
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA neutral porous organic polymer host for the recognition of anionic dyes in water†
Whitney S. Y.
Ong
 ,
Ronald A.
Smaldone
* and
Sheel C.
Dodani
*
,
Ronald A.
Smaldone
* and
Sheel C.
Dodani
*
Department of Chemistry and Biochemistry, The University of Texas at Dallas, 800 West Campbell Road, Richardson, TX 75080, USA. E-mail: sheel.dodani@utdallas.edu; ronald.smaldone@utdallas.edu
First published on 19th June 2020
Abstract
Neutral hosts for the recognition of anionic guests in water remain underdeveloped due to the inherent thermodynamic barrier for desolvation. To address this challenge, we have repurposed crosslinked porous organic polymers (POPs) as hosts. This polymer architecture affords a hydrophobic environment with a densely packed array of urea hydrogen bond donors to cooperatively promote anion desolvation and recognition in water. Using the principles of supramolecular design, we demonstrate through adsorption assays that the resulting Urea-POP-1 can recognize structurally different dyes containing phosphonate, sulfonate, and carboxylate anions in water. Moreover, when compared to Methyl-POP-1, a control POP lacking hydrogen bond donors, we find that the driving force for desolvation and adsorption of each dye is achieved through hydrophobic interactions with the POP backbone and, more importantly, cooperative hydrogen bonding interactions with the urea sidechains. This starting point sets the stage to exploit the modularity of our design to build a family of neutral polymer hosts with tunable pore sizes and anion preferences for fundamental investigations and targeted applications.
Introduction
Molecular recognition of anionic guests in water remains a key challenge in synthetic supramolecular chemistry.1–6 Drawing inspiration from Nature's ability to recognize anions in water, abiotic polymer hosts offer an attractive solution not only for fundamental host–guest chemistry, but also for biological and environmental applications.7–12 To achieve anion binding in water, organic polymers can rely on a combination of polar, cooperative interactions and the hydrophobic effect to overcome the energetic penalty for desolvation of the anion.2,4,5,11–15 In one widely explored approach, polymers can be functionalized with molecular receptors that are designed to be structurally rigid with densely packed arrays of donors tailored to the size, shape, and charge of a specific anion.12,16–18 Anion receptors with positively charged atoms undergo anion exchange to form electrostatic interactions with more electron rich or basic anions in water.19–21 Likewise, Lewis acidic elements can form coordinate bonds with anions in water.22–28 Alternatively, receptors with charge-neutral donors including polar hydrogen (–NH, –OH) and more hydrophobic –CH, halogen, chalcogen, or π-bonds can also be used.29–41 Even though polymers functionalized with receptors can be tailored to a specific anion, the receptor must be highly predesigned and can require complex synthetic routes. Polymers of this type can have limited utility for practical applications such as water purification or sensing, where scalability and ease of separation are important factors.As a complementary approach to expand our understanding of aqueous anion recognition for practical applications, we are developing neutral organic polymer hosts without predesigned receptors. Along these lines, we and others have recently demonstrated that a number of water soluble, neutral polyolefin polymers (e.g. PVP, PNIPAM, and PAAM) can be used for anion recognition in water.42–46 In parallel, we envisioned that neutral hydrogen bond donors could be encapsulated into rigidly structured polymer hosts, through the use of synthetically accessible porous organic polymers (POPs) (Fig. 1). POPs are highly crosslinked, amorphous hydrocarbon frameworks that can maintain permanent porosity owing to their rigid and inflexible backbones.47–49 Even though POPs are insoluble, these polymeric architectures have been used previously for binding a range of molecules and ions in water.50–67
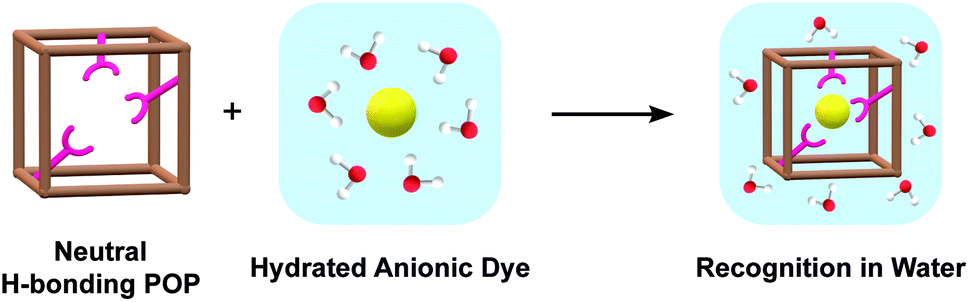 | ||
| Fig. 1 Cartoon schematic illustrating the approach used in this study to bind anionic dyes (yellow sphere) in water with a porous organic polymer host (brown cube) with neutral hydrogen (H) bonding functional groups (pink receptor). Water molecules are shown as red (oxygen atom) and white (hydrogen atom) spheres in the blue background to represent the aqueous environment. | ||
To test our approach, here we have prepared Urea-POP-1, a urea-functionalized POP host. Urea is commonly used as a neutral hydrogen bond donor that effectively provides parallel hydrogen bonds to cooperatively bind electron rich oxyanions to offset the energetic penalty of desolvation.68 Previous reports with urea containing hosts were carried out primarily in organic solvents or mixtures of organic solvents and water with a limited of number examples in pure water.69–80 We hypothesized that a densely packed array of confined urea sidechains, in combination with the hydrophobic microenvironment provided by the POP could cooperatively promote anion desolvation and recognition in water. As a proof-of-concept, we have systematically tested the ability of Urea-POP-1 as a supramolecular host to recognize organic dyes that contain phosphonate (R–PO32−), sulfonate (R–SO3−), and carboxylate (R–COO−) anions through adsorption assays.
