed for 2 at 575 nm (17390 cm−1, 2.16 eV), as compared to those of 1 and 3, at 645 nm (15500 cm−1, 1.92 eV) and 624 nm (16030 cm−1, 1.99 eV), respectively (Table 1).
Excited state properties
Emission is observed for 1 (λexc = 645 nm) and 2 (λexc = 585 nm) at 77 K in CH3CN with maxima at 830 nm and 750 nm, respectively, with excitation spectra that are consistent with the absorption profile of each complex (Fig. S9†). Similar results were previously reported for 3, with emission at 800 nm (λexc = 515 nm) under similar experimental conditions.36 No emission is observed from 1–3 at room temperature in CH3CN or DMF solutions. It should be noted, however, that luminescence has not been previously reported for other Rh2(II,II) complexes. The emission from 1–3 is attributed to the presence of a low-lying 3ML-LCT state, together with a deactivating metal-centered (MC) state(s) with electron density on the Rh2(σ*) orbital. In these complexes, it is hypothesized that the MC state(s) are located at a sufficiently high energy to avoid fast deactivation, afforded by the short distance of two rhodium atoms that raise the energy of the Rh2(σ*) MO.43 The onset (5% of maximum intensity) of the 77 K emission of each complex was used to estimate the energy of the triplet excited state, ET00, and the resulting values range from 1.49 eV in 1 to 1.67 in 2 (Table 2). As listed in Table 2, the triplet excited state reduction potentials of 1–3, E1/2[Rh2]*2+/+, range from +1.05 V in 1 to +1.32 V in 2, vs. Ag/AgCl, making them significantly better oxidants than the prototypical [Ru(bpy)3]Cl2 (bpy = 2,2′-bipyridine) complex, +0.82 V vs. Ag/AgCl in CH3CN.46The transient absorption spectrum of 1 collected upon excitation with 640 nm pulse (IRF = 12 ns) is shown in Fig. 3 and exhibits a sharp positive signal in the 390–550 nm region previously attributed to reduced bncn ligand,36 as well as bleach signals that correspond to the ground state absorption at ∼350 nm and in the 570–750 nm range. The ultrafast transient absorption spectra of 1 recorded following 650 nm excitation (IRF = 85 fs) is shown in Fig. S10,† and exhibit profiles that parallel those in Fig. 3 at longer timescales; similar spectral features are observed for 2 (Fig. S10†). The positive signals compare well to those previously assigned to the 1ML-LCT and 3ML-LCT states of 3.36 The 1ML-LCT states of 1–3 decay with lifetimes, 1τ, of 13–15 ps, whereas the corresponding triplet 3ML-LCT lifetimes, 3τ, were measured to be 33 ns in 1 and ∼8 ns in 2, respectively (Table 2 and Fig. S11†). The lifetime of 2 was estimated based on the lack of excited state signal observed in the nsTA experiment (IRF = 12 ns) and the remaining excited state absorption at 416 nm at 2.7 ns in the fsTA (Fig. S11b†). The 3ML-LCT state of 2, ET00 in Table 2, lies at a higher in energy than the same state in 1 and 3, placing it closer to the deactivating 3MC state, thus resulting in a shorter triplet lifetime in the former.