Results and discussion
Both Urea-POP-1 and Methyl-POP-1 were accessed through a modular synthetic route with simple building blocks (Fig. 2A and Scheme S1†). The [(2,5-dibromophenyl)methyl]urea monomer (1) was prepared in four high-yielding steps with minimal chromatography. Briefly, 2,5-dibromotoluene was brominated with N-bromosuccinimide and benzoyl peroxide (45% yield), followed by substitution with potassium phthalimide and subsequent deprotection with hydrazine (81% yield over two steps), and finally treated with potassium isocyanate to generate the urea (92% yield).81–84 Compound 1 and 2,5-dibromotoluene (2) were polymerized with 1,3,5-triethynylbenzene (3), a commonly used monomer for the preparation of porous organic polymers,47 using standard Sonogashira coupling reaction conditions to furnish Urea-POP-1 and Methyl-POP-1 as insoluble powders.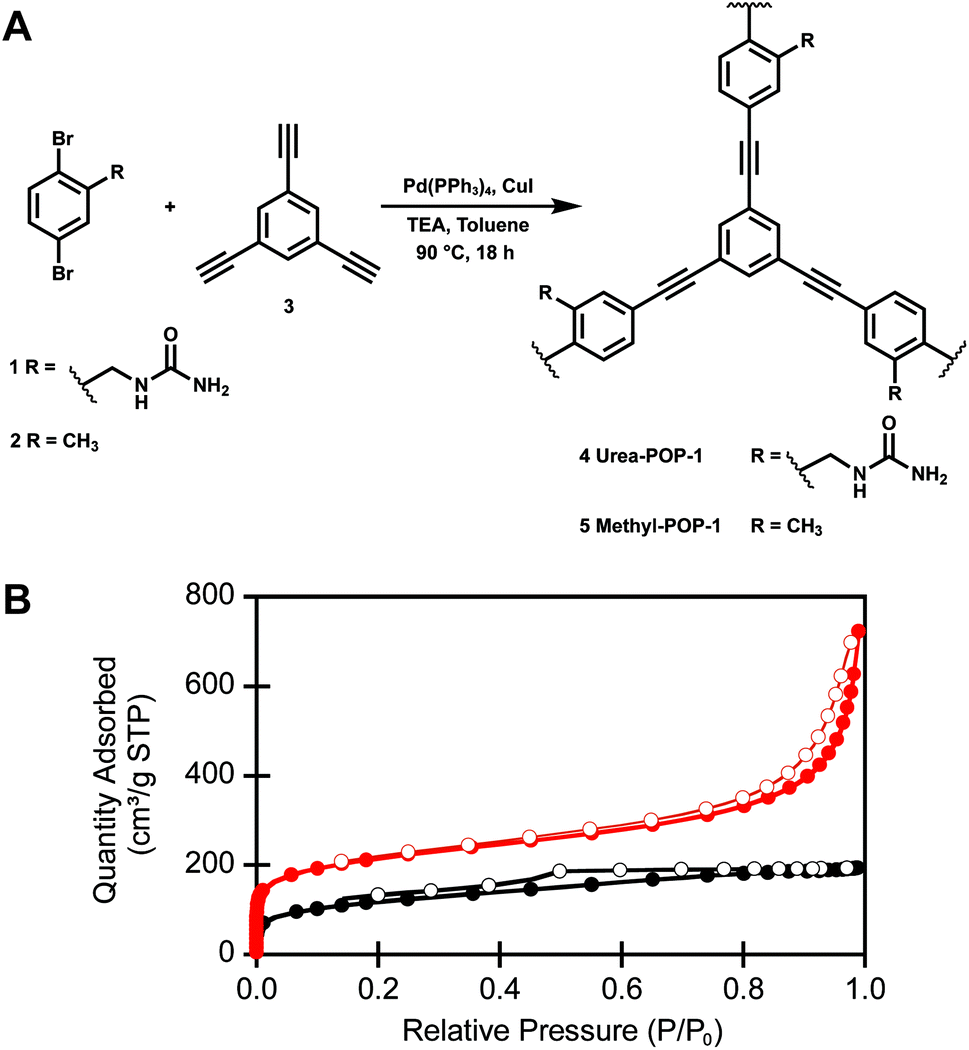 | ||
| Fig. 2 (A) Synthesis of Urea-POP-1 and Methyl-POP-1 through a Sonogashira polymerization. (B) Representative nitrogen adsorption (closed circles) and desorption (open circles) isotherms Urea-POP-1 (red) and Methyl-POP-1 (black). | ||
Characterization with infrared spectroscopy provided evidence for the extent of polymerization (Fig. S2†). For both POPs, the disappearance of the alkyne C–H stretch at 3276 cm−1 confirms that the starting 1,3,5-triethynylbenzene co-monomer was consumed. Interestingly, the N–H stretches corresponding to the urea functional group are broadened and centered at 3398 cm−1 in Urea-POP-1 relative to the N–H stretches at 3429 and 3338 cm−1 in the starting urea monomer (1), suggesting the presence of hydrogen bonding between the urea units within the POP.85 To determine the permanent porosity of the POPs, we measured the nitrogen adsorption isotherms of six independently prepared batches at 77 K (Fig. S3 and Table S1†). Although both POPs are permanently porous, on average Urea-POP-1 (870 ± 120 m2 g−1) has a larger Brunauer–Emmett–Teller (BET) surface area than Methyl-POP-1 (490 ± 70 m2 g−1) (Fig. 2B). Based on these isotherms, both POPs are microporous, as the majority of the total pore volume consists of pores with sizes between 10–20 Å. This is determined from the cumulative pore size measurements reported in Fig. S3.† Moreover, powder X-ray diffraction (PXRD) analysis does not reveal any uniform crystalline morphology indicating that both POPs are amorphous in nature (Fig. S4†).
We next tested if the urea sidechains in Urea-POP-1 could be used for the recognition of anionic guests in water. To do so, we selected dyes containing oxyanions commonly known to interact with urea-based hosts,69,74,78 including phosphonate (adenosine 5′-monophosphate or AMP, adenosine 5′-triphosphate or ATP, and riboflavin 5′-monophosphate or FMN), sulfonate (methyl orange, Lucifer Yellow CH, and bromophenol blue), and carboxylate anions (Rhodamine B, methyl red, Alizarin Yellow G, and fluorescein) (Fig. 3). Dye adsorption to the pores of the POP was monitored by measuring the optical intensity of the dye remaining in solution after incubation with varying concentrations of Urea-POP-1 and Methyl-POP-1 (Fig. 3 and S6–S15†). To ensure that recognition was driven by hydrogen bonding interactions, each dye was dissolved in ultrapure water (ca. pH 7) and diluted to 40 μM for detection within a dynamic, linear range. Moreover, at this concentration of dye, the number of urea sidechains available for binding was in 10 to 320-fold excess, corresponding to a range of 0.05 to 1.6 mg mL−1 of POP. As can be seen in Fig. 3, increasing concentrations of Urea-POP-1 promote greater adsorption of each dye, albeit to varying degrees depending on the anionic functional group. These differences were quantified by fitting the data to a binding model (see ESI† for details) to determine the amount of Urea-POP-1 at a saturation point where the relative absorbance or fluorescence signal of each dye decreased by 95% (Fig. S16 and Table S3†). At this saturation point, the adsorption of each dye to Urea-POP-1 was likely maximized given the number of excess urea sidechains, thus allowing for a better comparison between each anionic functional group. To explain the observed differences, we have considered the partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D value), overall charge, and any additional functional groups of each dye (Fig. 4, Table S3†).
D value), overall charge, and any additional functional groups of each dye (Fig. 4, Table S3†).