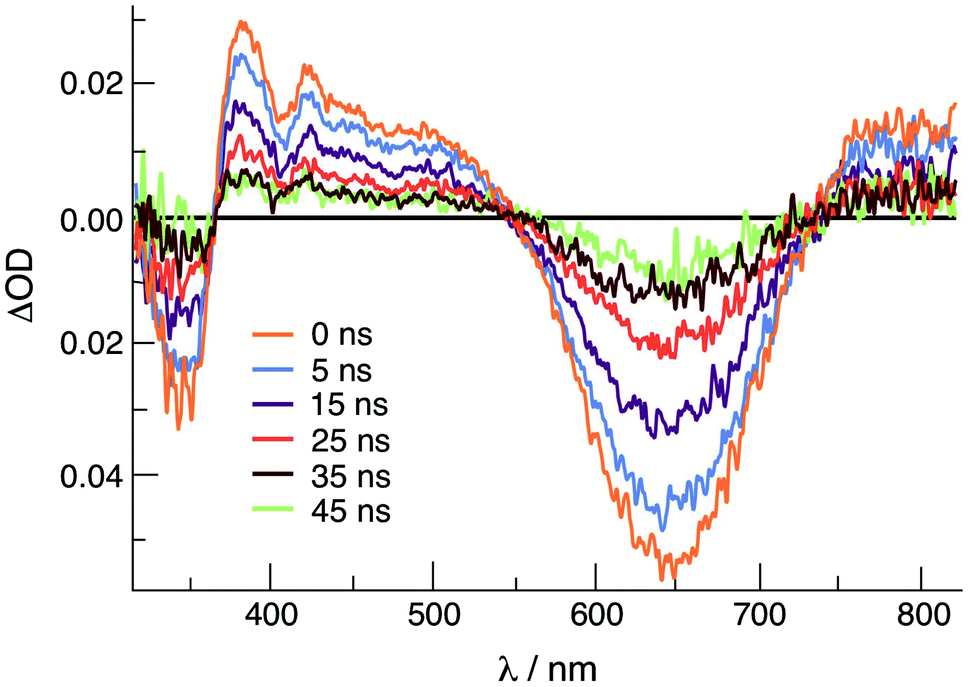 | ||
| Fig. 3 Transient absorption spectra of 1 at various delay times following excitation in CH3CN (640 nm, IFR ∼ 6 ns). | ||
Electro- and photocatalysis
In the presence of excess p-toluenesulfonic acid (TsOH), the application of a negative bias beyond the second reduction couple of 1 and 2 results in a catalytic current that is dependent on the concentration of acid (Fig. S12†). Bulk electrolysis measurements show that the catalytic current in these experiments is due to the production of H2. These results indicate that the doubly-reduced forms of complexes 1 and 2 is able to undergo H2 generation (Fig. S12†). Similar results were previously obtained for 3.36 The ability of 1 and 2 to undergo electrocatalytic H2 production in acidic solutions, together with the presence of 3ML-LCT excited states with nanosecond lifetimes that are powerful oxidants, led to the investigator of their potential as single-molecule photocatalysts. Importantly, this class of complexes absorbs light strongly from the ultraviolet to the near-IR range and possess excited states that can engage in charge transfer reactions, making them ideal systems to efficiently harvest sunlight for solar energy applications.33,34The irradiation of 64 μM of 1 with 655 nm light in DMF in the presence of 0.1 M TsOH and 30 mM BNAH (1-benzyl-1,4-dihydronicotinamide) as a sacrificial electron donor results in the evolution of H2 gas with a turnover number (TON) of 180 in 24 h, as shown in Fig. 4 (black symbols). No H2 is observed in the absence of any single component, complex, light, acid, or sacrificial donor (Table S2†). At early photolysis times, up to 4 h, the generation of H2 is linear resulting in an initial turnover frequency (TOF) of 18 h−1. These findings are similar to those previously observed for 3, which resulted in TON = 170 in 24 h and initial TOF = 28 h−1 (λirr = 670 nm).36Fig. 4 also shows that under these conditions, the production of H2 begins to plateau at longer irradiation times. In order to eliminate the possibility of the consumption of substrate and/or sacrificial donor as the cause of the deviation from linearity in Fig. 4, aliquots of 300 μL DMF solution containing 0.16 M BNAH and 0.53 M TsOH were added to the reaction mixture every 4 h (Fig. 4, red trace). It is evident from a comparison of the data in Fig. 4 that there is a marked improvement in the activity when additional substrate is present in solution, resulting in an initial TOF of 22 h−1 and TON = 250 after 24 h. However, it should be noted that deviations from linearity are still observed in the red curve in Fig. 4, which could be indicative of catalyst degradation over long irradiation times. In contrast, the irradiation of 180 μM of 2 under similar experimental conditions results in a TON value of only 33 after 24 h (λirr = 655 nm, 0.1 M TsOH, 30 mM BNAH). A higher concentration of 2 was used in order to compensate for the lower molar absorption of 2 at 655 nm. The lower activity of 2 compared to those of 1 and 3 can be explained by its less negative reduction potentials, as discussed below. The similarity in the activities of 1 and 3 can be rationalized by their nearly identical molar absorptivities at the irradiation wavelength, excited state lifetimes, and excited state redox potentials (Table 1).