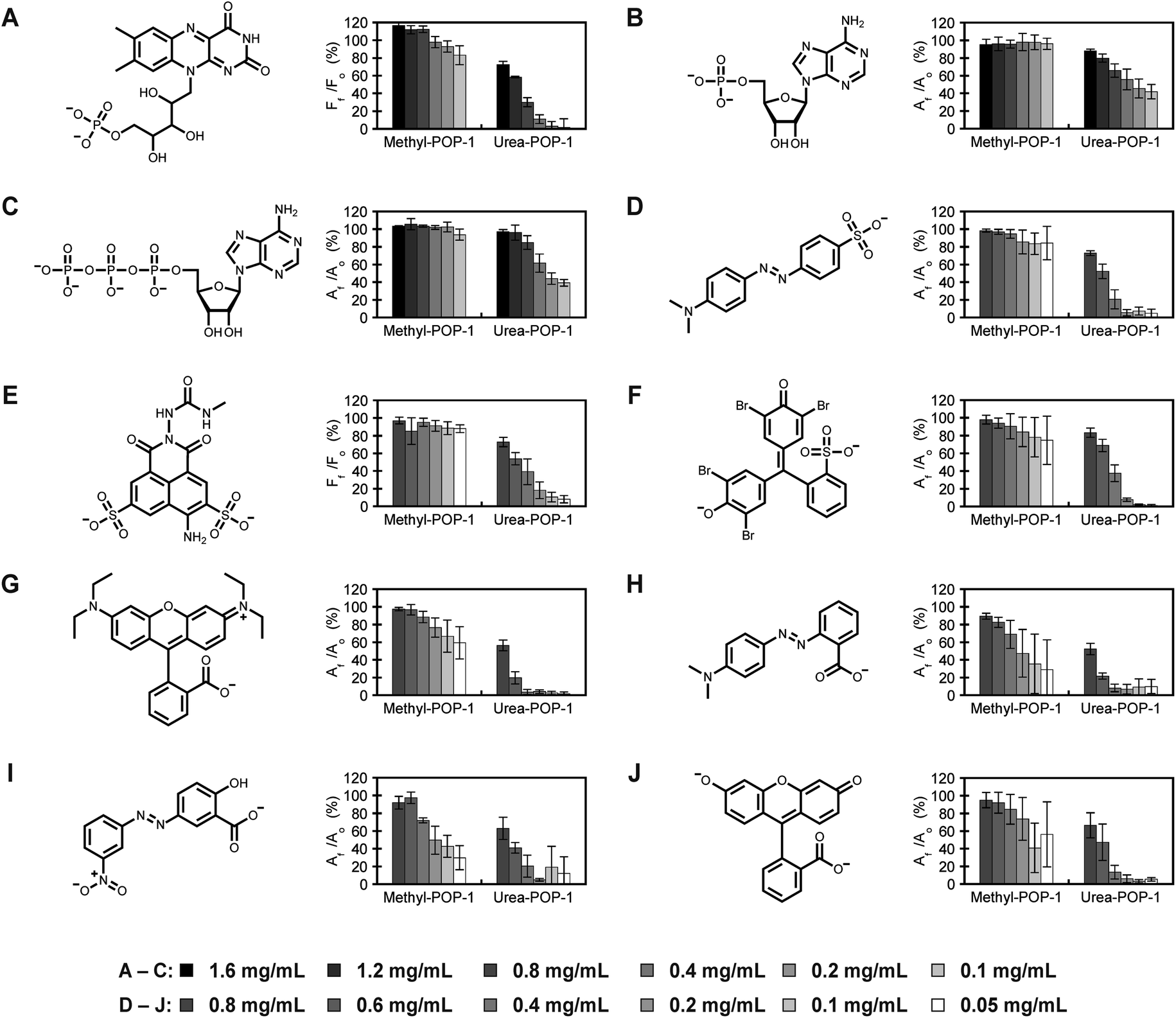 | ||
| Fig. 3 Urea-POP-1 selectively adsorbs dyes with anionic functional groups. The absorbance or fluorescence (AF or FF) of each dye relative to 40 μM dye only (A0 or F0) in the presence of increasing concentrations of Methyl-POP-1 and Urea-POP-1 is shown for (A) flavin mononucleotide, (B) adenosine monophosphate, (C) adenosine triphosphate, (D) methyl orange, (E) Lucifer Yellow CH, (F) bromophenol blue, (G) Rhodamine B, (H) methyl red, (I) Alizarin Yellow G, and (J) fluorescein. All measurements were carried out in ultrapure water (ca. pH 7). The overall charge on each dye is shown at pH 7. The average of at least three technical replicates from at least two different batches of each POP with standard deviation is reported for each dye. The absorbance or fluorescence spectra are shown in Fig. S6–S15.† Counterions for each dye are specified in the ESI.† | ||
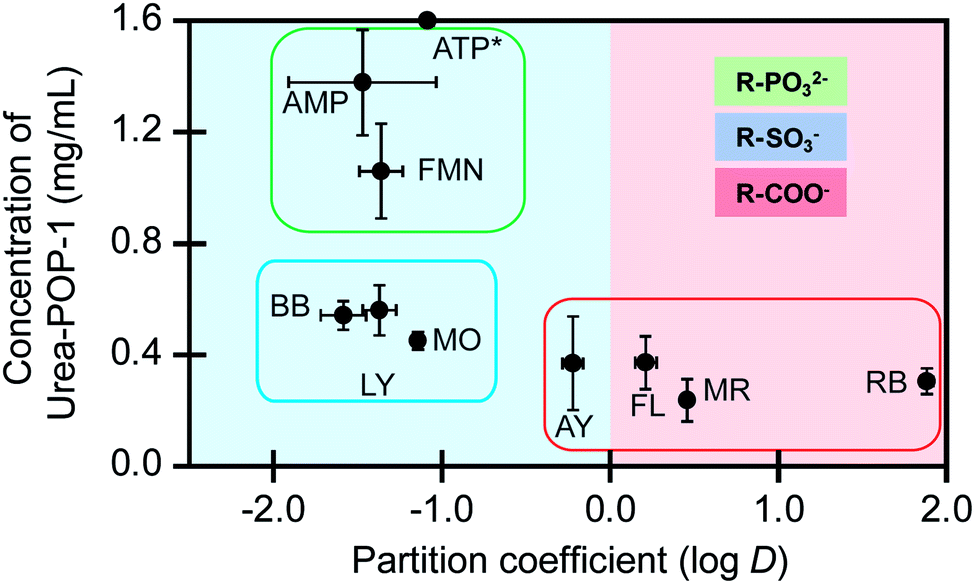 | ||
| Fig. 4 The amount of Urea-POP-1 required for adsorption is correlated to the anionic functional groups. The amount of Urea-POP-1 at the 95% saturation point for each dye versus the dye partition coefficient is shown. Dyes that contain phosphonate (R–PO32−), sulfonate (R–SO3−), and carboxylate (R–COO−) functional groups are grouped by the green, blue, and red boxes, respectively. The light blue background represents the preference of the dye to stay in water (hydrophilic), and the light red background represents the preference of the dyes to stay in octanol (hydrophobic). The dyes are abbreviated as follows: adenosine monophosphate (AMP), adenosine triphosphate (ATP), Alizarin Yellow G (AY), bromophenol blue (BB), flavin mononucleotide (FMN), fluorescein (FL), Lucifer Yellow CH (LY), methyl orange (MO), methyl red (MR), and Rhodamine B (RB). *For ATP the 95% saturation point could not be determined. For each dye, the average of at least three technical replicates from at least two batches of each POP with standard deviation is reported (Table S3†). | ||
As expected, the phosphonate dyes have negative logD values (−1.5 ± 0.4 to −1.08 ± 0.03) and readily partition into water over octanol, independent of the overall charge. In order to adsorb the dyes from water, more than 1 mg mL−1 of Urea-POP-1 is needed. Both FMN and AMP have an overall charge of −2, and both dyes are adsorbed at similar concentrations of Urea-POP-1 (1.1 ± 0.2 mg mL−1 and 1.4 ± 0.2 mg mL−1, respectively). For ATP, the saturation point could not be determined due to weak binding. Based on this, more than 1.6 mg mL−1 of Urea-POP-1 would be required for adsorption of ATP, which correlates with its increased negative charge from −2 to −4.