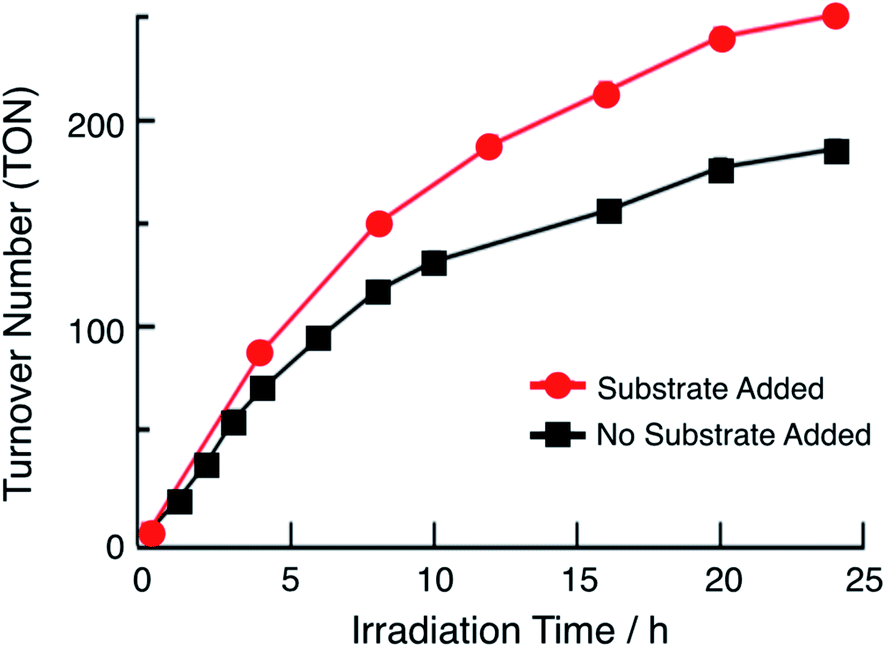 | ||
| Fig. 4 Plot of turnover number (TON) as a function of irradiation time (λirr = 655 nm) of 64 μM 1, 30 mM BNAH, 0.1 M TsOH in DMF without the addition of any acid or BNAH during the course of the reaction (black squares) and upon the addition of 1.5 mL of a DMF solution containing 0.16 M BNAH and 0.53 M TsOH every four hours (red circles). | ||
The addition of one and two equivalents of cobaltocene to a DMF solution of 1 results in the generation of the corresponding one- and two-electron reduced species, [1]− and [1]2−, respectively. The electronic absorption spectra of [1]− and [1]2− are shown in Fig. 5a, and the corresponding species for 2 are shown in Fig. S13.† The absorption spectra of the one- and two-electron reduced species for 1 and 2 are consistent with those obtained from spectroelectrochemistry in DMF at applied potentials slightly more negative than the first and second reduction couples of each complex, respectively (Fig. S14 and S15†). The lowest energy transition of [1]− is observed at 760 nm and the intensity of the absorption at λ > 600 nm decreases significantly in [1]2− (Fig. S14a†). It should be noted that in Fig. 5 the maximum of [1]− is slightly blue-shifted relative to that recorded in the spectroelectrochemistry in Fig. S14,† attributed to the incomplete conversion from 1 to [1]−. These results parallel those previously reported for [3]− and [3]2−,36 as well as those recorded herein for [2]− and [2]2−, as shown in Fig. S13 and S15† for the chemically and electrochemically reduced complex, respectively.