Of the three phosphonate dyes, only FMN adsorbs to the hydrophobic pores of Methyl-POP-1. At 1.6 mg mL−1 of Methyl-POP-1, the optical signal of FMN decreases by only 20% versus more than 95% with the same amount of Urea-POP-1. This suggests that the hydrophobicity of the dye could contribute, in part, to adsorption but not as significantly as the phosphonate anion hydrogen bonding with the urea sidechains.
Like the phosphonate dyes, the sulfonate dyes have negative logD values (−1.6 ± 0.1 to −1.14 ± 0.03), but an average of 60% less Urea-POP-1 is needed for adsorption with no significant adsorption to Methyl-POP-1. Within this series, methyl orange is adsorbed at 0.45 ± 0.03 mg mL−1, whereas Lucifer Yellow CH and bromophenol blue are adsorbed at similar concentrations of 0.56 ± 0.09 mg mL−1 and 0.54 ± 0.05 mg mL−1, respectively. Indeed, these results not only correlate with the number of sulfonate groups but also the overall negative charge. Specifically, methyl orange has one sulfonate group with an overall charge of −1, whereas Lucifer Yellow CH with an additional sulfonate group and bromophenol with a phenolate group both have an overall charge of −2.
On the other end, the carboxylate dyes have logD values ranging from −0.22 ± 0.06 to 1.88 ± 0.04 and partition more favorably into octanol. Given this, it is surprising that the average amount of Urea-POP-1 required for adsorption of the carboxylate dyes is only ∼35% less than that of the sulfonate dyes. Furthermore, even though the carboxylate dyes have different functional groups and overall charges, all of the dyes are adsorbed at similar concentrations of Urea-POP-1. Notably, all of the carboxylate dyes adsorb to Methyl-POP-1. This is consistent with the fact that the carboxylate dyes, relative to the phosphonate and sulfonate dyes, have more positive logD values. However, clear differences are observed between Urea-POP-1 and Methyl-POP-1, at concentrations as low as 0.1 mg mL−1, indicating that the carboxylate anions, in large part, preferentially adsorb within Urea-POP-1. These control experiments clearly show that hydrophobic interactions do occur between the dyes and the hydrophobic environment of the POP; however, it is not the primary driving force as the dyes do not adsorb to Methyl-POP-1 to the same extent as Urea-POP-1 – even when more hydrophobic dyes are used. Moreover, irrespective of the identity of the anionic functional group, adsorption is observed, suggesting that hydrogen bonding interactions do occur with the urea sidechains.