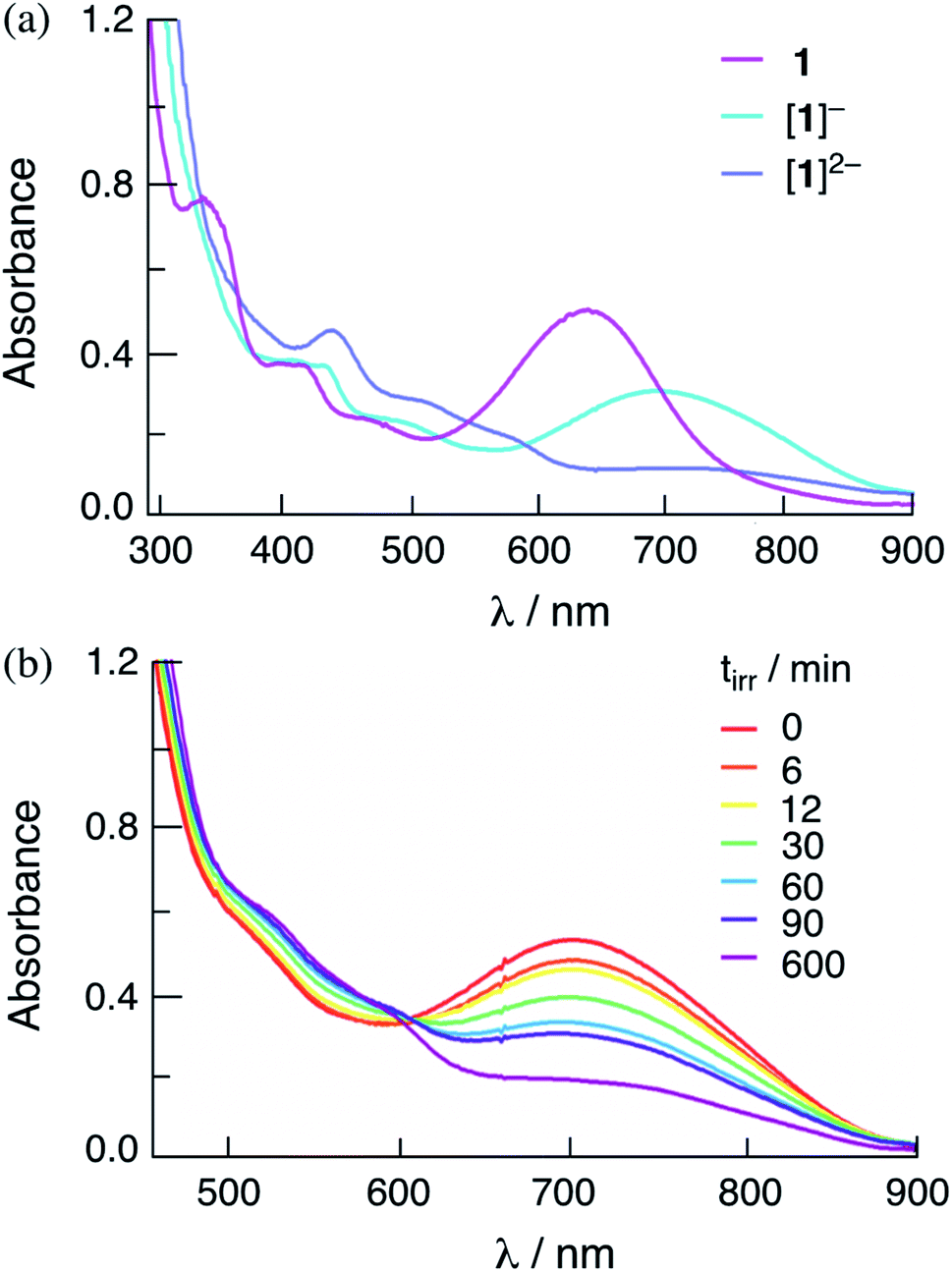 | ||
| Fig. 5 Electronic absorption spectra of (a) 1, [1]−, and [1]2− in DMF obtained through the addition of 1 eq. and 2 eq. of cobaltocene, respectively, to 1 and (b) changes to the electronic spectrum of [1]−, obtained by the addition of 1 eq. cobaltocene to 1, as a function of irradiation time in the presence of 30 mM BNAH in DMF (λirr = 655 nm). | ||
Importantly, Fig. 5b shows that the irradiation of solutions containing the chemically-generated one-electron reduced species, [1]− in DMF in the presence of 30 mM BNAH result in the quantitative production of the corresponding doubly-reduced complex, [1]2− (λirr = 655 nm). Similar results are observed for the irradiation of [2]− with excess BNAH in DMF (Fig. S16†). The reduction of [2]− under these conditions was occurred at significantly lower irradiation times as compared to that for [1]−, indicating that [2]− functions as a better excited state oxidant than [1]− under similar conditions, as expected from the more positive E1/2[Rh2]*2+/+ for 2 as compared to 1 (Table 2). These results parallel those from spectroelectrochemistry, where the generation of [1]− from 1 required a longer time period, 83 min, as compared to [2]− from 2, 3 min (Fig. S14 and S15†). No spectral changes are observed upon the irradiation of [1]− and [2]− in DMF in the absence of the sacrificial electron donor under similar conditions and no reactivity with BNAH is observed in the dark. These results indicate that [1]− and [2]− possess an excited state able to oxidize BNAH. The addition of excess TsOH to solutions containing chemically-generated [1]− and [2]− in DMF result in the regeneration of the corresponding initial complex, 1 and 2, respectively, as well as the production of H2 (Fig. S17†). Similar results were also observed for [1]2− and [2]2−, which also produce H2 and regenerate 1 and 2 when excess TsOH is added to the DMF solution (Fig. S17†). These observations suggest that both the one- and/or two-electron reduced species of these complexes can serve as intermediates in the photocatalytic process.
Control experiments of the photocatalysis show that no H2 product is detected in the absence of any one of the components, light, catalyst, BNAH, or acid (Table S2†). In addition, when 1 and 2 are irradiated in the presence of only BNAH, [1]− and [2]− are produced within 1 min, respectively (Fig. S18†). Fig. S18† also shows that continued irradiation of these solutions generates the corresponding doubly-reduced species, [1]2− (tirr ≥ 15 h) and [2]2− (tirr = 1 h). This difference in the photolysis times for the reduction 1 and 2 can be attributed to the more positive excited state reduction potential of 2 as compared to that of 1 (Table 2), resulting in a more negative driving force for the oxidation of BNAH by the latter. It is important to note that the addition of excess TsOH at the end of the photolysis resulted in spectral changes consistent with the regeneration of the starting catalyst in each case, 1 and 2, and the release of H2 confirmed by gas chromatography. These photochemical results parallel those discussed above for the chemically-generated one- and two-electron reduced catalysts.
The mechanism of photocatalysis was further explored through electronic absorption spectroscopy, as shown in Fig. S19.† Upon irradiation of the photocatalytic DMF solution of 1 (64 μM 1, 30 mM BNAH, 0.1 M TsOH, λirr = 655 nm), spectral changes consistent with the formation of [1]− were apparent within the first 5 min, followed by a slight decrease of the peak at 760 nm indicative of the production of a small amount of [1]2−. A significantly faster generation of [2]− was observed for 2, within 3 min of irradiation of the photocatalytic solution (180 μM 2, 30 mM BNAH, 0.1 M TsOH, λirr = 655 nm). This fact indicates that the relatively short 3ML-LCT lifetime of 2 does not negatively impact on the bimolecular reductive quenching by BNAH as compared to 1. The transient absorption of spectra of the chemically-generated one-electron reduced complexes, [1]− and [2]−, show that these species possess excited states with a short component with τ1 ∼ 3 ps in both complexes, as well as a longer decay, τ2, of 630 ps and 1.0 ns, respectively (Fig. S20†).
Together, the results presented herein can be used to propose the photocatalytic cycle for 1 and 2 illustrated in Fig. 6, using complex 1 as the example. The excitation of 1 with red light places the complex in its 3ML-LCT excited state, which is then reductively quenched by BNAH to form the corresponding singly reduced complex, [1]−. Since H2 evolution is observed when the chemically generated [1]− is treated with excess TsOH, it is proposed that at least some fraction of the sample is protonated during photolysis to generate the corresponding hydride, [1-H]. However, the accumulation of the singly-reduced complex upon irradiation of the photocatalysis solution points at a slow reaction of [1-H] to produce H2 (Fig. S19†), making this pathway a minor source of H2 under these conditions. Based on the production of H2 upon the treatment of the chemically-generated [1]2− with TsOH, it may be hypothesized that there is an equilibrium that results in the protonation of the doubly-reduced complex to generate [1-H]−, and that [1-H]− is able to then react rapidly with acid to produce H2 and regenerate 1. A parallel mechanism is proposed for 2. Complexes 1 and 2 feature an open axial coordination site, such that the formation of axial Rh-hydrides is possible, however, isolation of these species has not yet been possible.30,31 A similar photocatalytic mechanism for the production of H2 was recently proposed for 3 under similar experimental conditions.36