By design, the dense array of urea hydrogen bond donors and the hydrophobic pores should have favorable effects on the enthalpy of dye adsorption. This would be achieved by forming primary interactions with the anionic functional groups and secondary interactions such as cooperative cation effects. The desolvation of the anionic functional groups and the hydrophobic pores, although minimal for the latter since the POP host is insoluble, could provide significant entropic gains to the overall driving force for adsorption. Moreover, the extent to which the phosphonate, sulfonate, and carboxylate groups of the dyes are hydrated could provide an explanation for the differences in adsorption between the dyes within Urea-POP-1. In fact, the adsorption behavior of Urea-POP-1 correlates well with the Gibbs free energy of hydration (ΔhydG) and the basicity, particularly on the extreme end with the hydrogen phosphate anion (ΔhydG: HPO42− (−1366 kJ mol−1) > HCO2− (−395 kJ mol−1) > HSO4− (−330 kJ mol−1);86 basicity: RPO42− > RCO2− > RSO3− where R corresponds to a hydrogen or a phenyl group).69,87
Conclusions
To close, we have demonstrated that in the absence of a predesigned receptor, Urea-POP-1 can recognize structurally different dyes containing phosphonate, sulfonate, and carboxylate anions in water, whereas Methyl-POP-1, a control lacking hydrogen bond donors, cannot. Our data indicate that the preference of Urea-POP-1 is determined through interactions with the anionic functional groups rather than the partition coefficient, overall charge, or any additional functional groups of each dye. By design, the driving force for desolvation and adsorption of each dye is achieved through hydrophobic interactions with the POP backbone and, more importantly, cooperative hydrogen bonding interactions with the urea sidechains. With this systematic analysis, we showcase as a proof-of-concept that permanently porous polymer architectures can be readily functionalized with hydrogen bonding donors and repurposed as supramolecular hosts for the recognition of anionic guests in water. Beyond fundamental investigations, we envision that Urea-POP-1 and related neutral POP hosts could be applied for the removal of anionic pollutants including the organic dyes tested and associated countercations, thus bypassing the need for anion exchange and separation processes.88 Along these lines, future efforts will exploit the modularity of our starting design to build a family of polymer hosts with tunable ligands, pore sizes and anion preferences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. A. S acknowledges support from the Army Research Laboratory (W911NF-18-2-0035). S. C. D acknowledges support from The University of Texas at Dallas, the Welch Foundation (AT-1918-20170325), and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128923). We thank Mr. Darby Ball from the D'Arcy Lab at The University of Texas at Dallas for acquiring the high-resolution mass spectrum for this study. We thank Ms. Shashini D. Diwakara from the Smaldone lab for acquiring PXRD data for both POPs. We also thank members of the Dodani and Smaldone Labs for helpful discussions.References
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- E. A. Kataev and C. Müller, Tetrahedron, 2014, 70, 137–167 CrossRef CAS.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- P. S. Cremer, A. H. Flood, B. C. Gibb and D. L. Mobley, Nat. Chem., 2018, 10, 8–16 CrossRef CAS PubMed.
- P. A. Gale, E. N. W. Howe, X. Wu and M. J. Spooner, Coord. Chem. Rev., 2018, 375, 333–372 CrossRef CAS.
- S. Mangani and M. Ferraroni, Natural Anion Receptors: Anion Recognition by Proteins, in Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. Garcia-Espana, Wiley-VCH, New York, NY, 1997, pp. 63–78 Search PubMed.
- D. M. Roundhill and H. F. Koch, Chem. Soc. Rev., 2002, 31, 60–67 RSC.
- C. Gérente, Y. Andrès, G. McKay and P. Le Cloirec, Chem. Eng. J., 2010, 158, 593–598 CrossRef.
- M. R. Awual, A. Jyo, S. A. El-Safty, M. Tamada and N. Seko, J. Hazard. Mater., 2011, 188, 164–171 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- H. Wang, X. Ji, M. Ahmed, F. Huang and J. L. Sessler, J. Mater. Chem. A, 2019, 7, 1394–1403 RSC.
- P. Sokkalingam, J. Shraberg, S. W. Rick and B. C. Gibb, J. Am. Chem. Soc., 2016, 138, 48–51 CrossRef CAS PubMed.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- M. R. Sullivan, W. Yao, D. Tang, H. S. Ashbaugh and B. C. Gibb, J. Phys. Chem. B, 2018, 122, 1702–1713 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- B. M. Rambo, E. S. Silver, C. W. Bielawski and J. L. Sessler, Covalent Polymers Containing Discrete Heterocyclic Anion Receptors, in Anion Recognition in Supramolecular Chemistry, Topics in Heterocyclic Chemistry, ed. P. A. Gale and W. Dehaen, Springer, Berlin, Heidelberg, 2010, vol 24, pp. 1–37, https://link.springer.com/chapter/10.1007/ <?pdb_no 7081?>7081<?pdb END?>_2010_39 Search PubMed.
- A. Rostami and M. S. Taylor, Macromol. Rapid Commun., 2012, 33, 21–34 CrossRef CAS PubMed.
- E. Li, L. Lin, L. Wang, M. Pei, J. Xu and G. Zhang, Macromol. Chem. Phys., 2012, 213, 887–892 CrossRef CAS.