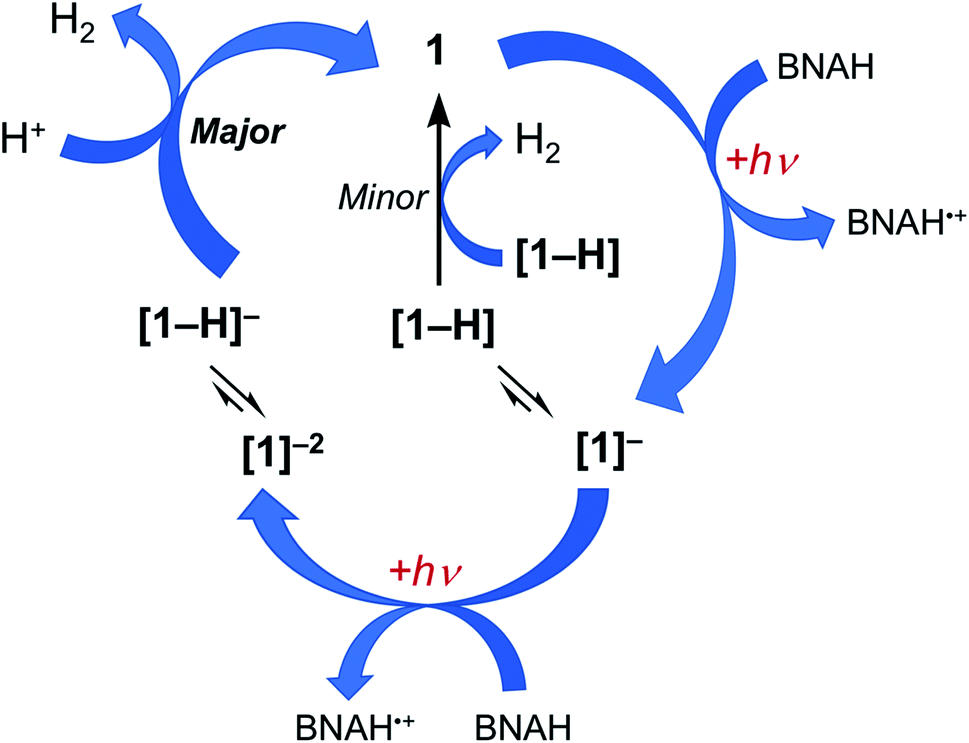 | ||
| Fig. 6 Proposed catalytic cycle of photocatalytic H2 production by 1, showing two possible pathways, one through the reaction of two molecules [1-H] (minor pathway) and the other through the protonation of [1-H]−. | ||
The photolysis experiments in the presence of BNAH show that the rates of reductive quenching steps of 1 and 2, as well as those of [1]− and [2]−, are not affected by the different excited state lifetimes of these species. However the amount of photocatalytic H2 generated is approximately 6-fold lower for 2 as compared to 1. Thus the hydrogen production activities were investigated by the successive linear sweep voltammograms of reaction mixtures in which the acid concentrations were increased. TsOH was used as a strong acid (pKMeCNa = 8.6),47 and a catalytic current is observed for both complexes after the second reduction peak, but 2 requires a larger overpotential as compared to 1 under the similar conditions (Fig. S12†). Salicylic acid (pKMeCNa = 16.7) and acetic acid (pKMeCNa = 23.5) were chosen as weaker acids to investigate the role of acid strength on the catalytic activity.47 Using salicylic acid, the catalytic current decreased for 2, however, complex 1 maintained high efficiency (Fig. S21†). In contrast, the catalytic performance for 2 was hindered significantly upon addition of acetic acid, whereas 1 remained fairly active but required a more negative potential, −1.2 V vs. Ag/AgCl (Fig. S21†). Considering the hydride intermediates required for H2 evolution shown in Fig. 6, the difference in reactivity between 1 and 2 can be explained as arising from [1-H]− being more hydridic than [2-H]−. In that case, [2-H]− would require a stronger acid than [1-H]− for the formation of H2 to be thermodynamically favorable. In fact, the first reduction potentials of 1 and 2 associated with the placement of an electron in the lowest unoccupied molecular orbitals (LUMO), can also be considered as the hydride ligand acceptor orbital that is able to affect the hydride donor ability of [1-H]− and [2-H]−. The bridging ligands in 2 are a more electron-withdrawing as compared to those in 1, resulting in a lower energy LUMO in the former, making 2 a poorer hydride donor and less catalytically active than 1. A similar trend was previously reported for Ni and Co catalyst.48–50
Conclusion
In summary, we report a series of single-molecule dirhodium photocatalysts that can generate hydrogen upon irradiation with 655 nm red light. Complex 1 reaches TON = 250 after 24 h of photolysis following two sequential reductive quenching steps in acidic solution. Complexes 1–3 process 3ML-LCT excited states with lifetimes that range from ∼8 ns to 33 ns at room temperature, are good excited state oxidants, and are emissive at 77 K. In addition, the ability of the complexes to absorb strongly from the ultraviolet to the near-IR extends their reactivity into the red, thus improving the utilization of the solar spectrum beyond traditional sensitizers. Importantly, 1–3 are able to store two electrons upon irradiation, which facilitates the two-electron photocatalytic generation of hydrogen. The substituents on the electron-donating ligands bridging ligands were used to tune the electronic properties of the complexes, including the excited state lifetime and excited state reduction potential, while retaining open coordination sites for catalysis. Photocatalysts with longer excited state lifetimes and the ability to oxidize electron donors in solution, as well as open site for hydride binding are expected to result in an increased efficiency of H2 production. Complexes 1 and 2 represent new additions to a new class of photocatalysts that act both as the light absorber and catalyst within the same chromophore, with absorption throughout the visible range for enhanced harnessing of the solar spectrum, making them useful for the direct conversion of solar energy into H2 fuel.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Support from the Department of Energy, Office of Science, Office of Basic Energy Sciences (DE-SC0020243) and The Ohio State University for partial support of this work and the Center for Chemical and Biophysical Dynamics (CCBD) for use of the ultrafast laser facility.References
- J. Kiwi and M. Grätzel, Nature, 1979, 281, 657–658 CrossRef CAS.
- (a) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS; (b) J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS.
- (a) D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed; (b) A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS; (c) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS.
- Y.-J. Yuan, Z.-T. Yu, D.-Q. Chen and Z.-G. Zou, Chem. Soc. Rev., 2017, 46, 603–631 RSC.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477–1488 RSC.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS.
- G. Reginato, L. Zani, M. Calamante, A. Mordini and A. Dessi, Eur. J. Inorg. Chem., 2020, 11–12, 899–917 CrossRef.
- O. Bettucci, T. Skaltsas, M. Calamante, A. Dessi, M. Bartolini, A. Sinicropi, J. Filippi, G. Reginato, A. Mordini, P. Fornasiero and L. Zani, ACS Appl. Energy Mater., 2019, 2, 5600–5612 CrossRef CAS.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- S. Kampouri and K. C. Stylianou, ACS Catal., 2019, 9, 4247–4270 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- K. R. Brereton, A. G. Bonn and A. J. M. Miller, ACS Energy Lett., 2018, 3, 1128–1136 CrossRef CAS.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef.
- S. Mozaffari, M. R. Nateghi and M. B. Zarandi, Renewable Sustainable Energy Rev., 2017, 71, 675–686 CrossRef CAS.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS.
- (a) S. J. Mora, E. Odella, G. F. Moore, D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2018, 51, 445–453 CrossRef CAS; (b) Y. Terazono, T. A. Moore, A. L. Moore and D. Gust, Multiporphyrin Arrays, 2012, 349–387 CAS.
- T. Stoll, M. Gennari, J. Fortage, C. E. Castillo, M. Rebarz, M. Sliwa, O. Poizat, F. Odobel, A. Deronzier and M. N. Collomb, Angew. Chem., Int. Ed., 2014, 53, 1654–1658 CrossRef CAS.
- Y. Halpin, M. T. Pryce, S. Rau, D. Dini and J. G. Vos, Dalton Trans., 2013, 42, 16243–16254 RSC.
- R. Katoh and A. Furube, J. Photochem. Photobiol., C, 2014, 20, 1–16 CrossRef CAS.
- M. K. Gish, A. M. Lapides, M. K. Brennaman, J. L. Templeton and T. J. Meyer, J. Phys. Chem. Lett., 2016, 7, 5297–5301 CrossRef CAS.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- M.-Q. Yang, M. Gao, M. Hong and G. W. Ho, Adv. Mater., 2018, 30, 1802894 CrossRef PubMed.
- (a) A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS; (b) A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639–1641 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- (a) N. Elgrishi, T. S. Teets, M. B. Chambers and D. G. Nocera, Chem. Commun., 2012, 48, 9474–9476 RSC; (b) D. C. Powers, S. J. Hwang, S. L. Zheng and D. G. Nocera, Inorg. Chem., 2014, 53, 9122–9128 CrossRef CAS PubMed.