- B. Gierczyk, M. Cegłowski and M. Zalas, PLoS One, 2015, 10, e0122891 CrossRef PubMed.
- Z. Wang and N. V. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1173–1182 CrossRef CAS.
- A. Ojida, Y. Mito-oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454–2463 CrossRef CAS PubMed.
- D. G. Smith, G. Law, B. S. Murray, R. Pal, D. Parker and K.-L. Wong, Chem. Commun., 2011, 47, 7347–7349 RSC.
- A. Dorazco-González, Organometallics, 2014, 33, 868–875 CrossRef.
- M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 CrossRef CAS PubMed.
- N. Malviya, M. Das, P. Mandal and S. Mukhopadhyay, Soft Matter, 2017, 13, 6243–6249 RSC.
- G. M. Peters, X. Chi, C. Brockman and J. L. Sessler, Chem. Commun., 2018, 54, 5407–5409 RSC.
- S.-Y. Huang, M. Qian and V. C. Pierre, Inorg. Chem., 2019, 58, 16087–16099 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- J.-M. Suk, M. K. Chae and K.-S. Jeong, Pure Appl. Chem., 2012, 84, 953–964 CAS.
- A. Schaly, R. Belda, E. García-España and S. Kubik, Org. Lett., 2013, 15, 6238–6241 CrossRef CAS PubMed.
- A. Dorazco-González, M. F. Alamo, C. Godoy-Alcántar, H. Höpfl and A. K. Yatsimirsky, RSC Adv., 2014, 4, 455–466 RSC.
- M. Lisbjerg, B. E. Nielsen, B. O. Milhøj, S. P. A. Sauer and M. Pittelkow, Org. Biomol. Chem., 2015, 13, 369–373 RSC.
- J. Y. C. Lim and P. D. Beer, Chem. Commun., 2015, 51, 3686–3688 RSC.
- M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54, 276–279 CrossRef CAS PubMed.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed.
- X. Ji, C. Guo, W. Chen, L. Long, G. Zhang, N. M. Khashab and J. L. Sessler, Chem.–Eur. J., 2018, 24, 15791–15795 CrossRef CAS PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119–4129 CrossRef CAS PubMed.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195–5206 RSC.
- S. Scheiner, M. Michalczyk and W. Zierkiewicz, Coord. Chem. Rev., 2020, 405, 213136 CrossRef CAS.
- E. R. Birnbaum, K. C. Rau and N. N. Sauer, Sep. Sci. Technol., 2003, 38, 389–404 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- R. Sadeghi and F. Jahani, J. Phys. Chem. B, 2012, 116, 5234–5241 CrossRef CAS PubMed.
- J. P. Kim, Z. Xie, M. Creer, Z. Liu and J. Yang, Chem. Sci., 2017, 8, 550–558 RSC.
- H. C. Kam, D. T. S. Ranathunga, E. R. Payne, R. A. Smaldone, S. O. Nielsen and S. C. Dodani, Supramol. Chem., 2019, 31, 514–522 CrossRef CAS.
- H. Bildirir, V. G. Gregoriou, A. Avgeropoulos, U. Scherf and C. L. Chochos, Mater. Horiz., 2017, 4, 546–556 RSC.
- S. Zhang, Q. Yang, C. Wang, X. Luo, J. Kim, Z. Wang and Y. Yamauchi, Adv. Sci., 2018, 5, 1801116 CrossRef PubMed.
- Y. Yuan and G. Zhu, ACS Cent. Sci., 2019, 5, 409–418 CrossRef CAS PubMed.
- H. Ren, T. Ben, F. Sun, M. Guo, X. Jing, H. Ma, K. Cai, S. Qiu and G. Zhu, J. Mater. Chem., 2011, 21, 10348–10353 RSC.
- X.-L. Lu, T.-Y. Zhou, D. Wu, Q. Wen, X. Zhao, Q. Li, Q. Xiang, J.-Q. Xu and Z.-T. Li, Chin. J. Chem., 2015, 33, 539–544 CrossRef CAS.
- S. Li, H. Han, X. Zhu, X. Jiang and X. Z. Kong, Chin. J. Polym. Sci., 2015, 33, 1196–1210 CrossRef CAS.
- Q. Tang, H.-M. Nie, C.-B. Gong, H.-D. Liu and K. Xiao, RSC Adv., 2015, 5, 3888–3893 RSC.
- S. Lee, G. Barin, C. M. Ackerman, A. Muchenditsi, J. Xu, J. A. Reimer, S. Lutsenko, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 7603–7609 CrossRef CAS PubMed.
- Y. Li, H. Wang, W. Zhao, X. Wang, Y. Shi, H. Fan, H. Sun and L. Tan, J. Appl. Polym. Sci., 2019, 136, 47987 CrossRef.
- A. Deshmukh, S. Bandyopadhyay, A. James and A. Patra, J. Mater. Chem. C, 2016, 4, 4427–4433 RSC.