- W. Wang, T. B. Rauchfuss, L. Bertini and G. Zampella, J. Am. Chem. Soc., 2012, 134, 4525–4528 CrossRef CAS.
- A. J. Huckaba, H. Shirley, R. Lamb, S. Guertin, S. Autry, H. Cheema, K. Talukdar, T. Jones, J. W. Jurss, A. Dass, N. I. Hammer, R. H. Schmehl, C. E. Webster and J. H. Delcamp, ACS Catal., 2018, 8, 4838–4847 CrossRef CAS.
- R. M. Bullock, A. M. Appel and M. L. Helm, Production of Hydrogen by Electrocatalysis: Making the H–H Bond by Combining Protons and Hydrides, Chem. Commun., 2014, 50, 3125–3143 RSC.
- (a) E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock and A. J. M. Miller, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed; (b) M. B. Chambers, D. A. Kurtz, C. L. Pitman, M. K. Brennaman and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 13509–13512 CrossRef CAS.
- Z. Li, N. A. Leed, N. M. Dickson-Karn, K. R. Dunbar and C. Turro, Chem. Sci., 2014, 5, 727–737 RSC.
- T. J. Whittemore, H. J. Sayre, C. Xue, T. A. White, J. C. Gallucci and C. Turro, J. Am. Chem. Soc., 2017, 139, 14724–14732 CrossRef CAS.
- T. J. Whittemore, A. Millet, H. J. Sayre, C. Xue, B. S. Dolinar, E. G. White, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2018, 140, 5161–5170 CrossRef CAS.
- (a) C. Xue, H. J. Sayre and C. Turro, Chem. Commun., 2019, 55, 10428–10431 RSC; (b) H. J. Sayre, A. Millet, K. R. Dunbar and C. Turro, Chem. Commun., 2018, 54, 8332–8334 RSC.
- T. J. Whittemore, C. Xue, J. Huang, J. C. Gallucci and C. Turro, Nat. Chem., 2020, 12, 180–185 CrossRef CAS PubMed.
- A. C. Hontz and E. C. Wagner, Org. Synth., 1951, 31, 48 CrossRef CAS.
- Z. Li, H. T. Chifotides and K. R. Dunbar, Chem. Sci., 2013, 4, 4470–4485 RSC.
- SAINT, V. 7.60 A, Bruker Analytical X-ray Instruments. Inc., Madison, WI, 2008 Search PubMed.
- Bruker, A. V., 1 and SAINT Version 7.34 a Data Collection and Processing Software, Bruker Analytical X-Ray Instruments. Inc., Madison, WI, USA, 2009 Search PubMed.
- J. D. Knoll, B. A. Albani and C. Turro, Chem. Commun., 2015, 51, 8777–8780 RSC.
- T. N. Rohrabaugh, Jr., K. A. Collins, C. Xue, J. K. White, J. J. Kodanko and C. Turro, Dalton Trans., 2018, 47, 11851–11858 RSC.
- H. T. Chifotides and K. R. Dunbar, Rhodium Compounds, in Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer-Verlag, New York, 2005, pp. 465–589 Search PubMed.
- L. P. He, C. L. Yao, M. Naris, J. C. Lee, J. D. Korp and J. L. Bear, Inorg. Chem., 1992, 31, 620–625 CrossRef CAS.
- M. B. Hursthouse, M. A. Mazid, T. Clark and S. D. Robinson, Polyhedron, 1993, 12, 563–565 CrossRef CAS.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef CAS.
- (a) A. Kütt, I. Leito, I. Kaljurand, L. Sooväli, V. M. Vlasov, L. M. Yaguspolskii and I. A. Koppel, J. Org. Chem., 2006, 71, 2829–2838 CrossRef; (b) F. Eckert, I. Leito, I. Kaljurand, A. Kütt, A. Klamt and M. Diedenhofen, J. Comput. Chem., 2009, 30, 799–810 CrossRef CAS.
- D. E. Berning, A. Miedaner, C. J. Curtis, B. C. Noll, M. C. Rakowski DuBois and D. L. DuBois, Organometallics, 2001, 20, 1832–1839 CrossRef CAS.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed.
- A. Koca, M. Özçeşmeci and E. Hamuryudan, Electroanalysis, 2010, 22, 1623–1633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1980420. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03114c |
| This journal is © The Royal Society of Chemistry 2020 |