- Y. He, T. Xu, J. Hu, C. Peng, Q. Yang, H. Wang and H. Liu, RSC Adv., 2017, 7, 30500–30505 RSC.
- S. Bi, Y. Li, S. Zhang, J. Hu, L. Wang and H. Liu, J. Mater. Chem. C, 2018, 6, 3961–3967 RSC.
- Y. Liu, Y. Cui, C. Zhang, J. Du, S. Wang, Y. Bai, Z. Liang and X. Song, Chem.–Eur. J., 2018, 24, 7480–7488 CrossRef CAS PubMed.
- S. Karak, K. Dey, A. Torris, A. Halder, S. Bera, F. Kanheerampockil and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 7572–7581 CrossRef CAS PubMed.
- Q. Sun, B. Aguila and S. Ma, Trends Chem., 2019, 1, 292–303 CrossRef.
- Y. Su, Y. Wang, X. Li, X. Li and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18904–18911 CrossRef CAS PubMed.
- J. Kamcev, M. K. Taylor, D. Shin, N. N. Jarenwattananon, K. A. Colwell and J. R. Long, Adv. Mater., 2019, 31, 1808027 CrossRef PubMed.
- Z.-W. Liu, C.-X. Cao and B.-H. Han, J. Hazard. Mater., 2019, 367, 348–355 CrossRef CAS PubMed.
- H.-J. Zhang, J.-H. Wang, Y.-H. Zhang and T.-L. Hu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1329–1337 CrossRef CAS.
- Q. Sun, B. Aguila, Y. Song and S. Ma, Acc. Chem. Res., 2020, 53, 812–821 CrossRef CAS PubMed.
- Y. Yuan, Y. Yang and G. Zhu, ACS Cent. Sci., 2020 DOI:10.1021/acscentsci.0c00311.
- E. Fan, S. A. Van Arman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369–370 CrossRef CAS.
- T. R. Kelly and M. H. Kim, J. Am. Chem. Soc., 1994, 116, 7072–7080 CrossRef CAS.
- T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Biomol. Chem., 2005, 3, 48–56 RSC.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- R. Sakai, A. Nagai, Y. Tago, S.-I. Sato, Y. Nishimura, T. Arai, T. Satoh and T. Kakuchi, Macromolecules, 2012, 45, 4122–4127 CrossRef CAS.
- V. Blažek Bregović, N. Basarić and K. Mlinarić-Majerski, Coord. Chem. Rev., 2015, 295, 80–124 CrossRef.
- M. M. Watt, J. M. Engle, K. C. Fairley, T. E. Robitshek, M. M. Haley and D. W. Johnson, Org. Biomol. Chem., 2015, 13, 4266–4270 RSC.
- J. Ho, V. E. Zwicker, K. K. Y. Yuen and K. A. Jolliffe, J. Org. Chem., 2017, 82, 10732–10736 CrossRef CAS PubMed.
- S. Byrne and K. M. Mullen, Supramol. Chem., 2018, 30, 196–205 CrossRef CAS.
- Z. Huang, C. Jia, B. Wu, S. Jansone-Popova, C. A. Seipp and R. Custelcean, Chem. Commun., 2019, 55, 1714–1717 RSC.
- X. Wu, P. Wang, P. Turner, W. Lewis, O. Catal, D. S. Thomas and P. A. Gale, Chem, 2019, 5, 1210–1222 CAS.
- S. Shinde, A. Incel, M. Mansour, G. D. Olsson, I. A. Nicholls, C. Esen, J. Urraca and B. Sellergren, J. Am. Chem. Soc., 2020 DOI:10.1021/jacs.0c00707.
- M. R. Golder, C. E. Colwell, B. M. Wong, L. N. Zakharov, J. Zhen and R. Jasti, J. Am. Chem. Soc., 2016, 138, 6577–6582 CrossRef CAS PubMed.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- H. Akbulut, T. Endo, S. Yamada and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1785–1793 CrossRef CAS.
- J. B. Ernst, N. E. S. Tay, N. T. Jui and S. L. Buchwald, Org. Lett., 2014, 16, 3844–3846 CrossRef CAS PubMed.
- D. J. Skrovanek, S. E. Howe, P. C. Painter and M. M. Coleman, Macromolecules, 1985, 18, 1676–1683 CrossRef CAS.
- Y. Marcus, Ions in Solution and their Solvation, John Wiley & Sons, Inc., Hoboken, NJ, 2015 Search PubMed.
- G. Rayner-Canham and T. Overton, Descriptive Inorganic Chemistry, W. H. Freeman and Company, New York, NY, 3rd edn, 2002 Search PubMed.
- B. Lellis, C. Z. Fávaro-Polonio, J. A. Pamphile and J. C. Polonio, Biotechnol. Res. Innov., 2019, 3, 275–290 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02941f |
| This journal is © The Royal Society of Chemistry 2020 |